

Abstract
Pleural effusion (PE), excess fluid in the pleural space, is often observed in lung cancer patients and also forms due to many benign ailments. Classifying it quickly is critical, but this remains an analytical challenge often lengthening the diagnosis process or exposing patients to unnecessary risky invasive procedures. We tested the analysis of PE using a multiplexed cytokeratin (CK) panel with targeted mass spectrometry–based quantitation for its rapid classification. CK markers are often assessed in pathological examinations for cancer diagnosis and guiding treatment course. We developed methods to simultaneously quantify 33 CKs in PE using peptide standards for increased analytical specificity and a simple CK enrichment method to detect their low amounts. Analyzing 121 PEs associated with a variety of lung cancers and noncancerous causes, we show that abundance levels of 10 CKs can be related to PE etiology. CK-6, CK-7, CK-8, CK-18, and CK-19 were found at significantly higher levels in cancer-related PEs. Additionally, elevated levels of vimentin and actin differentiated PEs associated with bacterial infections. A classifier algorithm effectively grouped PEs into cancer-related or benign PEs with 81% sensitivity and 79% specificity. A set of undiagnosed PEs showed that our method has potential to shorten PE diagnosis time. For the first time, we show that a cancer-relevant panel of simple-epithelial CK markers currently used in clinical assessment can also be quantitated in PEs. Additionally, while requiring less invasive sampling, our methodology demonstrated a significant ability to identify cancer-related PEs in clinical samples and thus could improve patient care in the future.
Introduction
Lung cancer is the most common cancer worldwide in terms of incidence and mortality [1]. It is classified into small cell lung cancer (SCLC) and non–small cell lung cancer (NSCLC, 85% of all cases), with NSCLC further subtyped most commonly into adenocarcinoma (ADC) and squamous cell carcinoma (SCC). A definite diagnosis in most cases occurs after highly invasive procedures that allow for visual assessment of the thoracic cavity and/or sampling of the tumor and histopathological analysis [2]. The hope is that an in-depth analysis of the pleural effusion (PE), the fluid that accumulates in the pleural space and is developed by 50% of lung cancer patients, could bring diagnostically valuable information and, at some level, replace the invasive procedures if they are unnecessary [3]. The less invasive procedure for its required removal (thoracentesis) and the proximity to the tumor make PE an ideal source of potential biomarkers, as has already been demonstrated [4]. PEs also develops due to benign ailments and can usually be separated into transudates (often caused by heart, renal, and liver failures) or exudates (caused often by infections, malignancy, and lung cancer) based on standard clinical laboratory parameters [3]. Determining the exudate etiology can be difficult. The diagnosis of a malignant PE (metastasis to the pleural cavity) requires a positive result of cytology in the sediment. Unfortunately, the sensitivity of this test is low, often below 60% [5]. Hence, patients with a negative cytology result but still suspected of having cancer sometimes undergo potentially unnecessary invasive procedures. Cancer patients with no malignant involvement of the pleura can also develop exudates (paramalignant PEs), further confounding a cancer diagnosis. Sensitive assays that could quickly guide patients with a cancer-related PE toward the invasive procedures that ultimately diagnose the cancer type and lead to early treatment commencement are favorably seen in the clinic.
Cytokeratin (CK) markers have been used in pathological examinations with immunohistochemistry for cancer classification for over 30 years [6], [7]. CKs are epithelial cytoskeletal proteins whose expression pattern differs between epithelial tissues and their developmental stages but is maintained when a malignant transformation occurs which is helpful in tumor differentiation [8], [9]. In lung cancer, profiles of CK-5/6, CK-7, CK-10/13, CK-14, CK-17, and CK-18 enable discrimination between SCC and ADC, and ADC and mesothelioma [6], [7], [10]. Additionally, CKs provide prognostic information and enable therapy monitoring (e.g., M30 and CYFRA 21-1 ELISAs which measure caspase-cleaved CK-18 and CK-19, respectively) [11], [12]. Furthermore, one of only a few FDA-approved protein tumor marker tests identifies circulating tumor cells of epithelial origin in blood using antibodies against CK-8, -18, and -19 [13]. Despite antibody-based tests being widely used with a reliable level of analytical sensitivity, cross-reactivity and poorly defined target issues have recently led to controversies and questions as to their specificity [14]. This includes cancer-related CK assays such as CYFRA 21-1 and CK-8/18 immunohistochemistry [15], [16]. Antibody-based tests, like ELISA, also do not easily multiplex and can suffer from phenomena which underreport high-target samples [13]. The targeted mass spectrometry (MS)–based method, multiple reaction monitoring (MRM) with stable-isotope-labeled standard (SIS) peptides, which is characterized by higher analytical specificity, high precision, and the possibility of measuring numerous proteins within a single rapid analysis, could improve assay performance. These advantages have made this method ideal for biomarker assessment and validation; it has seen an increase in use in clinical proteomics and has been deemed key for bridging biomedical discovery and clinical implementation as expressed in the 2012 Nature Methods “Method of the Year” article [17].
A few MS-based studies have been conducted to characterize PE proteomes and to identify potential protein biomarkers of lung cancer [4], [18], [19], [20], [21], [22], [23], [24], [25]. Two studies are worth mentioning as they verified a larger group of potential biomarkers thru MRM use or assessed the results on a large patient set [26], [27]. Using marker panels, they obtained diagnostically acceptable areas under the curve (AUC: 0.90-0.95) for differentiating malignant (ADC/NSCLC) from control PEs [tuberculosis (TB) and pneumonia]. However, paramalignant PEs were included in the control group or excluded. Similarly, studies have shown promise in determining malignant PEs using ELISAs for tumor markers common in clinical use, with an individual marker specificity and sensitivity ranging between 85% and 96% and between 25% and 55%, respectively [28]. A combination of four such markers showed an improvement over cytology [29], revealing that such less invasive testing may aid to select cytology-negative patients which require further invasive procedures.
In this work, we devised a simple precipitation-based CK enrichment method that allows the detection of low quantities of CKs in PE and determined peptide-specific MS parameters for their precise, specific, and sensitive multiplexed quantitation by MRM. Using these targeted assays for 33 different CK proteins, including some previously not associated with cancer, we screened a broad set of 121 PEs. We show which CKs are detectable and that levels of 10 CKs are related to PE etiology. For the first time, we reveal that simple-epithelial CKs are at significantly higher levels in cancer-related PEs and show that our method, while requiring less invasive sampling, can significantly identify cancer-related PEs in clinical samples and thus could shorten PE diagnosis time in the future.
Material and Methods
Clinical PE Specimens
The study was approved by the Bioethical Committee at the Regional Medical Chamber in Warsaw (KB/928/14). PE samples were obtained during thoracentesis and classified at the Mazovian Center of Pulmonary Disease and Tuberculosis Treatment. A written informed consent was obtained from each patient. Samples were deidentified and coded for the proteomic analyses and were handled according to Biosafety Level 2 practices. This is a preliminary study to show if and what CKs are present in PEs related to cancer, and therefore a predetermined sample size was not calculated as we were analyzing all samples accessible. Thus, we were working with a real representation of PEs in a clinical setting, including those that could not be diagnosed. We collected 121 PEs over a 21-month period. Forty-two PEs were from patients with a diagnosed form of cancer (cancer PEs), 47 were from patients with non–cancer-related diseases (benign PEs), and the etiology of 32 PEs could not be initially established (Table 1). Within the cancer PEs, 35 were from patients with lung cancer [28 NSCLC (15 ADC and 10 SCC), 7 SCLC] and 7 were from patients with other forms of cancer (2 pleural mesotheliomas, 1 prostate cancer, 1 breast cancer, 1 ovarian cancer, 1 nipple cancer, and 1 cancer of unknown origin). Out of the cancer PEs, five (12%) had positive cytology results, and additional videothoracoscopy done on select patients increased this sum to eight (19%) confirmed malignant PEs. The benign PE group included 24 pleural empyema PEs (an advanced stage of a bacterial infection, usually the complication of bacterial pneumonia), 6 TB PEs, 14 transudates [caused by heart failure (4), renal failure (1), and liver cirrhosis (1); 8 were not further subtyped], and 3 posttraumatic PEs. For data analysis, the cancer PE and benign PE groups were divided into subgroups: lung-cancer PEs (n = 35), other-cancer PEs (n = 7), bacterial-infection PEs (empyema and TB, n = 30), and control PEs (transudate and posttraumatic PEs, n = 17).
Table 1.
(A) Makeup of the PE Sample Set; (B) Characteristics of the Patients’ Population
| A | B | |||||||
|---|---|---|---|---|---|---|---|---|
| Cancer | 42 | Number of Patients | Median of Age (yr) | |||||
| Lung cancer | 35 | |||||||
| NSCLC | 28 | Cancer | 68 | |||||
| ADC | 15 | Female | 13 | |||||
| SCC | 10 | Male | 29 | |||||
| SCLC | 7 | Benign | 67 | |||||
| Nonlung cancer | 7 | Female | 5 | |||||
| Male | 42 | |||||||
| Benign | 47 | Unknown | 64 | |||||
| Bacterial | 30 | Female | 10 | |||||
| Empyema | 24 | Male | 22 | |||||
| Tuberculosis | 6 | |||||||
| Transudates | 14 | |||||||
| Posttraumatic | 3 | |||||||
| Unknown | 32 | |||||||
PEs were collected into tubes with the anticoagulant EDTA dipotassium salt dihydrate (Sigma-Aldrich, St. Louis, MO) (final concentration, 4.5 mM) and were stored on ice for a maximum of 2 hours before processing. PEs were centrifuged at 2000 ×g for 10 minutes at 4°C to remove cellular debris. The PEs were centrifuged again at 15,000 ×g for 10 minutes at 4°C, and the supernatants were stored at − 80°C until analysis. Total protein concentration was determined using the bicinchoninic acid (Thermo Fisher Scientific, Rockford, IL, USA) assay according to the manufacturer’s instructions for the microplate plate format.
Nano-LC-MRM-MS Assay Development, Optimization, and Analysis
Peptide selection for MRM analysis was performed according to the criteria described previously by manual selection and the aid of the PeptidePicker software [30]. In brief, chosen proteotypic peptides met the following conditions: sequence uniqueness in the human proteome; numerous observations in spectral libraries; length not exceeding 20 amino acids; and, if possible, not containing easily chemically modifiable residues or sequences prone to modifications. Peptides were excluded when they had low digestion efficiency, a high frequency of single-nucleotide polymorphisms, known posttranslational modifications (PTMs), or biological features affecting their measurement accuracy. The selected peptide sequences were synthesized as stable isotope–labeled standard peptides using isotopically labeled amino acids on the C-terminus: Arg 13C6; 15N4 (98% isotopic enrichment) or Lys 13C6; 15N2 (98% isotopic enrichment) by JPT Peptide Technologies GmbH (Berlin, Germany) as SpikeTides_L.
MRM analysis was performed as described previously with minor modifications [31]. Briefly, a Waters Xevo TQ MS (Waters, Milford, MA) coupled to a Waters nanoAcquity UPLC was used with peptides loaded onto a trap column and separated using a 60-minute LC run, with a gradient of acetonitrile changing from 1% to 10% from 0 to 10 minutes and from 10% to 50% from 10 to 40 minutes. Modified MS parameters included a capillary voltage of 3.5 kV and NanoFlow gas set at 2.0 bar.
To generate the highest ion signal, the optimization of peptide- and fragment-specific MRM settings was performed as previously described using SIS peptides [31]. In brief, the optimal precursor charge and optimal cone voltage were determined. Best b- and y-series fragment ions with their optimal collision energy voltages were selected. Five to three interference-free optimized transitions generating the highest signals were chosen. For vimentin, a standard MRM analysis without SIS was performed using nine transitions with calculated cone and collision energy voltages. Our analytical method targeted 37 proteins with 115 peptides in three 60-minute LC-MRM analyses per sample.
For MRM analysis with SIS peptides, the quantity of the endogenous peptide is reported as the peak area ratio which is the sum of the peak areas of all transitions for the endogenous peptide divided by the sum of the peak areas of transitions of its heavy standard. Equivalent SIS peptide amounts added to all samples enabled normalization of intensity data between samples in terms of MS signal fluctuations and postdigestion sample processing differences. For the analysis of vimentin, the sum of the peak areas of nine transitions for the endogenous peptide was presented as the relative amount. Signals for each sample were manually inspected to ensure correct peak detection, accurate integration, and interference-free transitions. MRM methods and data analyses were performed in Skyline (Ver. 3.5) [32].
Precipitation-Based CK Enrichment Method
A pooled sample of PEs, representative of the study population, was used to establish the optimal buffer pH for increased CK precipitation and to test the assay reproducibility. Ten different pH levels in a 3.7-M potassium chloride (KCl; Carl Roth GmbH, Karlsruhe, Germany) solution were tested: from pH 3.9 to 5.7, buffered using citrate; for pH 7.5 and 8.0, buffered using 0.1 M HEPES (Carl Roth). Citrate buffers (Carl Roth) had a final concentration of citric acid ranging from 7.4 to 50 mM and of trisodium citrate ranging from 42.6 to 85.2 mM. Each pH level was analyzed in triplicate. Three replicates were analyzed for the intraday reproducibility test, and five sample precipitations were performed on 5 different days using independent sets of reagents to establish the interday reproducibility.
PE Sample Analysis with CK Enrichment and MRM
To limit epidermal keratin contamination from dust, PE samples prior to trypsin digestion were handled in a HEPA-filter-laminar safety cabinet and prepared using filtered tips and ultrapure reagents (Sigma-Aldrich). Sample preparation and MRM analyses were randomized. After defrosting, PEs were centrifuged at 15,000 ×g for 5 minutes at 4°C. The supernatants were diluted with 10 mM ammonium bicarbonate (AmmBic, Sigma-Aldrich) to a final volume of 500 μl and a final protein concentration of 15 mg/ml. Samples were precipitated by adding 500 μl of 3.7 M KCl in citrate buffer, pH 5.1 (19.4 mM citric acid, 73.1 mM trisodium citrate), and incubating overnight at 5°C and then pelleted by centrifugation at 14,000 ×g for 10 minutes at 4°C. Pellets were resuspended in 40 μl of 8 M urea (Carl Roth) in 25 mM AmmBic and sonicated for 5 minutes. To inhibit protein carbamylation, 60 μl of 100 mM AmmBic was added. Samples were reduced with 5 μl of 50 mM tris(2-carboxyethyl)phosphine (in 100 mM AmmBic, Sigma-Aldrich) for 30 minutes at 60°C and alkylated with 5 μl of 100 mM iodoacetamide (in 100 mM AmmBic, Sigma-Aldrich) for 30 minutes at 37°C. The alkylation reagent was quenched by addition of 5 μl of 100 mM DTT (in 100 mM AmmBic, Carl Roth) for 30 minutes at 37°C. Finally, 224 μl of 100 mM AmmBic was added to decrease the final urea concentration to 0.94 M, and proteins were digested with 1.7 μg (20 μl) of sequencing-grade trypsin (Promega, Fitchburg, WI) for 16 hours at 37°C. To quench the digestion, samples were acidified with 0.1% formic acid (FA) to 1 ml, and a mixture of SIS peptides was added to give an amount of individual SIS ranging roughly from 8 to 480 fmol/μg for the SIS to be 10-fold above the average endogenous peptide concentration. Samples were desalted using Oasis HLB 1-ml (10 mg) cartridges (Waters). Samples were loaded, washed twice with 1.0 ml of water, and eluted in 200 μl of 50% acetonitrile, 0.1% FA. Fractions were evaporated to dryness using a SpeedVac (Labconco) and resuspended in 28 μl of 0.1% FA, and 3.8 μl was injected for nano-LC-MS-MRM analysis. Blanks (0.1% FA) were run between every sample.
ELISA Analysis of PEs for CK-18
To determine the CK-18 protein concentration in a set of PE samples, we used the M65 ELISA (Peviva, Stockholm, Sweden) according to the manufacturer’s instructions. Samples were measured in duplicate. PE samples with high levels of CK-18 were 60 times diluted, while the rest were diluted 10 times in Standard A as recommended by the manufacturer.
Statistical Analysis
The data were processed using SPSS Statistics (Ver. 22.0; IBM Corp., Armonk, NY). The nonparametric Mann-Whitney U test was performed for the protein level comparisons. Linear regression and a Pearson correlation analysis were used to compare the MRM and ELISA results. Hierarchical clustering of the MRM data was performed using the heatmap tool "heatmap.2" of the gplots package of the statistical environment R: A Language and Environment for Statistical Computing using the complete clustering and Euclidean distance methods [33]. Receiver operating characteristic (ROC) curves were performed in SPSS. The true-positive rate (sensitivity) was plotted against the false-positive rate (FPR) (1 − specificity), and the AUC values are reported with the 95% confidence interval (CI) as an estimate of diagnostic usefulness. Box-plots illustrating relative protein abundances were prepared in R version 3.2.2.
Random Forest Classifiers
Classification tasks were performed using a random forest algorithm implemented in Scikit-learn package, a machine learning algorithm in Python [34]. All classification performance measures (area under ROC curve, true- and false-positive ratios) were estimated using a 10-fold cross-validation procedure. Missing values were replaced using median imputation. To estimate adequate forest size, we observed how doubling the number of trees affects the performance of classifiers, starting with a size of 10. Increasing the size above 40 did not result in noticeable improvement in AUC values; therefore, we opted for a simpler model with 40 trees. Classifiers based only on geometric mean values of proteins were trained on a selected set of top five features according to information gain measure. However, when training on both individual peptide values and protein geometric means, to avoid selection of highly redundant feature sets, e.g., including geometric mean and multiple individual peptide features from one CK, we switched to greedy forward feature selection. For the classification of the independent PE sample set, the value reported as score is the fraction of trees that assigned a sample to a given group.
Results
MRM Assay and CK Enrichment Method Development
To screen the PEs for the presence of CKs, we targeted all the CKs expressed in simple epithelia, stratified squamous epithelia, suprabasal cells, and selected structural CKs [7], [8]. Specific MRM assays were developed to measure the level of 33 CKs through unique peptides. These included the CK-1, -1B, -2e, -2P, -3 to -6, -6A, -6B, -7 to -10, -12 to -20, -23, -24, -26 to -28, -39, -40, and -78 to -80 (Supplementary Table S-1). Seventeen of these are associated with some form of cancer, and 12 have been linked with lung cancer [7], [8], [9]. Multiple peptides were targeted per protein (90% of proteins with 3 or more peptides), culminating in a set of 102 peptides for which pairing SIS peptides were generated. These standards increased the analytical assay specificity by verifying the presence of endogenous peptides through coelution, and selection of interference-free MRM transitions. They also allowed to obtain accurate relative abundance information for each peptide and to normalize the sample set. Peptide-specific, optimal MRM-MS parameters were empirically determined to improve assay sensitivity (Supplementary Table S-2) as this was shown to increase the peptide signal by an average of 11-fold [35]. Our MRM method also assessed the levels of serum albumin, transferrin, actin, and vimentin using 13 peptides (Supplementary Tables S-1 and S-2). The first two proteins were included to show patient variation in the PE’s plasma content. Actin was included as a possible indicator of cell damage, occurring either during sampling or from the disease state, and vimentin was included due to links with lung cancer progression [36].
As initial attempts of PE analysis using standard methods revealed weak signal intensities for only a few CKs, we resorted to developing an enrichment method for CKs based on a salting-out effect by a high-concentration KCl buffer, which was previously used to enrich for intermediate filament proteins through their selective precipitation [37], [38]. Our method consisted of taking an equivalent amount of total protein from each PE sample and adding a buffered KCl-saturated solution. The resolubilized precipitate was subjected to tryptic digestion and MRM-MS analysis. Because proteins precipitate preferentially around their pI point, we tested 10 pH levels covering the entire range of pI points for the acidic (type I) and basic (type II) CKs [38]. The precipitation method significantly improved the detectability of many CKs (15- to 200-fold signal increase) compared with direct PE analysis approaches, and we observed the important cancer-related CK-7, -8, -18, and -19. The buffer at pH 5.1 resulted in roughly a two-fold enrichment for these CKs as compared with pH 4.9 and 5.3 (Figure 1A). Above pH 5.3, no practically quantifiable precipitate was obtained, and at pH below 4.5, nonspecific protein aggregation was observed without CK enrichment. CK-1, CK-2e, CK-9, and CK-10 were more enriched in the pH 5.3 buffer (Supplementary Figure S-1A). However, because these are less often associated with cancer and, in MS analysis, sometimes originate from dust contamination, we selected the pH 5.1 buffer as the best one for the cancer-related CKs and used it in further analyses. Vimentin, also an intermediate filament protein, was enriched in the pH 5.1 buffer, whereas actin was highest in the pH 5.3 buffer (Supplementary Figure S-1A). We additionally determined the optimal trypsin amount used in the digestion and the optimal on-column sample loading amount for maximal signal intensity (data not shown).
Figure 1.
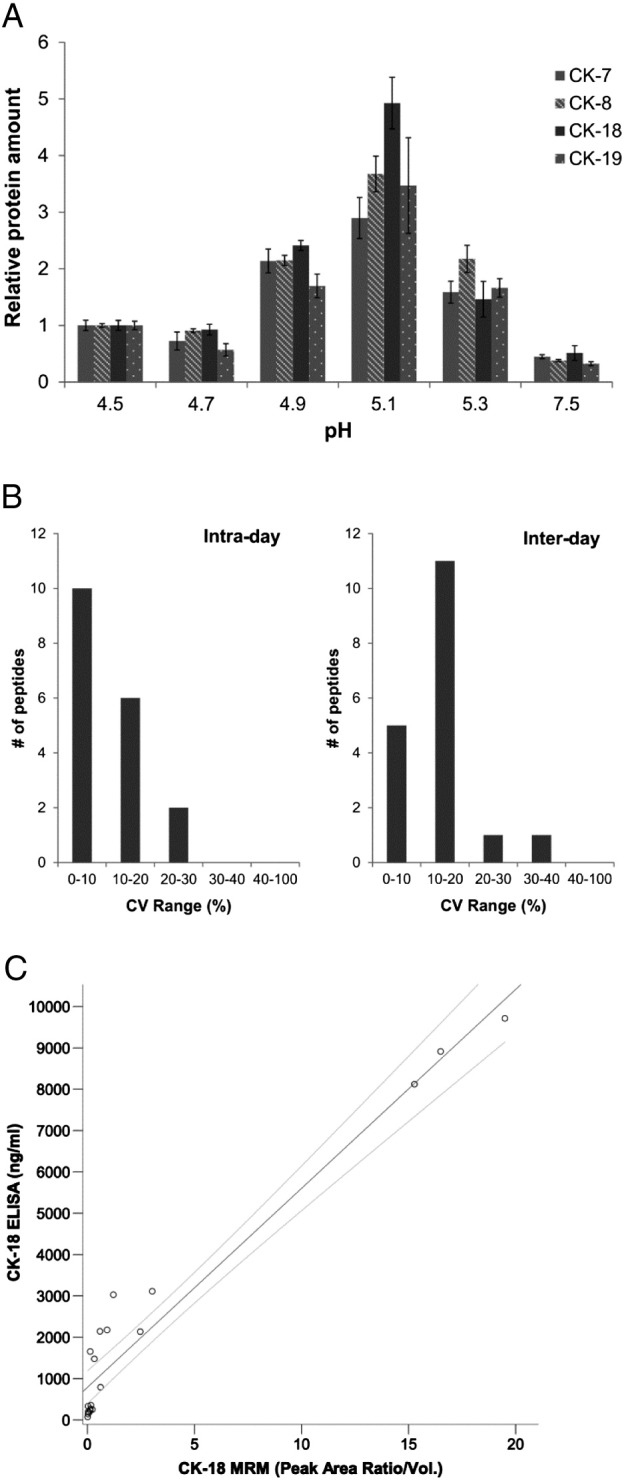
CK enrichment method development. (A) Optimization of pH of the CK precipitation buffer indicating relative CK protein levels per equivalent amount of total protein precipitated by the KCl buffer at different pH levels. Performed in triplicate. Errors bars are standard error of the mean (SEM). (B) The intra- and interday reproducibility assessment of the CK enrichment method as shown for multiple peptides of CK-7, -8, -18, and -19, and vimentin and actin. Three replicates were analyzed for the intraday test, and five replicates on 5 different days were performed for the interday test. (C) Comparison of CK-18 protein levels in 20 PE samples as measured by MRM and the CK-18 ELISA. Line indicates a linear regression with 95% CI (r = 0.97).
To prove that a precipitation step is feasible in an analytical technique that requires high precision, we tested the method reproducibility. The intra- and interday precision was assessed using replicates of a pooled PE sample for the MRM-MS measurement of peptides for the important cancer-related CK-7, -8, -18, and -19 and for vimentin and actin, which were later discovered to be relevant markers. The intraday reproducibility test (Figure 1B) showed that 89% of the peptides had a coefficient of variation (CV) below 20% and that the results were more peptide than protein dependent. We also performed a more rigorous interday reproducibility test where the sample was processed on 5 different days using an independent set of reagents. Again, 89% of the peptides had a clinically acceptable CV below 20%, although a higher proportion of peptides fell into the 10% to 20% CV range. We observed that the keratinizing epidermis CK-1, -2e, -9, and -10 associated with dust contamination had interday %CVs ranging over 40%, although in this pooled sample they were observed with a low signal intensity which in MRM analysis can contribute to low reproducibility (Supplementary Figure S-1B). We therefore decided to keep these CKs in the MRM panel taking dust-free precautions.
We compared our MS-based and therefore peptide-centric quantitation method to a protein-based measurement given by ELISA. Because established ELISAs for all detected CKs do not exist, we compared our method to the most relevant CK-18 ELISA (M65), commonly used in cancer research [39]. Twenty individual PE samples from a variety of diagnosed cases, spanning a wide concentration range of CK-18, were measured using both methods. The results were expressed in ng/ml from the ELISA reading and as a relative amount per PE volume for the MRM measurement (Figure 1C). A reasonable correlation was observed for the two methods with a linear correlation coefficient (r) of 0.97. The methods agree well at the highest concentrations of CK-18 (8-10 μg/ml) and less in the midrange levels (0.8-4 μg/ml) where the ELISA had slightly higher levels. Both methods were in good agreement for very low levels of CK-18 (~ 70 to 360 ng/ml). This comparison revealed that our method had a lowest level of detection (LOD; MRM signal to noise ratio > 10:1 and > 8000 counts endogenous signal) of around 250 ng/ml for the CK-18 protein.
The above results show that our developed analytical method, composed of the CK enrichment step, protein digestion, and the targeted MRM-MS analysis with SIS peptides, can be used for the precise relative measurement of specific CK levels in PE samples.
General Results of the MRM Analysis of 121 PEs
The sample total protein concentration within the PE set varied marginally [mean of 42 ± 9.6 (SD) mg/ml]; however, due to the transudate and exudate character of the PE, our analysis was carried out with normalization on a per total protein content basis. Protein levels were measured with up to four peptides taking the geometric mean of their levels normalized to SIS peptide levels. CK-6 could be measured using peptides specific for isoforms 6A and 6B or measuring all three isoforms (CK-6A, -6B, -6C) together (CK-6). An N-terminal peptide of CK-19 (CK-19-Nterm, amino acids 25-32) was observed to have a different level pattern across the sample set compared with the other three peptides from CK-19 and was treated separately in the data analysis. Analysis of the 121 PEs revealed the presence of 16 CKs, many of which have connections to cancer (14 CKs) and lung cancer (10 CKs, Figure 2). We detected CK-1, -2e, -4 to -10, -14 to -16, and -18 to -20 across the PE sample set (Figure 2). CK-4, CK-6A, and CK-20 were only detected in a few samples, and CK-6A, CK-14 to CK-16, and CK-20 were detected with low-intensity signals.
Figure 2.

Heatmap display and hierarchical clustering of the MRM data of observed CKs for all analyzed PE samples. Yellow indicates a higher abundance level. Displayed data are log transformed. The abundance is expressed on a relative scale for each protein/peptide, and therefore, intensities cannot be compared between targets. CKs marked with an asterisk have previously been associated with lung cancer. Individual samples are indicated by number and the type of PE as described in the text. Number and question mark denotes nondiagnosed PEs. NA indicates where MRM data could not be acquired.
Serum albumin and transferrin had a small abundance variability across the PE samples after the CK enrichment as well as in the neat PE sample analysis (CVs 23% and 39%, respectively). This was in contrast to the CKs, actin, and vimentin (CV from 77% to 584%) whose measured levels spanned a wide range across the sample set (e.g., 400-fold difference between highest expression and LOD sample for CK-18) (Figure 2). Interestingly, CK-8, CK-18, and CK-19, which are often coexpressed and pair together, also clustered together when the data set was hierarchically clustered. This also occurred for vimentin and actin, which showed an elevated presence in the empyema samples. A trend could also be observed on the clustered data for CK-8, -18, -19, -19-Nterm, -6, and -7, where an elevated level was nearly exclusively associated with cancer PEs.
Differences in Protein Levels between Cancer and Benign PE Groups
A comparison of all cancer PEs (n = 42) to all benign PEs (n = 47) revealed a statistically significant difference in the levels of CK-6, -7, -8, -18, -19, and -19-Nterm (Figure 3) and in the levels of actin and vimentin. The CKs exhibited higher levels in the cancer PE group, with CK-7 and the CK-19-Nterm fragment being the most significant. ROC curves generated based on CK-7 and CK-19-Nterm showed diagnostically useful AUCs of 0.75 (95% CI = 0.65-0.85) and 0.83 (95% CI = 0.74-0.91), respectively (Figure 4A). Simple-epithelial CK-7, CK-8, CK-18, and CK-19 were at high levels in many cancer PEs in contrast to the stratified-epithelial CK-6 which was also less significant. Many cancer PEs also had these CKs at baseline levels. Interestingly, the highest CK level samples did not correlate with the few verified malignant PEs (19% of the cancer PEs), which actually had low to mid levels of CKs.
Figure 3.

Significant differences in CK levels between benign PEs (n = 47) and cancer PEs (n = 42). Box-plots indicate the median and quartiles, with whiskers indicating the 1.5 interquartile range. Statistical significance is indicated using the Mann-Whitney U test, **P ≤ .01, *P < .05.
Figure 4.

ROC curves showing diagnostic efficacy of significant targets. (A) ROC curves for CKs whose levels significantly differ between cancer PEs (n = 42) and benign PEs (n = 47). (B) ROC curves for actin and vimentin whose levels significantly differ between the bacterial-infection PEs (n = 30) and all other PEs (n = 59). AUCs are indicated for each target.
Actin and vimentin levels were significantly higher in the benign PE group and the bacterial-infection PE group as revealed in a four-group comparison (Figures 4B and 5A). More specifically, actin and vimentin were elevated in the empyema PE group and possibly in the TB-PE group where three of six PEs had high levels (Supplementary Figure S-2). These proteins emphasized the empyema-PE group from other groups more than any other tested protein/peptide. The control-PE group had the lowest level of actin and vimentin out of all groups. The two proteins could distinguish the bacterial-infection PE group from the other samples with an AUC of 0.88 (95% CI = 0.80-0.97) for actin and with an AUC of 0.86 (95% CI = 0.77-0.96) for vimentin (Figure 4B).
Figure 5.

Significant differences in protein/peptide levels between lung-cancer PEs (n = 35), other-cancer PEs (n = 7), bacterial-infection PEs (empyema and TB, n = 30), and control PEs (posttraumatic and transudate PEs, n = 17). (A) Comparison of actin and vimentin levels. (B) Comparison of CK levels. Box-plots indicate the median and quartiles, with whiskers indicating the 1.5 interquartile range. Statistical significance is indicated using the Mann-Whitney U test, **P ≤ .01, *P < .05.
Comparing lung-cancer PEs and other-cancer PEs to the control PEs (transudates and posttraumatic PEs) shows that it is not just the overabundance of the bacterial-infection PEs that causes a significant difference in CK levels. Similarly, CK-7, CK-8, CK-18, CK-19, and CK-19-Nterm are at significantly higher levels in the lung-cancer PEs compared with the control PEs (Figure 5B). This difference is observed in spite of generally lower total protein concentrations in transudates within the control PE group (i.e., higher PE volume analyzed). CK-7 and CK-19-Nterm again showed the highest significance. Their amounts were also significantly lower in the bacterial-infection PEs. In this comparison, CK-6 was significantly higher only in the lung-cancer PEs when compared with the bacterial-infection PEs. The other-cancer PE (nonlung malignancies) group, composed of only seven cases, could not be distinguished from the lung-cancer PEs. However, CK-7 with CK-19-Nterm and CK-8 with CK-19-Nterm were significantly higher in the other-cancer PEs compared with the bacterial-infection PEs and the control PEs, respectively. In summary, the simple-epithelial CKs show an elevated level in the lung-cancer and other-nonlung-malignancy PEs as compared with the benign PEs irrespective of whether they are compared with the predominant transudates or the bacterial infection exudates together or separately.
Differences in CK Levels in Lung- and Other-Cancer PEs
Analyzing the data with all PE subtypes separated provides additional information on the CK profiles (Supplementary Figure S-3 and Table S-3). This revealed that CK-7 and CK-19-Nterm most often—and sometimes also CK-8, CK-18, and CK-19—were higher in the ADC, NSCLC, and other-cancer PEs compared with the TB, empyema, and control PEs. SCLC and SCC did not have this level of significant differences, showing lower levels of these CKs. Samples denoted NSCLC could not be clinically subclassified into ADC or SCC but were thought by the clinicians to be more likely an advanced form of ADC (personal communication). Although we only analyzed three such NSCLC PEs, these were often very high for the mentioned CKs. Interestingly, a comparison within lung cancer subtypes revealed significant differences in the levels of CK-2e, -7, -9, -10, and -19 between SCLC, ADC, and SCC (Figure 6). ADC had higher levels of CK-7 compared with SCLC and SCC and of CK-19 compared with SCC. In contrast, SCC had higher levels of CK-2e, -9, and -10 compared with ADC. We also observed very high abundance of CKs-4, -6, and -6A in some NSCLC and mesothelioma PEs and in a prostate-cancer PE.
Figure 6.

Significant differences in CK levels between lung cancer subtypes: SCLC (n = 7), ADC (n = 15), and SCC (n = 10). Box-plots indicate the median and quartiles, with whiskers indicating the 1.5 interquartile range. Statistical significance is indicated using the Mann-Whitney U test, **P ≤ .01, *P < .05.
A Classifier for PE Subtyping
The above data encouraged us to build a classifier tool that could combine the results of individual protein/peptide targets and with a multiplexed panel classify PE samples into their appropriate categories with increased power. Due to the nature of the eight PE groups, we first decided to use a nested dichotomy approach, building a set of binary classifiers and recursively splitting a larger group into two smaller subgroups (Figure 7). Although this is not necessary as random forest classifiers can handle multiclass problems directly, we believe that it is useful to look at the performance of each classifier individually. The classifiers were built on the geometric mean levels for all CKs, actin, and vimentin; the level of the CK-19-Nterm peptide; and the total protein concentration. The first classifier was able to discriminate the bacterial-infection PEs from all other PEs with an AUC of 0.92 ± 0.02 (Figure 7), with the top five contributing factors being (in order of weight) actin, vimentin, CK-19-Nterm, CK-7, and CK-19. The next classifier took the noninfected samples and split them into cancer PEs or control PEs with an AUC of 0.77 ± 0.04 based on the total protein concentration and the levels of CK-8, -19, and -18 and actin. Beyond this division level, the classifiers failed to provide high-significance subtyping, except for the moderate ability to differentiate the two PE groups of SCC and ADC from each other with an AUC of 0.65 ± 0.05 based on the top five CKs being CK-9, -7, -2e, -6A, and -5.
Figure 7.
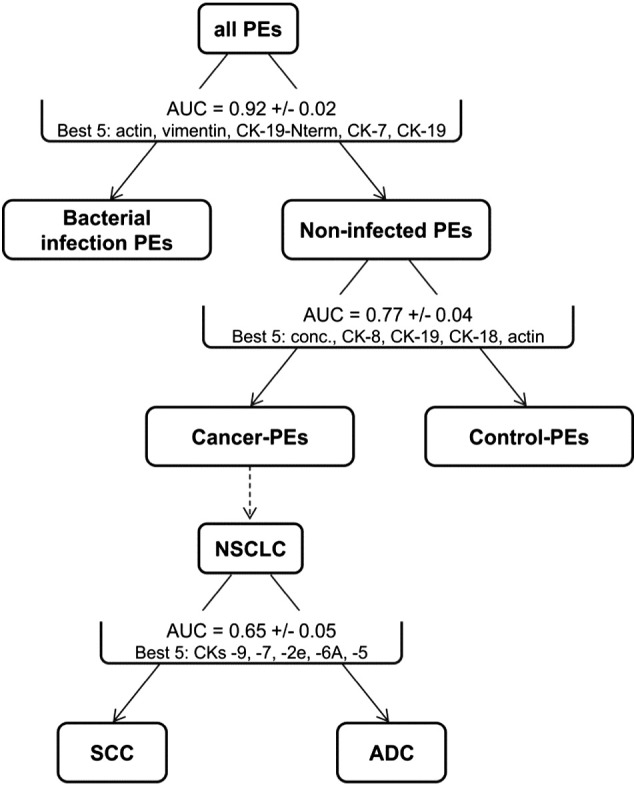
Performance of individual classifiers for PE subtyping. Classification tasks were performed using a random forest algorithm. Classification performance measure (AUC plus/minus error) was estimated using a 10-fold cross-validation procedure. Bacterial-infection PEs: empyema and TB. Control PEs: posttraumatic and transudate PEs. Cancer PEs: all lung- and other-cancer PEs. Best 5 indicates the top five contributing features used by the classifiers for the indicated separation.
The above results show that the most practical aspect of a classifier will be the differentiation of the cancer PEs versus benign PEs. For increased accuracy, a new two-group classifier was built with the above data plus additionally submitting all peptide-level values as independent factors as individual peptides might have their own behavior independent of the geometric mean protein level (e.g., CK-19-Nterm). The resulting classifier had an AUC for classifying cancer PEs from benign PEs of 0.87 (Figure 8A). At an FPR of 21%, this test has a sensitivity of 81% at correctly identifying patients with a cancer PE (Figure 8B). At an FPR set at 9%, for example, to exclude more noncancer patients being sent for invasive procedures, the classifier is predicted to correctly classify 50% of patients having some form of cancer-related PE. The top seven factors that were used for this separation in order of weight are CK-7_FET, CK-6_ADT, vimentin_ILL, protein concentration, CK-4_NLE, CK-19_geo, and vimentin_ISL [protein name_three amino acids of peptide or geometric mean (geo) of all peptides used].
Figure 8.
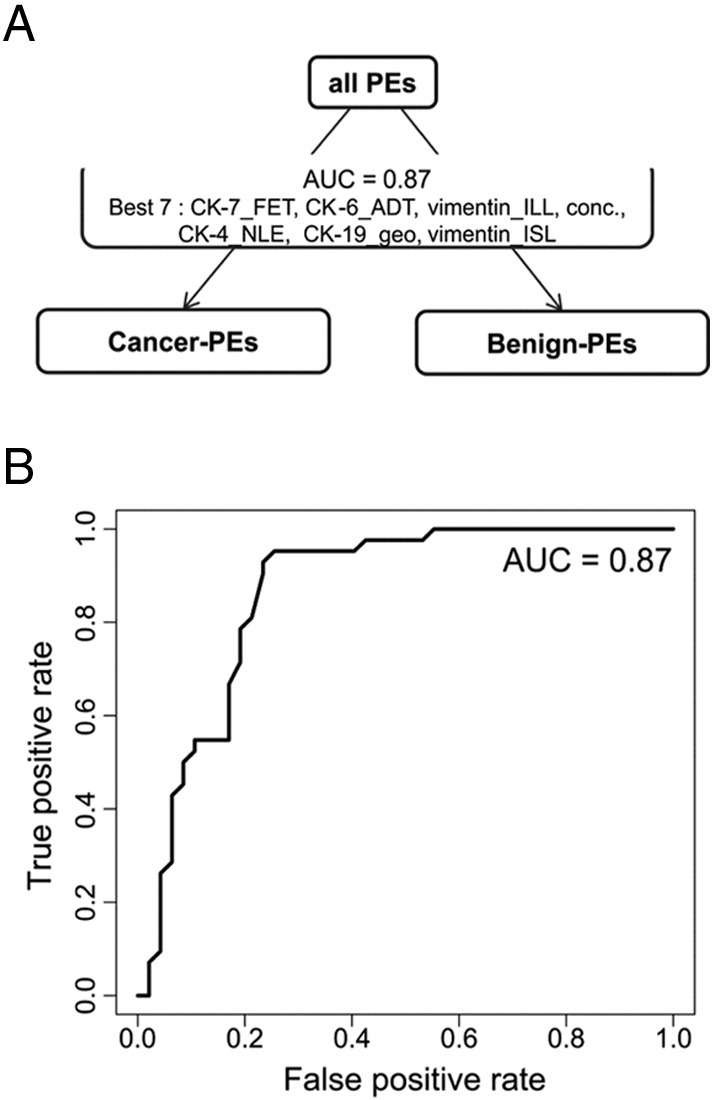
Performance of the two-group classifier for the cancer PE versus benign PE classification. (A) Classifier AUC for classifying cancer PEs from benign PEs and the top seven contributing features used for this separation. (B) ROC curve of the two-group classifier.
Analysis of Undiagnosed PEs Using the Two-Group Classifier
Using the two-group classifier, we analyzed data from the 32 PEs that could not be diagnosed at the initial clinical assessment. The classifier results for these independent samples could be compared for some PEs where more clinical data were obtained to derive a later diagnosis, whereas others remained with an unresolved etiology (Table 2). The classifier indicated one high-scoring sample (score = 1.0) with an FPR of 5% and four samples with scores ranging from 0.75 to 0.80 but with an FPR of 10% for being cancer PEs. For the highest-scoring sample, a later videothoracoscopy examination confirmed a malignant NSCLC case. For PEs with the higher FPR of 10%, one patient had a later diagnosis revealing pleurisy (inflammation of the pleura); another patient was diagnosed with bacterial pneumonia; and for the remaining two patients, there was no definite diagnosis. One patient (#91) with a score of 0.27 was later diagnosed with NSCLC based on a computed tomographic scan but with a paramalignant PE. All PE samples tested had negative cytology results, and the remainder of the patients (scores ≤ 0.7) had no evidence of cancer, with 10 remaining with an undetermined etiology of PE.
Table 2.
Classification Results of the Two-Group (Benign PE Versus Cancer PE) Classifier Performed on an Independent PE Sample Set
| Sample | Score | FPR = 5% | FPR = 10% | PE Type | Videothoracoscopy | Cytology of PE Sediment | Etiology of PE |
|---|---|---|---|---|---|---|---|
| 62 | 1 | + | Exudate | Cancer revealed | Negative | NSCLC (malignant) | |
| 9 | 0.8 | + | Exudate | NP | Negative | Pleurisy | |
| 15 | 0.77 | + | Exudate | NP | Negative | Bacterial pneumonia | |
| 106 | 0.77 | + | Exudate | NP | Negative | Unknown (suspicion of lupus) | |
| 75 | 0.75 | + | Exudate | NP | Negative | Unknown (diaphragmatic hernia) | |
| 69 | 0.7 | Exudate | NP | Negative | Pleurisy | ||
| 48 | 0.67 | Exudate | No evidence of cancer | Negative | Unknown | ||
| 77 | 0.62 | Exudate | No evidence of cancer | Negative | Unknown | ||
| 16 | 0.6 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 37 | 0.57 | Exudate | NP | Negative | Unknown (heart and renal failure) | ||
| 60 | 0.52 | Exudate | NP | Negative | Pleurisy | ||
| 81 | 0.52 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 33 | 0.5 | Exudate | NP | Negative | Unknown (suspicion of leukemia) | ||
| 12 | 0.45 | Exudate | NP | Negative | Unknown | ||
| 132 | 0.45 | Exudate | No evidence of cancer | Negative | Unknown | ||
| 53 | 0.42 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 40 | 0.32 | Transudate | NP | Negative | Heart and renal failure | ||
| 43 | 0.3 | Exudate | NP | Negative | Tuberculosis | ||
| 113 | 0.3 | Exudate | NP | Negative | Unknown (heart failure) | ||
| 127 | 0.3 | Exudate | No evidence of cancer | Negative | Unknown (renal failure) | ||
| 91 | 0.27 | Exudate | NP | Negative | NSCLC (paramalignant) | ||
| 30 | 0.25 | Exudate | NP | Negative | Unknown (tumor of left kidney) | ||
| 13 | 0.22 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 82 | 0.17 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 129 | 0.17 | Exudate | NP | Negative | Tuberculosis | ||
| 115 | 0.15 | Exudate | No evidence of cancer | Negative | Inflammation | ||
| 74 | 0.12 | Exudate | NP | Negative | Unknown (history of chest trauma) | ||
| 105 | 0.1 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 110 | 0.1 | Exudate | NP | Negative | Bacterial pneumonia | ||
| 121 | 0.07 | Transudate | NP | Negative | Liver failure and ascites | ||
| 20 | 0.05 | Exudate | NP | Negative | Pleurisy | ||
| 26 | 0.05 | Exudate | NP | Negative | Pleurisy |
Thirty-two patient PEs whose etiology could not be determined in the initial clinical assessment were analyzed and are presented with a later obtained diagnosis for some and other clinical data. Score and FPR are results of the classifier. NP, not performed.
Discussion
For the first time, we report on the development of targeted MS-based assays to simultaneously quantify a comprehensive set of 33 CKs and screen for these in a broad set of PEs. We reveal the detectable CKs and show that 10 CK abundance levels relate to PE etiology. To the best of our knowledge, this work is also the first to show the detection and quantitation of the important cancer-related simple-epithelial CKs (CK-7, -8, -18, and -19) as a multiplexed panel in PE. No MS-based or immunobased methods have demonstrated this before. Furthermore, no previous work has shown an increase of these CKs in the cancer PEs despite such evidence existing for solid lung tumor tissues. We believe that this is due to our CK enrichment method and the multiplexing capability of MRM analysis.
Initially, we analyzed PEs using in-solution digestion methods used for plasma analysis [40] because PEs consist two thirds of plasma proteins [19]. These targeted approaches, additional global-MS analyses, and alternate methods for trypsin-resistant proteins revealed weak signal intensities for only a few CKs, indicating that CK levels are low among the total protein PE content even in highly positive samples (data not shown). Published PE proteomic investigations, despite applying methodologies to reveal low-abundance biomarkers, largely identified only the high-abundance and potentially contaminating structural- and stratified-epithelial CKs [19], [20], [26], [27], [41]. Our CK enrichment method improved the assay sensitivity with a signal increase of up to 200-fold, which is comparable to antibody-based enrichment methods but without the large reagent cost. Furthermore, the enrichment works on a large population of CKs, i.e., versus requiring an antibody for each protein, and the method can be easily implemented into a routine sample preparation workflow.
Based on the ELISA, our method has a CK-18 LOD of 250 ng/ml, and we expect this to be similar for other CKs as they are structurally highly related. The CK-18 ELISA with an equally determined LOD (at signal to noise ratio > 10:1) of 44 ng/ml showed better analytical sensitivity. However, its analytical specificity is not guaranteed, and the test lacks the multiple levels of molecular specificity assessment that MRM provides. A contributing interference (e.g., other CKs) in the ELISA could cause the observed slightly higher CK-18 levels compared with our method for the midrange samples. The lowest sample measured by the ELISA had 73 ng/ml of CK-18, which is below the capability of our method; however, it has been demonstrated that newer-generation MS instruments could reach quantitation levels even in the range of 1 to 10 ng/ml for certain plasma proteins [42]. This could eliminate the need for CK enrichment, making the analysis more robust due to a simpler workflow. We plan to test this, additionally using SIS peptides characterized by amino acid analysis to obtain precise quantitation values defined in absolute units of ng/ml which will be more appropriate in a clinical application allowing for reference values to be set up and for comparisons of results across studies.
The analysis of 121 PEs revealed the presence of 16 CKs, many of which have connections in the literature to lung cancer (CK -5 to -8, -10, -14, -16, -18, and -19) as well as to other non-lung cancers (as for lung cancer plus: CK-1, -4, -15, and -20) [7], [9]. Some of these are used in pathological examinations for lung cancer classification and prognosis (CK-5/6, -7, -8/18, and -19) [7], [8], [11] or have been published as potential biomarkers in lung cancer research [43], [44]. Detected CK-2e and CK-9 are not often associated with cancer, but they can pair with the cancer-related CK-1 and CK-10. All four are expressed in stratified epithelia and thus can be potential dust contamination in MS analysis from the keratinizing epidermis. Due to the indefinite reproducibility test results caused by low signals for these CKs, we kept them in the MRM panel, taking dust-free precautions, and in the individual patient analysis, their signals were indeed increased in specific PEs, revealing significantly elevated levels of CK-2e, -9, and -10 in the SCC group compared with ADC.
CK-6, CK-7, CK-8, CK-18, and CK-19 are at significantly higher levels in cancer PEs. Still, many PEs diagnosed as cancer related had baseline levels of these CKs. Possibly, these are below the LOD of our method, and future instrumentation and improved methodology could allow their measurement, increasing the power of the assay. Nonetheless, these CKs show an elevated level in the lung-cancer and other-cancer PEs irrespective of whether they are compared with the predominant transudates or empyema PEs together or separately (Figures 3 and 5), indicating that we detected cancer-specific changes rather than what would be evoked in an advanced infection or in a transudate PE. Some PE types had a small number of samples and would need to be analyzed in a larger set to verify that the above CKs are low, e.g., posttraumatic and TB PEs. Traumatic injury, however, usually has an immediate diagnosis, and none of the six analyzed TB PEs had elevated CK levels, on average being lower than in the transudate or empyema PEs (Supplementary Figure S-3).
Elevated levels of the simple-epithelial CK-7, -8, -18, and -19 in the cancer PEs are in full agreement with solid lung tumor tissue analysis. Comparing normal lung tissue with lung ADC by 2D gel electrophoresis combined with Western and MS identification, Gharib et al. showed that the same four CKs are significantly increased in tumors which also correlated with their increased mRNA level [45]. Furthermore, in pathology, CK-8/18 and CK-19 are characteristic for several lung cancer types, whereas CK-7 is specific to ADC [7]. This indicates that our biofluid analysis is sensitive enough to detect changes occurring in the tumor tissue itself, and supports our strategy of devising an analytical method that can rely on less invasive sampling procedures. Release of CKs from tumor cells can occur during apoptosis or necrosis but also, as has been shown for CK-19, can be an active process releasing full-length protein from viable tumor cells, which in this case also correlated with bad prognosis [15].
CK-8, CK-18, and CK-19 compose three cancer biomarkers often applied in the clinical laboratory (tissue polypeptide antigen, tissue polypeptide specific antigen, and CYFRA 21-1) [11] for treatment monitoring and prognosis as their plasma levels indicate ongoing tumor cell activity [46]. This suggests an alternate use for our MRM panel yet to be evaluated.
Despite low numbers of PE samples in the lung cancer subtypes, like for the rare SCLC, we observed differences with some consistency to reports from lung cancer tissues. In histological typing, ADC has higher immunostaining rates for CK-7 and CK-18, whereas SCC is higher for CK-10/13 [10]. We observed that ADC had higher levels of CK-7 compared with SCLC and SCC and of CK-19 (highly related to CK-18) compared with SCC. SCC compared with ADC had elevated levels of CK-10 and additionally of CK-2e and CK-9, which are a binding partner and a biologically related CK to CK-10, respectively. Additionally, our classifier, although limited in its ability at differentiating the cancer subtypes, could distinguish ADC and SCC with a moderate AUC of 0.65 (based on CK-9, -7, -2e, -6A, and -5), making the avenue of cancer subtype differentiation encouraging. Future analysis with larger groups of these subtypes is a next step to verify this.
Gharib et al. observed that specific isoelectric point isoforms (likely due to PTMs) of the simple-epithelial CKs correlated with different clinical variables [45]. Interestingly, antibody cross-reactivity was observed for some isoforms of CK-7 and CK-8, highlighting the issue of inferior specificity of immunobased detection. This is especially relevant if biomarkers are specific peptides and not whole proteins. This might result from biological reasons as cancer-related PTMs (e.g., phosphorylation) or proteolytic activities from elastase or caspases (e.g., on CK-19) [11]. We observed that a specific peptide of CK-19 (CK-19-Nterm), located in the head region commonly involved with PTMs, behaved differently from the other CK-19 peptides. A phosphorylation site shown to modulate filament organization is located three amino acids away from this peptide’s trypsin cleavage site and could affect digestion efficiency [47]. This peptide alone differentiated the benign PEs and cancer PEs with an AUC of 0.83, which was much higher than that for the total CK-19 protein level (Figure 4A).
Bacterial-infection PEs could be excluded from our analysis as noncancer PEs with high confidence (AUC = 0.92) using a classifier that combines the actin and vimentin results with measurements of CK-19-Nterm, -7, and -19. Actin has been observed to be higher in TB and pneumonia PEs compared with malignant PEs in global proteomic analyses but was described as a potential marker (lowered) of malignant PEs with no connection to an infection [4], [21], [22]. To the best of our knowledge, the parallel changes in vimentin levels have also not been associated with PEs from infected individuals; rather, vimentin has been overexpressed in cancer and metastasis and recognized as a marker for the epithelial-to-mesenchymal transition [48]. But it has been demonstrated that macrophages, which are also the major host harboring Mycobacterium tuberculosis, in response to proinflammatory signaling secrete vimentin which is involved in bacterial killing, and this could explain the elevated levels [49], [50].
Our two-group classifier had a sensitivity of 81% and a specificity of 79% for identifying cancer-related PEs (AUC = 0.87). This is a very promising result comparable to published PE research where sensitivity and specificity ranged between 25% and 55% and between 85% and 100% (AUC = 0.65-0.95), respectively, in identifying malignant PEs using ELISA- or proteomic-based markers or panels thereof [26], [27], [28], [29]. Noteworthy is that, in these studies with high AUC values (> 0.9), classifications were performed only for PEs from malignant cases likely to have the highest marker levels [26], [27]. We believe that our analysis is an improvement as we considered both malignant and paramalignant PEs in the “cancer PE” group. For comparison, our best single marker had an AUC of 0.83 compared with that of Chen et al. whose best single marker had an AUC of 0.76 in such a grouping [26]. Our results are also on par with the FDA-approved test (CELLSEARCH) which partly uses CK antibodies [51] as it was used to improve on cytology assessment, differentiating malignant from benign PEs with an AUC of 0.86 and a sensitivity of 78% and specificity of 86% [52]. We had insufficient clinical data to exclude paramalignant PEs, and possibly, our classifier could score even higher in a malignant-centric approach. Comparing our results to cytology, which was positive for only 12% of the cancer PEs, and to videothoracoscopy, which further verified 19% of cancer PEs as definite malignant PEs, shows that our method, e.g., at 50% sensitivity and a 91% specificity for classifying cancer PEs, has potential at diagnosing cancer PEs not just associated with a metastasis to the pleura. The verified malignant PEs although scoring high in our classifier (> 0.8) and having increased levels of the simple-epithelial CKs were exceeded in both respects by many other cancer PEs, indicating that some malignant PEs might have also been missed due to the high false-negative rate of cytology and because videothoracoscopy was performed on only a few patients.
The selection of top factors for a differentiation in the two-group classifier was made based on a “greedy forward feature selection” method to avoid selection of markers with redundant behavior. This explains why the CK-19-Nterm peptide with the highest significance in the individual protein comparisons was later outperformed by similarly acting but slightly better differentiating peptides in the classifier. The classifier for SCC/ADC as expected from individual protein comparisons selected CK-7, -2e, and -9 as top factors but additionally chose CK-6A and -5, which alone showed no significance. This highlights the power of a multiplexed target panel and classifier tool which can use markers that individually are not differentially abundant but grouped together can be predictive.
Analyzing data using the two-group classifier from an unrelated set of 32 PEs, which could not be diagnosed at the time of sampling, scored one case at 1 with an FPR of 5% for being cancer related. Later, it was indeed confirmed to be caused by malignant NSCLC. Four samples diagnosed later as benign had moderate scores (0.75-0.8) but an FPR of 10%, suggesting a 5% FPR with a score > 0.8 as a cutoff for this classifier. Another NSCLC case did score only 0.27, but it had no involvement of the pleura (paramalignant PE). This independent sample set, although limited in cancer PEs, allowed to show that our classifier can identify an undiagnosed cancer-related PE missed by early standard clinical assessment. Thus, our analytical workflow can potentially shorten diagnosis time and guide patients to appropriate procedures. With refinement, in the future, such an assay could be an additional tool among existing clinical tests performed on patients presenting with PEs at first assessment.
Conclusions
This work integrates a collection of clinical samples and biochemical data obtained by advanced MS techniques for increased analytical specificity to capture a fingerprint of CK levels in the context of specific lung diseases. For the first time, we report on the quantitation of a wide panel of CKs in a multiplexed assay and screening a broad set of PEs that revealed the cancer-related increase of simple-epithelial CKs which are established tumor markers. These novel findings in PE, obtained by a less invasive procedure, corroborate observations in solid tumors. Using advanced statistical methods, we can significantly classify benign PEs versus cancer PEs and hope that this analytical workflow can be implemented into clinical applications in the future. Such a screening, following imaging diagnostics, in many clinical situations could precede the often difficult decision on the next steps involving highly invasive procedures. Also encouraging is the perspective of less invasive treatment monitoring, e.g., tumor stabilization versus progression in the case of lung cancer, as well as the analysis of other biofluids and epithelial cell–related cancers using our developed CK MRM panel.
Acknowledgements
This work was fully supported by the National Science Centre of Poland with the Opus 5 grant (2013/09/B/NZ2/02160). The authors would like to acknowledge the contribution of Dr. Andrzej Lewandowicz in the writing of the Opus 5 grant. The authors would like to thank Agata Malinowska for helpful discussions regarding the classifier tool. We would also like to thank the valuable contribution of the patients treated at the Mazovian Center of Pulmonary Disease and Tuberculosis Treatment.
Footnotes
This work was fully supported by the National Science Centre of Poland with the Opus 5 grant (2013/09/B/NZ2/02160).
The authors declare no conflict of interest.
This article contains Supplementary Figures S-1 to S-3 and Supplementary Tables S-1 to S-3: CK enrichment method development data for additional CKs, as well as actin and vimentin; MRM results for actin and vimentin for all eight analyzed PE groups; MRM results displayed for all eight analyzed PE groups for CK-7, -8, -18, -19, and -19Nterm; List of proteins and the corresponding peptides targeted by the MRM assay; List of all MRM transitions used including standard retention time, charge states, and settings for cone voltage and collision energy; Results of the nonparametric Mann-Whitney U test for the comparison of CK levels between all eight PE group combinations. Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.neo.2016.06.002.
Contributor Information
Dominik Domanski, Email: dom.domanski@ibb.waw.pl.
Michal Dadlez, Email: michald@ibb.waw.pl.
Appendix A. Supplementary data
Supplementary Figures S-1 to S-3: CK enrichment method development data for additional CKs, as well as actin and vimentin; MRM results for actin and vimentin for all eight analyzed PE groups; MRM results displayed for all eight analyzed PE groups for CKs -7, -8, -18, -19 and -19Nterm.
Supplementary Tables S-1 to S-3: List of proteins and the corresponding peptides targeted by the MRM assay; List of all MRM transitions used including standard retention time, charge states, and settings for cone voltage and collision energy; Results of the non-parametric Mann Whitney U-test for the comparison of CK levels between all eight PE group combinations.
Supplementary materials
References
- 1.Ferlay J, Soerjomataram II, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin D M, Forman D D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2014;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD. Classification of lung cancer. Semin Roentgenol. 2011;46(3):178–186. doi: 10.1053/j.ro.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 3.Porcel JM, Light RW. Diagnostic approach to pleural effusion in adults. Am Fam Physician. 2006;73(7):1211–1220. [PubMed] [Google Scholar]
- 4.Rodríguez-Piñeiro A M, Blanco-Prieto S, Sánchez-Otero N, Rodríguez-Berrocal F J, Páez de la Cadena M. On the identification of biomarkers for non–small cell lung cancer in serum and pleural effusion. J Proteomics. 2010;73(8):1511–1522. doi: 10.1016/j.jprot.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y C G, Light R W. 2004. Invited review series: pleural diseases management of malignant pleural effusions; pp. 148–156. [DOI] [PubMed] [Google Scholar]
- 6.Dubinski W, Leighl NB, Tsao M-S, Hwang DM. Ancillary testing in lung cancer diagnosis. Pulm Med. 2012;2012:1–8. doi: 10.1155/2012/249082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu PG, Weiss LM. Keratin expression in human tissues and neoplasms. Histopathology. 2002;40(5):403–439. doi: 10.1046/j.1365-2559.2002.01387.x. [DOI] [PubMed] [Google Scholar]
- 8.Moll R, Divo M, Langbein L. The human keratins: biology and pathology. Histochem Cell Biol. 2008;129(6):705–733. doi: 10.1007/s00418-008-0435-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011;30(2):127–138. doi: 10.1038/onc.2010.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nhung NV, Mirejovsky P, Mirejovsky T, Melinova L. Cytokeratins and lung carcinomas. Cesk Patol. 1999;35(3):80–84. [PubMed] [Google Scholar]
- 11.Barak V, Goike H, Panaretakis KW, Einarsson R. Clinical utility of cytokeratins as tumor markers. Clin Biochem. 2004;37(7):529–540. doi: 10.1016/j.clinbiochem.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Ulukaya E, Yilmaztepe A, Akgoz S, Linder S, Karadag M. The levels of caspase-cleaved cytokeratin 18 are elevated in serum from patients with lung cancer and helpful to predict the survival. Lung Cancer. 2007;56(3):399–404. doi: 10.1016/j.lungcan.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Füzéry AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10(1):13. doi: 10.1186/1559-0275-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker M. Blame it on the antibodies. Nature. 2015;521:274–275. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 15.Alix-Panabières C, Vendrell J-P, Slijper M, Pellé O, Barbotte E, Mercier G, Jacot W, Fabbro M, Pantel K. Full-length cytokeratin-19 is released by human tumor cells: a potential role in metastatic progression of breast cancer. Breast Cancer Res. 2009;11(3):R39. doi: 10.1186/bcr2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang K-L, Chao W-R, Han C-P. Anticytokeratin (CAM5.2) reagent identifies cytokeratins 7 and 8, not cytokeratin 18. Chest J. 2014;145(6):1441–1442. doi: 10.1378/chest.14-0168. [DOI] [PubMed] [Google Scholar]
- 17.Gillette MA, Carr SA. Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry. Nat Methods. 2013;10(1):28–34. doi: 10.1038/nmeth.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tyan Y-C, Wu H-Y, Su W-C, Chen P-W, Liao P-C. Proteomic analysis of human pleural effusion. Proteomics. 2005;5(4):1062–1074. doi: 10.1002/pmic.200401041. [DOI] [PubMed] [Google Scholar]
- 19.Pernemalm M, De Petris L, Eriksson H, Brandén E, Koyi H, Kanter L, Lewensohn R, Lehtiö J. Use of narrow-range peptide IEF to improve detection of lung adenocarcinoma markers in plasma and pleural effusion. Proteomics. 2009;9(13):3414–3424. doi: 10.1002/pmic.200800814. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Lian H, Jia Q, Wan Y. Proteome screening of pleural effusions identifies IL1A as a diagnostic biomarker for non–small cell lung cancer. Biochem Biophys Res Commun. 2015;457(2):177–182. doi: 10.1016/j.bbrc.2014.12.083. [DOI] [PubMed] [Google Scholar]
- 21.Sheng S, Zhu H. Proteomic analysis of pleural effusion from lung adenocarcinoma patients by shotgun strategy. Clin Transl Oncol. 2014;16(2):153–157. doi: 10.1007/s12094-013-1054-9. [DOI] [PubMed] [Google Scholar]
- 22.Wang Z, Wang C, Huang X, Shen Y, Shen J, Ying K. Differential proteome profiling of pleural effusions from lung cancer and benign inflammatory disease patients. Biochim Biophys Acta - Proteins Proteomics. 2012;1824(4):692–700. doi: 10.1016/j.bbapap.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Yu C-J, Wang C-L, Wang C-I, Chen C-D, Dan Y-M, Wu C-C, Wu Y-C, Lee I-N, Tsai Y-H, Chang Y-S. Comprehensive proteome analysis of malignant pleural effusion for lung cancer biomarker discovery by using multidimensional protein identification technology. J Proteome Res. 2011;10(10):4671–4682. doi: 10.1021/pr2004743. [DOI] [PubMed] [Google Scholar]
- 24.Mundt F, Johansson HJ, Forshed J, Arslan S, Metintas M, Dobra K, Lehtio J, Hjerpe A. Proteome screening of pleural effusions identifies galectin 1 as a diagnostic biomarker and highlights several prognostic biomarkers for malignant mesothelioma. Mol Cell Proteomics. 2014;13(3):701–715. doi: 10.1074/mcp.M113.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanchez-Otero N, Bielsa S, Porcel JM. Proteomics in pleural effusions. Plevra Bul. 2012;7(3):56–63. [Google Scholar]
- 26.Chen C-D, Wang C-L, Yu C-J, Chien K-Y, Chen Y-T, Chen M-C, Chang Y-S, Wu C-C, Yu J-S. Targeted proteomics pipeline reveals potential biomarkers for the diagnosis of metastatic lung cancer in pleural effusion. J Proteome Res. 2014;13(6):2818–2829. doi: 10.1021/pr4012377. [DOI] [PubMed] [Google Scholar]
- 27.Liu P-J, Chen C-D, Wang C-L, Wu Y-C, Hsu C-W, Lee C-W, Huang L-H, Yu J-S, Chang Y-S, Wu C-C. In-depth proteomic analysis of six types of exudative pleural effusions for nonsmall cell lung cancer biomarker discovery. Mol Cell Proteomics. 2015;14(4):917–932. doi: 10.1074/mcp.M114.045914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang Q-L, Shi H-Z, Qin X-J, Liang X-D, Jiang J, Yang H-B. Diagnostic accuracy of tumour markers for malignant pleural effusion: a meta-analysis. Thorax. 2008;63(1):35–41. doi: 10.1136/thx.2007.077958. [DOI] [PubMed] [Google Scholar]
- 29.Porcel JM, Vives M, Esquerda A, Salud A, Pérez B, Rodríguez-Panadero F. Use of a panel of tumor markers (carcinoembryonic antigen, cancer antigen 125, carbohydrate antigen 15-3, and cytokeratin 19 fragments) in pleural fluid for the differential diagnosis of benign and malignant effusions. Chest. 2004;126(6):1757–1763. doi: 10.1378/chest.126.6.1757. [DOI] [PubMed] [Google Scholar]
- 30.Mohammed Y, Domański D, Jackson AM, Smith DS, Deelder AM, Palmblad M, Borchers CH. PeptidePicker: a scientific workflow with web interface for selecting appropriate peptides for targeted proteomics experiments. J Proteomics. 2014;106:151–161. doi: 10.1016/j.jprot.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 31.Bakun M, Niemczyk M, Domanski D, Jazwiec R, Perzanowska A, Niemczyk S, Kistowski M, Fabijanska A, Borowiec A, Paczek L. Urine proteome of autosomal dominant polycystic kidney disease patients. Clin Proteomics. 2012;9(1):13. doi: 10.1186/1559-0275-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 2010;26(7):966–968. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.R Development Core Team, R . the R Foundation for Statistical Computing. ISBN: 3-900051-07-0; 2011. R: A Language and Environment for Statistical Computing Vienna, Austria:http://www.R-project.org/ Available online at. [Google Scholar]
- 34.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V. Scikit-learn: machine learning in python. J Mach Learn Res. 2012;12:2825–2830. [Google Scholar]
- 35.Kuzyk M A, Smith D, Yang J, Cross T J, Jackson A M, Hardie D B, Anderson N L, Borchers C H. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol Cell Proteomics. 2009;8(8):1860–1877. doi: 10.1074/mcp.M800540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kidd ME, Shumaker DK, Ridge KM. The role of vimentin intermediate filaments in the progression of lung cancer. Am J Respir Cell Mol Biol. 2014;50(1):1–6. doi: 10.1165/rcmb.2013-0314TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meiklejohn B, Nagle RB, McDaniel KM, Wilson GS. Improved methods for separation of human cytokeratins. Life Sci. 1990;47(7):637–645. doi: 10.1016/0024-3205(90)90576-d. [DOI] [PubMed] [Google Scholar]
- 38.Achtstaetter T, Hatzfeld M, Quinlan RA, Parmelee DC, Franke WW. Separation of cytokeratin polypeptides by gel electrophoretic and chromatographic techniques and their identification by immunoblotting. Methods Enzymol. 1986;134:355–371. doi: 10.1016/0076-6879(86)34102-8. [DOI] [PubMed] [Google Scholar]
- 39.Linder S, Olofsson MH, Herrmann R, Ulukaya E. Utilization of cytokeratin-based biomarkers for pharmacodynamic studies. Expert Rev Mol Diagn. 2010;10:353–359. doi: 10.1586/erm.10.14. [DOI] [PubMed] [Google Scholar]
- 40.Proc J L, Kuzyk M A, Hardie D B, Yang J, Smith D S, Jackson A M, Parker C E, Borchers C H. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J Proteome Res. 2010;9(10):5422–5437. doi: 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu CJ, Wang CL, Wang CI, Chen CD, Dan YM, Wu CC, Wu YC, Lee IN, Tsai YH, Chang YS. Comprehensive proteome analysis of malignant pleural effusion for lung cancer biomarker discovery by using multidimensional protein identification technology. J Proteome Res. 2011;10(10):4671–4682. doi: 10.1021/pr2004743. [DOI] [PubMed] [Google Scholar]
- 42.Domanski D, Percy AJ, Yang J, Chambers AG, Hill JS, Freue GVC, Borchers CH. MRM-based multiplexed quantitation of 67 putative cardiovascular disease biomarkers in human plasma. Proteomics. 2012;12(8):1222–1243. doi: 10.1002/pmic.201100568. [DOI] [PubMed] [Google Scholar]
- 43.Indovina P, Marcelli E, Pentimalli F, Tanganelli P, Tarro G, Giordano A. Mass spectrometry-based proteomics: the road to lung cancer biomarker discovery. Mass Spectrom Rev. 2012;32(2):129–142. doi: 10.1002/mas.21355. [DOI] [PubMed] [Google Scholar]
- 44.Lehtiö J, De Petris L. Lung cancer proteomics, clinical and technological considerations. J Proteomics. 2010;73(10):1851–1863. doi: 10.1016/j.jprot.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 45.Gharib TG, Chen G, Wang H, Huang C-C, Prescott MS, Shedden K, Misek DE, Thomas DG, Giordano TJ, Taylor JMG. Proteomic analysis of cytokeratin isoforms uncovers association with survival in lung adenocarcinoma. Neoplasia. 2002;4(5):440–448. doi: 10.1038/sj.neo.7900257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slesak B, Harlozinska-Szmyrka A, Knast W, Sedlaczek P, van Dalen A, Einarsson R. Tissue polypeptide specific antigen (TPS), a marker for differentiation between pancreatic carcinoma and chronic pancreatitis. A comparative study with CA 19-9. Cancer. 2000;89(1):83–88. doi: 10.1002/1097-0142(20000701)89:1<83::aid-cncr12>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 47.Zhou X, Liao J, Hu L, Feng L, Omary MB. Characterization of the major physiologic phosphorylation site of human keratin 19 and its role in filament organization. J Biol Chem. 1999;274(18):12861–12866. doi: 10.1074/jbc.274.18.12861. [DOI] [PubMed] [Google Scholar]
- 48.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci. 2011;68(18):3033–3046. doi: 10.1007/s00018-011-0735-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mor-Vaknin N, Punturieri A, Sitwala K, Markovitz DM. Vimentin is secreted by activated macrophages. Nat Cell Biol. 2003;5(1):59–63. doi: 10.1038/ncb898. [DOI] [PubMed] [Google Scholar]
- 50.Rajaram MVSS, Ni B, Dodd CE, Schlesinger LS. Macrophage immunoregulatory pathways in tuberculosis. Semin Immunol. 2014;26(6):471–485. doi: 10.1016/j.smim.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AGJ, Uhr JW, Terstappen LWMM. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10(20):6897–6904. doi: 10.1158/1078-0432.CCR-04-0378. [DOI] [PubMed] [Google Scholar]
- 52.Schwed Lustgarten DE, Thompson J, Yu G, Vachani A, Vaidya B, Rao C, Connelly M, Udine M, Tan KS, Heitjan DF. Use of circulating tumor cell technology (CELLSEARCH) for the diagnosis of malignant pleural effusions. Ann Am Thorac Soc. 2013;10(6):582–589. doi: 10.1513/AnnalsATS.201303-068OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S-1 to S-3: CK enrichment method development data for additional CKs, as well as actin and vimentin; MRM results for actin and vimentin for all eight analyzed PE groups; MRM results displayed for all eight analyzed PE groups for CKs -7, -8, -18, -19 and -19Nterm.
Supplementary Tables S-1 to S-3: List of proteins and the corresponding peptides targeted by the MRM assay; List of all MRM transitions used including standard retention time, charge states, and settings for cone voltage and collision energy; Results of the non-parametric Mann Whitney U-test for the comparison of CK levels between all eight PE group combinations.
Supplementary materials