

Abstract
Colorectal cancer (CRC) is the third leading cause of cancer death worldwide, which is consequence of multistep tumorigenesis of several genetic and epigenetic events. Since CRC is mostly asymptomatic until it progresses to advanced stages, the early detection using effective screening approaches, selection of appropriate therapeutic strategies and efficient follow-up programs are essential to reduce CRC mortalities. Biomarker discovery for CRC based on the personalized genotype and clinical information could facilitate the classification of patients with certain types and stages of cancer to tailor preventive and therapeutic approaches. These cancer-related biomarkers should be highly sensitive and specific in a wide range of specimen(s) (including tumor tissues, patients’ fluids or stool). Reliable biomarkers which enable the early detection of CRC, can improve early diagnosis, prognosis, treatment response prediction, and recurrence risk. Advances in our understanding of the natural history of CRC have led to the development of different CRC associated molecular and cellular biomarkers. This review highlights the new trends and approaches in CRC biomarker discovery, which could be potentially used for early diagnosis, development of new therapeutic approaches and follow-up of patients.
Keywords: Colorectal cancer, Biomarkers, Cancer diagnosis, Cancer therapy, Predictive marker, Prognostic marker
Core tip: Colorectal cancer (CRC) is one of the most common leading causes of cancer death in the world; therefore, any attempt in early diagnosis, selection of appropriate therapeutic strategies and efficient follow up can play an important role in reducing the disease related mortalities. Our review highlights the novel trends and approaches in CRC biomarker discovery, which are categorized as pathologic genetic or epigenetic changes within the tumor tissue as well as non-invasive biomarkers such as blood or stool based markers. These biomarkers could be used for the management of cancer patients.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide and which is considered for 10% of new cancer diagnoses[1,2]. More than 10% of patients diagnosed with CRC, already have reached to the advanced stages of disease and could show metastasis to the other tissues and organs. Furthermore, about 30% of diagnosed patients with early-stage CRC, have the potential to develop metastatic disease[3].
CRC can be gradually developed through an accumulation of different somatic or inherited changes within genome and epigenome. These pathologic changes lead to the transformation of colonic mucosa into invasive cancer[4]. There are three most important molecular pathways leading to CRC development: (1) Somatic or germ line derived genomic instability due to inactivation of several tumor suppressor genes such as APC, SMAD4 and TP53; aberrant DNA methylation, DNA repair defects induced by mutations in mismatch repair genes (MMR); (2) Mutational inactivation of tumor suppressor genes (e.g., APC, TP53, TGFb, and MMR genes); and (3) Over activation of oncogenic pathways including BRAF, RAS (KRAS and NRAS), Phosphatidyl inositol 3-kinase (PIK-3)[5]. The growth and proliferation of metastatic CRC (mCRC) mainly depends on two signaling pathways: the vascular endothelial growth factor (VEGF) and the epidermal growth factor receptor (EGFR) pathways[6].
The risk of CRC increases with the higher age, as well as by carrying certain inherited genetic mutations (familial adenomatous polyposis and hereditary non-polyposis CRC)[7], a personal or family history of colorectal neoplasia, or having inflammatory bowel disease (IBD)[8].
Cancer related molecular and cellular markers can be classified in four groups: (1) Diagnostic markers, used for risk stratification and early detection; (2) Prognostic markers, give an indication of the likely progression of the disease; (3) Predictive markers, predict treatment response; and (4) Surveillance markers, used to monitor disease recurrence[9].
The increasing disease associated morbidity and mortality, is in part due to a lack of efficient early detection; therefore, earlier diagnosis and more efficient treatment could play a key role in reducing CRC mortality. The conventional examination techniques such as colonoscopy, are invasive; therefore, can affect the patients’ willingness for participation in screening programs. Our increasing knowledge about molecular and cellular mechanisms of CRC development, could provide better objectives for management of the cancer patients using different potential biomarkers. Several marker classes have been evaluated for their use in CRC screening and have all shown potential in early phase biomarker studies (Figure 1). Importantly, non-invasive biomarkers derived from biological fluids (blood- or stool-based markers), due to their easy accessibility, could be considered as practical tools for CRC detection and monitoring (Figure 2A). Reliable biomarkers for early detection of CRC, could significantly improve patient prognosis, prediction of treatment response, and possible prediction of recurrence risk (Table 1). Figure 2B, illustrates the approved pipelines for CRC biomarker discovery and validation.
Figure 1.
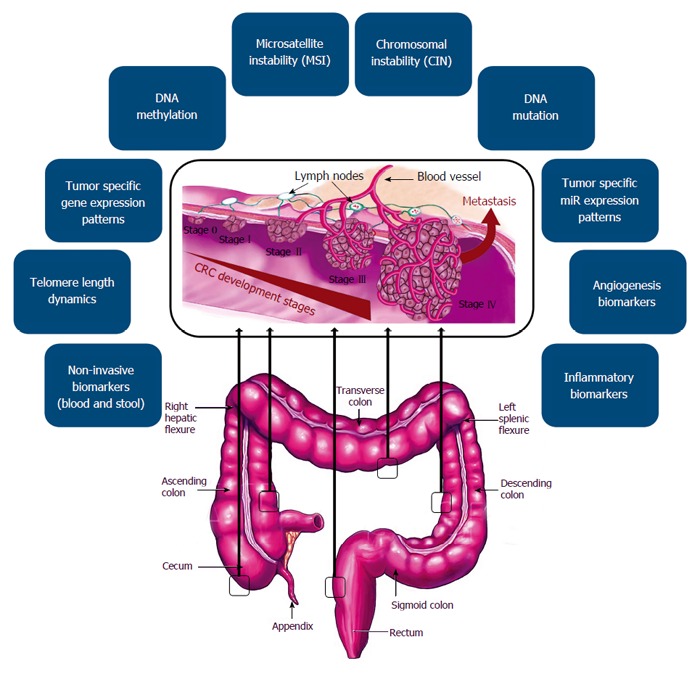
Different classes of colorectal cancer associated molecular and cellular biomarkers.
Figure 2.
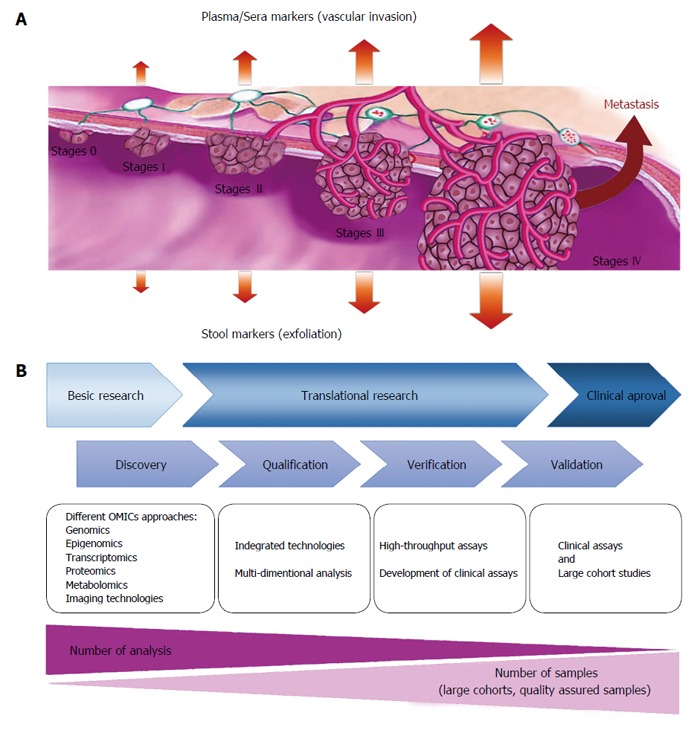
Schematic view of biomarker secretion during different stages of colorectal cancer development (blood and stool biomarkers) (A) and pipelines of biomarker discovery for colorectal cancer (B).
Table 1.
Associated biomarkers for colorectal cancer and their predictive value
|
Biomarkers |
Molecular basis | Predictive value | Detection method | |
| Category | Type | |||
| Pathological characteristics | Tumor stage | Diagnostic, prognostic and predictive markers | Diagnostic radiology and pathological/cytological examination | |
| Lymph node status | ||||
| Grade of differentiation | ||||
| Anatomy of invasion | ||||
| Proliferation markers | Ki67 | Nuclear antigen associated with proliferation | Diagnostic and prognostic markers | IHC |
| Cyclins | Regulation of cell cycle phase transition | Diagnostic and prognostic markers | IHC | |
| Chromosome abnormalities | p53 | Tumor suppressor gene which shows loss of function | Diagnostic, prognostic and predictive markers | IHC, RT-PCR, FISH |
| H-ras, K-ras, N-ras | Membrane-associated GTPase integral to signal transduction cascade, if mutated, causes increased cellular proliferation | Diagnostic, prognostic and predictive markers | IHC, RT-PCR, FISH | |
| Telomere length | Pathologic telomere length dynamics | Diagnostic and prognostic markers | RT-PCR, Flow cytometry | |
| Telomerase activity | Maintenance of telomeres and therefore chromosomal length | Diagnostic and prognostic markers | TRAP assay | |
| enables progression through successive cell cycles | ||||
| Hypoxia-regulated genes | HIF-1 | HIF-1 transcription factor complex stabilized in hypoxic conditions, leading to transcription of hypoxia-regulated genes | Diagnostic and prognostic markers | IHC |
| Glut-1 | Increased Glut-1 expression caused by malignant transformation and upregulated by hypoxia. Promotes switch to anaerobic glycolysis to support hypoxic tumor | Diagnostic and prognostic markers | IHC | |
| Angiogenesis | VEGF | Angiogenic growth factor | Prognostic and predictive markers | IHC, FISH, immunoassay |
| PD-ECGF | Angiogenic growth factor with thymidine phosphorylase activity | Prognostic and predictive markers | IHC | |
| Vascularity | New vasculature supports tumor growth | Prognostic and predictive markers | IHC staining for endothelial receptors e.g., CD31, CD34, von Willebrand (factor VIII) combined with measurement of ICD or MVD using digital image analysis techniques | |
| Epigenetics | Aberrant DNA hypermethylation | Inactivation of key tumor suppressor genes including APC, ATM, BMP3, CDKN2A, SFRP2, GATA4, GSTP1, HLTF, MLH1, MGMT, NDRG4, RASSF2A, SFRP2, TFPI2, VIM, and WIF1 | Diagnostic, prognostic and predictive markers | PCR-based methods and Pyrosequencing |
| Aberrant DNA hypomethylation | Lids to chromosomal instability and global loss of imprinting | Diagnostic and prognostic markers | PCR based methods and Pyrosequencing | |
| Tumor specific expression patterns | Gene expression patterns | Unique signature of the dysregulated genes/pathways at different forms and stages of CRC | Diagnostic, prognostic and predictive markers | Array-based methods, NGS, RT-PCR |
| MicroRNA expression patterns | Unique signature of the dysregulated microRNAs at different forms and stages of CRC | Diagnostic, prognostic and predictive markers | Array-based methods, NGS, RT-PCR | |
FISH: Fluorescent in-situ hybridization; HIF-1: Hypoxia inducible factor-1; HPLC: High pressure liquid chromatography; ICD: Intercapillary distance; IGF-1: Insulin growth factor-1; IHC: Immunohistochemistry; MVD: Microvessel density; NGS: Next generation sequencing; PD-ECGF: Plateletderived endothelial cell growth factor; RT-PCR: Reverse transcription–polymerase chain reaction; RIA: Radioimmunoassay; VEGF: Vascular endothelial growth factor.
MICROSATELLITE INSTABILITY
Microsatellites are repeating units of DNA sequences (usually 1-6bp in length) that can be found in both non-coding or protein coding sequences of DNA. Microsatellite instability (MSI) is defined as somatic alterations in microsatellite sequences due to the insertion or deletion of those repeat units leading to genomic instability and subsequently increasing the susceptibility for the malformations. Tumors with 10%-29% of unstable microsatellite loci are considered MSI-low (MSI-L) while tumors showing ≥ 30% of unstable microsatellite loci are known as MSI-high (MSI-H).
MSI generally results from inactivation of the MMR genes through aberrant promoter hypermethylation (80% of MSI CRCs) or mutations in the genes MLH1, MSH2, MSH6, and PMS2 (20% of MSI CRCs)[10]. Inactivation of these genes results in the accumulation of DNA replication errors in microsatellite sequences, importantly those are located in the exons of potential tumor suppressor genes. In sporadic CRC, 10%-15% of tumors display MSI-high. The MLH1 gene silencing due to the aberrant DNA methylation is responsible for the majority of sporadic CRC with MSI-high[11,12]. A recent meta-analysis assessing the prognostic importance of the MSI in > 7500 patients, could show MSI-H tumors had a better prognosis than MSI-L tumors[13].
Currently, in order to detect MSI in CRC, a specific microsatellite screening panel (Bat-25, Bat-26, MONO-27, NR-21, and NR-24) is used by different clinical laboratories. Therefore, MSI status can serve as a useful predictive and prognostic biomarker for the CRC.
CHROMOSOMAL INSTABILITY
Chromosomal instability (CIN) is defined as the presence of multiple structural or numerical chromosome changes (losses or gains of large portions or whole of the chromosomes) within tumor cells resulting karyotypic variability[14]. CIN is the most common form of genetic instability in CRC that present in about 65%-70% of CRCs. The consequence of chromosomal instability is numerical imbalance for chromosomes (aneuploidy), sub-chromosomal amplifications, and also loss of heterozygosity (LOH).
DNA MUTATIONS AS BIOMARKER
Recent data derived from many large-scale high-throughput DNA sequencing approaches, have identified different candidate genes which could have functional roles in the initiation and development of various human cancers[15,16]. Most of these mutations and genetic alterations occur at a relatively low frequency, while some are present in a high portion of tumors.
Analysis of somatic alterations in CRCs by the Cancer Genome Atlas included whole-genome sequencing that identified 24 significantly mutated genes. Commonly observed alterations enable a broad classification into (1) hypermutated tumors (about 15%), of which three-quarters show high-frequency MSI (MSI-H) and one-quarter have somatic mutations in MMR genes and polymerase-ε (POLE) mutations; and (2) nonhypermutated tumors (about 85%) with multiple somatic copy number alterations and aneuploidy that contain activating mutations in KRAS and PIK3CA and loss of heterozygosity of APC and TP53 tumor-suppressor genes[17].
KRAS GENE MUTATIONS
KRAS, a GTPase protein, is encoded by KRAS proto-oncogene, which is an early player in many biological pathways. Different point mutations in codons 12 and 13 of exon 2, or mutations in codon 61 of exon 3, lead to constitutive activation of RAS signaling pathway. Therefore, genetic disruption of the KRAS gene is one of the essential steps in development of many cancers including CRC.
KRAS is mutated in 30%-50% cases of CRC[18]. According to studies, the adverse impact of KRAS mutations on prognosis seems to be stronger in the distal compared with the proximal colon cancers[19]. More than 30% of CRCs carry mutations in exon 2 of KRAS, and an additional 15% of tumors were found to carry mutations at exons 3 and 4 of KRAS and also exons 2, 3, and exon 4 of NRAS gene[20-22].
Mutations detected in the KRAS gene is one of the most utilized predictive marker for response to the anti-EGFR antibody-based therapies using cetuximab and panitumumab[23]. In addition, approximately 60%-70% of metastatic CRC patients with no KRAS mutations, show poor response to EGFR antibody therapy, which highlights the contribution of additional mutations in different genes such as NRAS in resistance to anti-EGFR treatment[20]. Therefore, the expanded RAS (KRAS and NRAS) mutation screening is now recommended for all cancer patients before anti-EGFR antibody treatment.
BRAF GENE MUTATIONS
The Raf genes family includes three different serine/threonine kinases (ARAF, BRAF, and RAF1). These protein kinases can activate MEK family proteins (including MEK1 and MEK2), which can further phosphorylate ERK1 and ERK2 proteins. ERK controls cell cycle process via regulating enzymes such as Cyclin D1[24]. BRAF is the direct downstream effector of KRAS within the Ras/Raf/MAPK signaling pathway. BRAF gene mutation is reported to be associated with CRC development and also with the poor prognosis of patients[25,26]. Based on the previous studies, BRAF gene mutations is associated with aging, female gender, proximal colon location, poor differentiation, mucinous histology, infiltrating lymphocytes and advanced stage of disease[27]. BRAF mutations occur more frequently in MSI-H cases of CRC[28].
A subset (about 8%) of CRCs carry a point mutation (V600E) in the BRAF oncogene that is mutually exclusive with mutation in KRAS[29]. Patients whose tumors carry BRAFV600E mutations have been consistently shown to have a poor prognosis in the metastatic setting[30]. In contrast to patients with BRAFV600 mutant melanoma[31], CRCs which harbor BRAFV600E mutations were found to be resistant to inhibition of the BRAF/MEK/ERK signaling pathway by vemurafenib[32]. Resistance to vemurafenib was later found to be caused by feedback activation of EGFR when BRAF is inhibited[33]. This finding has led to ongoing clinical trials that investigate combinations of inhibitors of BRAF, EGFR, and MEK with or without chemotherapy[34].
TP53 GENE MUTATIONS
TP53 gene is a very important tumor suppressor which plays a role as a central regulator of different cellular processes including growth arrest and apoptosis, DNA damage, responses to stress, oxidative stress and aberrant proliferative signals[35]. TP53 stops cell cycle in damaged cells until alteration is properly repaired, otherwise it triggers the apoptosis cascade in those damaged cells.
TP53 protein dysfunction is one of the common hallmarks of human solid tumors which has been reported in more than 25% of adenomas, and in 50%-70% of patients with CRCs. TP53 dysfunction is also playing a critical role in the adenoma to carcinoma transition[36]. The majority (about 80%) of TP53 gene mutations are missense mutations leading to the synthesis of a dysfunctional protein with an abnormally long half-life. Those missense mutations mainly occur within five hotspot codons (175, 245, 248, 273, and 282)[37]. Different TP53 mutations are also reported in more than half of sporadic CRC patients[38].
APC/β-CATENIN MUTATIONS
Genetic disruption of APC leading to the activation of Wnt pathway, is one of the important early genetic event in colorectal tumorigenesis[39]. The APC gene product is a large protein that regulates development, chromosomal segregation, cellular differentiation, polarity, adhesion, migration, and also apoptosis. The APC protein is interacting with glycogen synthase kinase-3β (GSK-3β) and β-catenin (an important molecule of the Wnt pathway). Germline APC mutations are the cause for FAP (familial adenomatous polyposis)[40]. Somatic mutations in the APC gene are observed in 30%-70% of sporadic adenomas, and in around 70% of sporadic tumors[41]. These somatic mutations are mainly found between codons 1286 to 1513 (known as the mutation cluster region), while germline mutations are distributed throughout the entire gene[42]. As an alternative mechanism for APC gene dysfunction, APC promoter hypermethylation is also reported in more than 18% of primary colorectal carcinomas and adenomas[43].
DNA METHYLATION
The most widely studied epigenetic alteration in cancer is aberrant DNA methylation. DNA methylation is one of the important epigenetic modifications that can regulate gene expression. In humans, DNA methylation occurs at cytosine residues that precede guanines, called CpG dinucleotides (C-phosphodiester-G)[44,45].
Recently, along with a growing understanding of cellular epigenetic mechanisms, the role of pathologic epigenetics changes in human cancer became more visible[46]. It is becoming more evident that epigenetic events might be central to the cancer initiation and progression[44,45,47-52].
ABERRANT DNA HYPERMETHYLATION IN CRC
Hypermethylation of promoter regions leading to gene silencing, is frequently found in different types of human cancers[45,50]. Recently a third class of CRCs characterized by a high frequency of DNA hypermethylation has been reported. These cancers have been defined as having a “CpG island methylator phenotype (CIMP)”. Weisenberger et al[53] could show CIMP in CRC, is based on the methylation status of five genes (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1). CIMP-positive tumors exhibit unique clinicopathological and molecular features, including a predilection for proximal location in the colon, poor and mucinous histology and the presence of frequent KRAS and BRAF mutations[54]. Approximately, 80% of sporadic CRCs with MSI-H harbor biallelic hypermethylated MLH1 alleles[53]. The transition from normal mucosa to adenomatous polyp is marked by both genetic and epigenetic alterations, which dysregulate important molecular pathways[55]. These epigenetic alterations include hypermethylation of a variety of genes, such as SLC5A8, ITGA4, SFRP2, PTCH1, CDKN2A, HLTF, and MGMT, and some of these play a role in the initiation and progression of adenomas to CRC[56,57].
To date, a large number of hypermethylated genes including APC, ATM, BMP3, CDKN2A, SFRP2, GATA4, GSTP1, HLTF, MLH1, MGMT, NDRG4, RASSF2A, SFRP2, TFPI2, VIM, and WIF1, have been found in stool DNA assays for the early detection of CRC[58].
The SEPT9 gene methylation assay has recently been developed as a novel blood-based test for CRC (Epi proColon 2.0, Epigenomics AG, Germany). Septins are a family of conserved GTP-binding proteins which are scaffolding proteins during compartmentalization and cell division[59]. In human, there are 13 known septin genes (SEPT1 to SEPT13). All septins can form heteromeric complexes; within terminal position of these octamer protein family, SEPT9 plays an important role in subunit stabilization and polymerization[60]. Therefore, abnormal SEPT9 or no SEPT9 may seriously affect cytokinesis. The crucial role of SEPT9 in the septin complex may be a key factor in CRC carcinogenesis when the promoter region of the SEPT9 gene is aberrantly hypermethylated and the transcription is compromised[61]. To date, several independent clinical trials have proved the aberrant SEPT9 gene methylation as a specific biomarker for CRC early detection and screening.
GENOME-WIDE DNA HYPOMETHYLATION IN CRC
While DNA hypermethylation can silence tumor-suppressor genes, global DNA hypomethylation can also influence CRC development by inducing chromosomal instability and global loss of imprinting[62]. Genome-wide hypomethylation occurs within repetitive transposable DNA elements, such as the long interspersed nuclear element-1 (LINE-1) or short interspersed nucleotide elements (SINE or Alu) sequences in many cancers, including CRC[63]. LINE-1 hypomethylation inversely associates with MSI-H phenotype and/or CIMP in colorectal cancer[63].
TUMOR SPECIFIC GENE EXPRESSION PATTERNS
Gene expression analyses of tumor derived cells/tissues in comparison to the normal tissue, reviled to a better understanding of interplay between dysregulated genes and the affected pathways in colorectal neoplasms. CRC is a very heterogenic disease and patients with even the same type of cancer, often have dissimilar genetic/epigenetic defects in their tumors which indicates why those patients respond heterogeneously to the anticancer agents. The use of DNA-microarray and next generation sequencing (NGS) technology have made it possible to assess the expression of tens of thousands genes in a single experiment[64]. These gene-expression signatures can be used for the molecular classification of different tumors as well as cancer subtypes[65]. Those genes which their dysregulation is linked to CRC are summarized in the Figure 3.
Figure 3.
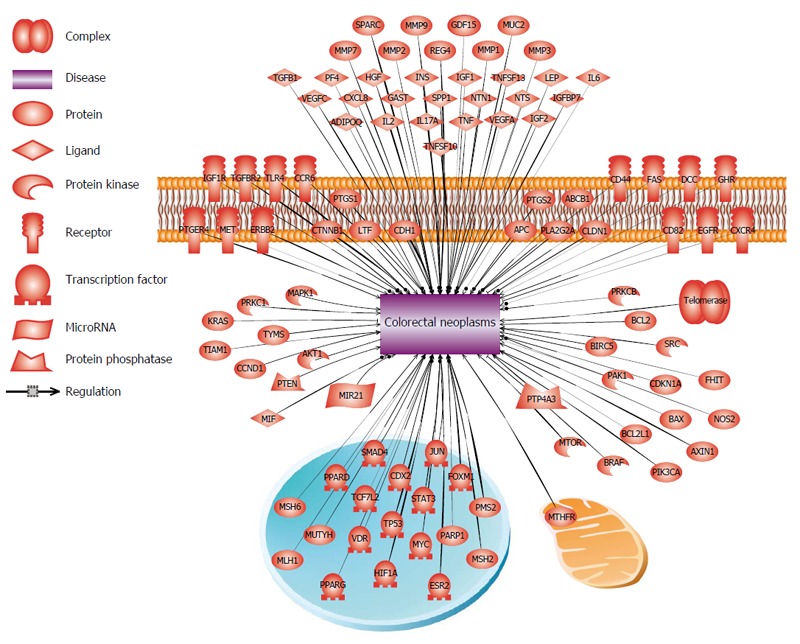
Genes associated to colorectal cancer development. Dysregulation of these genes as a single or in cooperation (due to the DNA mutation, epigenetic changes or as a consequence of change in the regulatory upstream genes/pathways), has been shown in different forms and stages of colorectal cancer.
Several groups have reported different molecular classifications of CRCs using gene expression data from NGS or expression arrays and in some cases, these subtypes could provide predictive values[66,67] or prognostic information[68,69]. Validation of a 23-gene microarray-based prognostic signature showed around 70% relapse predictive value for CRC patients[70]. High throughput gene expression profiling reveals tumor subtypes that show overlap with previous classification systems using MSI, CIN, CIMP or KRAS/BRAF mutation status[66], which cannot be identified only based on single mutations or epigenetic alterations.
TUMOR SPECIFIC MICRORNA EXPRESSION PATTERNS
Abnormalities and dysregulations in non-coding regulatory RNAs can also contribute to tumorigenesis and cancer development[71,72]. A class of small cellular RNAs, termed microRNAs (miRNAs), can lead to silencing of their cognate target genes. MicroRNAs are key players in regulating diverse cellular pathways.
It is known that some miRNAs can function either as tumor suppressors or oncogene[72,73], and expression profiling of microRNAs have revealed the characteristic signatures of these small regulatory RNAs in different cancers including colorectal cancer[71,74].
Recently, the interest for miRNA biomarker research in human cancer has increased due to the unique characteristics of miRNAs. First, miRNAs are remarkably stable under a variety of experimental and laboratory conditions. Second, due to their small size and the hairpin-loop structure, miRNAs are protected from RNase-mediated degradation, and thus are easily extractable from a wide variety of clinical specimens, including formalin-fixed paraffin embedded (FFPE) tissues, and a variety of body fluids including blood, saliva, urine, feces etc. Third, cell-free miRNAs are often protected from degradation because of being in high density lipoprotein particles, apoptotic bodies, microvesicles, and exosomes[75].
MiRNAs can have a critical role in invasion, migration and the progression of disease through epithelial mesenchymal transition (EMT) into metastases. Therefore, dysregulation of several miRNAs have been reported at different stages of CRC development and progress. More than 500 differentially expressed miRNAs have been associated with different stages of CRC. MiRNAs such as miR-20a, miR-21, miR-29a, miR-31, miR-34a, miR-92a, miR-200c, miR-215 and miR-375 were the most frequently dysregulated in CRC. Recent experimental analyses have validated a total of 530 miRNA-mRNA pairs in colorectal cancer, 200 unique miRNAs and 347 unique targets[76]. In BRAF mutated forms of CRC, a high expression of miR-31 was observed and suggested as a potential diagnostic/therapeutic biomarker[77]. A signature of miR-92a, miR-375 and miR-424 could discriminate invasive carcinoma from adenoma, representing a promising biomarker for the early diagnosis of CRC. The panel of six miRNA classifier (miR-21-5p, miR- 20a-5p, miR-103a-3p, miR-106b-5p, miR-143-5p, and miR-215) has potential to distinguish between high and low risk of disease progression[76].
The functional role of miR-34a and miR-200 family members have been shown in metastatic form of CRC. In addition, miR-34a can be used as an independent predictor of recurrence among patients[78]. Overexpression of miR-622 was induced by radiotherapy in rectal cancers, causing poor response to the therapy[79]. In metastatic CRC patients with wild-type KRAS that responded to anti-EGFR therapy, profiling the panel of miR-99a, let-7c and miR-125b could have a diagnostic potential[80]. In the other hand, reduced expression of miR-181a was associated with poor clinical outcome in CRC patients treated with anti-EGFR[81]. Whereas, down regulation of the miR-7, a direct regulator of EGFR, was reported as a prognostic biomarker for tumors were resistant to targeted anti-EGFR therapy[82].
TELOMERE LENGTH DYNAMICS
Telomeres, the ends of chromosomes in mammals, are composed of a 6-bp variable repeat sequence (TTAGGG), which is added on by the telomerase and has a crucial role in maintenance of chromosomal stability[83]. Telomeric DNA (tDNA) is progressively lost during each cell division due to the end replication mispairing, oxidative damage or other end processing events[84,85].
Telomerase activity and telomere length have a crucial role in malignant transformation. In the early stage of tumorigenesis, losing the telomere sequences (telomere shortening) limits cell proliferation, and telomerase activation protects the ends of chromosome and suppresses tumorigenesis. While, in the late stages of tumorigenesis, telomere shortening triggers instability in the genome, and telomerase activation induces immortalization of cancer cells[86,87]. Figure 4 summarizes the genes that regulate telomere length and also telomerase activity. Recent data proposed that the severe genomic instability exist in telomere crisis, accelerates secondary genetic alterations that lead to carcinogenesis and indeed highlights the implication of pathologic telomere length changes in cancer pathways. Therefore, telomere length dynamics can serve as a useful indicator and biomarker in risk assessment and prediction of different stages of cancer[88-92]. Telomere dysfunction has been indicated as a negative prognostic marker in solid tumors[90,93,94].
Figure 4.
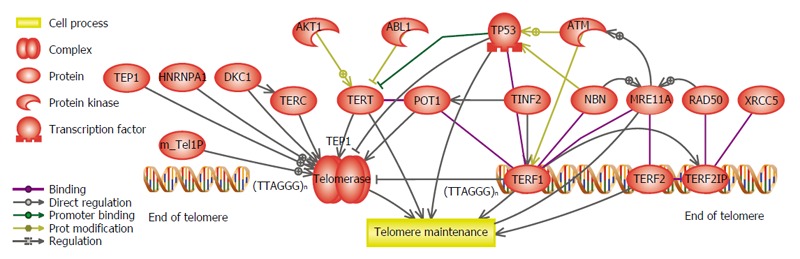
Genes which are involved in the telomere maintenance and telomerase activity pathway. Any dysregulation in those genes will lead to the pathologic telomere length dynamics during colorectal cancer development stages.
Several studies demonstrated telomeres are shorter in CRCs compared to the adjacent normal mucosa. In addition, malignant tumors have relatively shorter telomere length than benign tumors[95-98]. Since tumors in sporadic form of CRC appear relatively later compared with the hereditary nonpolyposis colorectal cancer (HNPCC), also the number of divisions between initiation and clinical presentation in sporadic may be more than HNPCC, therefore their base line telomere lengths at initiation are likely to be shorter in sporadic form of CRC[2,90,99].
The evidence that telomeres were shorter in CRCs, even in well-differentiated tumors, suggesting the telomere length shortening as an initial event in colorectal cancer which directly reflects pathologic cell proliferation[100,101].
ANGIOGENESIS BIOMARKERS
Angiogenesis, critical step in cancer progression, is a new blood vessels generation from endothelial precursor, which is mediated through a group of ligands and receptors that work together[102]. A group of glycoproteins, including the placental growth factor (PIGF) and VEGF family (VEGF-A, VEGF-B, VEGF-C, and VEGF-D), act as effectors of angiogenesis[102,103]. Several other factors including the PIGF, fibroblast growth factor (FGF), VEGF-C, VEGF-D, angiopoietin, hypoxia-inducible factor (HIF)-1α and HIF-2α, integrin, and platelet-derived growth factor, can overlap with the angiogenesis pathway, which makes the activation or inhibition outcomes very challenging[102].
The potential value for VEGF as a prediction or prognosis biomarker for metastatic CRC has been reported[104]. It is shown that the VEGF-1154G>A and VEGF405C>1 polymorphisms were associated with improved overall survival. This finding suggested that germline variants in VEGF-dependent and -independent angiogenesis can predict survival and treatment response to the first-line bevacizumab and oxaliplatin-based chemotherapy in patients with mCRC[105]. The importance of baseline levels of VEGF and soluble VEGFR-2 (sVEGFR-2) as predictive/prognostic biomarkers was shown in two independent phase-III studies evaluating the role of cediranib, an experimental angiogenesis inhibitor, in mCRC[106].
INFLAMMATORY BIOMARKERS
In patients with the inflammatory bowel diseases, chronic inflammation was suggested as a predisposing factor to CRC and the risk for developing CRC increases with longer duration of colitis. Therefore, anti-inflammatory therapies, such as 5-aminosalicylates, can significantly reduce the development of colorectal neoplasia in those patients[107].
C-reactive protein (CRP) is a very sensitive but non-specific systemic marker of inflammation. CRP is mainly produced in the liver in response to cytokines released during infection, trauma, surgery, burns, tissue infarction, advanced cancer, and chronic inflammatory conditions[108]. According to a recent study, the concentrations of CRP, might have a positive correlation to the CRC risk[109].
Interleukin-6 (IL6) secreted by the hematopoietic or non-hematopoietic cells, is a multifunctional cytokine and has a pro-inflammatory function via binding to a soluble IL6 receptor (sIL6R) or by acting on a transmembrane type 1 cytokine receptor[110]. IL6 upregulates several acute-phase proteins such as CRP, α1-antitrypsin, fibrinogen and serum amyloid A[111]. It is shown that IL6 is significantly augmented at the colonic tumor microenvironment[112].
TNFα is another pleiotropic cytokine which is secreted in response to any tissue damage or infection; some reports suggesting the role of TNFα in the pathogenesis of IBD[113]. The other inflammatory biomarker, macrophage inhibitory cytokine 1, is reported to be positively associated with CRC risk[114]. Certain inflammatory biomarkers from the CRC or tumor immune reaction, may lead to generation of individualized immune vaccines.
NON-INVASIVE BIOMARKERS
Blood biomarkers
As non-invasive biomarkers for the screening of colorectal cancer, particularly at early stage of the disease initiation, blood biomarkers could show very promising value. Theses blood-based markers could be also used to monitor therapeutic response in CRC patients as well as the detection of disease recurrence or relapse.
Circulating tumor cells: Circulating tumor cells (CTCs) are tumor cells (mainly cancer stem cells) derived from either primary tumors or metastases which are circulating in the peripheral blood. The presence of CTCs in blood is associated with progressive or metastatic disease. Therefore, can be used to monitor advanced stage disease in patients without other measurable surveillance markers.
The CTCs in colorectal cancer have epithelial origin with defined immunephenotype signature (CD45-, EpCAM+)[115]. Using the immunephenotype markers and sensitive cell sorting technologies, it is now possible to isolate and assess the complete genetic/epigenetic profiles of tumor derived CTCs. This holds promise for the development of more efficient personalized treatment to eliminate cancer stem cells in CRC patients.
Circulating cell-free DNA: The discovery of circulating cell-free DNA (ccf-DNA) could open up a new possibility for non-invasive analysis of tumor derived genetic material, as it can be isolated from human body fluids[116]. The potential diagnostic/prognostic values of quantifying ccf-DNA in CRC patients compared to the healthy controls, have been assessed in different studies[117]. The ccf-DNA levels in those patients, could show significantly increase within the advanced disease stages and fluctuated during chemotherapy period[118].
Many studies have investigated MSI or LOH within ccf-DNA from the plasma/sera of CRC patients as very valuable non-invasive biomarkers[119]. Different studies have reported the diagnostic/prognostic value of mutated genes within ccf-DNA[120]. The detection of mutated TP53 or KRAS genes in the ccf-DNA of CRC patients could predictive disease recurrence as well as the clinical outcome[121]. Aberrant DNA methylation can also have a diagnostic or prognostic value in the plasma/sera of CRC patients. Relying on aberrant DNA methylation markers, promoter methylation of several genes have reported as biomarkers for CRC including ALX4, APC, CDKN2A/P16h, FRP2, MLH1, NGFR, NEUROG1, P16, RUNX3, SEPT9, TMEFF2, TP53 and TPEF/HPP1[122,123].
Circulating microRNAs: Due to the stability of circulating microRNAs as well as the potential role of the microRNA dysregulation at different stages of carcinogenesis, they have the potential to serve as very promising non-invasive biomarkers for different types of human cancers[124].
As a well-characterized oncogenic miRNA, miR-21 is considered one of the promising non-invasive biomarkers for the early detection of CRC. Dysregulation of miR-21 occurs frequently at early stages of the adenoma-carcinoma sequence; miR-21 is one of the most highly expressed miRNAs in CRC; and miR-21 is highly secreted by cancer cells, which can be measured in exosomes or as free miRNAs in plasma or serum[125].
As another potential circulating microRNA, the elevated serum level of miR-92a has been reported for the advanced adenoma and CRC[126]. There are also other studies showing the elevated circulating levels of miR-17-3p and miR-29a[127]. Significantly higher expression of miR-17-92 cluster and miR-135 were found in CRC patients in comparison to healthy controls which could discriminate CRC with an overall sensitivity and specificity of 74% and 79%[128].
Plasma levels of miR-92 could distinguish patients with CRC from gastric cancer, IBD and healthy control subjects[129]. A high diagnostic accuracy has been shown by a panel of miR-409-3p, miR-7, and miR-93 in discriminating CRC from controls with more than 90% sensitivity and specificity[130].
Circulating proteins: Shed or secreted proteins from different cancer cells into the bloodstream, can be detected by different methodological approaches like enzyme-linked immunosorbent assay (ELISA) or chromatographic and mass spectrometric (MS) technologies. Different protein-based biomarkers have been reported for CRC. These protein markers include circulating carcinoembryonales antigen (CEA), carbohydrate antigens (CA19-9, CA50, CA72-4), soluble Fas ligand (FasL), p53, and VEGF[131]. Of these markers, CEA could show a strong prognostic impact in CRC patients[132].
STOOL BIOMARKERS
The presence of tumor biomarkers in stool can be attributed to secretion, exfoliation or leakage[133]. Stool-based markers come from vital and apoptotic colonocytes, shed into the colorectal lumen. Since the stool markers are directly derived from the tumor cells, assumed to be highly specific biomarkers for CRC. These stool biomarkers include stool DNA (sDNA), which can be used for checking the MSI, aberrant DNA methylation or somatic mutation for specific cancer related genes, miRNAs, protein biomarkers as well as secretory molecules and biochemical materials resulted from metabolism of cancer cell. Considering the off-site testing and non-invasiveness together with the low potential costs, stool biomarkers gets more reappraisal. Recent advances in laboratory techniques, could introduce modern generation of stool tests with higher sensitivity and specificity rates for different subgroups of the colorectal cancer. These stool biomarkers should be very sensitive with higher specificity because positive test results lead to unnecessary, potentially morbid, and costly colonoscopies[134].
Human DNA is less than 0.01% of total DNA in stool and the vast majority (99.99%) of sDNA is derived from intestine bacterial or dietary; therefore, one of the important technical challenge of sDNA testing is specific detection of methylated or mutated human DNA within a pool of nontarget DNA[135,136]. Several panels of methylated genes within sDNA have been reported for different stages of CRC, involving BMP3, CDH1, CDH13, CRBP1, CXCL12, ESR1, HLTF, ID4, IRF8, ITGA4, MINT1, MINT31, NDRG4, P14, P16, RUNX3, SFRP1, SFRP2, SLC5A8, and TIMP3[137]. These panels were differing in the marker selection, assay methods, and patient populations studied which many of them could not be further validated in the bigger cohorts.
Although several stool-based tests (fecal immunochemical test (FIT), guaiac fecal occult blood test (gFOBT), immunological fecal occult blood test (iFOBT), and sDNA test) are clinically available for the detection of CRC, the sensitivity and specificity for tests is not sufficient due to many factors such as inconvenience in sampling or misinterpretation. Recently, the US food and drug administration (FDA) has approved the Cologuard® test (Exact Sciences Corporation, United States). Cologuard® is a panel of multi-target sDNA test, which combines molecular tests for KRAS and β-catenin mutation, aberrantly methylated BMP3 and NDRG4 gene promoters and human hemoglobin immunochemical assay[138].
CONCLUSION
The fast growing knowledge of cancer biology, particularly in the field of genetic, epigenetic and molecular cell biology, could provide valuable objectives for the early detection of different malignancies including colorectal cancer. The poor outcome of advanced form of CRC, has prompted the need for reliable predictive and prognostic markers.
Circulating and stool-based tumor specific markers show promising potential as biomarkers. Additional biomarkers that have clinical applicability will continue to be proposed, tested, and developed from knowledge of the genetic and epigenetic changes in CRCs. This would facilitate more individualized treatment approaches, relying the specific molecular signature of tumors. Sequencing of individual cancer genomes that provide a comprehensive picture of driver mutations within a CRC, may become commonplace when costs for this technology are lower and rapid analytic systems are employed. This will be most beneficial with simultaneous development and use of therapies that can address the personalized findings.
Unfortunately, most of the identified biomarkers in different tumor studies, failed in the validation studies. The lack of consistency between biomarker panels within independent studies, highlights a major obstacle for the development of robust CRC biomarkers. For being able to use CRC associated biomarkers in clinical care of patients, large-scale studies are needed to identify optimal marker panels and validate those biomarkers in more cross-sectional and prospective cohort studies.
ACKNOWLEDGMENTS
We thank Dr. Hiten D. Mistry for his valuable comments and kindly proofreading the text.
Footnotes
Conflict-of-interest statement: There is no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Manuscript source: Invited manuscript
Peer-review started: March 22, 2016
First decision: May 12, 2016
Article in press: June 15, 2016
P- Reviewer: Lakatos PL, Morris DL S- Editor: Ma YJ L- Editor: A E- Editor: Wang CH
References
- 1.Koretz RL. Malignant polyps: are they sheep in wolves’ clothing? Ann Intern Med. 1993;118:63–68. doi: 10.7326/0003-4819-118-1-199301010-00011. [DOI] [PubMed] [Google Scholar]
- 2.González-González M, Garcia JG, Montero JA, Fernandez LM, Bengoechea O, Muñez OB, Orfao A, Sayagues JM, Fuentes M. Genomics and proteomics approaches for biomarker discovery in sporadic colorectal cancer with metastasis. Cancer Genomics Proteomics. 2013;10:19–25. [PubMed] [Google Scholar]
- 3.Böckelman C, Engelmann BE, Kaprio T, Hansen TF, Glimelius B. Risk of recurrence in patients with colon cancer stage II and III: a systematic review and meta-analysis of recent literature. Acta Oncol. 2015;54:5–16. doi: 10.3109/0284186X.2014.975839. [DOI] [PubMed] [Google Scholar]
- 4.Burt RW, Bishop DT, Lynch HT, Rozen P, Winawer SJ. Risk and surveillance of individuals with heritable factors for colorectal cancer. WHO Collaborating Centre for the Prevention of Colorectal Cancer. Bull World Health Organ. 1990;68:655–665. [PMC free article] [PubMed] [Google Scholar]
- 5.Markowitz SD, Bertagnolli MM. Molecular origins of cancer: Molecular basis of colorectal cancer. N Engl J Med. 2009;361:2449–2460. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmoll HJ, Van Cutsem E, Stein A, Valentini V, Glimelius B, Haustermans K, Nordlinger B, van de Velde CJ, Balmana J, Regula J, et al. ESMO Consensus Guidelines for management of patients with colon and rectal cancer. a personalized approach to clinical decision making. Ann Oncol. 2012;23:2479–2516. doi: 10.1093/annonc/mds236. [DOI] [PubMed] [Google Scholar]
- 7.Whiffin N, Hosking FJ, Farrington SM, Palles C, Dobbins SE, Zgaga L, Lloyd A, Kinnersley B, Gorman M, Tenesa A, et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet. 2014;23:4729–4737. doi: 10.1093/hmg/ddu177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawk ET, Limburg PJ, Viner JL. Epidemiology and prevention of colorectal cancer. Surg Clin North Am. 2002;82:905–941. doi: 10.1016/s0039-6109(02)00046-4. [DOI] [PubMed] [Google Scholar]
- 9.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 10.Ward R, Meagher A, Tomlinson I, O’Connor T, Norrie M, Wu R, Hawkins N. Microsatellite instability and the clinicopathological features of sporadic colorectal cancer. Gut. 2001;48:821–829. doi: 10.1136/gut.48.6.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, Jessup JM, Kolodner R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 12.Montazer Haghighi M, Radpour R, Aghajani K, Zali N, Molaei M, Zali MR. Four novel germline mutations in the MLH1 and PMS2 mismatch repair genes in patients with hereditary nonpolyposis colorectal cancer. Int J Colorectal Dis. 2009;24:885–893. doi: 10.1007/s00384-009-0731-1. [DOI] [PubMed] [Google Scholar]
- 13.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 14.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 15.Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 17.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santini D, Loupakis F, Vincenzi B, Floriani I, Stasi I, Canestrari E, Rulli E, Maltese PE, Andreoni F, Masi G, et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: implications for clinical practice. Oncologist. 2008;13:1270–1275. doi: 10.1634/theoncologist.2008-0181. [DOI] [PubMed] [Google Scholar]
- 19.Sinicrope FA, Mahoney MR, Yoon HH, Smyrk TC, Thibodeau SN, Goldberg RM, Nelson GD, Sargent DJ, Alberts SR. Analysis of Molecular Markers by Anatomic Tumor Site in Stage III Colon Carcinomas from Adjuvant Chemotherapy Trial NCCTG N0147 (Alliance) Clin Cancer Res. 2015;21:5294–5304. doi: 10.1158/1078-0432.CCR-15-0527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 21.Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann Oncol. 2011;22:1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 22.De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010;11:753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–345. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 24.Pruitt K, Der CJ. Ras and Rho regulation of the cell cycle and oncogenesis. Cancer Lett. 2001;171:1–10. doi: 10.1016/s0304-3835(01)00528-6. [DOI] [PubMed] [Google Scholar]
- 25.Fransén K, Klintenäs M, Osterström A, Dimberg J, Monstein HJ, Söderkvist P. Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis. 2004;25:527–533. doi: 10.1093/carcin/bgh049. [DOI] [PubMed] [Google Scholar]
- 26.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 27.Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B. BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer. 2006;5:2. doi: 10.1186/1476-4598-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de la Chapelle A, Palomaki G, Hampel H. Identifying Lynch syndrome. Int J Cancer. 2009;125:1492–1493. doi: 10.1002/ijc.24491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 30.Maughan TS, Adams RA, Smith CG, Meade AM, Seymour MT, Wilson RH, Idziaszczyk S, Harris R, Fisher D, Kenny SL, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;377:2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, Ribas A, Hogg D, Hamid O, Ascierto PA, et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. doi: 10.1016/S1470-2045(14)70012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopetz S, Chang GJ, Overman MJ, Eng C, Sargent DJ, Larson DW, Grothey A, Vauthey JN, Nagorney DM, McWilliams RR. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J Clin Oncol. 2009;27:3677–3683. doi: 10.1200/JCO.2008.20.5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–682. doi: 10.1016/j.ccr.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaeger R, Cercek A, O’Reilly EM, Reidy DL, Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et al. Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant metastatic colorectal cancer patients. Clin Cancer Res. 2015;21:1313–1320. doi: 10.1158/1078-0432.CCR-14-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 36.Leslie A, Carey FA, Pratt NR, Steele RJ. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89:845–860. doi: 10.1046/j.1365-2168.2002.02120.x. [DOI] [PubMed] [Google Scholar]
- 37.Béroud C, Soussi T. The UMD-p53 database: new mutations and analysis tools. Hum Mutat. 2003;21:176–181. doi: 10.1002/humu.10187. [DOI] [PubMed] [Google Scholar]
- 38.Wu W, Yang J, Feng X, Wang H, Ye S, Yang P, Tan W, Wei G, Zhou Y. MicroRNA-32 (miR-32) regulates phosphatase and tensin homologue (PTEN) expression and promotes growth, migration, and invasion in colorectal carcinoma cells. Mol Cancer. 2013;12:30. doi: 10.1186/1476-4598-12-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- 40.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 41.Otori K, Konishi M, Sugiyama K, Hasebe T, Shimoda T, Kikuchi-Yanoshita R, Mukai K, Fukushima S, Miyaki M, Esumi H. Infrequent somatic mutation of the adenomatous polyposis coli gene in aberrant crypt foci of human colon tissue. Cancer. 1998;83:896–900. doi: 10.1002/(sici)1097-0142(19980901)83:5<896::aid-cncr14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 42.Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- 43.Esteller M, Sparks A, Toyota M, Sanchez-Cespedes M, Capella G, Peinado MA, Gonzalez S, Tarafa G, Sidransky D, Meltzer SJ, et al. Analysis of adenomatous polyposis coli promoter hypermethylation in human cancer. Cancer Res. 2000;60:4366–4371. [PubMed] [Google Scholar]
- 44.Barekati Z, Radpour R, Lu Q, Bitzer J, Zheng H, Toniolo P, Lenner P, Zhong XY. Methylation signature of lymph node metastases in breast cancer patients. BMC Cancer. 2012;12:244. doi: 10.1186/1471-2407-12-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Radpour R, Barekati Z, Kohler C, Holzgreve W, Zhong XY. New trends in molecular biomarker discovery for breast cancer. Genet Test Mol Biomarkers. 2009;13:565–571. doi: 10.1089/gtmb.2009.0060. [DOI] [PubMed] [Google Scholar]
- 46.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 47.Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97:5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barekati Z, Radpour R, Kohler C, Zhang B, Toniolo P, Lenner P, Lv Q, Zheng H, Zhong XY. Methylation profile of TP53 regulatory pathway and mtDNA alterations in breast cancer patients lacking TP53 mutations. Hum Mol Genet. 2010;19:2936–2946. doi: 10.1093/hmg/ddq199. [DOI] [PubMed] [Google Scholar]
- 49.Radpour R, Barekati Z, Kohler C, Lv Q, Bürki N, Diesch C, Bitzer J, Zheng H, Schmid S, Zhong XY. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PLoS One. 2011;6:e16080. doi: 10.1371/journal.pone.0016080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radpour R, Barekati Z, Kohler C, Schumacher MM, Grussenmeyer T, Jenoe P, Hartmann N, Moes S, Letzkus M, Bitzer J, et al. Integrated epigenetics of human breast cancer: synoptic investigation of targeted genes, microRNAs and proteins upon demethylation treatment. PLoS One. 2011;6:e27355. doi: 10.1371/journal.pone.0027355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radpour R, Haghighi MM, Fan AX, Torbati PM, Hahn S, Holzgreve W, Zhong XY. High-throughput hacking of the methylation patterns in breast cancer by in vitro transcription and thymidine-specific cleavage mass array on MALDI-TOF silico-chip. Mol Cancer Res. 2008;6:1702–1709. doi: 10.1158/1541-7786.MCR-08-0262. [DOI] [PubMed] [Google Scholar]
- 52.Radpour R, Kohler C, Haghighi MM, Fan AX, Holzgreve W, Zhong XY. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene. 2009;28:2969–2978. doi: 10.1038/onc.2009.149. [DOI] [PubMed] [Google Scholar]
- 53.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 54.Toyota M, Ohe-Toyota M, Ahuja N, Issa JP. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. Proc Natl Acad Sci USA. 2000;97:710–715. doi: 10.1073/pnas.97.2.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013;14:16365–16385. doi: 10.3390/ijms140816365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qi J, Zhu YQ, Luo J, Tao WH. Hypermethylation and expression regulation of secreted frizzled-related protein genes in colorectal tumor. World J Gastroenterol. 2006;12:7113–7117. doi: 10.3748/wjg.v12.i44.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, Lutterbaugh J, Rerko RM, Casey G, Issa JP, et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci USA. 2003;100:8412–8417. doi: 10.1073/pnas.1430846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen WD, Han ZJ, Skoletsky J, Olson J, Sah J, Myeroff L, Platzer P, Lu S, Dawson D, Willis J, et al. Detection in fecal DNA of colon cancer-specific methylation of the nonexpressed vimentin gene. J Natl Cancer Inst. 2005;97:1124–1132. doi: 10.1093/jnci/dji204. [DOI] [PubMed] [Google Scholar]
- 59.Longtine MS, DeMarini DJ, Valencik ML, Al-Awar OS, Fares H, De Virgilio C, Pringle JR. The septins: roles in cytokinesis and other processes. Curr Opin Cell Biol. 1996;8:106–119. doi: 10.1016/s0955-0674(96)80054-8. [DOI] [PubMed] [Google Scholar]
- 60.Kim MS, Froese CD, Estey MP, Trimble WS. SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J Cell Biol. 2011;195:815–826. doi: 10.1083/jcb.201106131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee HS, Hwang SM, Kim TS, Kim DW, Park do J, Kang SB, Kim HH, Park KU. Circulating methylated septin 9 nucleic Acid in the plasma of patients with gastrointestinal cancer in the stomach and colon. Transl Oncol. 2013;6:290–296. doi: 10.1593/tlo.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 63.Ogino S, Kawasaki T, Nosho K, Ohnishi M, Suemoto Y, Kirkner GJ, Fuchs CS. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Int J Cancer. 2008;122:2767–2773. doi: 10.1002/ijc.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 65.van’t Veer LJ, Bernards R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008;452:564–570. doi: 10.1038/nature06915. [DOI] [PubMed] [Google Scholar]
- 66.De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, de Rooij LP, de Jong JH, de Boer OJ, van Leersum R, Bijlsma MF, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 67.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Budinska E, Popovici V, Tejpar S, D’Ario G, Lapique N, Sikora KO, Di Narzo AF, Yan P, Hodgson JG, Weinrich S, et al. Gene expression patterns unveil a new level of molecular heterogeneity in colorectal cancer. J Pathol. 2013;231:63–76. doi: 10.1002/path.4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10:e1001453. doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Y, Jatkoe T, Zhang Y, Mutch MG, Talantov D, Jiang J, McLeod HL, Atkins D. Gene expression profiles and molecular markers to predict recurrence of Dukes’ B colon cancer. J Clin Oncol. 2004;22:1564–1571. doi: 10.1200/JCO.2004.08.186. [DOI] [PubMed] [Google Scholar]
- 71.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 72.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 73.Slack FJ, Weidhaas JB. MicroRNAs as a potential magic bullet in cancer. Future Oncol. 2006;2:73–82. doi: 10.2217/14796694.2.1.73. [DOI] [PubMed] [Google Scholar]
- 74.Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 75.Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110:483–495. doi: 10.1161/CIRCRESAHA.111.247452. [DOI] [PubMed] [Google Scholar]
- 76.Weissmann-Brenner A, Kushnir M, Lithwick Yanai G, Aharonov R, Gibori H, Purim O, Kundel Y, Morgenstern S, Halperin M, Niv Y, et al. Tumor microRNA-29a expression and the risk of recurrence in stage II colon cancer. Int J Oncol. 2012;40:2097–2103. doi: 10.3892/ijo.2012.1403. [DOI] [PubMed] [Google Scholar]
- 77.Li Y, Zhang Z. Potential microRNA-mediated oncogenic intercellular communication revealed by pan-cancer analysis. Sci Rep. 2014;4:7097. doi: 10.1038/srep07097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao J, Li N, Dong Y, Li S, Xu L, Li X, Li Y, Li Z, Ng SS, Sung JJ, et al. miR-34a-5p suppresses colorectal cancer metastasis and predicts recurrence in patients with stage II/III colorectal cancer. Oncogene. 2015;34:4142–4152. doi: 10.1038/onc.2014.348. [DOI] [PubMed] [Google Scholar]
- 79.Liu H, Du L, Wen Z, Yang Y, Li J, Wang L, Zhang X, Liu Y, Dong Z, Li W, et al. Up-regulation of miR-182 expression in colorectal cancer tissues and its prognostic value. Int J Colorectal Dis. 2013;28:697–703. doi: 10.1007/s00384-013-1674-0. [DOI] [PubMed] [Google Scholar]
- 80.Huynh C, Segura MF, Gaziel-Sovran A, Menendez S, Darvishian F, Chiriboga L, Levin B, Meruelo D, Osman I, Zavadil J, et al. Efficient in vivo microRNA targeting of liver metastasis. Oncogene. 2011;30:1481–1488. doi: 10.1038/onc.2010.523. [DOI] [PubMed] [Google Scholar]
- 81.Ma Y, Zhang P, Wang F, Zhang H, Yang J, Peng J, Liu W, Qin H. miR-150 as a potential biomarker associated with prognosis and therapeutic outcome in colorectal cancer. Gut. 2012;61:1447–1453. doi: 10.1136/gutjnl-2011-301122. [DOI] [PubMed] [Google Scholar]
- 82.Ma W, Yu J, Qi X, Liang L, Zhang Y, Ding Y, Lin X, Li G, Ding Y. Radiation-induced microRNA-622 causes radioresistance in colorectal cancer cells by down-regulating Rb. Oncotarget. 2015;6:15984–15994. doi: 10.18632/oncotarget.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 84.Harley CB. Telomere loss: mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 85.Wright WE, Shay JW. Telomere dynamics in cancer progression and prevention: fundamental differences in human and mouse telomere biology. Nat Med. 2000;6:849–851. doi: 10.1038/78592. [DOI] [PubMed] [Google Scholar]
- 86.Hahn WC. Role of telomeres and telomerase in the pathogenesis of human cancer. J Clin Oncol. 2003;21:2034–2043. doi: 10.1200/JCO.2003.06.018. [DOI] [PubMed] [Google Scholar]
- 87.Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–179. doi: 10.1038/nrc2275. [DOI] [PubMed] [Google Scholar]
- 88.Zhang A, Wang J, Zheng B, Fang X, Angström T, Liu C, Li X, Erlandsson F, Björkholm M, Nordenskjörd M, et al. Telomere attrition predominantly occurs in precursor lesions during in vivo carcinogenic process of the uterine cervix. Oncogene. 2004;23:7441–7447. doi: 10.1038/sj.onc.1207527. [DOI] [PubMed] [Google Scholar]
- 89.Gisselsson D, Jonson T, Petersén A, Strömbeck B, Dal Cin P, Höglund M, Mitelman F, Mertens F, Mandahl N. Telomere dysfunction triggers extensive DNA fragmentation and evolution of complex chromosome abnormalities in human malignant tumors. Proc Natl Acad Sci USA. 2001;98:12683–12688. doi: 10.1073/pnas.211357798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muraki K, Nyhan K, Han L, Murnane JP. Mechanisms of telomere loss and their consequences for chromosome instability. Front Oncol. 2012;2:135. doi: 10.3389/fonc.2012.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haghighi MM, Aghagolzadeh P, Zadeh SM, Molaei M, Zali MR, Radpour R. Telomere shortening: a biological marker of sporadic colorectal cancer with normal expression of p53 and mismatch repair proteins. Genet Test Mol Biomarkers. 2014;18:236–244. doi: 10.1089/gtmb.2013.0436. [DOI] [PubMed] [Google Scholar]
- 92.Radpour R, Barekati Z, Haghighi MM, Kohler C, Asadollahi R, Torbati PM, Holzgreve W, Zhong XY. Correlation of telomere length shortening with promoter methylation profile of p16/Rb and p53/p21 pathways in breast cancer. Mod Pathol. 2010;23:763–772. doi: 10.1038/modpathol.2009.195. [DOI] [PubMed] [Google Scholar]
- 93.Bisoffi M, Heaphy CM, Griffith JK. Telomeres: prognostic markers for solid tumors. Int J Cancer. 2006;119:2255–2260. doi: 10.1002/ijc.22120. [DOI] [PubMed] [Google Scholar]
- 94.Heaphy CM, Baumgartner KB, Bisoffi M, Baumgartner RN, Griffith JK. Telomere DNA content predicts breast cancer-free survival interval. Clin Cancer Res. 2007;13:7037–7043. doi: 10.1158/1078-0432.CCR-07-0432. [DOI] [PubMed] [Google Scholar]
- 95.Hastie ND, Dempster M, Dunlop MG, Thompson AM, Green DK, Allshire RC. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 96.Takagi S, Kinouchi Y, Hiwatashi N, Chida M, Nagashima F, Takahashi S, Negoro K, Shimosegawa T, Toyota T. Telomere shortening and the clinicopathologic characteristics of human colorectal carcinomas. Cancer. 1999;86:1431–1436. [PubMed] [Google Scholar]
- 97.Gertler R, Rosenberg R, Stricker D, Friederichs J, Hoos A, Werner M, Ulm K, Holzmann B, Nekarda H, Siewert JR. Telomere length and human telomerase reverse transcriptase expression as markers for progression and prognosis of colorectal carcinoma. J Clin Oncol. 2004;22:1807–1814. doi: 10.1200/JCO.2004.09.160. [DOI] [PubMed] [Google Scholar]
- 98.Rial NS, Zell JA, Cohen AM, Gerner EW. Clinical end points for developing pharmaceuticals to manage patients with a sporadic or genetic risk of colorectal cancer. Expert Rev Gastroenterol Hepatol. 2012;6:507–517. doi: 10.1586/egh.12.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Meeker AK, Hicks JL, Gabrielson E, Strauss WM, De Marzo AM, Argani P. Telomere shortening occurs in subsets of normal breast epithelium as well as in situ and invasive carcinoma. Am J Pathol. 2004;164:925–935. doi: 10.1016/S0002-9440(10)63180-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Sullivan J, Risques RA, Mandelson MT, Chen L, Brentnall TA, Bronner MP, Macmillan MP, Feng Z, Siebert JR, Potter JD, et al. Telomere length in the colon declines with age: a relation to colorectal cancer? Cancer Epidemiol Biomarkers Prev. 2006;15:573–577. doi: 10.1158/1055-9965.EPI-05-0542. [DOI] [PubMed] [Google Scholar]
- 101.Raynaud CM, Jang SJ, Nuciforo P, Lantuejoul S, Brambilla E, Mounier N, Olaussen KA, André F, Morat L, Sabatier L, et al. Telomere shortening is correlated with the DNA damage response and telomeric protein down-regulation in colorectal preneoplastic lesions. Ann Oncol. 2008;19:1875–1881. doi: 10.1093/annonc/mdn405. [DOI] [PubMed] [Google Scholar]
- 102.Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ebos JM, Lee CR, Bogdanovic E, Alami J, Van Slyke P, Francia G, Xu P, Mutsaers AJ, Dumont DJ, Kerbel RS. Vascular endothelial growth factor-mediated decrease in plasma soluble vascular endothelial growth factor receptor-2 levels as a surrogate biomarker for tumor growth. Cancer Res. 2008;68:521–529. doi: 10.1158/0008-5472.CAN-07-3217. [DOI] [PubMed] [Google Scholar]
- 104.Tsai HL, Lin CH, Huang CW, Yang IP, Yeh YS, Hsu WH, Wu JY, Kuo CH, Tseng FY, Wang JY. Decreased peritherapeutic VEGF expression could be a predictor of responsiveness to first-line FOLFIRI plus bevacizumab in mCRC patients. Int J Clin Exp Pathol. 2015;8:1900–1910. [PMC free article] [PubMed] [Google Scholar]
- 105.Gerger A, LaBonte M, Lenz HJ. Molecular predictors of response to antiangiogenesis therapies. Cancer J. 2011;17:134–141. doi: 10.1097/PPO.0b013e318212db3c. [DOI] [PubMed] [Google Scholar]
- 106.Hoff PM, Hochhaus A, Pestalozzi BC, Tebbutt NC, Li J, Kim TW, Koynov KD, Kurteva G, Pintér T, Cheng Y, et al. Cediranib plus FOLFOX/CAPOX versus placebo plus FOLFOX/CAPOX in patients with previously untreated metastatic colorectal cancer: a randomized, double-blind, phase III study (HORIZON II) J Clin Oncol. 2012;30:3596–3603. doi: 10.1200/JCO.2012.42.6031. [DOI] [PubMed] [Google Scholar]
- 107.Itzkowitz SH. Cancer prevention in patients with inflammatory bowel disease. Gastroenterol Clin North Am. 2002;31:1133–1144. doi: 10.1016/s0889-8553(02)00047-x. [DOI] [PubMed] [Google Scholar]
- 108.Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–454. doi: 10.1056/NEJM199902113400607. [DOI] [PubMed] [Google Scholar]
- 109.Zhou B, Shu B, Yang J, Liu J, Xi T, Xing Y. C-reactive protein, interleukin-6 and the risk of colorectal cancer: a meta-analysis. Cancer Causes Control. 2014;25:1397–1405. doi: 10.1007/s10552-014-0445-8. [DOI] [PubMed] [Google Scholar]
- 110.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 111.Naka T, Nishimoto N, Kishimoto T. The paradigm of IL-6: from basic science to medicine. Arthritis Res. 2002;4 Suppl 3:S233–S242. doi: 10.1186/ar565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Becker C, Fantini MC, Wirtz S, Nikolaev A, Lehr HA, Galle PR, Rose-John S, Neurath MF. IL-6 signaling promotes tumor growth in colorectal cancer. Cell Cycle. 2005;4:217–220. [PubMed] [Google Scholar]
- 113.Craven B, Zaric V, Martin A, Mureau C, Egan LJ. Effect of genetic deletion or pharmacological antagonism of tumor necrosis factor alpha on colitis-associated carcinogenesis in mice. Inflamm Bowel Dis. 2015;21:485–495. doi: 10.1097/MIB.0000000000000303. [DOI] [PubMed] [Google Scholar]
- 114.Mehta RS, Song M, Bezawada N, Wu K, Garcia-Albeniz X, Morikawa T, Fuchs CS, Ogino S, Giovannucci EL, Chan AT. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J Natl Cancer Inst. 2014;106:dju016. doi: 10.1093/jnci/dju016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Akagi Y, Kinugasa T, Adachi Y, Shirouzu K. Prognostic significance of isolated tumor cells in patients with colorectal cancer in recent 10-year studies. Mol Clin Oncol. 2013;1:582–592. doi: 10.3892/mco.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Utting M, Werner W, Dahse R, Schubert J, Junker K. Microsatellite analysis of free tumor DNA in urine, serum, and plasma of patients: a minimally invasive method for the detection of bladder cancer. Clin Cancer Res. 2002;8:35–40. [PubMed] [Google Scholar]
- 117.Frattini M, Gallino G, Signoroni S, Balestra D, Battaglia L, Sozzi G, Leo E, Pilotti S, Pierotti MA. Quantitative analysis of plasma DNA in colorectal cancer patients: a novel prognostic tool. Ann N Y Acad Sci. 2006;1075:185–190. doi: 10.1196/annals.1368.025. [DOI] [PubMed] [Google Scholar]
- 118.Schwarzenbach H, Stoehlmacher J, Pantel K, Goekkurt E. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann N Y Acad Sci. 2008;1137:190–196. doi: 10.1196/annals.1448.025. [DOI] [PubMed] [Google Scholar]
- 119.Lazarev I, Leibovitch L, Czeiger D, Sion-Vardi N, Geffen DB, Douvdevani A, Ariad S. Cell-free DNA blood levels in colorectal cancer patients do not correlate with mismatch repair-proficiency. In Vivo. 2014;28:349–354. [PubMed] [Google Scholar]
- 120.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, Thornton K, Agrawal N, Sokoll L, Szabo SA, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14:985–990. doi: 10.1038/nm.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lefebure B, Charbonnier F, Di Fiore F, Tuech JJ, Le Pessot F, Michot F, Michel P, Frebourg T. Prognostic value of circulating mutant DNA in unresectable metastatic colorectal cancer. Ann Surg. 2010;251:275–280. doi: 10.1097/SLA.0b013e3181c35c87. [DOI] [PubMed] [Google Scholar]
- 122.Suzuki H, Yamamoto E, Maruyama R, Niinuma T, Kai M. Biological significance of the CpG island methylator phenotype. Biochem Biophys Res Commun. 2014;455:35–42. doi: 10.1016/j.bbrc.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 123.Tan SH, Ida H, Lau QC, Goh BC, Chieng WS, Loh M, Ito Y. Detection of promoter hypermethylation in serum samples of cancer patients by methylation-specific polymerase chain reaction for tumour suppressor genes including RUNX3. Oncol Rep. 2007;18:1225–1230. [PubMed] [Google Scholar]
- 124.Berger F, Reiser MF. Micro-RNAs as potential new molecular biomarkers in oncology: have they reached relevance for the clinical imaging sciences? Theranostics. 2013;3:943–952. doi: 10.7150/thno.7445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008;299:425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu GH, Zhou ZG, Chen R, Wang MJ, Zhou B, Li Y, Sun XF. Serum miR-21 and miR-92a as biomarkers in the diagnosis and prognosis of colorectal cancer. Tumour Biol. 2013;34:2175–2181. doi: 10.1007/s13277-013-0753-8. [DOI] [PubMed] [Google Scholar]
- 127.Huang Z, Huang D, Ni S, Peng Z, Sheng W, Du X. Plasma microRNAs are promising novel biomarkers for early detection of colorectal cancer. Int J Cancer. 2010;127:118–126. doi: 10.1002/ijc.25007. [DOI] [PubMed] [Google Scholar]
- 128.Perilli L, Vicentini C, Agostini M, Pizzini S, Pizzi M, D’Angelo E, Bortoluzzi S, Mandruzzato S, Mammano E, Rugge M, et al. Circulating miR-182 is a biomarker of colorectal adenocarcinoma progression. Oncotarget. 2014;5:6611–6619. doi: 10.18632/oncotarget.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Monzo M, Martínez-Rodenas F, Moreno I, Navarro A, Santasusagna S, Macias I, Muñoz C, Tejero R, Hernández R. Differential MIR-21 expression in plasma from mesenteric versus peripheral veins: an observational study of disease-free survival in surgically resected colon cancer patients. Medicine (Baltimore) 2015;94:e145. doi: 10.1097/MD.0000000000000145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pu XX, Huang GL, Guo HQ, Guo CC, Li H, Ye S, Ling S, Jiang L, Tian Y, Lin TY. Circulating miR-221 directly amplified from plasma is a potential diagnostic and prognostic marker of colorectal cancer and is correlated with p53 expression. J Gastroenterol Hepatol. 2010;25:1674–1680. doi: 10.1111/j.1440-1746.2010.06417.x. [DOI] [PubMed] [Google Scholar]
- 131.Hundt S, Haug U, Brenner H. Blood markers for early detection of colorectal cancer: a systematic review. Cancer Epidemiol Biomarkers Prev. 2007;16:1935–1953. doi: 10.1158/1055-9965.EPI-06-0994. [DOI] [PubMed] [Google Scholar]
- 132.Duffy MJ. Personalized treatment for patients with colorectal cancer: role of biomarkers. Biomark Med. 2015;9:337–347. doi: 10.2217/bmm.15.3. [DOI] [PubMed] [Google Scholar]
- 133.Osborn NK, Ahlquist DA. Stool screening for colorectal cancer: molecular approaches. Gastroenterology. 2005;128:192–206. doi: 10.1053/j.gastro.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 134.Dhaliwal A, Vlachostergios PJ, Oikonomou KG, Moshenyat Y. Fecal DNA testing for colorectal cancer screening: Molecular targets and perspectives. World J Gastrointest Oncol. 2015;7:178–183. doi: 10.4251/wjgo.v7.i10.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Diehl F, Schmidt K, Durkee KH, Moore KJ, Goodman SN, Shuber AP, Kinzler KW, Vogelstein B. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology. 2008;135:489–498. doi: 10.1053/j.gastro.2008.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zou H, Taylor WR, Harrington JJ, Hussain FT, Cao X, Loprinzi CL, Levine TR, Rex DK, Ahnen D, Knigge KL, et al. High detection rates of colorectal neoplasia by stool DNA testing with a novel digital melt curve assay. Gastroenterology. 2009;136:459–470. doi: 10.1053/j.gastro.2008.10.023. [DOI] [PubMed] [Google Scholar]
- 137.Coppedè F, Lopomo A, Spisni R, Migliore L. Genetic and epigenetic biomarkers for diagnosis, prognosis and treatment of colorectal cancer. World J Gastroenterol. 2014;20:943–956. doi: 10.3748/wjg.v20.i4.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.A stool DNA test (Cologuard) for colorectal cancer screening. JAMA. 2014;312:2566. doi: 10.1001/jama.2014.15746. [DOI] [PubMed] [Google Scholar]