

Abstract
Survival motor neuron (SMN) functions in the assembly of spliceosomal small nuclear ribonucleoproteins (snRNPs) that catalyze pre-mRNA splicing. Here, we used disruptions in Smn and two additional snRNP biogenesis genes, Phax and Ars2, to classify RNA processing differences as snRNP-dependent or gene-specific in Drosophila. Phax and Smn mutants exhibited comparable reductions in snRNAs, and comparison of their transcriptomes uncovered shared sets of RNA processing changes. In contrast, Ars2 mutants displayed only small decreases in snRNA levels, and RNA processing changes in these mutants were generally distinct from those identified in Phax and Smn animals. Instead, RNA processing changes in Ars2 mutants support the known interaction of Ars2 protein with the cap-binding complex, as splicing changes showed a clear bias toward the first intron. Bypassing disruptions in snRNP biogenesis, direct knockdown of spliceosomal proteins caused similar changes in the splicing of snRNP-dependent events. However, these snRNP-dependent events were largely unaltered in three Smn mutants expressing missense mutations that were originally identified in human spinal muscular atrophy (SMA) patients. Hence, findings here clarify the contributions of Phax, Smn, and Ars2 to snRNP biogenesis in Drosophila, and loss-of-function mutants for these proteins reveal differences that help disentangle cause and effect in SMA model flies.
Keywords: alternative splicing, alternative polyadenylation, αCOP, Abelson interacting protein, dUTPase, GARS, survival motor neuron, SMN, spinal muscular atrophy, SMA, phosphorylated adaptor for RNA export, PHAX, Ars2, Prp6, Prp8, RNA-sequencing, snRNA, snRNP biogenesis
INTRODUCTION
Survival motor neuron (SMN) protein functions in the assembly of Sm-class small nuclear ribonucleoproteins (snRNPs), core components of the spliceosome (Fischer et al. 2011; Matera and Wang 2014). Loss of SMN causes the neuromuscular disease spinal muscular atrophy (SMA) (Lefebvre et al. 1995), but the extent to which SMA is caused by disruptions in snRNP levels is not known. Loss of SMN causes transcriptome-wide changes in gene expression and RNA processing that are often assumed to stem from downstream disruptions in snRNP supply (Zhang et al. 2008, 2013; Garcia et al. 2013), but direct transcriptomic comparisons of SMA models with other targeted disruptions in snRNP biogenesis genes have, to date, not been performed.
The nuclear export of Sm-class small nuclear RNAs (snRNAs) is a key step in snRNP biogenesis. Sm-class snRNPs catalyze pre-mRNA splicing in the nucleus, but some steps of snRNP biogenesis occur in the cytoplasm, including assembly by SMN and associated SMN complex proteins (Matera and Wang 2014). The phosphorylated adaptor for RNA export protein (PHAX) mediates the nuclear export of newly transcribed snRNAs by linking them to the Exportin 1 (XPO1/CRM1) nuclear export machinery (Ohno et al. 2000). PHAX interacts with the 5′-terminal cap binding complex (CBC) and adjacent 5′-proximal RNA of RNA polymerase II transcripts, but it is displaced from longer mRNAs by the heterogeneous nuclear ribonucleoprotein C (C1/C2) tetramer (Ohno et al. 2000; McCloskey et al. 2012; Ohno 2012). Hence, PHAX functions to selectively export smaller snRNAs with lengths approximately 100 to 250 nucleotides (nt), making it an attractive target for genetically manipulating snRNP levels.
The nuclear export of Sm-snRNAs follows transcription termination. Prior to nuclear export, snRNAs are cotranscriptionally cleaved from downstream sequences. Unlike mRNAs, snRNAs are cleaved but not polyadenylated (Matera and Wang 2014). The Integrator is a multiprotein complex that cleaves snRNAs to form 3′-ends that lack a poly(A) tail (Baillat et al. 2005; Baillat and Wagner 2015). Proper 3′-end maturation of snRNAs is cotranscriptionally linked to upstream promoter sequences and 5′-ends (de Vegvar et al. 1986; Hernandez and Weiner 1986; Ezzeddine et al. 2011). Recently, the CBC-binding protein arsenite resistance protein 2 (ARS2/SRRT) was implicated in this crosstalk between snRNA 3′- and 5′-ends (Hallais et al. 2013). Knockdown of ARS2 or CBC proteins in mammalian cells increased readthrough of endogenous snRNAs and snRNA 3′-end reporters (Andersen et al. 2013; Hallais et al. 2013). In addition to snRNA 3′-end maturation, ARS2 contributes to the 3′-end maturation of replication-dependent histone mRNAs, at least in mammals, and it also has a conserved role in the regulation of microRNA biogenesis (Grigg et al. 2005; Lobbes et al. 2006; Yang et al. 2006; Gruber et al. 2009, 2012; Sabin et al. 2009). In this context, ARS2 is likely a less selective target than PHAX for disrupting the supply of snRNPs.
Observations in diverse eukaryotes suggest that perturbations in snRNP supply produce transcriptome-wide effects, particularly in alternative splicing and polyadenylation. Mutations in yeast genes encoding core spliceosomal factors disrupted the splicing of a subset of pre-mRNAs, including splicing events in highly expressed pre-mRNAs for ribosomal protein genes (RPGs) (Clark et al. 2002; Pleiss et al. 2007). Inversely, repressing the transcription of these highly expressed RPGs changed the alternative splicing patterns of other yeast pre-mRNAs, suggesting that spliceosomal accessibility controls pre-mRNA splicing patterns (Munding et al. 2013). In Drosophila, core splicing factors were uncovered by an RNAi screen for regulators of a predetermined set of alternative-splicing patterns (Park et al. 2004). In cultured human cells, knockdown of Sm-class snRNP-associated proteins led to numerous changes in alternative splicing (Saltzman et al. 2011). Furthermore, blocking U1 snRNP specifically with an antisense morpholino led to premature cleavage and polyadenylation of mRNAs in cells from diverse lineages (Kaida et al. 2010; Berg et al. 2012). These studies highlight that multiple strategies can be used to effectively disrupt snRNP supply and function, which creates an opportunity to use comparative transcriptomics to comprehensively categorize the snRNP dependence and/or spliceosomal accessibility requirements of disparate RNA processing events.
Here, we carried out transcriptome profiling of Drosophila Smn mutants, along with two additional snRNP biogenesis mutants in order to identify snRNP-dependent versus gene-specific changes in gene expression, premature cleavage and polyadenylation, and alternative splicing. Specifically, transposon insertion mutations in the fly orthologs of Phax and Ars2 were used for comparison with Smn null mutants (Ohno et al. 2000; Kitao et al. 2008; Laubinger et al. 2008; Gruber et al. 2009; Andersen et al. 2013; Hallais et al. 2013; Pabis et al. 2013). The Phax mutant was particularly useful in identifying an overlapping set of snRNP-dependent RNA changes in SMA model flies, and the analysis of the Ars2 mutants revealed a distinct set of RNA processing events that likely depend on the integrity of the CBC and 5′-cap. Additionally, alternative-splicing differences shared between the Phax and Smn mutants could be rescued at both the RNA and protein levels by expression of Phax or Smn transgenes, respectively. In combination with a modest restoration of steady-state snRNA levels, observations from these rescue lines corroborate the connection between snRNP levels and specific pre-mRNA splicing events. However, the link between these changes and SMA phenotypes remains unclear.
RESULTS
Phax and Smn mutants display similar steady-state snRNA decreases
RNA-sequencing (RNA-seq) of poly(A)-selected RNA was used to identify mRNA changes in snRNP biogenesis mutants, and Northern analysis coupled with RNA-seq of total ribosomal RNA-subtracted (rRNA−) RNA was used to quantify steady-state snRNA levels. Browser shots of Phax, Smn, and Ars2 gene loci confirmed the gene disruptions in the respective mutant lines (Fig. 1A; Garcia et al. 2013). In our previous RNA-seq analysis of Smn null mutants, publicly available modENCODE data were used to determine the developmental stage at which the mutants arrested (Garcia et al. 2013). By the same method, the Phax and Ars2 mutants exhibited a similar timing of developmental delay as did the Smn animals (Supplemental Fig. S1A,B). Using developmentally appropriate wild-type controls (Supplemental Fig. S2A), Northern blots demonstrated comparable, near twofold decreases in steady-state U1, U2, and U4 snRNAs in the Phax and Smn mutants (Fig. 1B,C). When assayed by Northern blotting, U5 snRNA levels were essentially unaffected in the Phax mutants (Fig. 1B,C), as were levels of nearly all of the snRNAs in the Ars2 mutants (Fig. 1B,C). When RNA-seq data were used to quantify snRNA levels, each of the three mutants appeared to exhibit snRNA deficits, but the extent varied by the method of normalization (Supplemental Fig. S2B,C). Only small (<15%) changes were observed in specific snRNA paralogs relative to the total (Supplemental Fig. S3), quantified by using reads covering isoform-specific nucleotides (Lu and Matera 2014). Overall, the Phax and Smn mutants displayed the most consistent decreases in steady-state snRNA levels.
FIGURE 1.

Analysis of three different snRNP biogenesis mutants. (A) Browser shots of larval RNA-seq reads at the three disrupted gene loci. Reads from Oregon-R (WT) versus PhaxSH/SH, SmnX7/D, or Ars2C/E snRNP biogenesis mutants are shown. Mapped read tracks were normalized to the median of the middle two quartiles of the mapped WT sequence read counts. (B) Northern blot of RNA from snRNP biogenesis mutants and WT larvae. RNA was extracted from WT larvae at 60 ± 2 h (60 h) post egg-laying, and mutant RNA was extracted at 74 ± 2 h to account for their delayed development (see text). (C) Quantification of Northern blot in B. Sm-class snRNAs were normalized to the Lsm-class U6 snRNA, and U6-normalized WT snRNA levels were set at 100. Asterisks are P-values from a Student's t-test: (*) P-value ≤0.05, (**) P-value ≤0.01, (***) P-value ≤0.001.
snRNP biogenesis mutants share a small number of gene expression changes
To identify shared and distinct changes in gene expression between snRNP biogenesis mutants, we used the TopHat/Cufflinks pipeline to quantify mRNA differences from our poly(A)-selected RNA-seq data (Trapnell et al. 2012). Raw reads from age-matched wild-type (WT) controls and modENCODE second (L2) and early third instar (L3 12h) developmental controls were integrated with sequencing data from the mutants into a single analytical pipeline. For comparison, we focused on genes whose expression changed uniformly and significantly (false discovery rate-adjusted P-value <0.05) relative to each of the controls (L2, L3 12h, and our WT). A relatively small number of genes and undifferentiated loci with multiple genes (186 out of ∼15,000) met these stringent criteria for differential expression (i.e., 186 loci were significantly different from all of the controls; total number from Supplemental Table S1), and most of these differences were mutant specific (Fig. 2A,C; Supplemental Tables S1–S4). A number of stress-responsive transcripts were among those that were specifically elevated in Smn mutants, though some of these were also up slightly in the Ars2 mutants (Fig. 2B). Phax mutants shared only 28 gene expression changes with Smn mutants (Fig. 2C; Supplemental Table S2). This small list of genes was enriched for genes involved in oxidation–reduction, as measured by DAVID gene ontology term analysis (P-value = 0.003) (Huang et al. 2007, 2009a,b). A notable gene expression change shared by the Phax and Smn mutants was a fivefold increase in the Activity-regulated cytoskeleton associated protein 1 (Arc1) transcript (Supplemental Table S2). Arc1 functions in a well conserved hyperlocomotion response to starvation, and hence elevated levels in Phax and Smn mutants might be an indirect consequence of a disruption in nutrient acquisition in these animals (Mattaliano et al. 2007). In summary, gene expression analysis uncovered a small number of overlapping changes that exceeded normal developmental fluctuations. The relationship of these changes to SMA etiology and/or pathology remains to be determined.
FIGURE 2.
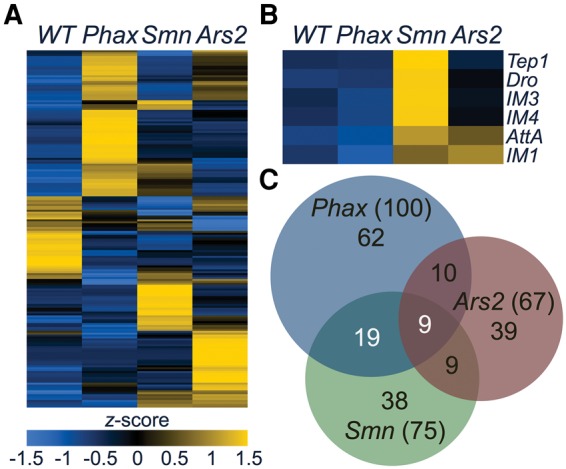
Gene expression differences in snRNP biogenesis mutants. (A) Heatmap comparison of Cuffdiff FPKM levels of differentially expressed transcripts. Heatmap colors were rescaled for each row and the rows were clustered based on pattern of gene expression between WT and mutants. (B) A heatmap of FPKMs from a set of stress-responsive transcripts in A. (C) Venn diagram of the overlap in gene expression differences in the snRNP biogenesis mutants. Phax = PhaxSH/SH, Smn = SmnX7/D, and Ars2 = Ars2C/E. Numbers in parentheses are totals.
snRNP biogenesis mutants exhibit a trend toward shorter mRNAs
In addition to its role in splicing, U1 snRNP has been reported to control mRNA length by preventing premature cleavage and polyadenylation (Kaida et al. 2010; Berg et al. 2012). To determine whether reduced U1 snRNA levels in our snRNP biogenesis mutants correlated with expression of shorter mRNAs, mapped RNA-seq reads were analyzed using the DaPars linear regression algorithm (Masamha et al. 2014; Xia et al. 2014). DaPars output is displayed as a percentage of distal poly(A) site usage index (PDUI) for each transcript, or as the difference in the PDUI value between wild-type and mutant (ΔPDUI). Pairwise comparisons of PDUI values for wild-type and mutants can be used to visualize changes in RNA length. Whereas the Phax and Smn mutants expressed transcripts that were both longer and shorter than their wild-type counterparts, there were greater numbers of shortened mRNAs (Fig. 3A–C; Supplemental Tables S5–S7). Focusing on differences that are greater than twofold, Phax and Smn mutants had a relatively small number of mRNA length changes in common with each other (34) (Supplemental Table S5). However, the changes were largely distinct from those observed in Ars2 animals, who had more than twice as many mRNA length changes as did the Phax and Smn mutants combined (Fig. 3D; Supplemental Table S8). The individual direction of length change, either longer or shorter, was also factored into the consideration of overlapping differences in Figure 3D. The overall trend of shorter transcripts in the mutants does not appear to be a consequence of developmental delay, as parallel DaPars analyses of modENCODE data sets of second to third instar larvae showed a trend toward shorter transcripts in the more mature animals (Supplemental Fig. S4). Among the few overlapping mRNA length changes between Phax and Smn, one of the largest and most apparent changes was a shortened mRNA for CG9662, a putative oligosaccharyl transferase complex subunit (ΔPDUI = −0.96, Supplemental Table S5). These results are generally consistent with the notion that reductions in U1 snRNP levels predispose mRNAs to premature cleavage and polyadenylation. Because the Ars2 mutants exhibited only a small decrease in steady state U1 levels (<10%; Fig. 1C), the mRNA length changes in these animals are more likely due to disruptions of the CBC, which are known to affect a subset of pre-mRNA termination events (Wong et al. 2007; Andersen et al. 2013; Hallais et al. 2013).
FIGURE 3.
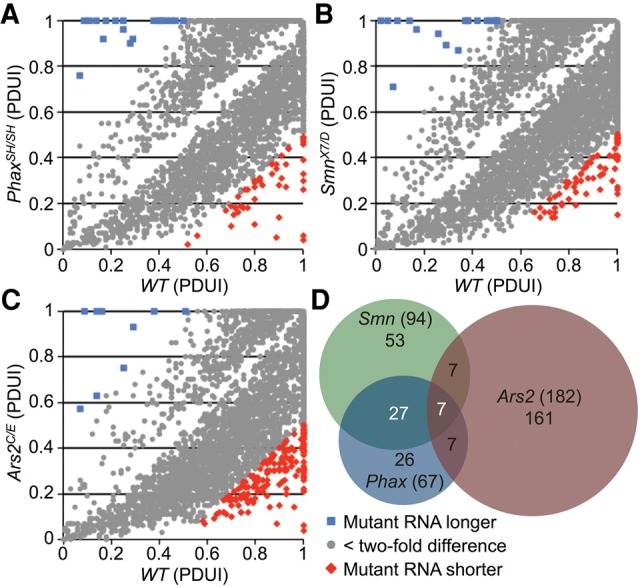
mRNA length changes in snRNP biogenesis mutants. Pairwise comparison of significant (false discovery rate adjusted P-value <0.05) differences in mRNA length between WT and PhaxSH/SH (A); WT and SmnX7/D (B); and WT and Ars2C/E (C). RNA length differences were extracted from mapped RNA-seq read files by the DaPars linear regression algorithm (see text). Percentage of distal poly(A) site usage index = PDUI. (D) Venn diagram of RNA length differences in snRNP biogenesis mutants labeled as in Figure 2.
Phax and Smn mutants share a set of alternative splicing pattern changes
Our previous RNA-seq analysis of Smn null animals revealed many small-amplitude alternative splicing differences (compared to wild-type controls), but it was unclear whether any of these changes might be due to decreases in snRNP levels (Garcia et al. 2013). To ascertain whether any of these splicing changes are snRNP-dependent, we used the mixture of isoforms (MISO) probabilistic framework to compare splicing differences in Smn mutants with those identified in Phax and Ars2 mutants (Katz et al. 2010). Simply put, the MISO algorithm estimates the expression of alternatively spliced isoforms in RNA-seq data, and reports it as a fraction of alternatively spliced events relative to the total (Katz et al. 2010). MISO denotes this fraction as the “percent spliced in” or “Psi” (Katz et al. 2010). Differences are reported here as an absolute change relative to the wild-type control, |deltaPsi|. Similar to our previous results with the Smn null animals, we found mostly small changes (less than twofold, |deltaPsi| <0.5) in alternative-splicing events in the Phax and Ars2 mutants (Fig. 4A–C; Supplemental Tables S9–S11). More than half of the splicing changes identified in the Smn null animals overlapped with changes in the Phax mutants, but neither the Phax nor Smn mutants had considerable overlap with those observed in the Ars2 mutants (Fig. 4D; Supplemental Table S11). Of the top 150 differences in the Ars2 background, close to 90% of these changes were located in the 5′-proximal intron (Supplemental Table S11). These changes likely reflect the association of Ars2 with the CBC and the role of this complex in promoter proximal splicing events (Laubinger et al. 2008) rather than Ars2-related changes in snRNPs. In contrast, the changes that were common to Phax and Smn mutants are more likely to be due to decreases in snRNP levels.
FIGURE 4.
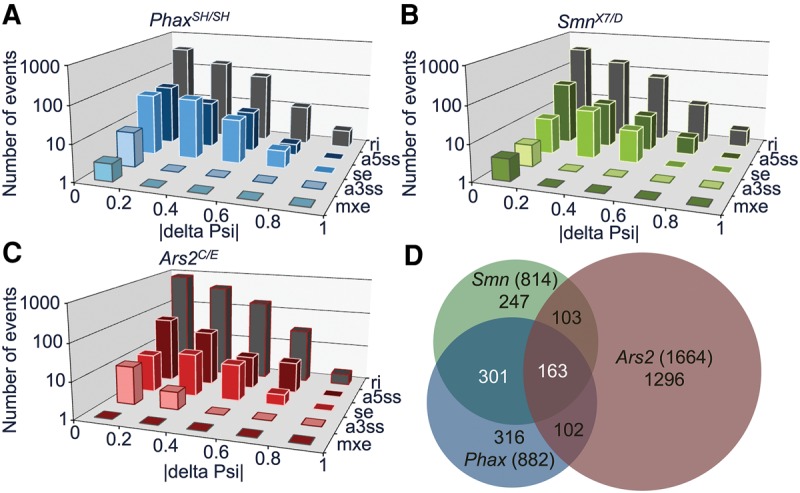
Alternative-splicing differences among snRNP biogenesis mutants. Distribution of significant alternative-splicing changes from small |delta Psi| near zero to large |delta Psi| near one: (A) PhaxSH/SH mutants relative to WT; (B) SmnX7/D mutants relative to WT; and (C) Ars2C/E mutants relative to WT. (D) Venn diagram of overlapping alternative-splicing changes in snRNP biogenesis mutants, labeled as in Figure 2.
Transgenic rescue of alternative splicing changes induced by loss of Phax or Smn
As described previously, Smn is essential for viability in the fly (Chan et al. 2003), and transgenic expression of Flag- or GFP-tagged SMN protein in an otherwise null mutant background fully rescues the larval lethality and other SMA-like phenotypes (Rajendra et al. 2007; Shpargel et al. 2009). Here, we found that Phax is also an essential gene, and that ectopic expression of Phax-GFP using the GAL4-UAS system rescued the lethality of PhaxSH/SH mutants (Supplemental Table S12). We next assayed (and confirmed by semi-quantitative RT-PCR) a number of the most apparent alternative splicing changes identified by RNA-seq (Supplemental Fig. S5). We then tested the ability of the Phax and Smn transgenes to rescue four of these splicing events by quantitative qRT-PCR (Fig. 5B,C). Ubiquitous expression of Phax using an Armadillo-GAL4 driver significantly rescued all of the splicing changes we tested (Fig. 5B). Transgenic expression of Smn, driven by its native promoter, also rescued these alternative-splicing events in Smn null animals (Fig. 5C). Together, the results confirm that the observed changes we identified by RNA-seq are indeed caused by loss of Phax or Smn expression.
FIGURE 5.
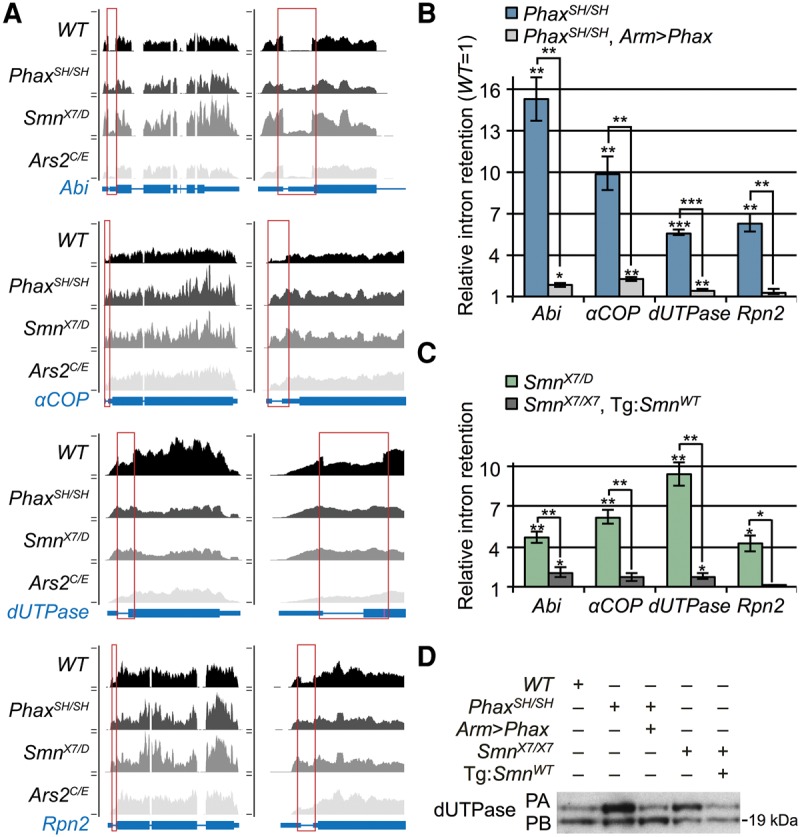
Transgenic rescue of alternative-splicing changes in Phax and Smn mutants. (A) UCSC browser shots of mapped read tracks for four of the top 15 identified alternative splicing changes. (B) qRT-PCR analysis of intron retention in PhaxSH/SH mutants versus PhaxSH/SH mutants expressing a UAS-Phax transgene from the armadillo promoter driven GAL4 (Arm > Phax). (C) qRT-PCR of intron retention in SmnX7/D mutants versus SmnX7/X7 mutants expressing a wild-type Smn transgene from its native promoter. For B and C, levels of WT intron retention were set at one. (D) Western blot for dUTPase. Alternate protein isoforms reflect alternative-splicing change in dUTPase mRNA. Wild-type Smn transgene in SmnX7/X7 background = Tg:SmnWT. Long dUTPase protein isoform = PA and short = PB.
One of the splicing changes we detected is expected to produce an alternative (larger) dUTPase protein isoform. Therefore, we assayed the impact of this alternative splicing change by Western blotting, using an antibody that targets both dUTPase isoforms. As shown in Figure 5D, the expected increase in the ratio of the long isoform relative to the short one was observed in the mutants, and this change was largely rescued in both transgenic lines. Thus, the transcriptomic changes we identified in SMA model animals can (and do) translate into detectable differences at the protein level.
An increase in steady-state snRNA levels correlates with rescue of alternative splicing
If the alternative splicing changes, observed in both Phax and Smn mutants, are truly caused by the decreased availability of snRNPs, then rescue of steady-state snRNA levels should mirror the rescue of these alternative splicing changes in the Phax and Smn transgenic lines. Northern blotting was used to determine whether the expression of Phax and Smn transgenes increased steady-state snRNA levels in the mutants (Supplemental Fig. S6A,B). Expression of the Phax transgene by the Armadillo driver significantly rescued U1 and U4 snRNA levels in the PhaxSH/SH mutant background (Supplemental Fig S6A,B). All of the snRNAs tested here were significantly rescued by expression of the wild-type Smn transgene in the Smn null background (Supplemental Fig. S6C,D). For the wild-type Smn rescue line, these observations differ from our previously published results (Praveen et al. 2012), wherein we detected a modest increase in snRNA levels in the rescue line. This difference likely reflects minor improvements to our Northern blotting protocol, specifically: using less total RNA per gel lane and more labeled probe in the hybridization reaction to ensure probe excess. The increases in steady-state snRNA levels that we observed in the Phax and Smn rescue lines correlated well with the phenotypic rescue at both the organismal (viability) and molecular (alternative splicing) levels.
Splicing changes shared between Phax and Smn mutants are also caused by knockdown of splicing factors Prp6 and Prp8
Although the correlation of snRNA levels with particular splicing events provides evidence that these events are acutely sensitive to disruption of the core spliceosomal machinery, it does not rule out other possibilities. For example, the covariance could be due to a direct interaction of splicing factors with Phax and SMN, or a result of indirect factors, such as altered developmental progression. In addition, snRNPs are well known to be stable RNPs, and we do not know at what point reductions in ongoing snRNP supply begin to impair spliceosomal function. To test whether the splicing changes in the Phax and Smn mutants are indeed linked to a reduction in snRNP supply and a corresponding disruption in spliceosome function, we depleted the tri-snRNP and U5-associated proteins Prp6 and Prp8 using RNA interference. As shown in Figure 6A–C, SMN depletion caused a slight (less than twofold) perturbation in the splicing events tested, but the Prp6 and Prp8 knockdowns disrupted the splicing of these transcripts to a much greater extent. Knockdown of Prp8 disrupted the splicing of the events identified above (Abi, αCOP, dUTPase, and Rpn2 intron 1 [int. 1]), but it did not significantly disrupt the splicing of a downstream intron in Rpn2, serving here as a control for broader spliceosomal dysfunction. The Prp6 and Prp8 knockdowns also altered dUTPase protein expression patterns to a greater extent than did knockdown of Smn (Fig. 6B). Steady-state snRNA levels were not significantly affected by any of these knockdowns, with the possible exception of U5 snRNA, which was slightly reduced (∼40%) in the Prp8 depletion (Fig. 6D,E). The lack of snRNA decreases in the SMN depleted samples is consistent with the well-known perdurance of snRNPs in cultured cells, and it likely mirrors the observed low-amplitude disruptions to splicing. Prp6 and Prp8 are core spliceosomal proteins, and their depletion is expected to severely compromise the spliceosome (Grainger and Beggs 2005; Will and Luhrmann 2011). Thus, the splicing changes seen in these knockdowns are perhaps not unexpected, but they nevertheless support the conclusion that the overlapping changes identified in the Phax and Smn null mutants are snRNP dependent.
FIGURE 6.
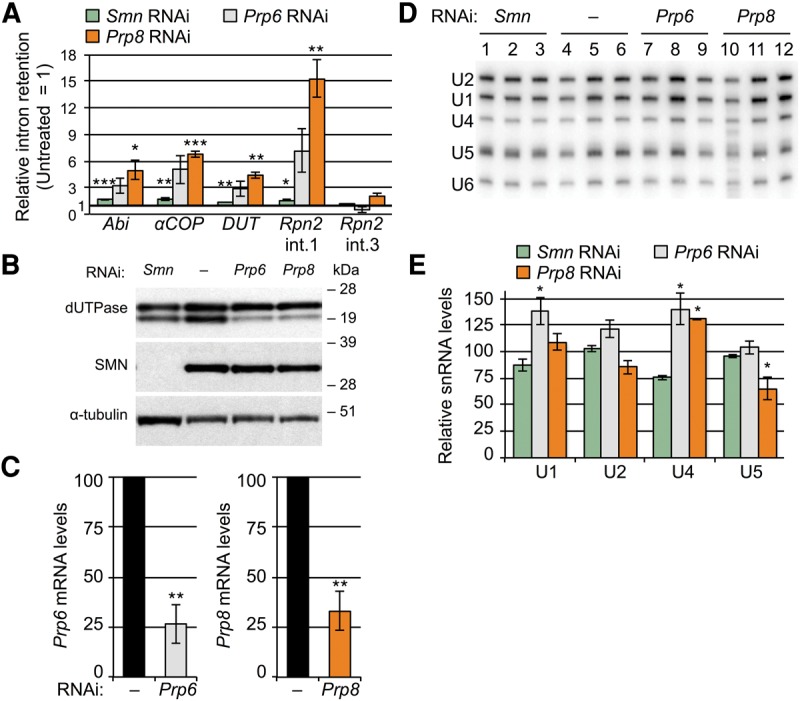
snRNP-specific protein knockdown in S2 cells. (A) qRT-PCR of untreated cells versus those treated with dsRNA for Smn, Prp6, or Prp8 mRNAs. (B) Western blot of dUTPase levels in RNAi-treated cells. Anti-SMN and anti-α-tubulin verify SMN knockdown and load, respectively. (C) qRT-PCR verification of Prp6 and Prp8 mRNA knockdowns. Levels in untreated cells were set at 100. (D) Northern blot of snRNA levels from S2 cell knockdowns, quantified in E. U6-normalized snRNA levels of WT were set at 100.
Hypomorphic Smn missense mutants rescue snRNP levels and do not display defects in alternative splicing
As mentioned, the snRNP biogenesis mutants described above are severe loss-of-function alleles. SMA is, however, a hypomorphic condition, as complete loss of SMN activity is lethal in all organisms tested. To better model the disease in the fly, we previously engineered an allelic series of transgenic animals that express Smn missense mutations known to cause SMA in humans (Praveen et al. 2012, 2014). In addition to the wild-type rescue transgene (SmnWT), we analyzed three hypomorphic lines that display intermediate (SMA type II) phenotypes: SmnT205I, SmnY107C, and SmnV72G (Praveen et al. 2014). These lines cover a range of phenotypic outcomes: SmnT205I is semi-viable (∼30% eclose as adults), SmnY107C is pharate-lethal (very few animals eclose), and SmnV72G mutants all die as pupae (Praveen et al. 2014). Importantly, in this system, the wild-type and mutant transgenes are integrated at the identical chromosomal locus and are expressed from the native promoter in an otherwise Smn null background (Praveen et al. 2012, 2014). Using qRT-PCR, we analyzed the alternative splicing patterns of the four transcripts described above. As shown in Figure 7A, none of the Smn missense lines exhibited a greater than twofold change (in either direction) relative to SmnWT. Consistent with this finding, the missense mutants also displayed little-to-no difference in steady-state snRNA levels, compared to the wild-type rescue line (Fig. 7B,C). To ensure that differences in developmental timing did not confound the analysis, animals were harvested after they began to display the wandering behavior that is characteristic of the late third instar stage of Drosophila development. We also measured snRNA levels of animals at slightly earlier and later developmental time points, and we found little difference between the mutant and the control lines (Supplemental Fig. S7). Steady-state snRNA differences in earlier animals may reflect subtle differences in developmental progression from wild-type controls, but these decreased levels were largely absent from the later pupal stage, which only exhibited significantly lower U4 levels in the single SmnY107C line.
FIGURE 7.
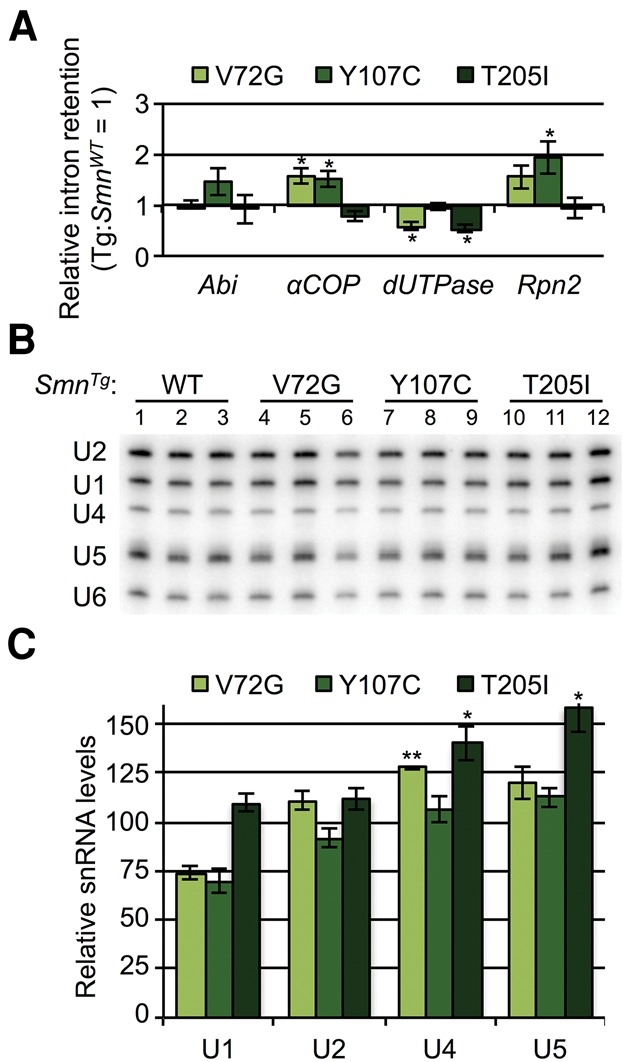
Analysis of steady-state snRNA levels and alternative splicing of target genes in SMN missense mutants. (A) qRT-PCR analysis of intron retention of flies expressing SMA patient-derived missense mutations. Transgenic animals of the following generalized genotype were used: SmnX7/X7, Flag-SmnTg/−, where Tg represents a WT, V72G, Y107C, or T205I transgene. Intron retention in the SmnWT transgenic rescue line was set at one. (B) Northern blots of snRNA levels in the missense mutant lines. (C) Quantification of snRNA levels from panel B. RNA levels in the SmnWT (WT) transgenic line were set at 100.
Smn missense mutants display limited overlap in splicing pattern changes
The large-amplitude changes identified in the severe loss-of-function mutants were not evident in the Smn hypomorphs; however, these lines could exhibit other splicing pattern or gene expression changes that might account for differences in their phenotypic outcomes. To identify RNA changes in the hypomorphic lines, we sequenced total (rRNA-subtracted) RNA from whole animals isolated ∼12 h after puparium formation, a developmental stage that Smn null animals never reach. The hypomorphs displayed numerous changes in their splicing patterns relative to the wild-type rescue line (1662 combined; Supplemental Tables S13–16). However, the missense mutants displayed few large amplitude (greater than twofold) changes (107/1662), and hardly any of these events were shared between them (42/1662) (Fig. 8A; Supplemental Table S13). Of the forty-two overlapping splicing changes, only six were previously identified as being snRNP-dependent (four) or Smn gene-specific (two) by comparison to data from the severe Phax and Smn loss-of-function lines (Fig. 8B). Browser shots of representative examples are shown in Figure 7C. We used qRT-PCR to validate one of the two Smn gene-specific changes. The scarface transcript (Supplemental Fig. S8; Supplemental Table S13) exhibited an increase in intron retention between the third and fourth exons in the SmnV72G transgenic line (deltaPsi = −0.61 and a >100-fold change by qRT-PCR). In the fly, Scarface functions as a negative regulator of JNK (c-Jun N-terminal kinase) stress signaling (Rousset et al. 2010). As measured by RNA-seq, the SmnY107C (deltaPsi = −0.23) and SmnT205I (deltaPsi = −0.07) mutants exhibited smaller changes; these events were below the limit of detection by qRT-PCR (Supplemental Fig. S8A; Supplemental Table S13). Consistent with the notion that this is not a snRNP-dependent change, an increase in scarface intron retention was not observed following knockdown of Prp8 (Supplemental Fig. S8B). Thus, RNA-seq profiling of hypomorphic, SMA-causing missense mutations is in agreement with the qRT-PCR data described above, and together these findings demonstrate that the splicing changes identified in the Smn null animals are not conserved among the intermediate SMA models.
FIGURE 8.
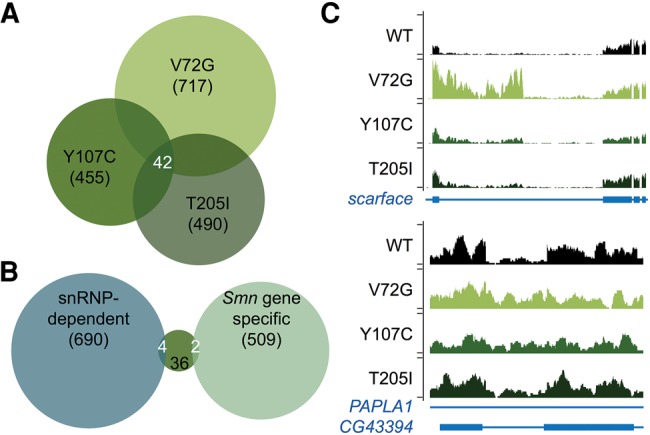
Alternative splicing changes in Smn missense mutants. (A) Venn diagram of overlapping alternative-splicing changes in Smn missense mutants. (B) Venn diagram of the overlap in shared splicing differences in the Smn missense mutants with either snRNP-dependent changes or Smn gene-specific changes categorized by comparison of the Phax and Smn total rRNA(−) RNA-seq. (C) Browser shot examples of an Smn gene-specific change in alternative-splicing in scarface (top) or a snRNP-dependent change in CG43394 within a PAPLA1 intron on the opposite strand (bottom). Genotypes are labeled by SmnTg, which was expressed in the SmnX7 null background.
Differential expression analysis reveals an activation of stress signaling in Smn missense mutant animals
In addition to changes in splicing patterns, our genome-wide analysis of RNA from the severe loss-of-function mutants revealed potential snRNP-dependent and Smn gene-specific changes in steady-state mRNA levels. Differential expression analysis of the Smn missense mutant lines revealed numerous changes in mRNA abundance in the most severe line, SmnV72G (599), but far fewer in the other less severe lines, SmnY107C (88) and SmnT205I (59) (Fig. 9A,B; Supplemental Table S17–S21). Despite the low number of changes in the least severe SmnT205I line, more than half of these overlapped with the other hypomorphic mutants (Fig. 9B). Surprisingly, many of the stress-responsive transcripts identified in the severe Smn null mutants were also among the mRNAs that were up-regulated in all three Smn transgenic lines, relative to the wild-type rescue line (Fig. 9C; Supplemental Table S18). As corresponding increases in mRNA levels were not found in the Phax mutants (Fig. 2B), these events were classified as Smn gene-specific. Thus, unlike the snRNP-dependent splicing changes (which are not conserved in the Smn hypomorphs), the intermediate models of SMA exhibited clear RNA signatures of stress that correlate with disease severity (Fig. 9C) and are independent of decreases in snRNP supply (Fig. 2B).
FIGURE 9.
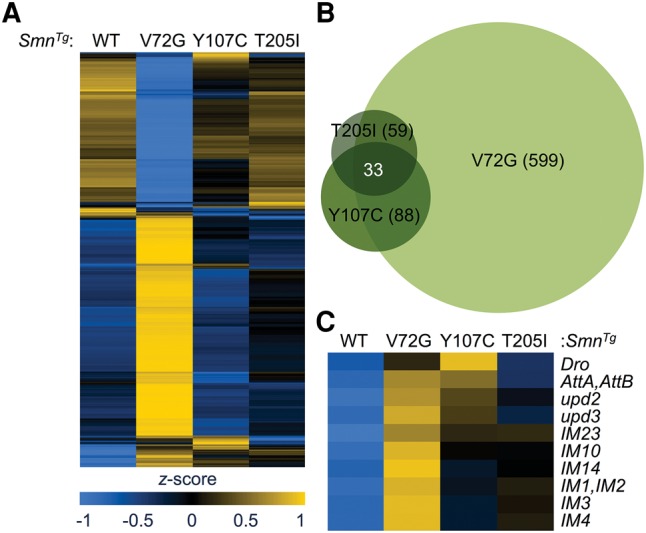
Differential gene expression in Smn hypomorphic animals. (A) Heatmap comparison of Cuffdiff FPKM levels of differentially expressed transcripts. Heatmap colors were rescaled for each row and the rows were clustered based on pattern of gene expression between SmnWT rescue line and the Smn missense mutants. (B) Venn diagram of overlapping differences in mRNA levels. (C) A heatmap of FPKMs from a set of stress-responsive transcripts. Genotypes are labeled by SmnTg, which was expressed in the SmnX7 null background.
DISCUSSION
Connecting SMA pathology to SMN's function in snRNP assembly and downstream expression of specific mRNA isoforms remains a necessary benchmark for defining how splicing changes might contribute to disease etiology. Therefore, a first step toward uncovering the molecular mechanisms that elicit SMA outcomes would be to separate snRNP-dependent phenotypes from those that are SMN specific. Transcriptomic profiling of Smn and Phax mutants enabled us to identify a robust set of snRNP-dependent alternative splicing changes in SMA model flies. We show that reductions in steady-state snRNA levels correlate with the appearance of these alternative-splicing events, and that rescue of snRNA levels in transgenic animals correlates with the restoration of normal splicing. Furthermore, depletion of core splicing factors (Prp8 or Prp6) essentially bypasses the snRNP depletion step, causing many of the same alternative-splicing changes. Together, these observations support a close connection between snRNP levels and proper gene expression in vivo. Whether or not disruptions to this ubiquitously required process contribute to SMA phenotypes is less clear.
Ars2, 5′ exons, and U1 telescripting
Changes in splicing and mRNA length in Phax and Smn animals were largely distinct from the more abundant changes in the Ars2 mutants (Figs. 3D, 4D). Ars2 is a component of the 5′ cap binding complex (CBC), and this association may underlie the transcriptomic changes that we identified in this mutant (Gruber et al. 2009; Gonatopoulos Pournatzis and Cowling 2014). The Ars2 splicing changes were mostly in 5′-proximal introns, and this finding is consistent with CBC mutants in yeast, Xenopus, and plants (Gonatopoulos Pournatzis and Cowling 2014). The association of Ars2 with the CBC likely also underlies the numerous mRNA shortening events we observed. These findings are consistent with previous studies in yeast and human cells that suggest the CBC protects a subset of weak poly(A) signals from premature cleavage and termination (Wong et al. 2007; Andersen et al. 2013; Hallais et al. 2013). Importantly, both types of Ars2 dependent mRNA processing events were largely distinct from the snRNP-dependent changes observed in the Phax and Smn mutants.
The distinct mRNA processing changes in Ars2 mutants may also reflect species-specific differences in the role of the CBC and Ars2. Unlike ARS2 knockdown experiments in mammalian cells (Gruber et al. 2012; Hallais et al. 2013), we did not observe defects in snRNA or histone mRNA processing. This finding is consistent with the observation that Ars2 protein is dispensable for proper histone mRNA maturation in Drosophila cells (Sabath et al. 2013), and it suggests that aspects of Ars2 function are not conserved between vertebrates and invertebrates.
Our identification of a large number of mRNA shortening events that are shared between Phax and Smn mutants supports a recently discovered role for the U1 snRNP in the protection of pre-mRNAs from premature cleavage and polyadenylation (PCPA). PCPA was previously observed by subtractive hybridization PCR, coupled with high-throughput sequencing of RNA from cells treated with antisense oligomers targeting the 5′-end of U1 snRNA (Kaida et al. 2010; Berg et al. 2012). This protective role for U1 snRNPs was termed “telescripting” (Kaida et al. 2010; Berg et al. 2012) and our study is the first to validate this process outside of cell culture models. Although we observed an overall trend toward shorter transcripts in our data sets (Fig. 3), we found only a few large-magnitude (greater than twofold) changes (Supplemental Table S2). This is perhaps unsurprising, given that treatment of cultured cells with relatively high concentrations of anti-U1 oligomers provides an acute and nearly complete inhibition of U1 snRNA function, whereas the mutations in Phax and Smn do not perturb U1 to the same extent in the mutant animals.
RNA signatures of disease
Alternative splicing depends on the regulated accessibility of pre-mRNAs to the spliceosome. Studies in a variety of eukaryotes have shown that changing this accessibility, either directly or indirectly, alters the splicing patterns of specific pre-mRNAs (Clark et al. 2002; Park et al. 2004; Pleiss et al. 2007; Saltzman et al. 2011; Munding et al. 2013). The extent to which snRNP levels contribute to normal splicing regulation and/or disease pathology is still being worked out; however, changes in the pattern of alternative-splicing have been reported in various SMA model systems (Zhang et al. 2008, 2013; Bäumer et al. 2009; Lotti et al. 2012). The splicing changes reported in these SMA models are typically assumed to stem directly from loss of SMN and snRNPs, but such changes are often difficult to distinguish from broad, indirect effects of disease pathology or developmental delay (Bäumer et al. 2009; Garcia et al. 2013). By identifying overlapping splicing changes in Phax and Smn mutants, we are now able to distinguish between snRNP-dependent changes and gene-specific effects.
Many of the alternative splicing changes shared by Phax and Smn occur in genes of known importance to neuromuscular biology. For example, the mammalian ortholog of the αCOP protein has a demonstrated interaction with SMN, and it colocalizes with SMN in neuronal projections (Peter et al. 2011; Custer et al. 2013). This interaction was recently shown to be required for neurite outgrowth in mammalian cell and zebrafish models of SMA (Li et al. 2015). Orthologs of the Abi protein appear to have important roles in neurogenesis via the regulation of actin dynamics (Proepper et al. 2007; Lin et al. 2009; Liebau et al. 2011). Dominant mutations in GARS cause the distal neuropathy Charcot–Marie–Tooth disease type 2D, and recent observations suggest that GARS may colocalize with SMN in human cells (Motley et al. 2010; Drew et al. 2011; Grice et al. 2015). Mouse models of SMA exhibit disruptions to ubiquitin homeostasis, and evidence of SMN interactions with proteasomal components, including Psmd1, the mouse ortholog of Drosophila Rpn2 (Aghamaleky Sarvestany et al. 2014; Wishart et al. 2014).
Despite our finding of splicing disruptions in genes of known importance to SMA and neuromuscular biology in severe loss-of-function mutants, these splicing changes were absent from less severe models of SMA. Splicing patterns are not conserved from flies to humans, and we would not expect to find similar splicing disruptions in mammalian models of SMA. Finding these genes at the top of our data set may merely reflect the sensitive nature of the splicing of genes that are important for neuromuscular biology, and the fact that transcriptome diversity is greatest in neuronal tissues, including the brain (Mohr and Hartmann 2014).
Although we found evidence for both snRNP-dependent and SMN-specific RNA changes in SMA models, it remains to be determined which of these, if any, contributes to disease pathology. Our analysis of severe and intermediate SMA models suggests that snRNP-independent, Smn gene-specific RNA changes may play a bigger role in SMA pathology than previously envisioned. The SMN-specific activation of stress signaling pathways is conserved in SMA models (Wu et al. 2011; Genabai et al. 2015) and correlates well with disease severity in Smn hypomorphs (Fig. 8C). This activation of stress signaling appears to be a common signature of neuromuscular disease (Deshpande and Rodal 2016).
Based on our finding of a potential activation in stress signaling in fly models of SMA, an important question moving forward will be to understand how loss of SMN causes this stress response. By text mining with the stress signatures identified in the SMA models described here, we found that mutations in the Activating transcription factor 3 (Atf3) gene cause a similar dysregulation in immune and metabolic homeostasis (Rynes et al. 2012). Among other things, this signaling pathway appears to regulate the cytosolic formation of SMN-containing U-bodies in human cells in response to bacterial pathogens (Tsalikis et al. 2015). If SMN does indeed intersect with a normal host cell response to bacteria, this would provide a parsimonious model for the activation of stress signaling in SMA models, and another avenue to explore potential connections to neuromuscular disease.
In conclusion, a direct comparison of snRNP biogenesis mutants revealed that defects in snRNP supply cause complex changes to the transcriptome. However, an analysis of three different Smn hypomorphic lines showed that RNA processing changes are unlikely to be the primary drivers of SMA pathophysiology. Our studies support a model of SMA etiology wherein general disruptions at the mRNA level and specific disruptions at the SMN protein–protein level are responsible for SMA phenotypes. In future experiments, the key will be to uncover the tissue specific molecular activities of SMN that are important for neuromuscular function. Ongoing work with these and other intermediate models of SMA should help clarify the relative contributions of the aforementioned factors to SMA pathology and etiology.
MATERIALS AND METHODS
Fly mutants and husbandry
Stocks were cultured on molasses and agar at room temperature (25 ± 1°C) in half-pint bottles. Oregon-R was used as the wild-type allele. The SmnX7 microdeletion allele was a gift from S. Artavanis-Tsakonis (Harvard University, Cambridge, USA) (Chang et al. 2008). Transposon insertion alleles SmnD (f01109), Ars2C (c00735), and Ars2E (e01128) were obtained from the Exelixis collection at Harvard Medical School (Rajendra et al. 2007; Sabin et al. 2009). The transposon insertion allele PhaxSH (P{lacW}PhaxSH0641) was a gift from S. Hou (Oh et al. 2003).
The wild-type and mutant Smn transgenes are expressed using the native Smn promoter. Transgenic constructs were integrated into the PhiC31 landing site located at 86F8, which was previously recombined into an SmnX7 background (Praveen et al. 2012, 2014). For RNA-seq analysis, Smn transgenes in the SmnX7 null background were crossed to SmnX7 animals, and trans-heterozygous animals were put into TRIzol <12 h after pupation, as brown prepupa. For qRT-PCR and developmental northerns, Smn transgenes in the SmnX7 background were self-crossed, and homozygous animals were put into TRIzol at 72–77 h post egg laying or <12 h after pupation before air bubble migration.
For exogenous expression of a wild-type Phax transgene, an intron-less Phax cDNA was subcloned into pBID-UASC-GV (Wang et al. 2012), and subsequently injected and integrated into the 51C1 PhiC31 landing site by BestGene Inc. The UAS:Phax-mVenus transgene at 51C1 was subsequently recombined into the PhaxSH background. The armadillo promoter-GAL4 driver P{GAL4-arm.S}11 (Sanson et al. 1996) was also recombined into the PhaxSH mutant background.
RNA-seq and bioinformatics analyses
RNA was isolated from 74 ± 2-h-old wild-type and snRNP biogenesis mutant larvae by homogenization in TRIzol (Invitrogen), according to the manufacturer's protocol. Staged animals were isolated from either the wandering third instar stage of development or just before bubble migration in early tan pre-pupae (<12 h after puparium formation). RNA was DNased with Amplification Grade DNase I (Invitrogen) and TURBO DNase (Ambion). A TruSeq RNA Sample Preparation Kit v2 (Illumina) was used for: poly(A)-enrichment or substituted here with Ribo-Zero (Epicentre) rRNA subtraction, barcoding for multiplexing, and cDNA library preparation. Paired end (2 × 50) sequencing was performed on an Illumina HiSeq 2000 platform. TopHat and Cufflinks were used for gene expression analyses, according to the bioinformatic pipeline from Trapnell et al. (2012). DaPars was used to identify differences in RNA length and alternative polyadenylation (Masamha et al. 2014; Xia et al. 2014). MISO (Katz et al. 2010) was used to measure changes in annotated alternative-splicing events. For comparison, we used a “strong” Bayes factor cutoff for our poly(A)-selected RNA-seq analysis (Jeffreys 1961). We used a “positive” Bayes factor as our cutoff for the total rRNA(−) RNA-seq analysis (Kass and Raftery 1995). For developmental comparison, all publicly available RNA-seq data from L2 to L3 puff stage 7–9 were downloaded from the NCBI Sequence Read Archive (SRA) and converted to fastq format with the SRA toolkit (Graveley et al. 2011) (http://www.modencode.org/). The heatmap was generated with the “pheatmap” package from bioconductor (http://www.bioconductor.org).
Northern blotting
Northern analyses were performed using standard protocols (Garcia et al. 2009). Briefly, RNA was isolated as indicated above and ∼300 ng of total RNA was separated on 10% Novex TBE Urea gels (Invitrogen), transferred to GeneScreen Plus charged nylon membranes (PerkinElmer), dried, and UV cross-linked. Northern blots were probed with 5′-end-labeled oligonucleotides for the respective snRNAs, labeled with T4 polynucleotide kinase (NEB) and [γ-32P]ATP (PerkinElmer). The snRNA-specific oligonucleotide probe sequences are listed in Supplemental Table S22. Hybridizations were performed at 65°C, and sequential washes in 2× SSC and 0.33× SSC were performed at 60°C. Damp blots were exposed to storage phosphor screens (GE Healthcare) and analyzed with a Typhoon TRIO+ (GE Healthcare). Bands were quantified using ImageQuant TL (GE Healthcare), and P-values for three biological replicates were determined with a one-tailed Student's t-test for unequal variance of two samples.
RT-PCR analysis
Semi-quantitative RT-PCR was performed with intron flanking oligos, using the 2× Apex Taq Master Mix (Genesee Scientific). Quantitative RT-PCR experiments were carried out as in Praveen et al. (2012). Briefly, random hexamers were used to prime the reverse transcription of isolated RNA according to the manufacturers protocol SupersScript III (Invitrogen). Real-time PCR reactions of cDNA were carried out on a 7900HT Fast Real-time PCR machine (Applied Biosystems), according to the Maxima SYBR Green/Rox qPCR Master Mix (2×) (Thermo Scientific) two-step protocol. Oligo pairs amplified cDNAs from intron inclusive versus spliced mature mRNAs. Three biological replicates were tested for each genotype. The ΔΔCt method was used to quantify differences, and P-values were determined with a one-tailed Student's t-test for unequal variance of two-samples. Gene-specific primer sequences are listed in Supplemental Table S22.
Western blotting
Western analyses were performed using standard protocols and modifications to the methods of Praveen et al. (2012). Briefly, larval protein lysates were prepared by crushing the animals in lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 1% deoxycholate) with 5× protease inhibitor cocktail (Invitrogen) and clearing the lysate by centrifugation. The anti-dUTPase antibody was a generous gift from B.G. Vértessy, and it was used at a dilution of 1 to 50,000 (Békési et al. 2004). Affinity purified anti-dSMN was used at a 1 to 2500 dilution (Praveen et al. 2012). Anti-α-tubulin antibody (Sigma) was used at a 1 to 50,000 dilution.
Cell culture and RNAi
The S2 cell line was obtained from the Drosophila Genome Resource Center and cultivated according to published protocols (Rogers and Rogers 2008). Gene-specific RNAi knockdown was also performed as described in Rogers and Rogers (2008). Briefly, S2 cells were maintained in Sf-900 II SFM media (Gibco) supplemented with 1× Pen Strep (Gibco). For RNAi, 1 mL of (1 × 106 cells/mL) was aliquoted into each well of a six-well tissue culture plate. Double-stranded RNA (dsRNA) was in vitro transcribed overnight (∼16 h) from PCR templates with the T7 MEGAscript kit (Invitrogen), and dsRNA products were subsequently treated with TURBO DNase (Ambion), according to the respective manufacturers’ protocols. Oligonucleotide sequences for amplifying the PCR templates are listed in Supplemental Table S22. PCR templates were amplified from the following plasmids: Smn, pAFW-SMN; Prp6/CG6841, cDNA clone OFa18909 (GenScript); or Prp8, cDNA clone OFa09689 (GenScript). Purified dsRNA was phenol–chloroform extracted, ethanol precipitated, resuspended in RNase-free water (Ambion), and quantified by spectrophotometer. Approximately 15 μg of dsRNA was added to 1 × 106 cells per each well of a six-well plate. The dsRNA was added once each day for three successive days. On the fourth day, cells were pelleted and resuspended in TRIzol (Invitrogen). RNA and proteins were extracted according to the manufacturer's protocol.
DATA DEPOSITION
The poly(A)-RNA-seq of wild-type Oregon-R and Smn null animals were deposited in the NCBI Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE49587 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE49587). The additional data discussed in this publication are accessible through GEO Series accession number GSE81121 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE81121).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge T.K. Rajendra for his contributions during the early phases of this study. We thank A. Malinová, D. Staněk, and S.L. Rogers for their assistance with the S2 cell knockdown experiments, and M.P. Meers for critical reading of the manuscript. We also thank C. Jones and P. Mieczkowski of the UNC High Throughput Sequencing Facility (HTSF). Their helpful discussions on platform choice, sequencing strategy, sample preparation, and sample submission were instrumental to the success of the project. P. Mieczkowski performed the sequencing, and A. Brandt from the HTSF prepared TruSeq cDNA libraries. This work was supported by a grant (to A.G.M.) from the National Institute of General Medical Sciences (R01-GM118636). E.L.G. was supported in part by a National Cancer Institute postdoctoral fellowship (T32 CA009156), administered by J. Pagano and the Lineberger Comprehensive Cancer Center.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057208.116.
REFERENCES
- Aghamaleky Sarvestany A, Hunter G, Tavendale A, Lamont DJ, Llavero Hurtado M, Graham LC, Wishart TM, Gillingwater TH. 2014. Label-free quantitative proteomic profiling identifies disruption of ubiquitin homeostasis as a key driver of Schwann cell defects in spinal muscular atrophy. J Proteome Res 13: 4546–4557. [DOI] [PubMed] [Google Scholar]
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. 2013. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 20: 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Wagner EJ. 2015. Integrator: surprisingly diverse functions in gene expression. Trends Biochem Sci 40: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, Shiekhattar R. 2005. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123: 265–276. [DOI] [PubMed] [Google Scholar]
- Bäumer D, Lee S, Nicholson G, Davies JL, Parkinson NJ, Murray LM, Gillingwater TH, Ansorge O, Davies KE, Talbot K. 2009. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet 5: e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Békési A, Zagyva I, Hunyadi-Gulyás É, Pongrácz V, Kovári J, Nagy ÁO, Erdei A, Medzihradszky KF, Vértessy BG. 2004. Developmental regulation of dUTPase in Drosophila melanogaster. J Biol Chem 279: 22362–22370. [DOI] [PubMed] [Google Scholar]
- Berg MG, Singh LN, Younis I, Liu Q, Pinto AM, Kaida D, Zhang Z, Cho S, Sherrill-Mix S, Wan L, et al. 2012. U1 snRNP determines mRNA length and regulates isoform expression. Cell 150: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YB, Miguel-Aliaga I, Franks C, Thomas N, Trulzsch B, Sattelle DB, Davies KE, van den Heuvel M. 2003. Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet 12: 1367–1376. [DOI] [PubMed] [Google Scholar]
- Chang HCH, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, et al. 2008. Modeling spinal muscular atrophy in Drosophila. PLoS One 3: e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark TA, Sugnet CW, Ares MJ. 2002. Genomewide analysis of mRNA processing in yeast using splicing-specific microarrays. Science 296: 907–910. [DOI] [PubMed] [Google Scholar]
- Custer SK, Todd AG, Singh NN, Androphy EJ. 2013. Dilysine motifs in exon 2b of SMN protein mediate binding to the COPI vesicle protein α-COP and neurite outgrowth in a cell culture model of Spinal Muscular Atrophy. Hum Mol Genet 22: 4043–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande M, Rodal AA. 2016. The crossroads of synaptic growth signaling, membrane traffic, and neurological disease: insights from Drosophila. Traffic 17: 87–101. [DOI] [PubMed] [Google Scholar]
- de Vegvar HE, Lund E, Dahlberg JE. 1986. 3′ end formation of U1 snRNA precursors is coupled to transcription from snRNA promoters. Cell 47: 259–266. [DOI] [PubMed] [Google Scholar]
- Drew AP, Blair IP, Nicholson GA. 2011. Molecular genetics and mechanisms of disease in distal hereditary motor neuropathies: insights directing future genetic studies. Curr Mol Med 11: 650–665. [DOI] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucl Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezzeddine N, Chen J, Waltenspiel B, Burch B, Albrecht T, Zhuo M, Warren WD, Marzluff WF, Wagner EJ. 2011. A subset of Drosophila integrator proteins is essential for efficient U7 snRNA and spliceosomal snRNA 3′-end formation. Mol Cell Biol 31: 328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Englbrecht C, Chari A. 2011. Biogenesis of spliceosomal small nuclear ribonucleoproteins. WIREs RNA 2: 718–731. [DOI] [PubMed] [Google Scholar]
- Garcia EL, Onafuwa-Nuga A, Sim S, King SR, Wolin SL, Telesnitsky A. 2009. Packaging of host mY RNAs by murine leukemia virus may occur early in Y RNA biogenesis. J Virol 83: 12526–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia EL, Lu Z, Meers MP, Praveen K, Matera AG. 2013. Developmental arrest of Drosophila survival motor neuron (Smn) mutants accounts for differences in expression of minor intron-containing genes. RNA 19: 1510–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genabai NK, Ahmad S, Zhang Z, Jiang X, Gabaldon CA, Gangwani L. 2015. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum Mol Genet 24: 6986–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos Pournatzis T, Cowling VH. 2014. Cap-binding complex (CBC). Biochem J 457: 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceosome. RNA 11: 533–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice SJ, Sleigh JN, Motley WW, Liu JL, Burgess RW, Talbot K, Cader MZ. 2015. Dominant, toxic gain-of-function mutations in gars lead to non-cell autonomous neuropathology. Hum Mol Genet 24: 4397–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigg SP, Canales C, Hay A, Tsiantis M. 2005. SERRATE coordinates shoot meristem function and leaf axial patterning in Arabidopsis. Nature 437: 1022–1026. [DOI] [PubMed] [Google Scholar]
- Gruber JJ, Zatechka DS, Sabin LR, Yong J, Lum JJ, Kong M, Zong WX, Zhang Z, Lau CK, Rawlings J, et al. 2009. Ars2 links the nuclear cap-binding complex to RNA interference and cell proliferation. Cell 138: 328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber JJ, Olejniczak SH, Yong J, La Rocca G, Dreyfuss G, Thompson CB. 2012. Ars2 promotes proper replication-dependent histone mRNA 3′ end formation. Mol Cell 45: 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallais M, Pontvianne F, Andersen PR, Clerici M, Lener D, Benbahouche NEH, Gostan T, Vandermoere F, Robert MC, Cusack S, et al. 2013. CBC-ARS2 stimulates 3′-end maturation of multiple RNA families and favors cap-proximal processing. Nat Struct Mol Biol 20: 1358–1366. [DOI] [PubMed] [Google Scholar]
- Hernandez N, Weiner AM. 1986. Formation of the 3′ end of U1 snRNA requires compatible snRNA promoter elements. Cell 47: 249–258. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, et al. 2007. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35: W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- Jeffreys H. 1961. Theory of probability. 3rd ed. Clarendon Press, Oxford. [Google Scholar]
- Kaida D, Berg MG, Younis I, Kasim M, Singh LN, Wan L, Dreyfuss G. 2010. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature 468: 664–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass RE, Raftery AE. 1995. Bayes factors. JASA 90: 773–795. [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao S, Segref A, Kast J, Wilm M, Mattaj IW, Ohno M. 2008. A compartmentalized phosphorylation/dephosphorylation system that regulates U snRNA export from the nucleus. Mol Cell Biol 28: 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubinger S, Sachsenberg T, Zeller G, Busch W, Lohmann JU, Ratsch G, Weigel D. 2008. Dual roles of the nuclear cap-binding complex and SERRATE in pre-mRNA splicing and microRNA processing in Arabidopsis thaliana. Proc Natl Acad Sci 105: 8795–8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. 1995. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80: 155–165. [DOI] [PubMed] [Google Scholar]
- Li H, Custer SK, Gilson T, Hao le T, Beattie CE, Androphy EJ. 2015. α-COP binding to the survival motor neuron protein SMN is required for neuronal process outgrowth. Hum Mol Genet 24: 7295–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau S, Steinestel J, Linta L, Kleger A, Storch A, Schoen M, Steinestel K, Proepper C, Bockmann J, Schmeisser MJ, et al. 2011. An SK3 channel/nWASP/Abi-1 complex is involved in early neurogenesis. PLoS One 6: e18148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TY, Huang CH, Kao HH, Liou GG, Yeh SR, Cheng CM, Chen MH, Pan RL, Juang JL. 2009. Abi plays an opposing role to Abl in Drosophila axonogenesis and synaptogenesis. Development 136: 3099–3107. [DOI] [PubMed] [Google Scholar]
- Lobbes D, Rallapalli G, Schmidt DD, Martin C, Clarke J. 2006. SERRATE: a new player on the plant microRNA scene. EMBO Rep 7: 1052–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao LT, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Matera AG. 2014. Developmental analysis of spliceosomal snRNA isoform expression. G3 (Bethesda) 5: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. 2014. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510: 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matera AG, Wang Z. 2014. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 15: 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattaliano MD, Montana ES, Parisky KM, Littleton JT, Griffith LC. 2007. The Drosophila ARC homolog regulates behavioral responses to starvation. Mol Cell Neurosci 36: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCloskey A, Taniguchi I, Shinmyozu K, Ohno M. 2012. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science 335: 1643–1646. [DOI] [PubMed] [Google Scholar]
- Mohr C, Hartmann B. 2014. Alternative splicing in Drosophila neuronal development. J Neurogenet 28: 199–215. [DOI] [PubMed] [Google Scholar]
- Motley WW, Talbot K, Fischbeck KH. 2010. GARS axonopathy: not every neuron's cup of tRNA. Trends Neurosci 33: 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munding EM, Shiue L, Katzman S, Donohue JP, Ares MJ. 2013. Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol Cell 51: 338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SW, Kingsley T, Shin HH, Zheng Z, Chen HW, Chen X, Wang H, Ruan P, Moody M, Hou SX. 2003. A P-element insertion screen identified mutations in 455 novel essential genes in Drosophila. Genetics 163: 195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M. 2012. Size matters in RNA export. RNA Biol 9: 1413–1417. [DOI] [PubMed] [Google Scholar]
- Ohno M, Segref A, Bachi A, Wilm M, Mattaj IW. 2000. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell 101: 187–198. [DOI] [PubMed] [Google Scholar]
- Pabis M, Neufeld N, Steiner MC, Bojic T, Shav-Tal Y, Neugebauer KM. 2013. The nuclear cap-binding complex interacts with the U4/U6.U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. RNA 19: 1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci 101: 15974–15979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter CJ, Evans M, Thayanithy V, Taniguchi-Ishigaki N, Bach I, Kolpak A, Bassell GJ, Rossoll W, Lorson CL, Bao ZZ, et al. 2011. The COPI vesicle complex binds and moves with survival motor neuron within axons. Hum Mol Genet 20: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleiss JA, Whitworth GB, Bergkessel M, Guthrie C. 2007. Transcript specificity in yeast pre-mRNA splicing revealed by mutations in core spliceosomal components. PLoS Biol 5: e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K, Wen Y, Matera AG. 2012. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep 1: 624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praveen K, Wen Y, Gray KM, Noto JJ, Patlolla AR, Van Duyne GD, Matera AG. 2014. SMA-causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in Drosophila. PLoS Genet 10: e1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proepper C, Johannsen S, Liebau S, Dahl J, Vaida B, Bockmann J, Kreutz MR, Gundelfinger ED, Boeckers TM. 2007. Abelson interacting protein 1 (Abi-1) is essential for dendrite morphogenesis and synapse formation. EMBO J 26: 1397–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra TK, Gonsalvez GB, Walker MP, Shpargel KB, Salz HK, Matera AG. 2007. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol 176: 831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SL, Rogers GC. 2008. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat Protoc 3: 606–611. [DOI] [PubMed] [Google Scholar]
- Rousset R, Bono-Lauriol S, Gettings M, Suzanne M, Speder P, Noselli S. 2010. The Drosophila serine protease homologue Scarface regulates JNK signalling in a negative-feedback loop during epithelial morphogenesis. Development 137: 2177–2186. [DOI] [PubMed] [Google Scholar]
- Rynes J, Donohoe CD, Frommolt P, Brodesser S, Jindra M, Uhlirova M. 2012. Activating transcription factor 3 regulates immune and metabolic homeostasis. Mol Cell Biol 32: 3949–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabath I, Skrajna A, Yang XC, Dadlez M, Marzluff WF, Dominski Z. 2013. 3′-End processing of histone pre-mRNAs in Drosophila: U7 snRNP is associated with FLASH and polyadenylation factors. RNA 19: 1726–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabin LR, Zhou R, Gruber JJ, Lukinova N, Bambina S, Berman A, Lau CK, Thompson CB, Cherry S. 2009. Ars2 regulates both miRNA- and siRNA-dependent silencing and suppresses RNA virus infection in Drosophila. Cell 138: 340–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman AL, Pan Q, Blencowe BJ. 2011. Regulation of alternative splicing by the core spliceosomal machinery. Genes Dev 25: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP. 1996. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383: 627–630. [DOI] [PubMed] [Google Scholar]
- Shpargel KB, Praveen K, Rajendra TK, Matera AG. 2009. Gemin3 is an essential gene required for larval motor function and pupation in Drosophila. Mol Biol Cell 20: 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsalikis J, Tattoli I, Ling A, Sorbara MT, Croitoru DO, Philpott DJ, Girardin SE. 2015. Intracellular bacterial pathogens trigger the formation of U small nuclear RNA bodies (U bodies) through metabolic stress induction. J Biol Chem 290: 20904–20918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JW, Beck ES, McCabe BD. 2012. A modular toolset for recombination transgenesis and neurogenetic analysis of Drosophila. PLoS One 7: e42102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Luhrmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart TM, Mutsaers CA, Riessland M, Reimer MM, Hunter G, Hannam ML, Eaton SL, Fuller HR, Roche SL, Somers E, et al. 2014. Dysregulation of ubiquitin homeostasis and β-catenin signaling promote spinal muscular atrophy. J Clin Invest 124: 1821–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CM, Qiu H, Hu C, Dong J, Hinnebusch AG. 2007. Yeast cap binding complex impedes recruitment of cleavage factor IA to weak termination sites. Mol Cell Biol 27: 6520–6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CY, Whye D, Glazewski L, Choe L, Kerr D, Lee KH, Mason RW, Wang W. 2011. Proteomic assessment of a cell model of spinal muscular atrophy. BMC Neurosci 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Donehower LA, Cooper TA, Neilson JR, Wheeler DA, Wagner EJ, Li W. 2014. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat Commun 5: 5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Liu Z, Lu F, Dong A, Huang H. 2006. SERRATE is a novel nuclear regulator in primary microRNA processing in Arabidopsis. Plant J 47: 841–850. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pinto AM, Wan L, Wang W, Berg MG, Oliva I, Singh LN, Dengler C, Wei Z, Dreyfuss G. 2013. Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc Natl Acad Sci 110: 19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.