

Abstract
Previous work has shown that Suc1/Cks proteins can promote the hyperphosphorylation of primed Cdk1 substrates through the formation of ternary Cdk1-Cks-phosphosubstrate complexes. This raises the possibility that Cks proteins might be able to both facilitate and interfere with hyperphosphorylation through a mechanism analogous to the prozone effect in antigen-antibody interactions, with sub-stoichiometric Cks promoting the formation of Cdk1-Cks-phosphosubstrate complexes, and suprastoichiometric Cks promoting instead the formation of Cdk1-Cks and Cks-phosphosubstrate complexes. We tested this hypothesis through a combination of theory, proof-of-principle experiments with oligonucleotide annealing, and experiments on the interaction of Xenopus cyclin B1-Cdk1-Cks2 with Wee1A in vitro and in Xenopus extracts. Our findings help explain why both Cks under-expression and overexpression interfere with cell cycle progression, and provide new insight into the regulation of the Cdk1 system.
eTOC Blurb
Ha et al. find that Cks2 can both promote and repress Cdk1 phosphorylation of Wee1A in a biphasic manner through its bivalent adaptor-like function. This phenomenon is related to the prozone effect in antigen-antibody interaction and to squelching in transcription.
INTRODUCTION
Mitotic cyclin-dependent kinase complexes consist of three proteins: the Cdk1 catalytic subunit, the allosteric activator cyclin B, and a third small protein referred to as Suc1 in S. pombe, Cks1 in S. cerevisiae, and Cks1 or 2 in vertebrates. The Cdk and cyclin subunits are well-known and well-studied; the Cks subunit, less so. Cks proteins are present throughout the eukaryotic kingdom and are well conserved (~50% amino acid identity between human Cks1 and its S. pombe and S. cerevisiae homologs).
Despite decades of study, the exact functions of the Cks proteins are not completely clear (Pines, 1996). Cks proteins are essential in fission yeast (Hayles et al., 1986) and in mice (Martinsson-Ahlzen et al., 2008). Cks1 is not required for viability in budding yeast, but Cks1-null cells do exhibit multiple abnormalities (Yu and Reed, 2004). In S. pombe, loss of function Suc1 mutations result in strains that are compromised for cell cycle progression (Basi and Draetta, 1995; Hayles et al., 1986; Moreno et al., 1989), but curiously, Suc1 overexpression results in a similar phenotype (Hayles et al., 1986). Similar conclusions have been drawn from biochemical studies of Suc1 and the Xenopus laevis Suc1 homolog Cks2, the main isoform present in Xenopus extracts (Wuhr et al., 2014). Depleting Cks2 from extracts diminishes the Cdk1-dependent hyperphosphorylation of Cdc25C, Cdc27 (APC3), Myt1, and Wee1A, and Cks2 promotes the hyperphosphorylation of these proteins in vitro (Patra and Dunphy, 1996, 1998; Patra et al., 1999). But adding excess Cks proteins to Xenopus also diminishes substrate hyperphosphorylation (Dunphy and Newport, 1989). This raises the question of what exactly Cks proteins do, and why the phenotypes of Cks deficiency and excess are similar.
An important step toward understanding Cks function came from structural studies of Cks proteins (Arvai et al., 1995; Bourne et al., 1995; Bourne et al., 2000; Bourne et al., 1996; Endicott et al., 1995; Parge et al., 1993). Cks proteins possess a surface-exposed pocket lined with highly conserved cationic residues, and this pocket sometimes co-crystallizes with either sulfate or phosphate (Arvai et al., 1995; Parge et al., 1993). This raises the possibility that Cks proteins bind phosphoepitopes, and could help Cdk complexes to interact with proteins that have already been primed by a first phosphorylation, either by the Cdk itself or by some other priming kinase. Indeed, Patra and Dunphy hypothesized that Xenopus Cks2 acts as a docking factor to promote the full hyperphosphorylation of substrates like Cdc25C, and that supraphysiological concentrations of Cks2 disrupt these interactions (Patra and Dunphy, 1996). Consistent with this hypothesis, the same authors showed that Cks2-containing Cdk1 complexes bind more strongly to hyperphosphorylated Cdc27 than to hypophosphorylated Cdc27 (Patra and Dunphy, 1998).
Recent studies of the budding yeast Cks1 protein have demonstrated that Cks1 can, in fact, act as a docking factor, promoting the interaction of Cdk1-cyclin complexes with primed substrates through direct interaction with the phosphoepitope (Koivomagi et al., 2013; McGrath et al., 2013). The S. cerevisiae Cks1 protein was shown to bind to phosphopeptides derived from the Cdk1 substrates Cdc6 and Sic1 with affinities on the order of 10 μM, with the optimal primary sequence for Cks1 binding being ΦXTP, with Φ being either a bulky hydrophobic residue or a proline (McGrath et al., 2013). Structural studies showed that the phosphate from a Cdc6-derived phosphopeptide did, as long suspected, bind in the putative phosphate-binding pocket of Cks1. Furthermore, the multisite phosphorylation of Cdc6 and Sic1 was promoted by the presence the ΦXTP sites, supporting the idea that an initial priming phosphorylation of a Cks1-binding TP site promotes additional phosphorylations at SP and TP sites in the vicinity of the priming site (Koivomagi et al., 2013; McGrath et al., 2013). The authors hypothesized that Cks1 functions as a specificity factor, and that through its interaction with particular primed TP sites, it restricts the activity of Cdk1 to specific nearby SP/TP sites in its substrates (Koivomagi et al., 2013; McGrath et al., 2013).
Another consequence of this priming mechanism is the possibility that it could contribute cooperativity to a multisite phosphorylation reaction. Multisite phosphorylation has the potential to generate a highly switch-like, ultrasensitive input-output relationship, and in simple models of multisite phosphorylation, ultrasensitivity is greatest if the last phosphorylations are more favorable than the early ones (Ferrell and Ha, 2014; Gunawardena, 2005; Huang and Ferrell, 1996; Markevich et al., 2004; Wang et al., 2010). Priming is one mechanism for promoting such later phosphorylation reactions and thus generating high ultrasensitivity. On the other hand, priming can make a multisite phosphorylation reaction more processive (Koivomagi et al., 2013; McGrath et al., 2013), and processivity in the phosphorylation and/or dephosphorylation reactions tends to work against the generation of an ultrasensitive response.
Here we examine another potentially important consequence of the binding of Cks proteins to both Cdk1 and substrate phosphoepitopes. In general, if two proteins (e.g. Cdk1 and the substrate) can both bind to an adaptor protein (e.g. Cks2) with sufficiently high affinity, then optimal concentrations of the adaptor should promote formation of the ternary Cdk1-Cks2-substrate complex, whereas higher-than-optimal concentrations should promote the formation of binary Cdk1-Cks2 and Cks2-substrate complexes at the expense of ternary complex formation. This phenomenon is analogous to the prozone effect, where excess antigen inhibits the formation of cross-linked antigen-antibody complexes (Bray and Lay, 1997; Heidelberger and Kendall, 1929). In other contexts, related phenomena go by different names. For example, in transcription, a similar sequestration-based phenomenon is generally referred to as “squelching” (Cahill et al., 1994; Guertin et al., 2014; Natesan et al., 1997; Prywes and Zhu, 1992), and in cascade biochemistry, the term “combinatorial inhibition” is sometimes used to describe a similar phenomenon (Levchenko et al., 2000).
Thus, it seemed plausible that the biphasic effects of Cks proteins on Cdk1 function could be due to a prozone-type mechanism. To test this hypothesis, we begin by exploring the theory of ternary complex formation, following the leads of Bray and Lay and Levchenko and colleagues (Bray and Lay, 1997; Levchenko et al., 2000), to see under what circumstances a biphasic response to Cks2 would be expected. We then test the theory by constructing a synthetic model of adaptor-mediated squelching from deoxyoligonucleotides with predictable binding behaviors. Finally, we show that the binding of cyclin B1-Cdk1 to phosphorylated Wee1 is promoted by equimolar concentrations of Cks2 and inhibited by higher concentrations of Cks2, in agreement with an adaptor-mediated squelching mechanism. These findings rationalize why, in vivo, both Cks deficiency and Cks excess can interfere with Cdk1 signaling.
RESULTS
Theory: The Prozone Effect in Ternary Complex Formation
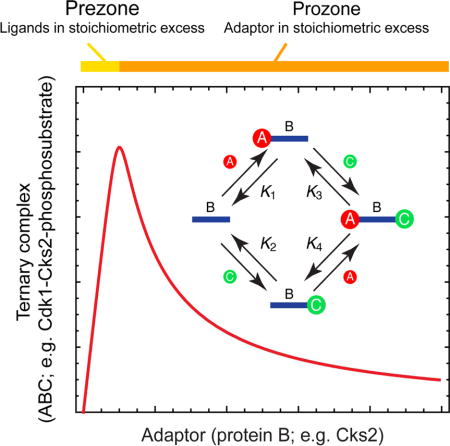
Bray and Lay examined a variety of protein oligomerization schemes through numerical simulations, looking for topologies that were capable of giving rise to a prozone effect (Bray and Lay, 1997). One of the simplest schemes they examined, and the one that is most directly applicable to the Cdk1-Cks2-phosphosubstrate system, involves the association of a bivalent adaptor (like Cks2, corresponding to species B in Figure 1A) with two monovalent binding partners (here Cdk1 and the phosphosubstrate, corresponding to species A and C in Figure 1A). We derived algebraic expressions (in the Supplementary Information) for the equilibrium concentrations of each of the individual state variables (A, B, C, AB, BC, or ABC) as a function of the parameters (the equilibrium constants and the total concentrations Atot, Btot, and Ctot). These equations were then used to examine how the binding curves depend upon the affinities and stoichiometries, with the aim of seeing under what circumstances a prozone effect is maximized or minimized.
Figure 1. Equilibrium Binding of Two Ligands, A and C, to a Bivalent Adaptor B.
(A) A schematic view of the four binding reactions leading to dimeric complexes and the full ternary complex. The K’s are the equilibrium constants.
(B) Calculated equilibrium concentration of the reactants (A and C, dashed curve), the binary complexes (AB and BC, dotted curve), and the ternary complex (ABC, solid curve) as a function of the total concentration of the adaptor B. Parameter values were Atot = Ctot = 1 and K1 = K2 = K3 = K4 = 0.01.
(C) The equilibrium concentration of ABC as a function of Btot for various assumed values of all four K’s. In each case we assumed Atot = Ctot = 1.
(D) The equilibrium concentration of ABC as a function of Btot for various assumed values of Atot and Ctot. The dashed curves represent the high affinity limit (K1 = K2 = K3 = K4 → 0) and the solid curves represent K1 = K2 = K3 = K4 = 0.01.
We began by assuming that the concentrations of Atot and Ctot were equal to 1 and all of the equilibrium constants were equal to 0.01; i.e., the binding reactions were all running close to saturation. As the assumed amount of Btot increased from 0 to 1, the equilibrium concentrations of A and C decreased approximately linearly and the equilibrium concentration of ABC increased approximately linearly (Figure 1B). However, once the assumed amount of Btot exceeded 1, the equilibrium concentration of ABC began to fall and the system transitioned toward producing dimeric AB and BC in preference to trimeric ABC (Figure 1B). The biphasic response of ABC to Btot is analogous to the “prezone” and “prozone” responses in an antibody-antigen interaction (Bray and Lay, 1997; Heidelberger and Kendall, 1929).
Optimal Affinities and Stoichiometries
Next we systematically varied the parameters of the model. First, we increased or decreased all of the binding affinities together, keeping the assumed concentrations of Atot and Ctot equal to 1. In the limit of infinitely high binding affinities, the equilibrium concentration of ABC increased linearly with Btot for Btot ≤ 1 and then decreased inversely with Btot for Btot > 1 (Figure 1C and Supplementary Information). As the assumed affinities decreased, the peak decreased in height and the position of the peak shifted to higher Btot concentrations, and the prozone effect became less prominent. Nevertheless there was still a discernible biphasic response even at relatively high K values. For example, when all four K values were taken to be 1, a maximal response was obtained when Btot ≈ 2, and the response when Btot was 10 had fallen to approximately 47% of maximum (Figure 1C).
The optimal concentration of Btot depended upon the assumed concentrations of Atot and Ctot.. In the high affinity limit, the optimal concentration of Btot was equal to the concentrations of Atot and Ctot. (Figure 1D). Thus the molar ratio of the adaptor to its binding partners determines whether or not squelching occurs. In the high affinity limit, if the concentrations of Atot and Ctot. were assumed to be different from each other, the amount of ternary complex peaked at the lower of the Atot and Ctot concentrations and then began to fall above the higher of the two concentrations.
Testing the Theory with Oligonucleotide Annealing
We next set out to test the theory described above through in vitro binding experiments. While the motivation for this work came from protein-protein interactions, we chose to carry out these experiments with oligonucleotides. The theory described above applies equally well to DNA annealing as it does to protein-protein interactions. Moreover, it is relatively easy to generate high affinity interactions, where the prozone effect would be most prominent, with oligonucleotides, and it is easy to assess complex formation through electrophoretic mobility shift assays.
Two Components and Binary Complex Formation
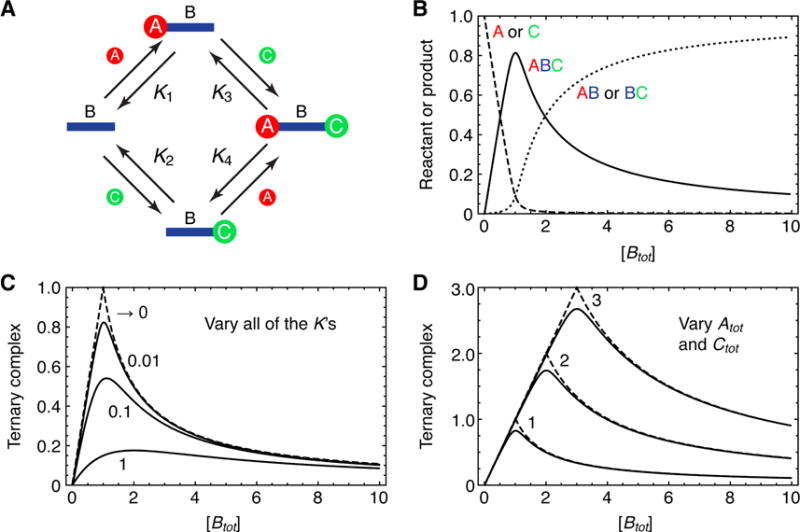
We started with a simple two-component system as shown in Figure 2A. A 42 nt oligonucleotide designated Adaptor-42, and its complement, Input-42, were chosen to minimize the formation of secondary structure and self-dimers, and to afford stable binding during the room temperature mobility shift assays (Jullien and Herman, 2011). The concentration of Input-42 was held at 100 nM or 1000 nM, and the concentration of Adaptor-42 was varied over a wide range. The equilibrium level of binary complex formation was then assessed by electrophoretic gel shift analysis and densitometry.
Figure 2. The Prozone Effect in the Binding of Two Oligonucleotides to a Complementary “Adaptor” Oligonucleotide.
(A) Schematic depiction of the two-component annealing reaction.
(B–G) Experimental data for the equilibrium binding of various concentrations of Adaptor-42 to 100 nM (B–D) or 1000 nM (E–G) Input-42. Panels B and E show the binding reaction products separated on a TBE polyacrylamide gel and stained with SYBR gold. Panels C, D, F, and G show the quantified levels of the binary complex as averages ± S.E. for 3 experiments. Panels C and F are linear plots; panels D and G are semi-log plots. The solid curves show the theoretical binding based on Eq. 12 in the Supplementary Information and the calculated free energy of binding (IDT-Biophysics, 2015; Owczarzy et al., 2011) of Input-42 for Adaptor-42.
(H) Schematic depiction of the three-component annealing reaction.
(I–N) Experimental data for the equilibrium binding of various concentrations of Adaptor-42 to 100 nM or 1000 nM Input-14 and Input-27. Panels I and L show the binding reaction products separated on a TBE polyacrylamide gel and stained with SYBR gold. Panels J, K, M, and N show the quantified levels of the ternary complex as averages ± S.E. for 3 experiments. Panels J and M are linear plots; panels K and N are semi-log plots. The solid curves show the theoretical binding based on Eqs. 14 and 17 in the Supplementary Information, assuming infinite affinity, no cooperativity (c = 1), and no skewing (s = 1), with no adjustable parameters.
As shown in Figure 2B–G, the amount of binary complex increased linearly with Adaptor-42 until the concentration of Adaptor-42 equaled that of Input-42 (100 nM or 1000 nM). Notably there was no decrease in binary complex formation at high concentrations of Adaptor 42. The linear increase in binding up to a maximum is what would be expected given the high affinity of Input-42 for Adaptor-42 (see Supplementary Information).
Three Components and Binary vs. Ternary Complex Formation
We then turned to a three-component system, with one oligonucleotide functioning as the adaptor (Adaptor-42) and two complementary functioning as ligands (Input-14 and Input-27). To minimize the possibility of interaction between the two input ligands, their binding sites on Adaptor-42 were separated by one nucleotide gap. The basic scheme for the formation of binary and ternary complexes by these binding partners is shown in Figure 2H. Again the concentration of the two input oligos was held at either 100 nM or 1000 nM, and the concentration of Adaptor-42 was varied over a range.
At a fixed concentration of Input-14 and -27 of 100 nM, the amount of ternary complex formed increased with the concentration of Adaptor-42 until Adaptor-42 reached 100 nM. At that point the amount of ternary complex began to fall (Figure 2J, K), and the amount of binary complex grew, eventually exceeding the concentration of ternary complex (Figure 2I). Similar results were obtained when Input-14 and Input-27 were fixed at 1000 nM, except that the binding did not begin to decrease until Adaptor-42 exceeded 1000 nM (Figure 2L–N). Thus, a prozone-type response was obtained, with the response being maximal when the concentrations of the inputs and the adaptor were equal.
To compare the experimental responses to theory, we assumed the equilibrium constants for the four binding-dissociation reactions shown in Figure 2H were all high. We also assumed, because of the gap between the binding sites for the two input oligos, that the binding was non-cooperative, and assumed, for simplicity, that there was no skewing in the binding (see Supplementary Information). The resulting theoretical binding curves are shown in the solid curves in Figure 2J, K, M, and N. The theoretical curves fit reasonably well with the observed data, with the amount of the ternary complex increasing linearly as the concentration of Adaptor-42 increased from zero to the optimal concentration (100 or 1000 nM), and then falling in proportion to Adaptor-42 above the optimal concentration. These experimental findings validate the theory.
Is There a Prozone Effect in the Interaction of Cks2 with Cyclin B1-Cdk1 and Wee1A?
We then turned to the question of whether a prozone-like effect might be observed in the interaction of cyclin B1-Cdk1 with particular substrate proteins via the intermediacy of Cks2. The substrate we chose to examine in detail was Wee1A, the embryonic isoform of the conserved Cdk1-inactivator Wee1. There were several reasons for this choice. First, Wee1A is a plausible Cks2-binding protein. Wee1A receives complex regulatory inputs, including positive regulation through the binding of 14-3-3 proteins to a C-terminal phosphorylation site (Lee et al., 2001), and a series of phosphorylations at at least five Ser/Thr-Pro sites located in the first 150 amino acids of the Wee1A N-terminus (Kim et al., 2005) that negatively regulate Wee1A. Two of the N-terminal phosphorylation sites fit the consensus defined by McGrath et al. for optimal binding of phosphopeptides to yeast Cks1 (PIT53P and PFT150P) (McGrath et al., 2013). In addition, the budding yeast homolog of Wee1A (Swe1) appears to bind to Cdk1 and to phosphorylate Cdk1 in a Cks-dependent fashion (McGrath et al., 2013). And, finally, Patra and Dunphy had shown that Cks2 promotes Wee1A phosphorylation, as well as the phosphorylation of Cdc27, Cdc25, and Myt1, both in Xenopus extracts and in vitro (Patra and Dunphy, 1998; Patra et al., 1999). Thus it seemed plausible that Cks2 would recognize primed Wee1A and promote its ultimate hyperphosphorylation and inactivation.
Second, quantitative studies of Wee1A’s steady-state response to Cdk1 in Xenopus egg extract have shown that the phosphorylation of T150, a site whose phosphorylation is required for inactivation of Wee1A is highly ultrasensitive, with a Hill coefficient of ~3.5 (Kim and Ferrell, 2007). Ultrasensitivity is particularly favored if the first phosphorylations promote the last, or if the few dephosphorylations promote the last; that is, if there is cooperativity in the phosphorylation and/or dephosphorylation of Wee1A (Ferrell and Ha, 2014; Kim and Ferrell, 2007). The ability of Cks2 to bind to specific phosphothreonine epitopes could provide this cooperativity.
We therefore tested whether Wee1A could bind Cks2, whether such binding required Wee1A phosphorylation, and if so, whether there was a prozone effect in the Cks2-mediated interaction of Wee1A with cyclin B1-Cdk1.
Hyperphosphorylated Wee1A binds More Strongly to Cks2 than Does Hypophosphorylated Wee1A
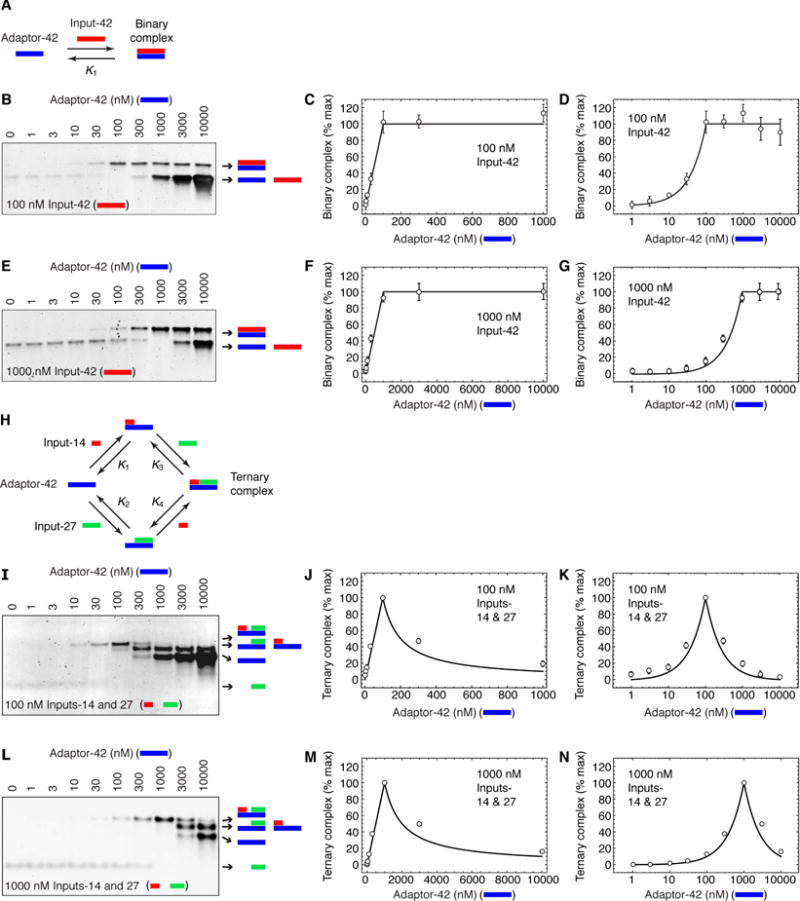
Hyperphosphorylated Wee1A was generated by incubating purified recombinant Wee1A in vitro with purified recombinant cyclin B1-Cdk1 complexes (Figure 3A). For comparison, we mock-phosphorylated the OP11-Wee1A mutant, which has alanine mutations at the eleven potential Ser/Thr-Pro phosphorylation sites and is not phosphorylated by Cdk1 (Figure 3A) (Kim et al., 2005). The reaction products were then incubated with excess recombinant GST-Cks2, and the Cks2-associated proteins on glutathione beads. As shown in Figure 3B, the glutathione beads pulled down Cks2 and cyclin B1-Cdk1 from both the wild-type and OP11-Wee1A phosphorylation reactions, but pulled down Wee1A and pT150-Wee1A (Wee1A phosphorylated at a critical late phosphorylation site, Thr 150) only from the wild-type phosphorylation reactions. These studies indicate that hyperphosphorylated Wee1A interacts more strongly with Cks2 than does hypophosphorylated Wee1A. Similar findings were reported for Xenopus Cdc27 (Patra and Dunphy, 1998).
Figure 3. Dependence of Cks2-Wee1A Binding on Wee1A Phosphorylation.
(A, B) GST-Cks2 pulls down phosphorylated Wee1A but not mock-phosphorylated OP11-Wee1A. (A) Schematic view of the experiment. The concentrations of the Wee1A, Δ65-cyclin B1, and Cdk1AF were ~20 nM; the concentration of GST-Cks2 was ~300 nM. (B) Immunoblots (for total Wee1A, pT150-Wee1, and Cdk1AF) and Ponceau S staining (for GST-Cks2).
(C–E) GST-Cks2 pulls down hyperphosphorylated Wee1A better than hypophosphorylated Wee1A. (C) Schematic view of the experiment. Protein concentrations were: Δ65-cyclin B1, ~200 nM; Cdk1AF, ~200 nM; GST-Cks2, ~300 nM; and Wee1A, various concentrations, as shown. (D) Wee1A, Cdk1, and GST-Cks2 in the GST pull downs, from one experiment. (E) Quantitative Wee1A pulldown data as averages ± S.E. for 4 experiments.
To further test this idea and to quantify the effect of the phosphorylation, we incubated various concentrations of recombinant Wee1A with cyclin B1-Cdk1 in the presence or absence of ATP, and then added GST-Cks2 and pulled down complexes with glutathione beads (Figure 3C). As shown in Figures 3D and E, the half-maximal binding was estimated to be attained with 120 ± 5 nM (mean ± S.E., from non-linear regression), and for hypophosphorylated Wee1A, the fitted value was 526 ± 39 nM.
To test the relevance of these in vitro measurements to the situation in cells, where numerous other Cdk1-interacting proteins are present that might, through competition or crowding, greatly alter the effective binding constants (Sadaie et al., 2014), we carried out binding experiments in crude Xenopus egg extracts. We prepared interphase extracts and M-phase arrested CSF extracts, added purified recombinant GST-Cks2, incubated the extracts for 45 min at room temperature, and then pulled down GST-Cks2 and any associated proteins on glutathione beads. As shown in Figure 4A, GST-Cks2 pulled down endogenous Cdk1 from either type of extract, and pulled down cyclin B2 and Wee1A from M-phase extracts, but pulled down only trace amounts of cyclin B2 and Wee1A from interphase extracts. None of these proteins were detected in control GST pull downs (Figure 4A).
Figure 4. Co-Precipitation of Hyperphosphorylated Wee1A with Cyclin B1-Cdk1 from Xenopus Egg Extracts.
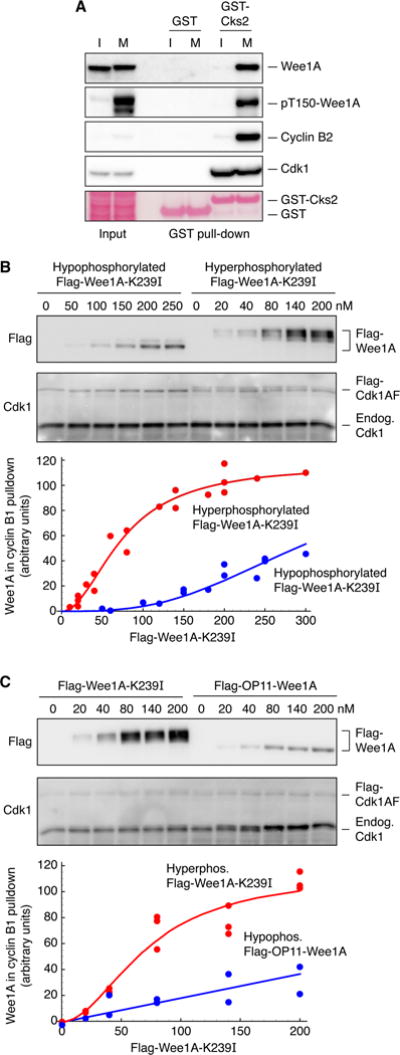
(A) Pull-down of endogenous proteins from Xenopus laevis interphase (I) or M-phase (M) extracts with recombinant GST-Cks2 proteins. The concentrations of GST and GST-Cks2 were approximately 500 nM.
(B) Complex formation as a function of Wee1A concentration for hypo- and hypophosphorylated Flag-Wee1-K239I. Hypo- and hyperphosphorylated Wee1A were generated by incubating recombinant catalytically-inactive Flag-Wee1-K239I in Xenopus egg extracts, supplemented with Cdk1AF (230 nM) and GST-Δ90-cyclin B1 (90 nM), for 3 min (hypophosphorylated) or 10 min (hyperphosphorylated). Phosphorylation and dephosphorylation were quenched with EDTA (25 mM) and okadaic acid (1 μM); GST-Δ90-cyclin B-Cdk1 was pulled down on glutathione beads and the associated Flag-Wee1A was assessed by blotting with Flag antibody. The blots shown are from one experiment. The plotted data are combined from three independent experiments and are fitted to a Hill curve using Mathematica 10.
(C) Complex formation as a function of Wee1A concentration for hyperphosphorylated Flag-Wee1-K239I and OP11-Wee1A. The Wee1A proteins were incubated in mitotic extracts supplemented with Cdk1AF (230 nM) and GST-Δ90-cyclin B1 (90 nM) for 10 min to induce maximal phosphorylation. Phosphorylation and dephosphorylation were quenched with EDTA (25 mM) and okadaic acid (1 μM); GST-Δ90-cyclin B-Cdk1 was then pulled down on glutathione beads and the associated Flag-Wee1A was assessed by blotting with Flag antibody. Blots are from one experiment, and the plotted data are combined from two independent experiments.
To determine whether the association of and Cdk1 with Wee1 in extracts depended upon the phosphorylation state of Wee1A, and to gauge the strength of the association, we incubated various concentrations of kinase-minus Wee1A-K239I with M-phase extract (interphase extract pre-incubated with 90 nM GST-Δ90-cyclin B1 and 230 nM Flag-Cdk1AF) for 3 min (to yield hypophosphorylated Wee1A) or 10 min (to yield hyperphosphorylated Wee1A), quenched the phosphorylation and dephosphorylation with EDTA (to inhibit all kinases) plus okadaic acid (to inhibit the relevant phosphatases), and then pulled down the GST-Δ90-cyclin B and any associated proteins by incubation with glutathione beads on ice. As shown in Figure 4B, the hyperphosphorylated Wee1A was pulled down by GST-Δ90-cyclin B1 better than the hypophosphorylated Wee1A was. The EC50 value for the binding of hyperphosphorylated Wee1A to cyclin B1-Cdk1-Cks2 was estimated to be 75 ± 6 nM, and for hypophosphorylated Wee1A 271 ± 9 nM, within a factor of two of the values measured in vitro.
Finally, we carried out similar experiments to compare the cyclin B1-Cdk1-Cks2 binding of hyperphosphorylated Wee1A to OP11-Wee1A. Various concentrations of Flag-OP11-Wee1A and Flag-KM-Wee1A were incubated with M phase extracts containing 90 nM GST-Δ90-cyclin B1 and 230 nM Flag-Cdk1AF, the reaction was stopped after 10 min, and the GST-Δ90-cyclin B1 and associated proteins were pulled down on glutathione beads. As shown in Figure 4C, half-maximal binding of kinase-minus Wee1A-K239I and OP11-Wee1A were obtained at Wee1A concentrations of 72 ± 9 and 279 ± 29 nM, again comparable to the values obtained in vitro.
Thus, both in vitro and in extracts, hyperphosphorylated Wee1A was found to bind to Cks2 (in vitro) and cyclin B1-Cdk1 (presumably via Cks2) with an apparent affinity of ~100 nM, and hypophosphorylated Wee1A bound several-fold less strongly. For comparison, the concentrations of Cdk1 and Wee1A in Xenopus eggs have been estimated to be ~230–270 nM and ~20–55 nM, respectively (Mueller et al., 1995; Pomerening et al., 2005; Walter et al., 2000; Wuhr et al., 2014). These affinities and concentrations are compatible with the possibility that the interaction of Wee1A with cyclin B1-Cdk1 might be squelched by a modest excess of Cks2, through a prozone effect.
Can Cks2 Bind Phosphorylated Wee1A in the Absence of Cyclin B1-Cdk1?
A prozone effect would require that Cks2 be able to form a binary complex with hyperphosphorylated Wee1A in the absence of cyclin B1-Cdk1 (Figure 5A). The data presented in Figure 3 suggest that this is in fact the case, since excess Cks2 was able to pull down hyperphosphorylated Wee1A. To test this idea more rigorously, we engineered mutations into Cks2 that would be expected to interfere with Cdk1 binding but not phosphosubstrate binding, or, as a control, to interfere with phosphosubstrate binding but not Cdk1 binding. The Cks2-R20A protein has a mutation at a conserved arginine residue in the anion-binding pocket of Cks2 (Figure 5B) and might compromise phosphosubstrate binding (Bourne et al., 2000; Koivomagi et al., 2013). As shown in Figure 5C, Cks2-R20A still bound to Cdk1, but did not bind to either hypo- or hyperphosphorylated Wee1A (lanes 5 and 9). The Cks2-E63Q mutant replaces a charged residue at the Cks2-Cdk1 interface with a polar residue (Figure 5B), and has been shown to compromise Cks function (Bourne et al., 1996). As shown in Figure 5C, Cks2-E63Q did not bind to Cdk1, but did bind to hyperphosphorylated Wee1A (lane 10). These data indicate that Cks2 can bind to Cdk1 without binding to Wee1A, and to hyperphosphorylated Wee1A without binding to Cdk1. This means that a prozone effect is possible.
Figure 5. A Prozone Effect in the Formation of Cyclin B1-Cdk1-Cks2-Wee1A Complexes.
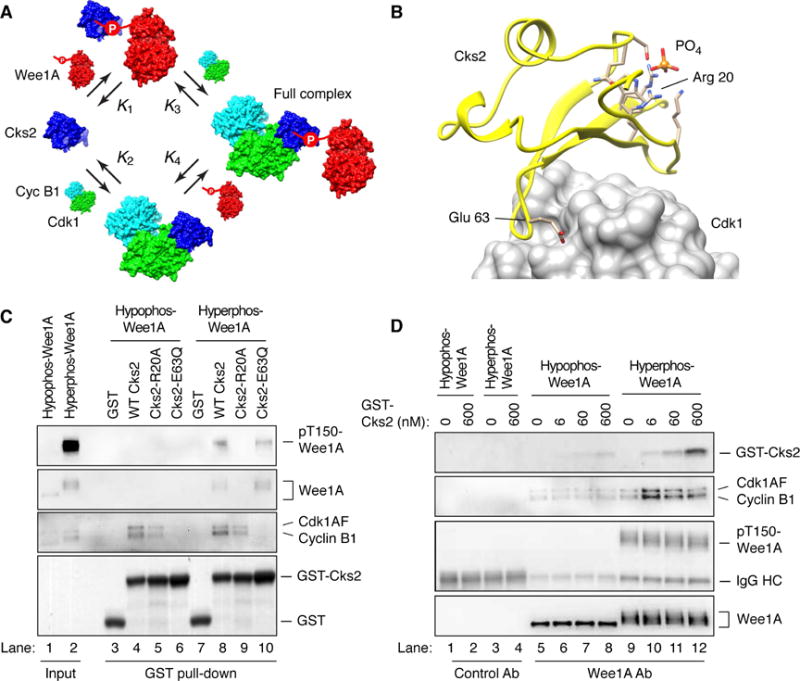
(A) Schematic view of the four binding reactions between Cks2, Wee1A, and (stable) cyclin B1-Cdk1.
(B) Residues in Cks2 critical for the binding of phosphopeptides (Arg 20) and Cdk1 (Glu 63). The structures are based on crystal structures of two related proteins, Cks1 (1DKS) and Cdk1 (1BUH and 4EOQ) and rendered in UCSF Chimera (Pettersen et al., 2004). Amino acid side chains are shown for the residues in the phosphate-binding pocket of Cks2.
(C) Binding of wild-type GST-Cks2 and two Cks2 mutants (R20A and E63Q) to cyclin B1-Cdk1 and hyperphosphorylated Wee1A in vitro. The input and GST pull-downs were blotted for total Wee1A, pT150-Wee1A, Flag (for Cdk1 and cyclin B1), and stained with Ponceau S (for GST-Cks2 and GST). Approximate concentrations were: Wee1A, 20 nM; cycB1/Cdk1AF, 20 nM; and GST proteins (GST, GST-Cks2 WT, GST-Cks2-R20A, GST-Cks2-E63Q), 300 nM.
(D) Biphasic effect of Cks2 on the binding of hyperphosphorylated Wee1A to cyclin B1-Cdk1. Active recombinant Δ65-cyclin B1-Cdk1AF was incubated with Wee1A with or without ATP to produce hypo- and hyperphosphorylated Wee1A. GST-Cks2 and Wee1A antibody-coated beads were added, and the associated proteins were pulled down and detected by immunoblotting. Approximate concentrations were: cyclin B1-Cdk1AF, 200 nM; Wee1A, 250 nM; and GST-Cks2 WT, 0 – 600 nM as shown.
A Prozone Effect in the Binding of Cyclin B1-Cdk1 to Phosphorylated Wee1A
To look for a prozone effect in the interaction of cyclin B1-Cdk1 with Cks2 and Wee1A, hypophosphorylated and hyperphosphorylated Wee1A proteins were generated and immobilized onto protein A beads coated with anti-Wee1A antibody. The immobilized Wee1A was then incubated with cyclin B1-Cdk1 in the presence of different concentrations of Cks2 (plus no ATP). The Wee1A beads were then pulled down and the associated proteins were assessed. As shown in Figure 5D, hypophosphorylated Wee1A interacted weakly with cyclin B1-Cdk1 and the interaction did not change much as the concentration of Cks2 increased (second blot, lanes 5–8, and Figure S1). In contrast, for hyperphosphorylated Wee1A, there was a biphasic response to Cks2, with the binding of cyclin B1-Cdk1 to the Wee1A beads first increasing and then decreasing (second blot, lanes 9–12, and Figure S1). Thus, the binding of hyperphosphorylated Wee1A to cyclin B1-Cdk1 depends upon the presence of an optimal concentration of Cks2; too much or too little decreases binding.
Effects of Cks2 on the phosphorylation of Wee1A by Cdk1AF
To test the functional relevance of these binding studies, we examined the dependence of Wee1A phosphorylation on the ratio of Cks2 to kinase (cyclin B1-Cdk1AF, which cannot be enzymatically inactivated by Wee1A) and substrate (Wee1A). As shown in Figure 6A, when cyclin B1-Cdk1AF was present in excess of Cks2, Cks2 promoted the production of T150-phosphorylated Wee1A. In contrast, once the concentration of Cks2 exceeded that of the Cdk1AF and Wee1A, the T150 phosphorylation decreased. Similar results were found using Cks2 without a GST tag, and using human Cdk1 in place of Xenopus Cdk1 (Supplementary Figure S2). Cks2 had neither a stimulatory nor an inhibitory effect on the phosphorylation of the model Cdk1 substrate histone H1 (Figure 6B), indicating that Cks2 affected the interaction of Cdk1 with particular substrates, but did not affect the intrinsic activity of the cyclin B1-Cdk1 complex. A mutant Cks2 protein defective for interaction with both Cdk1 and Wee1A, Cks2-R20A/E63Q, had no apparent effect on Wee1A T150 phosphorylation (Figure 6C). These results suggest that the formation of the full cyclin B1-Cdk1-Cks2-pWee1A complex is important for the phosphorylation of Wee1A at the critical T150 site. These results also rationalize why Cks proteins can both promote (Patra and Dunphy, 1996, 1998; Patra et al., 1999) and inhibit (Dunphy and Newport, 1989) the full phosphorylation of specific Cdk1 substrates.
Figure 6. A Prozone Effect in the Phosphorylation of Wee1A in Vitro.
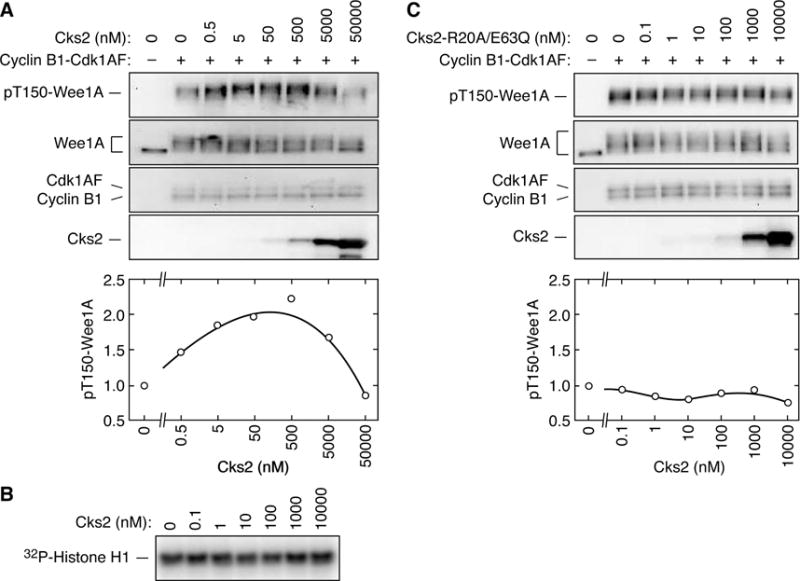
(A) Biphasic effect of Cks2-WT on Wee1A T150 phosphorylation.
(B) Lack of effect of Cks2-WT on histone H1 phosphorylation. Phosphorylation was assessed by autoradiography.
(C) Lack of effect of Cks2-R20A/E63Q on Wee1 T150 phosphorylation.
For each panel, active recombinant Δ65-cyclin B1-Cdk1AF (~50 nM) was incubated with Wee1A (~50 nM) for 15 min in the presence of various concentrations of mutant or wild-type GST-Cks2 as shown. The reaction products were then analyzed by immunoblotting.
Mutual Inhibition between Cyclin B1-Cdk1 and Wee1A
In Figure 6 we used the Cdk1-AF mutant, which cannot be feedback-inhibited by Wee1A. This simplifies the situation, and allows Wee1A to act like a canonical downstream target of Cdk1, rather than as a protein that is both substrate and regulator of Cdk1. Here we asked how Cks2 would affect the interplay between the two proteins when each protein was capable of regulating the other.
To this end we incubated complexes of cyclin B1 and wild-type Cdk1 with Wee1A at different concentrations of Cks2 (Figure 7). As shown in Figure 7A, in the absence of Cks2, there was maximal Y15 phosphorylation of Cdk1 and minimal T150 phosphorylation of Wee1A. This indicated that Cks2 is not required for the phosphorylation and inhibition of Cdk1 by Wee1A (Figure 7A, B). As the Cks2 concentration rose through the prezone range (Cks2 = 0 to 300 nM, the concentration of cyclin B1-Cdk1 in the assay), the amount of Wee1A T150 phosphorylation began to rise. Presumably this is because more and more full cyclin B1-Cdk1-Cks2-pWee1A complexes were formed, and these complexes are more capable than free cyclin B1-Cdk1 at fully phosphorylating Wee1A (Figure 7A, B). As the concentration of Cks2 rose above the concentration of cyclin B1-Cdk1, both the Cdk1 Y15 phosphorylation and Wee1A T150 phosphorylation decreased dramatically (Figure 7A). Consistent with these findings, the activity of Cdk1 toward histone H1 increased at the concentrations of Cks2 that caused the Cdk1 Y15 phosphorylation to decrease. These findings are consistent with the scheme shown in Figure 7B. We assume that Y15 phosphorylation is inefficient in both the full cyclin B1-Cdk1-Cks2-pWee1A complexes and in the interaction of cyclin B1-Cdk1-Cks2 with Cks2-Wee1A complexes, whereas Wee1A T150 phosphorylation is inefficient in the interaction of cyclin B1-Cdk1-Cks2 with Cks2-Wee1A complexes but efficient in the full cyclin B1-Cdk1-Cks2-pWee1A complexes.
Figure 7. The Effect of Cks2 on the Mutual Inhibition of Cyclin B1-Cdk1 and Wee1A.
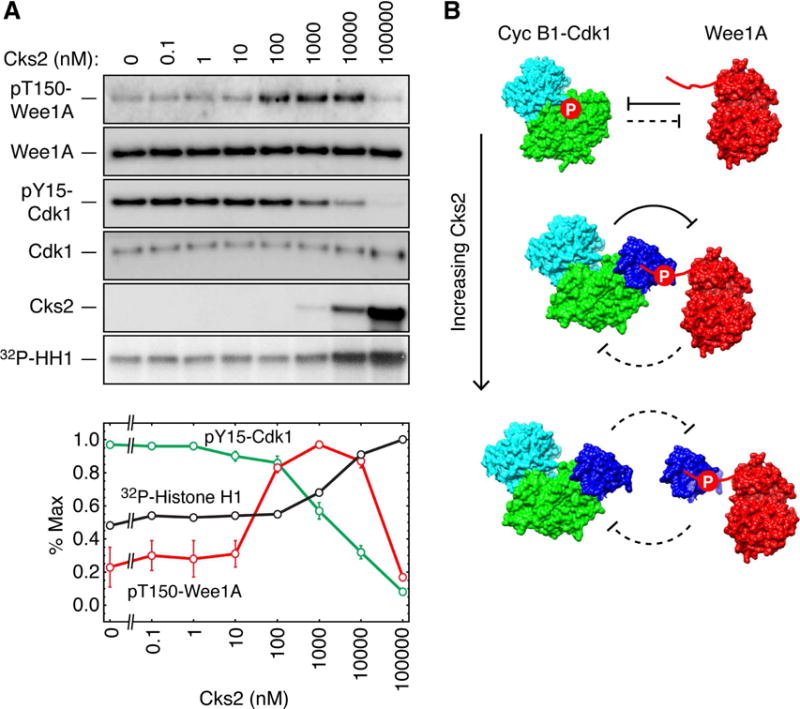
(A) Effect of Cks2 on Wee1A T150 phosphorylation and Cdk1 Y15 phosphorylation. Active recombinant Δ65-cyclin B1-Cdk1 (~300 nM) was incubated with Wee1A (~30 nM) for 15 min in the presence of various concentrations of wild-type GST-Cks2 as shown. The reaction products were then analyzed by immunoblotting. The blots are from one experiment. HH1 denotes histone H1. The graph shows the quantitated Cdk1 and Wee1A phosphorylation levels from two experiments.
(B) Schematic depiction of the interplay between Cdk1 and Wee1A at various ratios of the kinases to Cks2. When no Cks2 is added, Cdk1 Y15 phosphorylation is maximal and Wee1A T150 phosphorylation is minimal. As the Cks2 concentration increases through the prezone range (Cks2 = 0–300 nM) and full cyclin B1-Cdk1-Cks2-Wee1A complexes are formed, the amount of Wee1A T150 phosphorylation rises and the amount of Cdk1 Y15 phosphorylation falls slightly. In the prozone range (Cks2 > 300 nM), phosphorylation of both proteins falls. Data are means ± S.E. from 4 (for the 100,000 nM data) or 6 (for all other data) experiments.
Taken together, these findings show that the dominant direction of inhibition in the Cdk1-Wee1A system is dictated by the concentration of Cks2. When the system is in the prezone regime, Wee1A dominates over Cdk1; when the system enters the prozone regime, Cdk1 begins to dominate over Wee1A; and at the highest ratios of Cks2 to Wee1A and cyclin B1-Cdk1, neither kinase regulates the other efficiently.
DISCUSSION
Bray and Lay previously explored the equilibrium relationship between protein concentration and complex formation for various oligomeric complexes, through numerical solution of the equilibrium equations, and showed that a prozone-like effect can occur when one of the proteins (the equivalent of an adaptor) can bind its ligands either individually or at the same time (Bray and Lay, 1997). Similar findings were shown by Levchenko and co-workers in their computational studies of yeast mating pheromone signaling (Levchenko et al., 2000). Here we have extended this computational work by deriving analytical expressions for the equilibrium formation of various monomeric, dimeric, and ternary species in a system consisting of an adaptor (species B) and two ligands (species A and C). We show that the equilibrium concentration of the ternary ABC complex depends upon the stoichiometric ratio of the adaptor to its ligands. In the limiting case of high affinity binding, a stoichiometric ratio of 1:1:1 produces the maximal amount of ABC complex (Figure 1).
We then tested this theory by examining the binding of two oligonucleotides (Input-14 and Input-27, corresponding to the A and C species in the theory) to a complementary oligonucleotide (Adaptor-42, the B species). We found that this system exhibits a biphasic, bell-shaped dependence of the amount of the ternary ABC complex as a function of the concentration of B, with maximal complex formation obtained at the predicted 1:1:1 stoichiometric ratio (Figure 2). The experimental data were well accounted for by the theory, with no adjustable parameters.
Next we examined whether this conceptual framework applied to the interaction of cyclin B1-Cdk1 complexes with Cks2 and phosphorylated Wee1A, as suggested by previous work on the interaction of budding yeast Cks1 with phosphorylated Sic1, Cdc6 and Swe1 proteins (Koivomagi et al., 2013; McGrath et al., 2013), and on previous work on the Xenopus Cks2 protein (Patra and Dunphy, 1996, 1998; Patra et al., 1999). We found that hyperphosphorylated Xenopus Wee1A binds to the relevant Cks protein, Cks2, several-fold more strongly than does hypophosphorylated Wee1A (Figures 3 and 4), consistent with the longstanding hypothesis that Cks proteins promote the interaction of Cdk1 complexes with particular phosphoproteins (Arvai et al., 1995; Koivomagi et al., 2013; McGrath et al., 2013; Parge et al., 1993; Patra and Dunphy, 1996, 1998; Patra et al., 1999). This priming and cooperativity helps explain the ultrasensitivity observed in the multisite phosphorylation of Wee1A (Ferrell and Ha, 2014; Kim and Ferrell, 2007). Cdc25C exhibits an even higher degree of ultrasensitivity in its regulation by Cdk1, and, as previously noted, priming and cooperativity probably contributes to that ultrasensitivity as well (McGrath et al., 2013; Trunnell et al., 2011).
We also found that the relationship between the concentration of Cks2 and the amount of cyclin B1-Cdk1-Cks2-pWee1 complex formation is biphasic (Figure 5), with sub-stoichiometric amounts of Cks2 promoting full complex formation and suprastoichiometric amounts squelching it. Likewise, we found that the phosphorylation of Wee1A at T150, a functionally-critical late phosphorylation site (Kim et al., 2005), depends upon Cks2 in a biphasic fashion when Cdk1AF is used, so that Wee1A is acting like a “normal” downstream target of Cdk1 rather than as a protein that acts both upstream and downstream of Cdk1 (Figure 6). These findings rationalize why both decreases and increases in the expression levels of Cks proteins can compromise cell cycle progression (Dunphy and Newport, 1989; Hayles et al., 1986; Patra and Dunphy, 1996; Patra et al., 1999).
Finally, we found that Cks2 has a dramatic effect on the mutual inhibition of Cdk1 and Wee1A in vitro (Figure 7). At zero or low concentrations of Cks2, Wee1A wins out over cyclin B1-Cdk1. As the concentration of Cks2 increases, Wee1A begins to be inhibited by cyclin B1, presumably through the formation of full cyclin B1-Cdk1-Cks2-Wee1A complexes. At higher concentrations of Cks2, Cdk1 wins out over Wee1A, and at the highest Cks2 concentrations, each kinase is compromised in its regulation of the other. This is presumably due to the predominance of the less productive partial complexes (cyclin B1-Cdk1-Cks2 and Cks2-Wee1A). Of course, in vivo the system includes not just the two kinases and Cks2, but also opposing phosphatases whose relative activities will affect the two kinases’ phosphorylation and inactivation of each other. Nonetheless, the ability of Cks2 to push the mutual inhibition of Cdk1 and Wee1A in favor of Wee1A at low concentrations and in favor of Cdk1 at higher concentrations should contribute to the overall behavior of the system in vivo as well as in vitro.
The general phenomenon examined here, which is analogous to the prozone effect and to transcriptional squelching, is sometimes regarded as mainly being significant in that it can produce misleading phenotypes in overexpression studies. For example, the JIP1 scaffold protein, which interacts with kinases in the Jun kinase (JNK) cascade, can inhibit the function of the JNK cascade when overexpressed (Dickens et al., 1997), but at physiological concentrations JIP1 is thought to promote JNK activation (Yasuda et al., 1999). The same is true for the KSR proteins, facilitators of Ras/Raf/ERK signaling that when overexpressed can have opposite effects (Cacace et al., 1999; Roy et al., 2002).
However, it is possible that the prozone effect could be important for normal signal processing as well. Whether it does or does not depends upon the stoichiometric ratios of the proteins, their affinities, and any cooperativity (positive or negative) involved in the interaction (see Supplementary Information). In the case of the Xenopus Cdk1 system, we have some quantitative information along these lines. In a recent mass spectrometry study, the Xenopus Cks proteins were estimated to be expressed at concentrations in (slight) excess of Cdk1 (Cks1B, 45 nM; Cks2, 303 nM, Cdk1, 269 nM; (Wuhr et al., 2014)). Previous immunoblotting experiments yielded similar estimates (Cks2, 560 nM (Patra and Dunphy, 1996); Cdk1, 230 nM (Pomerening et al., 2005)). Thus, Cdk1 and the Cks proteins are present at similar concentrations, with there being perhaps a ~100–300 nM excess of Cks. If free Cks and Cdk1-bound Cks bind equally well to a phosphosubstrate produced during early mitosis, the initial rate of the substrate’s second phosphorylation would be ~50–75% of the maximal rate, where the maximal rate would be achieved once the phosphosubstrate concentration rose to the concentration of free Cks. This relatively subtle effect could be multiplied if the substate’s full hyperphosphorylation depended upon multiple non-processive rounds of Cdk1-Cks binding. Alternatively, if free Cks binds the phosphosubstrate more strongly than Cdk1-Cks does (i.e. there is negative cooperativity in the formation of the Cdk1-Cks-phosphosubstrate complex), the substrate’s hyperphosphorylation could be strongly squelched by any excess free Cks.
The same principles might be a feature of other regulatory processes that involve adaptor proteins, provided that (1) the affinity of the adaptor for its binding partners is reasonably high, and (2) the system changes from being in adaptor-excess to binding partner-excess in the course of its normal regulation. Given how frequently adaptor proteins are found in cell signaling processes, it will be interesting to see how common this mechanism for generating biphasic responses and thresholds is.
EXPERIMENTAL PROCEDURES
DNA Annealing
Oligonucleotides that have been developed for electrophoretic mobility shift assays (Jullien and Herman, 2011) were used with some variations. A 14-mer oligonucleotide (5′- GTGCCCTGGTCTGG-3′) derived from mouse SINE B1, a 27-mer oligonucleotide (5′- TGTCTTCCTGAATATGAATAAGAAATA -3′) derived from rat prolactin promoter and a 42-mer oligonucleotide (5′- GTGCCCTGGTCTGGATGTCTTCCTGAATATGAATAAGAAATA -3′) containing both mouse SINE B1 and rat prolactin promoter sequences were synthesized, and denoted Input-14, Input-27, and Input-42, respectively. A 42-mer oligonucleotide (5′- TATTTCTTATTCATATTCAGGAAGACATCCAGACCAGGGCAC-3′) complementary to the Input-42 oligonucleotide was synthesized and denoted Adaptor-42.
For annealing, oligonucleotides were mixed in an annealing buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 50 mM NaCl) and a heating-cooling step was performed in a thermocycler (Eppendorf). First, oligonucleotide mixtures were heated to 94°C for 2 min, cooled down to 70°C at a fast speed (3°C/sec), and then slowly cooled down to 18°C (at 1°C/min). The aim was to maximize the selectivity of the on-target, high affinity binding reactions over off-target, lower-affinity binding reactions, and to allow the binding reactions to equilibrate before they are locked in place by slow dissociation rates.
The mixture was then separated by a native gel electrophoresis (a 15% Tris/borate/EDTA gel (Bio-Rad) with Tris/borate/EDTA buffer in a constant voltage of 100 V for 2 hours) at room temperature, stained with SYBR® Gold nucleic acid staining solution (Invitrogen), and visualized with a ChemiDoc™ MP gel imaging system (Bio-Rad). The intensities of bands of interest were quantified with Image J software (NIH).
Preparation of Recombinant Cks2 Proteins in Bacteria
A gene encoding Xenopus laevis Cks2α (NCBI Locus NM_001088329) was synthesized (Integrated DNA Technologies) and subcloned into the BamHI and ZhoI sites in a pGEX4T-2 expression vector. Mutations at Arg 20 (R20A), Glu 63 (E63Q), or both residues were engineered using a QuickChange™ site-directed mutagenesis kit (Stratagene). The oligonucleotides used to produce the point mutations were as follows: R20A, 5′-CACGGATGAACACTTCGAGTACGCACATGTTATGTTACCCAAAGAGT-3′; E63Q, 5′-GGTCCATTATATGATTCATGAACCACAGCCGCACATTCT-3′. Mutations were verified by DNA sequencing. Cks2 N-terminal GST fusion proteins were expressed in bacteria with 0.1 mM IPTG induction and purified using glutathione-Sepharose as described in the GST purification manual (GE Healthcare). For some experiments, N-terminal GST was cleaved with thrombin (Sigma) and free thrombin was cleared with p-aminobenzamidine-agarose (Sigma) as described in manufacturer’s manual. Purified Cks2 proteins were further concentrated using Amicon (Millipore).
Preparation of Recombinant Wee1 Proteins in Insect Cells
N-terminally Flag-tagged Xenopus laevis Wee1A protein (Flag-Wee1A-WT), a kinase-dead form of Wee1A (Flag-Wee1A- K239I) and Flag-Wee1A-OP11 protein that has all eleven Ser/Thr-Pro sites mutated to Ala-Pro were expressed by infecting Sf9 cells with baculoviruses encoding wild-type, Wee1A-239I and Wee1A-OP11 as described previously (Kim and Ferrell, 2007). Cell pellets were lysed with Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 10 mM 2-mercaptoethanol, 0.1% NP-40, 10 μg/ml leupeptin, 10 μg/ml chymostatin, and 10 μg/ml pepstatin and were affinity-purified on anti-flag M2 agarose (Sigma). 3X-Flag peptides were used to elute Wee1A proteins as described in manufacturer’s manual (Sigma).
Preparation of the Recombinant Cdk1-Cyclin B1 Complex in Insect Cells
Active Cdk1-cyclin B1 complex was prepared as described previously (Kumagai and Dunphy, 1995). Sf9 cells infected with baculoviruses containing either a non-inhibitable Xenopus laevis Cdk1 (Flag-His-Cdk1-T14A/Y15F, denoted Flag-His-Cdk1AF) or wild-type Xenopus laevis Cdk1 were washed with ice-cold Tris buffered-saline (TBS; 10 mM Tris-HCl pH 7.5, 150 mM NaCl) and lysed in HEPES buffered-saline (HBS; 10 mM HEPES-NaOH pH 7.5, 10 mM NaCl) plus 1 mM EGTA, 1 mM phenylmethylsulfonylfluoride, 10 μg/ml leupeptin, 10 μg/ml chymostatin, and 10 μg/ml pepstatin with a Dounce homogenizer. After adjusting the NaCl concentration to 150 mM, cell lysates were centrifuged at 16,000×g for 20 min. Supernatants containing Flag-His-Cdk1AF were stored at −80°C.
Baculovirus encoding non-degradable Xenopus laevis cyclin B1 (Flag-His-Δ65-cyclin B1) was infected into Sf9 cells. Cells were first washed with ice-cold TBS and then lysed with ice-cold HBS plus 5 mM EGTA, 1 mM phenylmethylsulfonylfluoride, 0.5% Triton X-100, 10 μg/ml leupeptin, 10 μg/ml chymostatin, and 10 μg/ml pepstatin, sonicated on ice and centrifuged at 16,000×g for 20 min. The resulting supernatants were incubated with Ni-NTA agarose (Qiagen) at 4°C for 30 min and the r esulting beads were washed first with HBS plus 0.5% NP-40 and then with HBS alone. To assemble the active Cdk1-cyclin B1 complex, Flag-His-Δ65-cyclin B1 immobilized on Ni-NTA agarose was incubated with Sf9 cell lysate containing either Flag-His-Cdk1AF or WT-Cdk1 as described above in the presence of 0.5 mM ATP and 10 mM MgCl2 at 22°C for 20 min. After washing with HBS, the His-tagged proteins were eluted with 200 mM imidazole in HBS.
Phosphorylation of Wee1A by Cdk1-Cyclin B1, and Phosphorylation of Cdk1-Cyclin B1 by Wee1A
Recombinant Wee1A proteins (50 nM) were mixed with either Xenopus laevis Cdk1AF-Δ65-cyclin B1 complex (Cdk1AF has non-phosphorylatable residues at Thr 24 (Ala) and Tyr 15 (Phe) and cannot be phosphorylated and inactivated by Wee1-family kinases; Δ65-cyclin B1 is not degraded by the anaphase-promoting complex/cyclosome) (50 nM), Xenopus laevis Cdk1 (wild-type)-Δ65-cyclin B1 complex (300 nM), or recombinant human Cdk1-cyclin B1 complex (1 unit, New England Biolabs) in a kinase assay buffer (5 mM Tris pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.1 mg/mL ovalbumin, 1 μM okadaic acid, 100 μM ATP) at 22°C for 15 min. To test the effect of Cks2, different concentrations of Cks2 (0 to 50 μM) were added. The phosphorylation reactions were stopped by addition of SDS sample buffer.
Interaction of Wee1A with Cks2
Bacterial pellets containing recombinant GST-Cks2 proteins were lysed in bacterial lysis buffer (10 mM HEPES-NaOH pH 7.5, 150 mM NaCl, 1 mM phenylmethylsulfonylfluoride, 1% Triton X-100), clarified and incubated with glutathione-Sepharose (GE Healthcare) in a cold room for 30 min. Recombinant Wee1A proteins were incubated with either recombinant Xenopus laevis Cdk1AF-Δ65-cyclin B1 complex (as described above) or recombinant human Cdk1-cyclin B1 complex (New England Biolabs) in the absence of Cks2. Kinase reactions were performed in a kinase assay buffer (5 mM Tris pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.1 mg/mL ovalbumin, 1 μM okadaic acid, 100 μM ATP) at 22°C for 45 min as des cribed previously with a minor modification (Patra et al., 1999).
The resulting immobilized GST-Cks2 was incubated with phosphorylated recombinant Wee1A proteins in a binding buffer (50 mM HEPES pH 7.8, 10 mM NaCl) at 4°C for 2 hrs.
Interaction of Wee1A with Cdk1-Cyclin B1
Recombinant Wee1A proteins were phosphorylated by Cdk1-cyclin B1 in the absence of Cks2 as described above. The resulting mixtures were incubated with anti-Wee1A antibody immobilized on Protein A agarose in the presence of different amounts of Cks2 in a binding buffer (50 mM HEPES pH 7.8, 10 mM NaCl) at 4°C for 2 hrs.
Cdk1 Kinase Assay (H1 Kinase Assay)
The activity of recombinant Cdk1-cyclin B1 complex was measured with histone H1 as a model substrate as described elsewhere (Murray, 1991). In brief, different concentrations of Cks2 protein (0–10 μM) and histone H1 (0.5 mg/mL, Millipore) were added into kinase assay buffer (5 mM Tris pH 7.5, 10 mM MgCl2, 1 mM DTT, 0.1 mg/mL ovalbumin, 1 μM okadaic acid, 100 μM ATP) plus γ-32P-ATP. Protein samples were resolved in a Bis-Tris gel with MES running buffer (Bio-Rad) and transferred to a PVDF membrane. Phosphorylation of histone H1 was quantified by autoradiography.
Western Blotting
All protein samples were subjected to SDS polyacrylamide electrophoresis and transferred to PVDF membranes. Blotting membranes were either stained with Ponceau S or probed with anti-Flag (Sigma), anti-Wee1A (Invitrogen), anti-GST (Santa Cruz Biotechnology), anti-cyclin B2 (Santa Cruz Biotechnology), anti-Cdk1 pY15 (Cell Signal Transduction) or anti-Cdk1 (Cell Signal Transduction) antibodies. Rabbit serum against the synthetic peptide corresponding to VNINPFpTPESY in Xenopus laevis Wee1A was affinity-purified and then used as anti-pThr150-Wee1A antibody (Kim et al., 2005). All blots were probed with a horseradish peroxidase-conjugated secondary antibody and detected with Immobilon Western Chemiluminescent HRP substrate (Millipore). A ChemiDoc™ MP gel imaging system (Bio-Rad) and Image J software (NIH) were used for the visualization and quantification, respectively. Care was taken to ensure that the immunoblotting signal varied linearly with protein concentration.
Preparation of Xenopus Egg Extracts
For the experiments shown in Figure 4BC, Xenopus interphase-arrested extracts were prepared as described previously (Murray, 1991) by dejellying unfertilized eggs in 2% cysteine, and activating them with 0.4 μg/ml calcium ionophore A23187 (Sigma) in the presence of 10 μg/ml cycloheximide. To drive interphase extracts into a stable mitotic state, GST-Δ90-cyclin B or Δ65-cyclin B1 was added at a concentration of 90–100 nM unless indicated otherwise. Demembranated sperm chromatin was routinely added to the extracts at a concentration of 500 sperm/μl and stained with 4′,6-diamidino-2-phenylindole (DAPI) to monitor progression into mitosis.
For the experiment shown in Figure 4A, CSF extracts were prepared similarly (Murray, 1991), except that the eggs were not activated with A23187 and the extracts were not treated with cycloheximide. A portion of the CSF extract was driven into interphase by incubation with CaCl2 (0.4 mM).
Supplementary Material
Highlights.
-
-
Cks2 regulates the phosphorylation of Wee1A by Cdk1 in a biphasic manner.
-
-
This biphasic regulation accounts for Cks overexpression phenotypes.
-
-
Cks2 changes the balance in the mutual inhibition of Cdk1 and Wee1A.
Acknowledgments
We thank Tek Hyung Lee and Lendert Gelens for comments on the paper. This work was supported in part by grants from the National Institutes of Health (R01 GM046383 and P50 GM107615) and from the National Research Foundation of Korea (NRF-2011-357-C00151, 2012K1A1A2045441, and 2012M3A9B9036680).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Arvai AS, Bourne Y, Williams D, Reed SI, Tainer JA. Crystallization and preliminary crystallographic study of human CksHs1: a cell cycle regulatory protein. Proteins. 1995;21:70–73. doi: 10.1002/prot.340210109. [DOI] [PubMed] [Google Scholar]
- Basi G, Draetta G. p13suc1 of Schizosaccharomyces pombe regulates two distinct forms of the mitotic cdc2 kinase. Mol Cell Biol. 1995;15:2028–2036. doi: 10.1128/mcb.15.4.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Arvai AS, Bernstein SL, Watson MH, Reed SI, Endicott JE, Noble ME, Johnson LN, Tainer JA. Crystal structure of the cell cycle-regulatory protein suc1 reveals a beta-hinge conformational switch. Proc Natl Acad Sci U S A. 1995;92:10232–10236. doi: 10.1073/pnas.92.22.10232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Watson MH, Arvai AS, Bernstein SL, Reed SI, Tainer JA. Crystal structure and mutational analysis of the Saccharomyces cerevisiae cell cycle regulatory protein Cks1: implications for domain swapping, anion binding and protein interactions. Structure. 2000;8:841–850. doi: 10.1016/s0969-2126(00)00175-1. [DOI] [PubMed] [Google Scholar]
- Bourne Y, Watson MH, Hickey MJ, Holmes W, Rocque W, Reed SI, Tainer JA. Crystal structure and mutational analysis of the human CDK2 kinase complex with cell cycle-regulatory protein CksHs1. Cell. 1996;84:863–874. doi: 10.1016/s0092-8674(00)81065-x. [DOI] [PubMed] [Google Scholar]
- Bray D, Lay S. Computer-based analysis of the binding steps in protein complex formation. Proc Natl Acad Sci U S A. 1997;94:13493–13498. doi: 10.1073/pnas.94.25.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace AM, Michaud NR, Therrien M, Mathes K, Copeland T, Rubin GM, Morrison DK. Identification of constitutive and ras-inducible phosphorylation sites of KSR: implications for 14-3-3 binding, mitogen-activated protein kinase binding, and KSR overexpression. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill MA, Ernst WH, Janknecht R, Nordheim A. Regulatory squelching. FEBS Lett. 1994;344:105–108. doi: 10.1016/0014-5793(94)00320-3. [DOI] [PubMed] [Google Scholar]
- Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Dunphy WG, Newport JW. Fission yeast p13 blocks mitotic activation and tyrosine dephosphorylation of the Xenopus cdc2 protein kinase. Cell. 1989;58:181–191. doi: 10.1016/0092-8674(89)90414-5. [DOI] [PubMed] [Google Scholar]
- Endicott JA, Noble ME, Garman EF, Brown N, Rasmussen B, Nurse P, Johnson LN. The crystal structure of p13suc1, a p34cdc2-interacting cell cycle control protein. EMBO J. 1995;14:1004–1014. doi: 10.1002/j.1460-2075.1995.tb07081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE, Jr, Ha SH. Ultrasensitivity part II: multisite phosphorylation, stoichiometric inhibitors, and positive feedback. Trends Biochem Sci. 2014;39:556–569. doi: 10.1016/j.tibs.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin MJ, Zhang X, Coonrod SA, Hager GL. Transient estrogen receptor binding and p300 redistribution support a squelching mechanism for estradiol-repressed genes. Mol Endocrinol. 2014;28:1522–1533. doi: 10.1210/me.2014-1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena J. Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc Natl Acad Sci U S A. 2005;102:14617–14622. doi: 10.1073/pnas.0507322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayles J, Aves S, Nurse P. suc1 is an essential gene involved in both the cell cycle and growth in fission yeast. EMBO J. 1986;5:3373–3379. doi: 10.1002/j.1460-2075.1986.tb04653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger M, Kendall FE. A quantitative study of the precipitin reaction between type III pneumococcus polysaccharide and purified homologous antibody. J Exp Med. 1929;50:809–823. doi: 10.1084/jem.50.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CYF, Ferrell JE., Jr Ultrasensitivity in the mitogen-activated protein kinase cascade. PNAS. 1996;93:10078–10083. doi: 10.1073/pnas.93.19.10078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IDT-Biophysics. DNA Thermodynamics & Hybridization. 2015 http://biophysics.idtdna.com, accessed December 1, 2015.
- Jullien N, Herman JP. LUEGO: a cost and time saving gel shift procedure. Biotechniques. 2011;51:267–269. doi: 10.2144/000113751. [DOI] [PubMed] [Google Scholar]
- Kim SY, Ferrell JE., Jr Substrate competition as a source of ultrasensitivity in the inactivation of Wee1. Cell. 2007;128:1133–1145. doi: 10.1016/j.cell.2007.01.039. [DOI] [PubMed] [Google Scholar]
- Kim SY, Song EJ, Lee KJ, Ferrell JE., Jr Multisite M-phase phosphorylation of Xenopus Wee1A. Mol Cell Biol. 2005;25:10580–10590. doi: 10.1128/MCB.25.23.10580-10590.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M, Ord M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ER, Rubin SM, Loog M. Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol. 2013;20:1415–1424. doi: 10.1038/nsmb.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG. Control of the Cdc2/cyclin B complex in Xenopus egg extracts arrested at a G2/M checkpoint with DNA synthesis inhibitors. Mol Biol Cell. 1995;6:199–213. doi: 10.1091/mbc.6.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell. 2001;12:551–563. doi: 10.1091/mbc.12.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci U S A. 2000;97:5818–5823. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markevich NI, Hoek JB, Kholodenko BN. Signaling switches and bistability arising from multisite phosphorylation in protein kinase cascades. J Cell Biol. 2004;164:353–359. doi: 10.1083/jcb.200308060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinsson-Ahlzen HS, Liberal V, Grunenfelder B, Chaves SR, Spruck CH, Reed SI. Cyclin-dependent kinase-associated proteins Cks1 and Cks2 are essential during early embryogenesis and for cell cycle progression in somatic cells. Mol Cell Biol. 2008;28:5698–5709. doi: 10.1128/MCB.01833-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath DA, Balog ER, Koivomagi M, Lucena R, Mai MV, Hirschi A, Kellogg DR, Loog M, Rubin SM. Cks confers specificity to phosphorylation-dependent CDK signaling pathways. Nat Struct Mol Biol. 2013;20:1407–1414. doi: 10.1038/nsmb.2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Hayles J, Nurse P. Regulation of p34cdc2 protein kinase during mitosis. Cell. 1989;58:361–372. doi: 10.1016/0092-8674(89)90850-7. [DOI] [PubMed] [Google Scholar]
- Mueller PR, Coleman TR, Dunphy WG. Cell cycle regulation of a Xenopus Wee1-like kinase. Mol Biol Cell. 1995;6:119–134. doi: 10.1091/mbc.6.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Meth Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- Natesan S, Rivera VM, Molinari E, Gilman M. Transcriptional squelching re-examined. Nature. 1997;390:349–350. doi: 10.1038/37019. [DOI] [PubMed] [Google Scholar]
- Owczarzy R, You Y, Groth CL, Tataurov AV. Stability and mismatch discrimination of locked nucleic acid-DNA duplexes. Biochemistry. 2011;50:9352–9367. doi: 10.1021/bi200904e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parge HE, Arvai AS, Murtari DJ, Reed SI, Tainer JA. Human CksHs2 atomic structure: a role for its hexameric assembly in cell cycle control. Science. 1993;262:387–395. doi: 10.1126/science.8211159. [DOI] [PubMed] [Google Scholar]
- Patra D, Dunphy WG. Xe-p9, a Xenopus Suc1/Cks homolog, has multiple essential roles in cell cycle control. Genes Dev. 1996;10:1503–1515. doi: 10.1101/gad.10.12.1503. [DOI] [PubMed] [Google Scholar]
- Patra D, Dunphy WG. Xe-p9, a Xenopus Suc1/Cks protein, is essential for the Cdc2-dependent phosphorylation of the anaphase- promoting complex at mitosis. Genes Dev. 1998;12:2549–2559. doi: 10.1101/gad.12.16.2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra D, Wang SX, Kumagai A, Dunphy WG. The xenopus Suc1/Cks protein promotes the phosphorylation of G(2)/M regulators. J Biol Chem. 1999;274:36839–36842. doi: 10.1074/jbc.274.52.36839. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pines J. Cell cycle: reaching for a role for the Cks proteins. Curr Biol. 1996;6:1399–1402. doi: 10.1016/s0960-9822(96)00741-5. [DOI] [PubMed] [Google Scholar]
- Pomerening JR, Kim SY, Ferrell JE., Jr Systems-level dissection of the cell-cycle oscillator: bypassing positive feedback produces damped oscillations. Cell. 2005;122:565–578. doi: 10.1016/j.cell.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Prywes R, Zhu H. In vitro squelching of activated transcription by serum response factor: evidence for a common coactivator used by multiple transcriptional activators. Nucleic Acids Res. 1992;20:513–520. doi: 10.1093/nar/20.3.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy F, Laberge G, Douziech M, Ferland-McCollough D, Therrien M. KSR is a scaffold required for activation of the ERK/MAPK module. Genes Dev. 2002;16:427–438. doi: 10.1101/gad.962902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie W, Harada Y, Matsuda M, Aoki K. Quantitative in vivo fluorescence cross-correlation analyses highlight the importance of competitive effects in the regulation of protein-protein interactions. Mol Cell Biol. 2014;34:3272–3290. doi: 10.1128/MCB.00087-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunnell NB, Poon AC, Kim SY, Ferrell JE., Jr Ultrasensitivity in the regulation of Cdc25C by Cdk1. Mol Cell. 2011;41:263–274. doi: 10.1016/j.molcel.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter SA, Guadagno SN, Ferrell JE., Jr Activation of Wee1 by p42 MAPK in vitro and in cycling Xenopus egg extracts. Mol Biol Cell. 2000;11:887–896. doi: 10.1091/mbc.11.3.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Nie Q, Enciso G. Nonessential sites improve phosphorylation switch. Biophys J. 2010;99:L41–43. doi: 10.1016/j.bpj.2010.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhr M, Freeman RM, Jr, Presler M, Horb ME, Peshkin L, Gygi SP, Kirschner MW. Deep proteomics of the Xenopus laevis egg using an mRNA-derived reference database. Curr Biol. 2014 doi: 10.1016/j.cub.2014.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu VP, Reed SI. Cks1 is dispensable for survival in Saccharomyces cerevisiae. Cell Cycle. 2004;3:1402–1404. doi: 10.4161/cc.3.11.1208. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.