

Abstract
CFTR, the cystic fibrosis (CF) gene, encodes for the CFTR protein that plays an essential role in anion regulation and tissue homeostasis of various epithelia. In the gastrointestinal (GI) tract CFTR promotes chloride and bicarbonate secretion, playing an essential role in ion and acid–base homeostasis. Cftr has been identified as a candidate driver gene for colorectal cancer (CRC) in several Sleeping Beauty DNA transposon-based forward genetic screens in mice. Further, recent epidemiological and clinical studies indicate that CF patients are at high risk for developing tumors in the colon. To investigate the effects of CFTR dysregulation on GI cancer, we generated ApcMin mice that carried an intestinal-specific knockout of Cftr. Our results indicate that Cftr is a tumor suppressor gene in the intestinal tract as Cftr mutant mice developed significantly more tumors in the colon and the entire small intestine. In Apc+/+ mice aged to ~ 1 year, Cftr deficiency alone caused the development of intestinal tumors in >60% of mice. Colon organoid formation was significantly increased in organoids created from Cftr mutant mice compared with wild-type controls, suggesting a potential role of Cftr in regulating the intestinal stem cell compartment. Microarray data from the Cftr-deficient colon and the small intestine identified dysregulated genes that belong to groups of immune response, ion channel, intestinal stem cell and other growth signaling regulators. These associated clusters of genes were confirmed by pathway analysis using Ingenuity Pathway Analysis and gene set enrichment analysis (GSEA). We also conducted RNA Seq analysis of tumors from Apc+/+ Cftr knockout mice and identified sets of genes dysregulated in tumors including altered Wnt β-catenin target genes. Finally we analyzed expression of CFTR in early stage human CRC patients stratified by risk of recurrence and found that loss of expression of CFTR was significantly associated with poor disease-free survival.
INTRODUCTION
CFTR, a cyclic adenosine monophosphate-regulated anion channel, is expressed at high levels in various epithelia, including the mucosa of the intestinal tract. CFTR protein is expressed in the apical membrane of enterocytes where it acts as the dominant ion channel transporter in the intestinal crypt epithelium. Essential functions of CFTR include chloride and bicarbonate secretion, and maintenance of fluid homeostasis.1 Within the small intestine, CFTR expression is the strongest in the duodenum, including high expression in the mucus and bicarbonate-secreting Brunner’s glands.1 In the large intestine, CFTR expression is considered moderate. Along the crypt–villi axis, CFTR expression is highest in the crypts of the small intestine and near the crypt bases of the large intestine. Mutations in the CFTR gene are causative for cystic fibrosis (CF),2 the most common autosomal recessive disorder in Caucasians. CF patients develop a range of dysfunctions in the gastrointestinal (GI) tract, including deficient anion (Cl− and HCO−3) and fluid transport; altered cellular pH; an altered luminal environment with impaired secretion, release and clearance of mucus leading to meconium ileus and obstruction in the distal ileum and proximal large intestine; and abnormal bacterial colonization, microbial dysbiosis and abnormal innate immune responses that lead to chronic inflammation.1
Downregulated expression and function of ion channels and transporters is observed in virtually all cancers.3 Thus, with its highly important role in the normal physiology of various organs such as the GI tract, disruption of CFTR function and/or dysregulation of CFTR expression is associated with a wide array of cancers including esophageal, breast, gastric, hepatobiliary, gall bladder, prostate, pancreatic, small intestine and colorectal cancers (CRC).4–11 Furthermore, a 20-year study in CF patients revealed an increased risk of digestive tract cancers, with large numbers of cancers detected in the small intestine, colon and biliary tract.6 Downregulation of CFTR mRNA gene expression was also included in a prognostic predictor gene set for poor disease-free survival (DFS) in CRC.12 Finally, in a recent study conducted at the University of Minnesota early colon screening of adult CF patients revealed a high incidence of colon tumors, especially in males.13
Mouse models of Cftr deficiency have proven invaluable in understanding the role of CFTR in the normal physiology of the GI tract and in disease pathogenesis as human CF patients and Cftrfl/fl-Villin-Cre knockout mice share very similar intestinal disease pathophysiology.14 In studies of CRC, Cftr was identified as a common insertion site candidate GI tract cancer gene in multiple Sleeping Beauty (SB) DNA transposon-based forward mutagenesis screens in mouse intestine. In an Apc wild-type screen, Cftr mutations were found in eight tumors (6%)15 where SB transposon insertions were found in both the forward and reverse strand orientation, consistent with a well-accepted model that the SB transposon acts to disrupt function of the gene (Supplementary Figure S1). An SB screen on the ApcMin background found Cftr mutations in 102 tumors (23%), ranking it #70 of 641 common insertion site genes.16 Most recently, Cftr was identified as a common insertion site gene in a set of SB screens in Apc-, SMAD4-and p53-deficient mice.17 Finally, an SB screen in a p53R27OH mutant background identified Cftr mutations in 12 tumors (20%) (TK Starr, personal communication).
To confirm the role of CFTR in GI cancer we examined CFTR deficiency in targeted mouse models and in human CRC patients. We measured GI cancer phenotypes in ApcMin mice carrying an intestinal-specific knockout of Cftr. We also examined the tumor phenotype of Apc+/+ Cftrfl/fl-Villin-Cre mice that were aged to ~ 1 year. We performed global gene expression profiling using RNA isolated from normal colon and small intestine of Cftrfl/fl-Villin-Cre mice plus tumors from ApcMin Cftrfl/fl-Villin-Cre mice and Apc+/+ Cftrfl/fl-Villin-Cre mice. We identified gene sets and potential signaling pathways associated with the loss of Cftr. Finally, we examined the correlation between CFTR mutations and maintenance of CFTR expression and DFS in CRC patients.
RESULTS
Lethality phenotype of Cftr mutant mice
The intestinal-specific conditional Cftr knockout mice employed in our study18 exhibited phenotypes previously attributed to Cftr constitutive and intestinal-specific knockout mice,19,20 including inflammation in the proximal large intestine, reduced growth and body weight, and perinatal lethality due to intestinal obstruction caused by mucus accumulation.14,18,21 These phenotypes were strongest in male mice. Less than 15% of the expected Cftrfl/fl-Villin-Cre mice were observed at weaning, and survival of these mice post-weaning was only made possible by the addition of an oral osmotic laxative to the drinking water, resulting in >50% survival, which is significantly higher than in Cftr constitutive knockout mice. Cftrfl/fl-Villin-Cre mice aged to ~ 1 year also exhibited increased length and size of the distal small intestine as has been observed previously.14
Loss of Cftr enhances tumor multiplicity in ApcMin mice
To investigate the role of Cftr in mouse GI cancer, Cftrfl/fl and Villin-Cre transgenic alleles were introgressed into the ApcMin model of GI cancer generating ApcMin test mice that were either wild type, heterozygous or homozygous mutant for Cftr. ApcMin Cftr mutant mice developed significantly more tumors than ApcMin Cftr wild-type mice. This phenotype was observed in both Cftr heterozygous and Cftr homozygous mutants, with the tumor phenotype strongest in Cftr homozygous mutants. The increase in tumor multiplicity occurred in both the small intestine (SI) and the colon (Table 1). The rectum of some ApcMin Cftrfl/fl-Villin-Cre mice (~15%) demonstrated adenomatous rectal hyperplasia and regions of focal invasion (Figure 1). Histological examination of an animal with rectal prolapse (Figure 1a) showed locally extensive adenomatous hyperplasia with disorganization, irregular tortuosity and branching of crypts (Figure 1b); focal invasion of the submucosa was present suggestive of malignant transformation (Figures 1b and c). As all test mice were initially scheduled for killing at 120 days and the ApcMin Cftrfl//fl−Villin-Cre mice became moribund as early as 75 days (mean ~ 90 days) it was not possible to conduct an accurate comparative analysis of tumor sizes; however, tumors in the Cftrfl/fl-Villin-Cre mice appeared to be larger than expected at ~ 90 days.
Table 1.
Cftr deficiency enhances intestinal tumor multiplicity in ApcMin mice
| Class | Gender | N | Age | Colon | Colon incidence | SI-1 | SI-2 | SI-3 | SI-4 | Total tumors |
|---|---|---|---|---|---|---|---|---|---|---|
| Cftr+/+ | F | 55 | 120 | 1.5 ±1.9 | 56% | 9.2 | 21.1 | 53.7 | 42.4 | 127.9 ±47 |
| Cftrfl/+-Villin-Cre | F | 22 | 118 | 4.6 ±3.7a | 95% | 10.3 | 20.6 | 65.8 | 52.1 | 153.4 ±47a |
| Cftrfl/fl-Villin-Cre | F | 13 | 91 | 8.9 ±6.7a | 100% | 20.0 | 30.0 | 71.6 | 76.1 | 206.6 ±75a |
| Cftr+/+ | M | 92 | 120 | 3.3 ±3.4 | 85% | 7.1 | 11.2 | 46.0 | 37.7 | 105.3 ±43 |
| Cftrfl/+-Villin-Cre | M | 22 | 110 | 4.4 ±2.4 | 91% | 10.8 | 19.3 | 55.5 | 50.0 | 140.0 ±65a |
| Cftrfl/fl-Villin-Cre | M | 11 | 91 | 8.5 ±6.9a | 100% | 16.5 | 19.8 | 66.7 | 73.2 | 184.7 ±30a |
P-values: <0.05; two-sided P-values for tumor counts were determined by use of the Wilcoxon rank sum test comparing gender and age-matched classes produced in the same genetic crosses. Mice were generated in crosses between females that were Cftrfl/fl, Cftrfl/+ or Cftrfl/+-Villin-Cre × males that were ApcMin Cftrfl/fl, ApcMin Cftrfl/+ or ApcMin Cftrfl//+ Villin-Cre. Cftr+/+ mice included mice with the genotypes Cftr+/+, Cftrfl/+ and Cftrfl/fl. Cftr heterozygous mice had the genotype Cftrfl/+-Villin-Cre+ and Cftr intestinal-specific knockout mice had the genotype Cftrfl/fl-Villin-Cre. All mice were on the C57BL/6 genetic background. SI-1 through SI-4 represent four equal sized segments of the small intestine, proximal to distal, with SI-1 containing the duodenum and proximal jejunum, SI-2 containing the remainder of the jejunum, and SI-3 and SI-4 containing the ileum. ApcMin Cftr+/+, ApcMin Cftrfl/+-Villlin-Cre and ApcMinCftrfl/fl-Villin-Cre mice were killed at 120 days of age or when moribund and tumors were counted in formalin-fixed tissues.
Figure 1.

Tumor in the rectum of ApcMin Cftrfl/fl-Villin-Cre mouse. Diffuse mucosal thickening in prolapsed rectal mucosa is shown in (a) (bar =5 mm). There is adenomatous hyperplasia with disorganization, irregular tortuosity and branching of crypts with focal invasion into the submucosa (arrow) (panel b, bar = 200 μm); focal invasion of the submucosa by a proliferating tubule is indicated by an arrow (panel c, bar =100 μm).
Loss of Cftr causes intestinal tumorigenesis in Apc+/+ mice
Upon aging a cohort of Apc+/+ Cftrfl/fl-Villin-Cre mice to 12 months, we found that loss of Cftr alone could induce tumor formation in both the colon and small intestine, with a penetrance >60% (multiplicity 1.6; Table 2), compared with zero tumor incidence in control mice. Representative intestinal tumors were examined by light microscopy; in all cases they were diagnosed as adenomas with occasional microadenomas (Figure 2). These tumors were found throughout the intestinal tract with the highest number found in the proximal SI.
Table 2.
Intestinal tumorigenesis in Apc+/+ Cftr knockout mice
| Class | N | Mean age (days) | Tumor penetrance (%) | Tumor multiplicity | Total tumors in colon | Total tumors in small intestine |
|---|---|---|---|---|---|---|
| Cftr+/+ | 15 | 360 | 0 | 0 | 0 | 0 |
| Cftrfl/fl-Villin-Cre | 18 | 375 | 61 | 1.6 | 2 | 15 |
C57BL/6-Apc+/+ Cftrfl/fl-Villin-Cre+ mice and a contemporaneous control set of C57BL/6 Cftr wild-type mice, of roughly equal numbers by gender, were aged to ~ 1 year, then killed. Tumors were counted in formalin-fixed tissues from throughout the intestinal tract using a dissecting microscope. A subset of tumors was removed fresh frozen for RNA analysis and another subset was removed as formalin-fixed tissues for histopathological and immunohistochemical analysis. As has been observed before, no tumors were found in C57BL/6 Cftr wild-type control mice.
Figure 2.
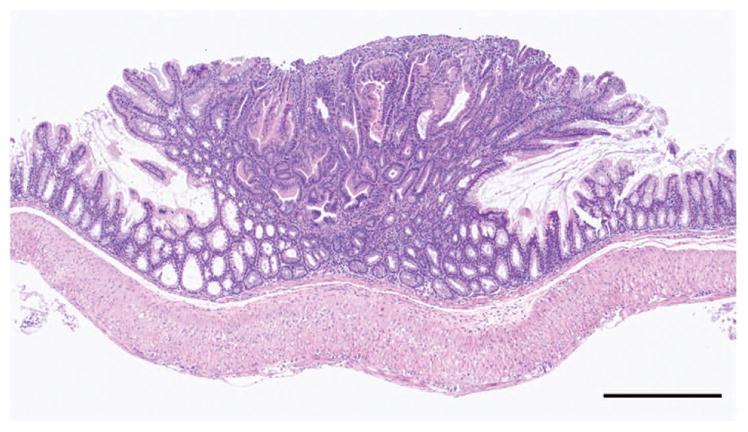
Tumor in the small intestine of Apc+/+ Cftrfl/fl-Villin-Cre mouse. A polypoid adenoma (bar = 500 μm).
Loss of Cftr alters gene expression in normal colon and SI
Microarray gene expression studies in the colon and the proximal SI of Apc+/+ Cftr+/+ and Apc+/+ Cftrfl/fl-Villin-Cre mice identified more than 100 genes that showed a >1.5-fold change in expression (with a P-value <0.05) in the SI and 58 in the colon. The top up-and downregulated genes are listed in Table 3 (SI) and Table 4 (colon) (see Supplementary Table S1 for full list). Expression of 12 of these genes, from both SI (Figure 3a) and colon (Figure 3b), was confirmed by reverse transcriptase–PCR. Among top gene clusters identified are immune cell homeostasis (Reg3g, Reg3b), mucins, inflammation (Retnlb, S100A11), growth regulation and cell signaling (Areg), migration and invasion, lipid metabolism, stress response and detoxification (nine cytochrome p450 genes), water homeostasis (Aqp4) and stem cell regulation (Aldh1a1, Phlda1). Ingenuity Pathway Analysis (IPA) on the microarray data sets of normal SI and colon in wild-type and Cftr-deficient samples identified several major networks. In the SI these include genes involved in drug metabolism–small-molecule biochemistry–lipid metabolism and molecular transport–GI disease–inflammatory disease (Supplementary Figures S2A and B). In the colon, major networks included molecular transport–lipid metabolism–small-molecule biochemistry and cellular growth/proliferation–renal and urological system development and function (Supplementary Figures S2C and D). Among top biological functional groups are altered genes that play important roles in immune trafficking. We have previously reported functional overlap between the ion channel genes Cftr and Kcnq1 in the intestinal crypt.22 In our current analysis, gene set enrichment analysis (GSEA) of microarray data from normal colon and normal SI showed overlapping changes in gene expression in Kcnq1 and Cftr-deficient mice (Supplementary Figures S3A and B). Moreover, GSEA also showed overlap between both Cftr and Kcnq1 in the Mucin 2 protective pathway (Supplementary Figure S3C) as gene expression changes in Kcnq1 and Cftr-deficient mice overlap with those in Muc2 deficient mice.22
Table 4.
Top upregulated and downregulated genes identified by microarray in normal colon of Apc+/+ Cftrfl/fl-Villin-Cre mice
| Top upregulated genes | Top downregulated genes |
|---|---|
| Defa6 | Sycn |
| Defa3 | Gsdmc |
| Slpi | Clps |
| Reg3g | Fxyd4 |
| Clca4 | Ces1d |
| Defa-rs1 | Rn18s |
| Lyz1/Lyz2 | Cyp4b1 |
| C3 | Actb |
| Reg3a | Prlr |
| Clca6 | Pbld |
Figure 3.

(a) Quantitative reverse transcriptase PCR (qRT–PCR) gene expression analysis of mouse normal small intestine of Apc+/+ Cftrfl/fl-Villin-Cre mice. Each gene sample was run in triplicate and gene expression was normalized to the expression of 18S. Data are presented as the mean fold change ±s.d. Each bar represents the mean and s.e. of multiple experiments that measured fold differences in the mRNA expression in proximal small intestine tissue isolated from adult (~100 days) littermate and gender matched pairs of Apc+/+ Cftrfl/fl-Villin-Cre and Cftr+/+ mice. mRNAs were isolated from 1 cm sections of the proximal small intestine from the same region for all mice. Villi were removed from the tissue prior to processing. Four replicates of each assay were performed for each matched pair of mRNAs and these sets of assays were repeated at least two times for each pair of mRNAs. At least two matched pairs of mRNAs were tested for each gene with most genes tested in at least three matched pairs of mRNAs. To be included in this figure genes met the flowing criteria: (1) the mean fold difference was at least 1.5; and (2) each gene showed a change in gene expression in the same direction in each matched pair of mRNAs. In all cases the direction of changes in gene expression confirmed microarray data. *P<0.05. (b) qRT–PCR gene expression analysis of mouse normal colon of Apc+/+ Cftrfl/fl-Villin-Cre mice. Samples were analyzed in triplicate and normalized to 18S ribosomal RNA. Data are presented as the mean fold change ± s.d. Each bar represents the mean and s.e. of multiple experiments that measured fold differences in the mRNA expression of whole colon tissue isolated from adult (~100 days), littermate and gender matched pairs of Apc+/+ Cftrfl/fl-Villin-Cre and Cftr+/+ mice. RNA was isolated from 1 cm sections from the same region of distal colon. Four replicates of each assay were performed for each matched pair of mRNAs and these sets of assays were repeated at least two times. At least two matched pairs of mRNAs were tested for each gene with most genes tested in at least three matched pairs of mRNAs. To be included in this figure genes met the flowing criteria: (1) the mean fold difference was at least 1.5; and (2) each gene showed a change in gene expression in the same direction in each matched pair of mRNAs. In all cases the direction in changes in gene expression confirmed microarray data. *P<0.05.
Loss of Cftr alters gene expression in tumors
mRNAs from adenomas from the SI and colon of Apc+/+ Cftrfl/fl-Villin-Cre mice were analyzed by RNA Seq and compared with mRNAs from adenomas isolated from SI and colon from ApcMin Cftr+/+ and ApcMin Cftrfl/fl-Villin-Cre mice, along with mRNAs from Apc+/+ Cftr+/+ and Apc+/+ Cftrfl/fl-Villin-Cre normal colon and SI. A full list of all genes that changed two-fold is included in Supplementary Table S2. Analysis of Apc+/+ Cftrfl/fl-Villin-Cre tumors identified several clusters of genes that are distinct from the Cftrfl/fl-Villin-Cre sets identified in normal tissues. The most notable clusters were upregulation of Wnt/β-catenin target genes (Ccnd1, CD44, Axin2, Lgr5, Mmp7, Wnt10A and Ptgs2; Supplementary Table S12) and altered expression of genes associated with the intestinal stem cell (ISC) compartment (Lgr5, CD44, Aldh1a1, Aqp4, Ascl2 and Hopx; Supplementary Table S13). GSEA comparing Apc+/+ Cftrfl/fl-Villin-Cre tumors with all other classes identified gene sets associated with EphB2 ISCs and WNT target genes23–26 (Supplementary Figure S4), with a strong overlap in core genes enriched in ApcMin Cftr+/+ tumors (Supplementary Table S14). A further confirmation for a role for Cftr in the stem cell compartment was demonstrated by an enhanced 2.3-fold colon organoid growth in Cftrfl/fl-Villin-Cre mice (Supplementary Figure S5A). These organoids demonstrated no differences in morphology compared with controls (Supplementary Figure S5B). GSEA of Apc+/+ Cftrfl/fl-Villin-Cre tumors also found a strong enrichment for ISC genes that are dysregulated in ApcMin Cftr+/+ tumors (Supplementary Figure S6), further supportive of these tumors developing via a dysregulated Wnt/β-catenin signaling pathway (Supplementary Figure S7). Interestingly, GSEA also found an overlap in ApcMin Cftr+/+ tumors for genes that are differentially expressed in normal colon and SI of Cftr knockout tissues. To confirm the upregulation of β-catenin in tumors from Apc+/+-Cftrfl/fl-Villin-Cre mice we conducted immunohistochemistry and found upregulation of cytoplasmic β-catenin and enhanced nuclear localization (Supplementary Figure S8). Finally, IPA analysis was conducted to compare changes in gene expression in Cftr-deficient tumors and Cftr-deficient normal tissues to identify overlapping pathways that are further activated in tumors. Significant overlap was observed in the LXR/RXR (lipid metabolism) and toll-like receptor (innate immune response) pathways, with the strongest Z-scores linked to genes involved in NF-κB inflammatory responses, including members of the IL1β and its receptor family (Il1β, I1rl1, Il1rrn, St2l), Ptgs2 and Tnfrsr11B (an upstream activator of NF-κB) (Supplementary Figures S9A and B). Low CFTR expression is associated with poor DFS in CRC patients We analyzed DFS in a set of stage II CRC samples (AMC-AJCCII-90),27 stratified by CFTR-high and CFTR-low mRNA expression. The Kaplan–Meier analysis and log-rank test were used to correlate CFTR expression with DFS. We found that in the complete AMC90 set the 3-year relapse free survival was 85% in the CFTR high-expressing group compared with 56% in the CFTR low-expressing group (P = 0.02, log-rank test; Figure 4a). Moreover, in the MSS subset the 3-year survival was 85% in the CFTR high-expressing group compared with 38% in the CFTR low-expressing group (P = 0.002, log-rank test) (Figure 4b). In addition, the multivariate analysis showed that CFTR expression is an independent prognostic determinant (Table 5). Further, based on the mouse tumor pathway analyses, we examined the expression levels of several genes involved in inflammatory/innate response pathways in the CFTR low-expressing group and found a significant correlation with higher levels of IL1β, XBP1 and NFKB2 in the CFTR low tumors, similar to that observed in the mouse tumors (Supplementary Figure S10).
Figure 4.

Kaplan–Meier estimate of disease-free survival (DFS) in the AMC-AJCCII-90 set between CFTR low and CFTR high-expressing tumors. Disease-free survival within the AMC-AJCCII-90 set stratified by CFTR expression of the tumor in the complete set (a) and in the MSS subset (b). The 3-year disease-free survival for the complete set (a) was 85% in the CFTR high group and 56% in the CFTR low group. In the MSS set (b) the 3-year disease-free survival was 85 and 38% in the CFTR high group and CFTR low group, respectively. P-values are based on log-rank test.
Table 5.
Cox regression analysis. Univariate (top) and multivariate (bottom) regression analysis of prognostic features in the AMC-AJCCII-90 set
| P-value | Hazard ratio | 95% CI | |
|---|---|---|---|
| Univariate analysis | |||
| Location | 0.884 | 1.07 | 0.430–2.662 |
| Differentiation | 0.580 | ||
| Differentiation (1) | 0.297 | 1.643 | 0.646–4.179 |
| T-stage | 0.320 | 0.534 | 0.155–1.840 |
| MSI | 0.968 | 1.021 | 0.368–2.836 |
| CFTR expression | 0.028 | 2.775 | 1.114–6.910 |
| Multivariate analysis | |||
| Location | 0.909 | 1.063 | 0.374–3.021 |
| Differentiation | 0.808 | ||
| Differentiation (1) | 0.514 | 1.398 | 0.512–3.820 |
| T-stage | 0.379 | 0.57 | 0.163–1.996 |
| MSI | 0.209 | 2.289 | 0.628–8.340 |
| CFTR expression | 0.022 | 3.584 | 1.205–10.664 |
The univariate and multivariate analysis both show that CFTR expression is an important prognostic determinant in the AMC-AJCCII-set.
DISCUSSION
Several overlapping phenomena related to the pathophysiology of CFTR deficiency in the GI tract have been proposed to explain the susceptibility to CRC. Foremost is the inflammatory landscape in the GI tract observed in CF patients, a phenotype that is modeled well in Cftr intestinal-specific knockout mice. In both human CF patients and Cftr knockout mice there is impaired mucosal barrier function, including disrupted tight junctions,28 that leads to compromised resistance to bacterial colonization,29 infection, and abnormal innate and adaptive immune responses and inflammation, factors that together may promote oncogenesis.1,21,30,31 Increased epithelial permeability is also a major factor in bicarbonate loss in the inflamed colon.32 At the molecular level, CFTR has been reported to be a negative regulator of the proinflammatory NFKB-mediated innate immune responses, including IL8,33 and the COX2-PGE2 positive feedback loop in inflammation.34
Consistent with this, our microarray studies in normal mouse SI and colon, along with IPA and GSEA pathway analyses, showed dysregulation of signaling pathways involving immune mediation and inflammation. RNA Seq analyses of Cftr-deficient intestinal tumors confirmed an altered proinflammatory gene expression profile in both normal Cftr-deficient tissues and in Cftr-deficient tumors. An example are the numerous members of the S100A gene family found to be upregulated, including S100A4, −A6, −A9, −A11, −A14 and −A16, all genes associated with inflammation and progression of human cancers. Another example is the 13-fold increase in expression of the proinflammatory oncogenic Ptgs2 gene (Supplementary Table S12). Finally, Cftr deficiency in tumors caused the upregulation of numerous proinflammatory chemokines, cytokines and their receptors, and members of the NF-kB signaling pathway. Examples included several CCL chemokines (e.g., CCL2 (MCP-1)), five CXC chemokine receptors and their ligands, four CCR chemokine receptors (e.g., CCR5), eight members of the TNF family, three toll-like receptor (TLR) genes and the Rel transcription factor (Supplementary Table S2).
Previous studies in KO mice found an overlap in gene sets from Cftr, Kcnq1 and Muc2 deficient mice.22 This was confirmed by GSEA (Supplementary Figures S3A and B) of normal Cftrfl/fl-Villin-Cre tissues. For example, several members of the regenerating islet-derived gene (Reg3) family, which are involved in innate immune responses in the intestinal tract, were dysregulated in all three KO mouse models22,35 (and Tables 3 and 4). CFTR and KCNQ1 closely cooperate in maintaining ion and fluid homeostasis in the intestine, thereby facilitating proper processing of MUC2, and preventing mucus dehydration and related mucus plugging, intestinal obstruction and resultant inflammation. In particular, mucus and Muc2 are important for immunomodulatory signaling.36 In Cftr knockout mice Muc2 protein is secreted but remains attached to the epithelium, and thus unable to form a mucus gel. This defect is proposed to be due to poor mucus folding caused by the absence of bicarbonate.14,37,38 Therefore, regulation of chloride and bicarbonate secretion is critical to intestinal homeostasis. Chloride secretion is a complex mechanism that involves several ion transporters, including K+ and Cl− channels. Potassium ion recycling, via KCNQ1, across the basolateral membrane is the electrochemical driving force for Cl− to be secreted across the apical membrane through CFTR. Chloride secretion in turn also identifies CFTR and KCNQ1 as important regulators of water homeostasis. Aquaporins are water channels that play very important roles in normal intestinal function and several water channel genes, including aquaporins (Aqp1, Aqp4), were found to be dysregulated in microarray studies in Kcnq122 and Cftrfl/fl-Villin-Cre mice (Supplementary Table S1). Notably, Aqp4 was found to be significantly downregulated (by 85%) in tumors arising in Apc+/+ Cftrfl/fl-Villin-Cre mice (Supplementary Table S13).
Table 3.
Top upregulated and downregulated genes identified by microarray in the normal small intestine of Apc+/+ Cftrfl/fl-Villin-Cre mice
| Top upregulated genes | Top downregulated genes |
|---|---|
| Reg3g | Cfd |
| Reg3a | Scd |
| Amy2a | Defa1 |
| Retnlb | Igk-V8 |
| Anxa4 | Cyp2C9 |
| B3galt5 | Lpl |
| Muc3a | Slc5a4b |
| Cela3a | Adipoq |
| Defa3 | Reg3g |
| Cldn4 | Aldh1a1 |
Both Kcnq1 (in situ hybridization25) and Cftr (single-cell gene expression studies39) have been implicated in regulation of the ISC compartment. For Cftr, single-cell analysis of colon cell heterogeneity reported that Cftr was part of a gene signature for a stem/progenitor cell subset of EpCAMhigh/CD44+ cells that was Lgr5+/Ascl2+ and which also included Aqp1.39 These findings have now been supported by our RNA Seq analysis of tumors from Apc+/+ Cftrfl/fl-Villin-Cre mice, which found a number of canonical ISC genes dysregulated (Supplementary Table S13); by GSEA (Supplementary Figure S4 and Supplementary Table S14) of the same data sets; and by enhanced organoid growth in Cftrfl/fl-Villin-Cre colon organoids (Supplementary Figure S5). How CFTR may alter ISC dynamics is unknown but there is strong evidence that the microenvironment is a key regulator of ISCs, including their capacity for malignant transformation.40,41
Model of Cftr-deficient tumorigenesis
Tumors that developed in Apc+/+ Cftrfl/fl-Villin-Cre mice were found to express elevated levels of Wnt/β-catenin target genes indicating that CFTR-deficient tumors likely arise from activation of β-catenin. This finding was supported by GSEA of RNA Seq data from these tumors (Supplementary Figure S6) that found a significant overall enrichment for genes dysregulated in ApcMin Cftr+/+ tumors, in particular in Wnt target genes (Supplementary Figure S7) and upregulation of cytoplasmic and nuclear β-catenin in the tumors (Supplementary Figure S8). Accordingly, a model for tumor development in both humans and mice deficient for CFTR would include long-term chronic inflammation associated with microbial dysbiosis, leading to altered innate (Supplementary Figures S9 and S10) and adaptive immune responses, and dysregulation of the stem cell compartment. Loss of CFTR may cooperate with defects in other ion channels genes, ultimately leading to activation of β-catenin. This model is similar to that proposed for tumor development in AOM-DSS-treated mice, which is also characterized by chronic inflammation and activation of β-catenin.
The identification of CFTR as a prognostic predictor of DFS in CRC patients is fully consistent with epidemiological6,7 and clinical13 data indicating that CF patients are at an increased risk for CRC. Our finding of a correlation between CFTR mRNA expression and DFS for CRC is also consistent with at least one other study that included downregulation of CFTR expression in a prognostic predictor set for CRC.12 It is also consistent with a report that CFTR was a signature gene and a biomarker for a type of transit amplifying CRC associated with good DFS.42 It was also reported that CFTR expression was lower in the stem cell-like CRC subset and lowest in the inflammatory CRC class.
The data are also supportive of functional links between CFTR, KCNQ1 and MUC2 as both Kcnq1 and Muc2 have been shown to act as tumor suppressors in the GI tract of mice,22,43,44 similar to Cftr. Notably, maintenance of KCNQ1 protein expression is associated with increased DFS in stage IV CRC patients,22 like CFTR, and KCNQ1 mRNA and protein expression is associated with DFS in stage II and III CRC patients (RJA Fijneman and L Vermeulen, personal communication).
The stratification of human CRCs into high and low expression of CFTR in our study is consistent with potential epigenetic silencing of CFTR in these cancers, which is supported by reports that the promoter of CFTR is GC rich and that CFTR is frequently hypermethylated in both cancer cell lines (promyelocytic leukemia)45 and primary non-small-cell lung cancer samples.46 It is important to note that ~ 4% of the Caucasian population carry CFTR mutations. Therefore, epigenetic silencing could be one means for LOH at the CFTR locus.
Therapies under development to treat complications in the GI tract of CF patients, such as correctors and potentiators, may be effective in preventing or treating GI tract cancers.3,47,48 Further studies to understand the mechanisms underlying CFTR’s tumor resistance can lead to its application in the clinic as a prognostic predictor and therapeutic target in CRC.
MATERIALS AND METHODS
Mice
C57BL/6J, C57BL/6-Villin-Cre and C57BL/6J-ApcMin mice have been maintained in our mouse colony. C57BL/6 Cftrfl10 mice18 were obtained from Drs Mitchell Drumm & Craig Hodges (CWRU). To bypass early lethality49 a commercially available osmotic laxative was added to the drinking water (Gavilyte-C; Gavis Pharmaceuticals, Somerset, NJ, USA). The genotype of the Apc,50 Villin-Cre50 and Cftr18 loci were determined by PCR assays. All animal experiments were conducted per protocol approved by the UM IACUC.
Tumor multiplicity analysis
Scoring of formalin-fixed intestinal tumor tissues blinded to genotype was performed as previously described.50 Two-sided P-values for tumor counts were determined by use of the Wilcoxon rank sum test comparing gender and age-matched classes produced in the same genetic crosses.
Histopathology
Histopathological analysis of tumors and adjoining normal tissue was performed on tissues that were fixed in 10% neutral buffered formalin, routinely processed into paraffin, sectioned at a thickness of 4 μm, stained with hematoxylin and eosin and examined using light microscopy. All tissues were analyzed blind to the genotype of the samples by an A.C.V.P.-certified veterinary pathologist (MGO’S) from the University of Minnesota Masonic Cancer Comparative Pathology Shared Resources facility, and using the standardized nomenclature of the 2003 Consensus Report and Recommendations for pathology of mouse models of intestinal cancer as published.50,51
Immunohistochemistry for β-catenin
Four-micrometer formalin-fixed, paraffin-embedded sections of small intestine tumors were deparaffinized and rehydrated, followed by antigen retrieval using 10 mM Citrate buffer pH 6.0 in a steamer. Immunohistochemistry for β-catenin was performed on a Dako Autostainer using a rabbit anti-human β-catenin monoclonal antibody (Abcam, Cambridge, MA, USA; catalog # AB32572) as primary antibody (after blocking endogenous peroxidase and application of a protein block). Detection was achieved using a rabbit EnVision+ Kit (catalog K4011; Dako, Carpinteria, CA, USA) with diaminobenzidine (Dako) as the chromogen. Mayer’s hematoxylin (Dako) was used as the counterstain. Small intestinal adenomas from ApcMin mice were used as a positive control tissue, and for negative control slides the primary antibody was substituted with Negative Control Rabbit IgG (Biocare, Tempe, AZ, USA).
Epithelial RNA processing for illumina bead arrays
Mouse SI and colon tissues were opened longitudinally, and rinsed in phosphate-buffered saline. For the SI, villi were removed from the distal quarter of the proximal quarter, and tissue was then flash frozen in liquid nitrogen. Tissue was transferred from liquid nitrogen to pre-cooled RNALaterIce (Ambion/Life Technologies, Grand Island, NY, USA) at −80 °C for 30 min, and then was transferred to −20 °C for at least 48 h. Immediately before RNA isolation, tissue in either RNALater or RNALaterIce was placed in 2–4 ml of RLT/14.3 M β-mercaptoethanol buffer. Tissue was then homogenized using an IKA Ultra-Turrax T25 digital homogenizer (Fisher Scientific, Pittsburgh, PA, USA). RNA isolation was then conducted using an RNeasy Mini Kit with an additional DNAse Digestion step (Qiagen, Valencia, CA, USA). For the colon, epithelial RNA processing and isolation were performed as previously described.49 RNA sample concentrations were measured using a Nanodrop-1000 Spectrophotometer (Thermo Scientific, Waltham, MA, USA). RNA was stored at −80 °C.
Illumina bead microarray and data analysis
RNA labeling, microarray hybridization and scanning were performed at the University of Minnesota Genomics Center using Illumina MouseWG-6 v2.0 Expression BeadChips (Illumina, San Diego, CA, USA), according to the manufacturer’s instructions. Differential expression of genes was quantified by the moderated t-statistic. The BeadArray data was processed via R/Bioconductor and packages lumi and limma. Background-corrected log2-transformed data was normalized via quantile normalization from the lumi package. Data were further analyzed using GeneData Expressionist Software (GeneData Inc., San Francisco, CA, USA); genes that showed statistically significant differences in expression between groups were determined using the two-group test t-test and analysis of variance. Gene expression data have been submitted to the Gene Expression Omnibus (GEO), Accession number SuperSeries GSE76096 SubSeries GSE75996.
Quantitative reverse transcriptase–PCR
Quantitative reverse transcriptase-PCR was performed as previously described.22 Significant differences in expression between groups were determined using the two-group t-test.
List of primers used
All primers were designed using Primer-BLAST (National Center for Biotechnology Information) and obtained from Integrated DNA Technologies (Coralville, IA, USA).
| Forward | Reverse | |
|---|---|---|
| SI | ||
| Cfd | 5′-GATTCTGGGTGGCCAGGAG-3′ | 5′-CACCTGCACAGAGTCGTCAT-3′ |
| Reg3a | 5′-CTGGTCTGCCAGAAGAGACC-3′ | 5′-CCATCCACCTCCATTGGGTT-3′ |
| Scd1 | 5′-CTGAACACCCATCCCGAGAG-3′ | 5′-GTGGTGGTGGTCGTGTAAGA-3′ |
| Areg | 5′-GGTCTTAGGCTCAGGCCATTA-3′ | 5′-CGCTTATGGTGGAAACCTCTC-3′ |
| Muc3 | 5′-CAGCAGAGAATAGAGGACGGA-3′ | 5′-CTGCTTTGCGCTCTTCTTGGA-3′ |
| Retnlb | 5′-GGAAGCTCTCAGTCGTCAAGA-3′ | 5′-GCACATCCAGTGACAACCAT-3′ |
| Colon | ||
| Reg3b | 5′-CATGTGAGGTGAAGTTGCCC-3′ | 5′-GTCCCTTGTCCATGATGCTC-3′ |
| Clps | 5′-CGAGTGCTCCCCAAAGAC-3′ | 5′-AGGCAGATGCCATAGTTGGT-3′ |
| Cd177 | 5′-CTGAAATGCCAGCATGGGAC-3′ | 5′-GTGCAGCCTTTCGTGAGAAC-3′ |
| Duoxa2 | 5′-GCTCTGCCACTTCCGCCTGG-3′ | 5′-AATCACCACCGCCCCTCCGA-3′ |
| Clca6 | 5′-AAGGCTTCCATAATGTTCATGC-3′ | 5′-ACTTCCCAAGTGCTTCTGTAAT-3′ |
| Slpi | 5′-GGACTGTGGAAGGAGGCAAA-3′ | 5′-CCCAGTCAGTACGGCATTGT-3′ |
RNA Seq
RNA Seq was performed at the UMGC as described previously.22 RNA was isolated from 12 comparison groups: Apc+/+ Cftrfl/fl-Villin-Cre normal colon, Apc+/+ Cftr+/+ normal colon, Apc+/+ Cftrfl/fl-Villin-Cre normal SI, Apc+/+ Cftr+/+ normal SI, Apc+/+ Cftrfl/fl-Villin-Cre colon tumors, Apc+/+ Cftrfl/fl-Villin-Cre SI tumors, ApcMin Cftrfl/fl-Villin-Cre colon tumors, ApcMin Cftrfl/+-Villin-Cre colon tumors, ApcMin Cftr+/+ colon tumors, ApcMin Cftrfl/fl-Villin-Cre SI tumors, ApcMin Cftrfl/+-Villin-Cre SI tumors and ApcMin Cftr+/+ SI tumors. The extracted RNA samples were sequenced using the Illumina HiSeq 2500 platform, v.4 SBS chemistry, 125 bp paired-end reads. All raw reads that passed the CASAVA 1.8P/F filter were directly deposited at the Minnesota Supercomputing Institute, where the data were analyzed. Sequence quality was assessed via fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Since the raw sequence reads were of sufficient quality, no data trimming or filtering was necessary. Hence, the raw paired-end reads were mapped to mouse genome (mm10 assembly) using Tophat 2.0.13 with the iGenomes reference (UCSC mouse mm10 annotation, downloaded from http://cufflinks.cbcb.umd.edu/igenomes.html). Final mapping percentages of the samples range from 89 to 93%. The mapped samples were then analyzed via Cuffdiff 2.2.1 (default parameters using the iGenomes UCSC mm10 GTF file previously mentioned) to quantify the expression level of each known gene in units of FPKM (Fragment mapped per kilobase of exons per million mapped reads). In order to minimize the potential confounding factors, cuffdiff analysis was only carried out between sample pairs that were prepared and sequenced in batches. The results were all ranked by the absolute value of fold. Gene expression data have been submitted to the Gene Expression Omnibus (GEO), Accession number SuperSeries GSE76096 SubSeries GSE76095.
Pathway analysis
IPA
Bead Array Data
Genes showing a 1.5-fold change in expression (microarray; Supplementary Figure S2) or two-fold (RNA Sea; Supplementary Figure S9) changes in expression were analyzed using IPA.52
GSEA
GSEA v. 2.0, http://www.broad.mit.edu.gsea/.53 Default parameters were used except that gene-set permutation was used due to the small number of samples.
Bead Array
GSEA compared the entire ranked Cftrfl/fl-Villin-Cre gene expression data sets from the normal colon and proximal small intestine to gene signatures derived from Kcnq1 KO SI and colon22 and from Muc2 KO SI.43
RNA Seq
Entire ranked gene expression data sets comparing indicated Cftr-deficient tissues were compared with published gene signatures for ISC and WNT target genes,24,26 and to gene sets derived from ApcMin-Cftr+/+ tumors.
Kaplan–Meier survival plots
Briefly, the AMC-AJCCII-90 set (GSE33113) contains 90 patients with stage II colon cancer that underwent intentionally curative surgery at the Academic Medical Center Amsterdam in Amsterdam, the Netherlands. To test the prognostic relevance of CFTR expression in tumor tissue, we analyzed the correlation between CFTR expression and DFS. Via scan-modus of the R2 platform (http://hgserver1.amc.nl/) we determined optimal cutoff for low and high expression. Survival curves for DFS were calculated with the Kaplan–Meier analysis and were compared by the log-rank test in GraphPad Prism 5. Multivariate analysis of prognostic relevance was evaluated by Cox regression analysis in IBM SPSS statistics version 20. For all tests, a P-value of <0.05 was considered significant.
Colon organoids
Five pairs of male littermate-matched C57Bl/6J Cftrfl/fl and Cftrfl/fl Villin-Cre+ mice were killed between 8 and 12 weeks of age. Colons were removed, cut open and washed in cold phosphate-buffered saline. Colon organoids were then cultured using the protocol of Sato et al.54 following the plating of 500 crypt bottoms per well in triplicate per sample. Significance (P<0.05) was determined by one-tailed Mann–Whitney test.
Supplementary Material
Acknowledgments
This work was supported by the grant from the National Cancer Institute (NCI) to RC and DL (NCI R01 CA134759-01A1); grant from the Mezin-Koats Colon Cancer Research fund to TS; grant from NCI to support the University of Minnesota Masonic Cancer Center (NCI P30-CA775598); grant from the Whiteside Institute for Clinical Research to RC and PS; grant from the University of Minnesota Foundation to RC, TS and DL; grant from Essentia Health Systems to RC; grant from the AICR (14–1164) to LV; grants from the Dutch Cancer Society (UVA2011–4969, UVA2014–7245) to LV; NWO gravitation grant to JL; grants from the National Cancer Institute to TS (NCI 5R00CA151672-04 and P30-CA77598). We also wish to thank Drs Mitchell Drumm and Craig Hodges of CWRU for providing the Cftr mutant mice.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013;3:a009753. doi: 10.1101/cshperspect.a009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 3.Pedersen SF, Stock C. Ion channels and transporters in cancer: pathophysiology, regulation, and clinical potential. Cancer Res. 2013;73:1658–1661. doi: 10.1158/0008-5472.CAN-12-4188. [DOI] [PubMed] [Google Scholar]
- 4.Neglia JP, FitzSimmons SC, Maisonneuve P, Schöni MH, Schöni-Affolter F, Corey M, et al. The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N Engl J Med. 1995;332:494–499. doi: 10.1056/NEJM199502233320803. [DOI] [PubMed] [Google Scholar]
- 5.McWilliams R, Highsmith WE, Rabe KG, de Andrade M, Tordsen LA, Holtegaard LM, et al. Cystic fibrosis transmembrane regulator gene carrier status is a risk factor for young onset pancreatic adenocarcinoma. Gut. 2005;54:1661–1662. doi: 10.1136/gut.2005.074534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst. 2013;105:122–129. doi: 10.1093/jnci/djs481. [DOI] [PubMed] [Google Scholar]
- 7.Maisonneuve P, FitzSimmons SC, Neglia JP, Campbell PW, III, Lowenfels AB. Cancer risk in nontransplanted and transplanted cystic fibrosis patients: a 10-year study. J Natl Cancer Inst. 2003;95:381–387. doi: 10.1093/jnci/95.5.381. [DOI] [PubMed] [Google Scholar]
- 8.Gelfond D, Borowitz D. Gastrointestinal complications of cystic fibrosis. Clin Gastroenterol Hepatol. 2013;11:333–342. doi: 10.1016/j.cgh.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Zhang JT, Jiang XH, Xie C, Cheng H, Da Dong J, Wang Y, et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim Biophys Acta. 2013;1833:2961–2969. doi: 10.1016/j.bbamcr.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 10.Xie C, Jiang XH, Zhang JT, Sun TT, Dong JD, Sanders AJ, et al. CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator (uPA) in prostate cancer. Oncogene. 2013;32:2282–2291. 2291.e1–7. doi: 10.1038/onc.2012.251. [DOI] [PubMed] [Google Scholar]
- 11.Sun TT, Wang Y, Cheng H, Xiao HZ, Xiang JJ, Zhang JT, et al. Disrupted interaction between CFTR and AF-6/afadin aggravates malignant phenotypes of colon cancer. Biochim Biophys Acta. 2014;1843:618–628. doi: 10.1016/j.bbamcr.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 12.Oh SC, Park YY, Park ES, Lim JY, Kim SM, Kim SB, et al. Prognostic gene expression signature associated with two molecularly distinct subtypes of colorectal cancer. Gut. 2012;61:1291–1298. doi: 10.1136/gutjnl-2011-300812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Billings JL, Dunitz JM, McAllister S, Herzog T, Bobr A, Khoruts A. Early colon screening of adult patients with cystic fibrosis reveals high incidence of adenomatous colon polyps. J Clin Gastroenterol. 2014;48:e85–e88. doi: 10.1097/MCG.0000000000000034. [DOI] [PubMed] [Google Scholar]
- 14.Wilke M, Buijs-Offerman RM, Aarbiou J, Colledge WH, Sheppard DN, Touqui L, et al. Mouse models of cystic fibrosis: phenotypic analysis and research applications. J Cyst Fibros. 2011;10(Suppl 2):S152–S171. doi: 10.1016/S1569-1993(11)60020-9. [DOI] [PubMed] [Google Scholar]
- 15.Starr TK, Allaei R, Silverstein KA, Staggs RA, Sarver AL, Bergemann TL, et al. A transposon-based genetic screen in mice identifies genes altered in colorectal cancer. Science. 2009;323:1747–1750. doi: 10.1126/science.1163040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43:1202–1209. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeda H, Wei Z, Koso H, Rust AG, Yew CC, Mann MB, et al. Transposon mutagenesis identifies genes and evolutionary forces driving gastrointestinal tract tumor progression. Nat Genet. 2015;47:142–150. doi: 10.1038/ng.3175. [DOI] [PubMed] [Google Scholar]
- 18.Hodges CA, Cotton CU, Palmert MR, Drumm ML. Generation of a conditional null allele for Cftr in mice. Genesis. 2008;46:546–552. doi: 10.1002/dvg.20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 20.Xu Z, Gupta V, Lei D, Holmes A, Carlson E, Gruenert DC. In-frame elimination of exon 10 in Cftrtm1Unc CF mice. Gene. 1998;211:117–123. doi: 10.1016/s0378-1119(98)00090-0. [DOI] [PubMed] [Google Scholar]
- 21.De Lisle RC, Mueller R, Boyd M. Impaired mucosal barrier function in the small intestine of the cystic fibrosis mouse. J Pediatr Gastroenterol Nutr. 2011;53:371–379. doi: 10.1097/MPG.0b013e318219c397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Than BL, Goos JA, Sarver AL, O’Sullivan MG, Rod A, Starr TK, et al. The role of KCNQ1 in mouse and human gastrointestinal cancers. Oncogene. 2014;33:3861–3868. doi: 10.1038/onc.2013.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Sousa E, Melo F, Colak S, Buikhuisen J, Koster J, Cameron K, et al. Methylation of cancer-stem-cell-associated Wnt target genes predicts poor prognosis in colorectal cancer patients. Cell Stem Cell. 2011;9:476–485. doi: 10.1016/j.stem.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 24.Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8:511–524. doi: 10.1016/j.stem.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Muñoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Van der Flier LG, Sabates-Bellver J, Oving I, Haegebarth A, De Palo M, Anti M, et al. The intestinal Wnt/TCF signature. Gastroenterology. 2007;132:628–632. doi: 10.1053/j.gastro.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 27.De Sousa E, Melo F, Wang X, Jansen M, Fessler E, Trinh A, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med. 2013;19:614–618. doi: 10.1038/nm.3174. [DOI] [PubMed] [Google Scholar]
- 28.De Lisle RC. Disrupted tight junctions in the small intestine of cystic fibrosis mice. Cell Tissue Res. 2014;355:131–142. doi: 10.1007/s00441-013-1734-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clarke LL, Gawenis LR, Bradford EM, Judd LM, Boyle KT, Simpson JE, et al. Abnormal Paneth cell granule dissolution and compromised resistance to bacterial colonization in the intestine of CF mice. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1050–G1058. doi: 10.1152/ajpgi.00393.2003. [DOI] [PubMed] [Google Scholar]
- 30.Norkina O, Kaur S, Ziemer D, De Lisle RC. Inflammation of the cystic fibrosis mouse small intestine. Am J Physiol Gastrointest Liver Physiol. 2004;286:G1032–G1041. doi: 10.1152/ajpgi.00473.2003. [DOI] [PubMed] [Google Scholar]
- 31.Munck A. Cystic fibrosis: evidence for gut inflammation. Int J Biochem Cell Biol. 2014;52:180–183. doi: 10.1016/j.biocel.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Juric M, Xiao F, Amasheh S, May O, Wahl K, Bantel H, et al. Increased epithelial permeability is the primary cause for bicarbonate loss in inflamed murine colon. Inflamm Bowel Dis. 2013;19:904–911. doi: 10.1097/MIB.0b013e3182813322. [DOI] [PubMed] [Google Scholar]
- 33.Vij N, Mazur S, Zeitlin PL. CFTR is a negative regulator of NFkappaB mediated innate immune response. PLoS One. 2009;4:e4664. doi: 10.1371/journal.pone.0004664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Jiang XH, Chen H, Guo JH, Tsang LL, Yu MK, et al. CFTR negatively regulates cyclooxygenase-2-PGE(2) positive feedback loop in inflammation. J Cell Physiol. 2012;227:2759–2766. doi: 10.1002/jcp.23020. [DOI] [PubMed] [Google Scholar]
- 35.Burger-van Paassen N, Loonen LM, Witte-Bouma J, Korteland-van Male AM, de Bruijn AC, van der Sluis M, et al. Mucin Muc2 deficiency and weaning influences the expression of the innate defense genes Reg3β, Reg3γ and angiogenin-4. PLoS One. 2012;7:e38798. doi: 10.1371/journal.pone.0038798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shan M, Gentile M, Yeiser JR, Walland AC, Bornstein VU, Chen K, et al. Mucus enhances gut homeostasis and oral tolerance by delivering immunoregulatory signals. Science. 2013;342:447–453. doi: 10.1126/science.1237910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schütte A, Ermund A, Becker-Pauly C, Johansson ME, Rodriguez-Pineiro AM, Bäckhed F, et al. Microbial-induced meprin β cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc Natl Acad Sci USA. 2014;111:12396–12401. doi: 10.1073/pnas.1407597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia MA, Yang N, Quinton PM. Normal mouse intestinal mucus release requires cystic fibrosis transmembrane regulator-dependent bicarbonate secretion. J Clin Invest. 2009;119:2613–2622. doi: 10.1172/JCI38662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nat Rev. 2011;474:318–326. doi: 10.1038/nature10212. [DOI] [PubMed] [Google Scholar]
- 41.Isella C, Terrasi A, Bellomo SE, Petti C, Galatola G, Muratore A, et al. Stromal contribution to the colorectal cancer transcriptome. Nat Genet. 2015;47:312–319. doi: 10.1038/ng.3224. [DOI] [PubMed] [Google Scholar]
- 42.Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013;19:619–625. doi: 10.1038/nm.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Velcich A, Yang W, Heyer J, Fragale A, Nicholas C, Viani S, et al. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science. 2002;295:1726–1729. doi: 10.1126/science.1069094. [DOI] [PubMed] [Google Scholar]
- 44.Yang K, Popova NV, Yang W, Lozonschi I, Tadesse S, Kent S, et al. Interaction of Muc2 and Apc on Wnt signaling and in intestinal tumorigenesis: potential role of chronic inflammation. Cancer Res. 2008;68:7313–7322. doi: 10.1158/0008-5472.CAN-08-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Denamur E, Chehab FF. Methylation status of CpG sites in the mouse and human CFTR promoters. DNA Cell Biol. 1995;14:811–815. doi: 10.1089/dna.1995.14.811. [DOI] [PubMed] [Google Scholar]
- 46.Son JW, Kim YJ, Cho HM, Lee SY, Lee SM, Kang JK, et al. Promoter hypermethylation of the CFTR gene and clinical/pathological features associated with non-small cell lung cancer. Respirology. 2011;16:1203–1209. doi: 10.1111/j.1440-1843.2011.01994.x. [DOI] [PubMed] [Google Scholar]
- 47.Collawn JF, Fu L, Bartoszewski R, Matalon S. Rescuing ΔF508 CFTR with trimethylangelicin, a dual-acting corrector and potentiator. Am J Physiol Lung Cell Mol Physiol. 2014;307:L431–L434. doi: 10.1152/ajplung.00177.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roth EK, Hirtz S, Duerr J, Wenning D, Eichler I, Seydewitz HH, et al. The K+ channel opener 1-EBIO potentiates residual function of mutant CFTR in rectal biopsies from cystic fibrosis patients. PLoS One. 2011;6:e24445. doi: 10.1371/journal.pone.0024445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clarke LL, Gawenis LR, Franklin CL, Harline MC. Increased survival of CFTR knockout mice with an oral osmotic laxative. Lab Anim Sci. 1996;46:612–618. [PubMed] [Google Scholar]
- 50.Fijneman RJ, Anderson RA, Richards E, Liu J, Tijssen M, Meijer GA, et al. Runx1 is a tumor suppressor gene in the mouse gastrointestinal tract. Cancer Sci. 2012;103:593–599. doi: 10.1111/j.1349-7006.2011.02189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124:762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 52.IPA Ingenuity Systems. [Accessed 2 February 2013]; Available at http://www.ingenuity.com.
- 53.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.