

Abstract
The protein Chibby (Cby) is an antagonist of the Wnt signaling pathway, where it inhibits the binding between the transcriptional coactivator β‐catenin and the Tcf/Lef transcription factors. The 126 residue Cby is partially disordered; its N‐terminal half is unstructured while its C‐terminal half comprises a coiled‐coil domain. Previous structural analyses of Cby using NMR spectroscopy suffered from severe line broadening for residues within the protein's C‐terminal half, hindering detailed characterization of the coiled‐coil domain. Here, we use hydrogen/deuterium exchange‐mass spectrometry (HDX‐MS) to examine Cby's C‐terminal half. Results reveal that Cby is divided into three structural elements: a disordered N‐terminal half, a coiled‐coil domain, and a C‐terminal unstructured extension consisting of the last ∼ 25 residues (which we term C‐terminal extension). A series of truncation constructs were designed to assess the roles of individual structural elements in protein stability and Cby binding to TC‐1, a positive regulator of the Wnt signaling pathway. CD and NMR data show that Cby maintains coiled‐coil structure upon deletion of either disordered region. NMR and ITC binding experiments between Cby and TC‐1 illustrate that the interaction is retained upon deletion of either Cby's N‐terminal half or its C‐terminal extension. Intriguingly, Cby's C‐terminal half alone binds to TC‐1 with significantly greater affinity compared to full‐length Cby, implying that target binding of the coiled‐coil domain is affected by the flanking disordered regions.
Keywords: Chibby, intrinsically disordered protein, Thyroid Cancer 1, HDX‐MS, NMR, ITC, coiled‐coil
Abbreviations
- Cby
Chibby
- CD
circular dichroism
- HDX‐MS
hydrogen/deuterium exchange‐mass spectrometry
- NMR
nuclear magnetic resonance
- TC‐1
Thyroid Cancer 1.
Introduction
The Wnt/β‐catenin pathway, a signaling cascade whose roles include embryonic development and stem‐cell self‐renewal, has been linked to a number of human diseases upon its dysregulation, including cancer.1, 2, 3 Human Chibby (Cby), an evolutionarily conserved protein, plays a critical role in this pathway by functioning as an antagonist of the transcriptional co‐activator β‐catenin, the key mediator of Wnt signaling.4 Cby achieves its antagonistic activity via two mechanisms. First, Cby interacts with β‐catenin, forming a complex that prevents β‐catenin from binding to and activating TCF/LEF transcription factors.4 Secondly, Cby/β‐catenin forms a tripartite complex with 14‐3‐3 proteins to promote β‐catenin's export from the nucleus, further suppressing Wnt signaling.5, 6
Besides binding to β‐catenin and 14‐3‐3, Cby participates in the Wnt signaling pathway by interacting with several other partners including nuclear export receptor CRM1,5 importin‐α,5, 7 and Thyroid Cancer 1 (TC‐1, also known as C8orf4).8, 9 In particular, TC‐1, which was originally identified as a gene highly expressed in thyroid cancer, positively regulates Wnt signaling through its interaction with Cby.10 By competing with β‐catenin for binding to Cby, TC‐1 enables free β‐catenin to up‐regulate Wnt target genes.9 Recent studies show that TC‐1 is also associated with ovarian carcinoma,11 tongue squamous cell carcinomas,12 and lung cancer,13 all likely through the Wnt/β‐catenin pathway.
In addition to these binding partners, Cby interacts with several proteins outside of Wnt signaling such as CEP164,14 NBPF1, clusterin,15 polycystin‐2, and GM130.16 Even though numerous Cby interactors have been identified, the molecular details of most of these interactions remain largely unknown. It has been suggested that the ability of Cby to bind multiple targets is facilitated by its partially disordered nature.17 In our previous work and that of others,7, 17 biophysical and biochemical techniques, which include circular dichroism (CD) spectropolarimetry, nuclear magnetic resonance (NMR) spectroscopy, cross‐linking, and gel filtration experiments, were used to characterize the structure of Cby. The results demonstrated that the N‐terminal half of Cby is largely unstructured, whereas its C‐terminal is α‐helical and harbors a predicted coiled‐coil domain.7, 17 The coiled‐coil domain allows for the self‐association of Cby in solution, where it exists predominantly as a homodimer.7 These distinct structural elements of Cby are important for mediating its interactions with various targets. For example, in Wnt/β‐catenin signaling, the disordered N‐terminus of Cby binds to the 14‐3‐3 proteins (upon phosphorylation at residue 20 by the kinase Akt)5, 18 and the nuclear export receptor CRM1,5 whereas its C‐terminal half is responsible for binding to β‐catenin,4 TC‐1,9 and importin‐α.5, 7
Proteins containing both unstructured regions and coiled‐coil motifs are widespread. As in the case of Cby, utilization of both types of structural elements may be critical to their biological function.19, 20, 21, 22 For instance, disordered regions can tune the affinity for protein interaction partners.23 Increasing or decreasing these affinities may be accomplished by different mechanisms.23, 24 Examples include providing enhanced stability to the protein or its complex,25 as well as disorder‐to‐order transitions upon interaction26 or autoinhibition.27 In this work, we sought to determine how the disordered regions of Cby affect the stability and target recognition of its coiled‐coil domain.
NMR spectroscopy is a valuable tool to dissect the structural details and dynamics of partially disordered proteins.28, 29, 30 However, in the case of Cby, its characterization by NMR spectroscopy has proven to be a challenge. Our previous work showed that most of the NMR resonances for Cby's C‐terminal half are too broad to be observed under non‐denaturing conditions, likely due to the monomer/dimer conformational exchange or formation of higher oligomeric forms.17 This behavior is not unique to Cby, as severe NMR broadening has been seen in other proteins that are comprised of long disordered regions and coiled‐coil motifs.31, 32, 33
Hydrogen/deuterium exchange‐mass spectrometry (HDX‐MS) is another widely used technique to analyze protein folding, structure, dynamics, and interactions.34, 35, 36, 37, 38 Backbone amide hydrogens within disordered regions will exchange rapidly upon D2O exposure, in comparison to residues in well‐folded, rigid regions, where slow D2O uptake is mediated by conformational fluctuations that lead to transient opening of hydrogen bonds. In this work, we combined HDX‐MS, NMR spectroscopy, CD, and isothermal titration calorimetry (ITC) to investigate the interplay of different Cby structural elements during target (TC‐1) recognition.
Results
Identifying the coiled‐coil region of Cby using HDX‐MS
Owing to previous difficulties encountered during NMR‐based characterization attempts of full‐length Cby,17 we resorted to HDX‐MS. The primary goal of these measurements was to delineate the coiled‐coil dimerization domain of Cby. Figure 1 shows deuterium uptake curves for the Cby peptic peptides. Deuteration was quasi‐instantaneous for the N‐terminal half of Cby, that is, deuterium levels of >90% were observed already for the first experimental time point of 1 min. Similarly, residues 102‐126, which make up the very C‐terminal end of Cby, exhibited extremely rapid deuterium uptake. Strong HDX protection is observed in peptides that are near (residues 53‐59) and within (residues 60‐90) the predicted boundaries of Cby's coiled‐coil domain (residues 68‐102),7, 39 with nearly no exchange in the range of residues 91‐98.
Figure 1.
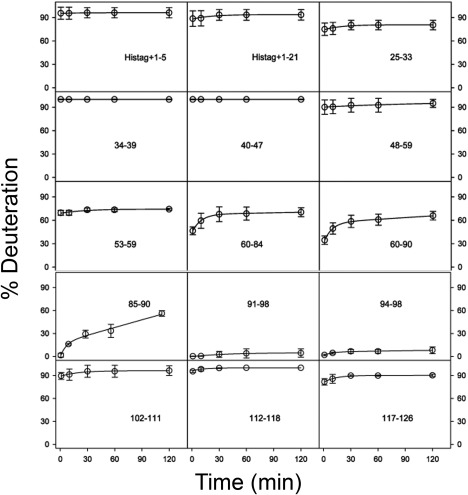
HDX kinetics of peptides in Cby. The 28‐residue His‐tag and Cby residue numbers are indicated in each panel. Lines are bi‐exponential fits. Standard deviations of triplicate measurements are shown as error bars.
For visualization of these data, the deuteration percentages were mapped onto a structural model of dimeric Cby for t = 60 min (Fig. 2). In this model, the coiled‐coil is presented as residues 73‐100, corresponding to four turns of heptad repeats. However, we should point out that the N‐terminal boundary of the coiled‐coil domain is not clearly defined in our HDX data since only relatively large peptic fragments (60‐84 and 60‐90) were obtained for this region. In contrast, it is clear that the coiled‐coil ends around residues 98, as the significant protection observed is immediately succeeded by rapid deuterium uptake in the 102‐111 peptic fragment. For the rest of the model, Cby's N‐termini and very C‐termini (which will be denoted as its C‐terminal extension from now on) are displayed in a largely extended state.
Figure 2.
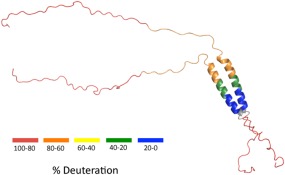
HDX‐MS data of Fig. 1 mapped onto a structural model of Cby for t = 60 min. The 126‐residue Cby is shown as a dimer. Residues 1‐72 are shown in a highly extended state, the coiled‐coil is comprised of residues 73‐100 and residues 101‐126 are displayed in a disordered form. The colouring represents a range of deuteration percentages. Peptide mapping did not cover the grey regions.
The disordered N‐terminal half and C‐terminal extension of Cby
As identified by HDX‐MS, Cby contains two disordered regions of different length flanking the coiled‐coil motif. We sought to determine whether these two unstructured segments have differing effects on the stability of the coiled‐coil structure. A number of truncated Cby constructs were generated for this purpose (Fig. 3). First, we tested if Cby's C‐terminal extension plays a role in the formation of the coiled‐coil by truncating the last 20 residues of Cby (CbyΔC20). A CD spectrum of CbyΔC20 in 10 mM ammonium acetate, pH 5.0 is highly similar to that of full‐length Cby [Fig. 4(A)]. Deconvolution of the spectra using the DichroWeb software44 gives 30% helical, 21% strand, and 49% disordered/turn structure for the full‐length Cby and 41% helical, 17% strand, and 42% disordered/turn for CbyΔC20. The data indicate that the C‐terminal 20 residues are not structured and that the helical content of Cby is maintained upon deletion of this disordered extension. Notably, the CD spectrum of a synthetic construct comprising the C‐terminal 25 residues of Cby (CbyC25), shows that the C‐terminal extension is largely disordered but does have α‐helical propensity (deconvolution: 42% helical, 12% strand, and 46% turns/disordered) [Fig. 4(C)].
Figure 3.
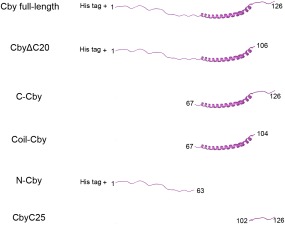
Cby constructs used in this study.
Figure 4.
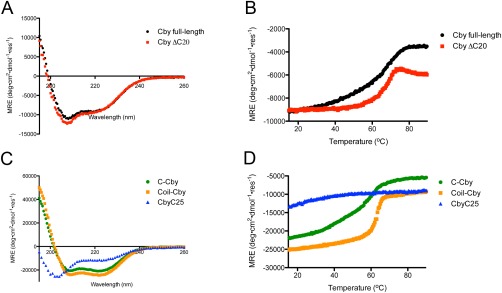
(A) CD spectra of full‐length Cby and CbyΔC20. Displayed spectra are the average of 5 scans. (B) Thermal melts of full‐length Cby and CbyΔC20 monitored using CD at 222 nm. (C) CD spectra of C‐Cby, Coil‐Cby, and CbyC25. Displayed spectra are the average of 5 scans. (D) Thermal melts of C‐Cby, Coil‐Cby, and CbyC25 monitored using CD at 222 nm.
Next, we investigated the effect of C‐terminal truncation on the thermal stability of the coiled‐coil motif. Figure 4(B) shows the CD thermal denaturation profiles of wild‐type Cby and CbyΔC20. For the full‐length Cby, the ellipiticity at 222 nm gradually becomes less negative as the temperature increases from 20 to 50°C, while the signal remains steady for the truncated CbyΔC20 over this temperature range. Once the temperature reaches ∼60°C, both constructs display a significant increase (becoming less negative) in ellipiticity, signifying the melting of the coiled‐coil. A thermal melt of the CbyC25 peptide shows a gradual loss of signal at 222 nm in the 20‐50°C range, which likely explains the differences between the thermal denaturation profiles of wild‐type Cby and CbyΔC20 in that temperature range [Fig. 4(D)].
While the thermal denaturation of the full‐length Cby is reversible, as the CD spectrum of the protein at 25°C is identical before and after the melt, CbyΔC20 precipitates upon denaturation. Also, during optimization of the CbyΔC20 purification strategy, we observed that the truncated version tends to aggregate in the presence of NaCl at low concentrations (50 mM) while full‐length Cby remains soluble in 100 mM NaCl up to a protein concentration of 200 μM. Taken together, these observations suggest that the C‐terminal extension is important for Cby's solubility, consistent with the prevalence of charged side chains in this region (Supporting Information Fig. 1).
An overlay of the 1H‐15N HSQC NMR spectra of full‐length Cby and CbyΔC20 is shown in Figure 5. Similar to the full‐length protein, only peaks from the N‐terminal half of CbyΔC20 can be observed in the HSQC spectrum. Consistent with the CD data, the NMR spectrum suggests that Cby's coiled‐coil structure remains intact upon deletion of the C‐terminal extension, rendering the peaks from this region unobservable by NMR. Notably, truncation of the C‐terminal extension of Cby does not induce significant chemical shift changes or intensity attenuation to the signals originating from the N‐terminal half, indicating that there are no long‐range interactions between the N‐ and C‐terminal disordered regions of Cby.
Figure 5.
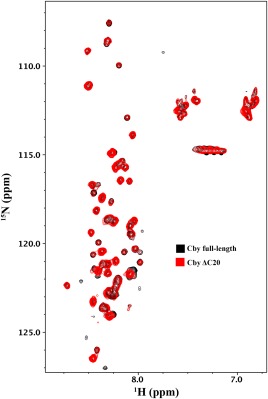
1H‐15N HSQC NMR spectrum of full‐length Cby (black) overlayed with a 1H‐15N HSQC of CbyΔC20 (red).
Next, we sought to characterize isolated C‐terminal half of Cby in the absence of the disordered N‐terminus. A CD spectrum reveals that C‐Cby (residues 67‐126) is largely helical [Fig. 4(C)]. The deconvoluted procedure reports 64% helical, 3% strand, and 33% disordered/turn. The [θ]222/[θ]208 ratio is ≈ 1, which is indicative of coiled‐coil structure.41 A thermal melt of C‐Cby, using CD monitored at 222 nm, displays a profile similar to that of full‐length Cby, where a broad transition is observed between 20°C and 70°C [Fig. 4(D)]. Like full‐length Cby, C‐Cby could completely refold after thermal denaturation. A HSQC spectrum collected for isolated C‐Cby only contains a few broad peaks, indicating that most of the NMR signals from the coiled‐coil motif and C‐terminal extension in this construct remain largely unobservable (data not shown).
After extensive efforts, we managed to purify the coiled‐coil domain of Cby (Coil‐Cby, residues 67‐104), despite its significantly lower solubility relative to the other constructs. A HSQC spectrum of Coil‐Cby has no detectable peaks, while the deconvoluted CD spectrum returns 80% helical and 20% disordered/turn and the [θ]222/[θ]208 ratio ≈ 1 [Fig. 4(C)]. Its thermal denaturation profile, like all Cby constructs comprising the coiled‐coil domain, displays a sharp reduction in signal between 60‐70°C [Fig. 4(D)]. Similar to CbyΔC20, Coil‐Cby aggregated upon thermal denaturation.
Disordered regions of Cby tune its binding affinity to TC‐1
Focusing on the Cby/TC‐1 interaction, we next investigated to what extent target recognition of Cby is modulated by specific structural elements, that is, the coiled‐coil domain, or the N‐ and C‐terminal disordered regions. TC‐1 functions as a positive regulator of Wnt signaling by competing with β‐catenin in binding the C‐terminal half of Cby.9 Previous structural characterization of TC‐1 by our lab established that the predominantly disordered TC‐1 contains three regions with α‐helical propensity in its C‐terminal half and that these regions are responsible for mediating the interactions with Cby.8
Upon titration of unlabelled CbyΔC20 to 15N‐labelled TC‐1 (2:1), decreases in peak intensity were observed for residues within TC‐1's C‐terminal half [Fig. 6(B)]. The peaks that displayed intensity losses matched the intensity losses observed when full‐length Cby was titrated to 15N‐labelled TC‐1 [Fig. 6(A,C)]. A titration of the CbyC25 peptide, which comprises the C‐terminal extension, to TC‐1 did not result in any intensity changes [Supporting Information Fig. 2(A)]. Together, these results suggest that the C‐terminal extension is likely expendable for Cby's interaction with TC‐1.
Figure 6.
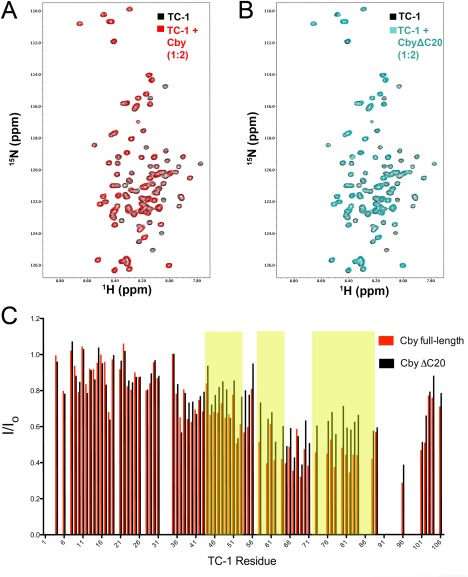
(A) Overlay of 1H‐15N HSQC spectra of TC‐1 in the absence (black) and presence (red) of full‐length Cby at a 1:2 ratio (TC‐1 60 μM:Cby 120 μM). (B) Overlay of 1H‐15N HSQC spectra of TC‐1 in the absence (black) and presence (cyan) of CbyΔC20 at a 1:2 ratio (TC‐1 60 μM:CbyΔC20 120 μM). (C) Intensity ratios (I/Io) for assigned TC‐1 amide resonances in the presence (I) or absence (Io) of either full‐length Cby or CbyΔC20. Yellow shaded areas correspond to the three regions on TC‐1 with high‐helical propensity as determined by chemical shift analysis.8
Next, the individual halves of Cby, N‐Cby, and C‐Cby, were titrated with 15N‐labelled TC‐1. As expected, titrating N‐Cby did not induce any changes in the HSQC spectrum of TC‐1 [Supporting Information Fig. 2(B)]. Conversely, titrating C‐Cby led to the broadening of most TC‐1 amide resonances [Fig. 7(A,B)]. The drops in peak intensities were most severe in the C‐terminal half of TC‐1, where many signals were broadened beyond detection.
Figure 7.
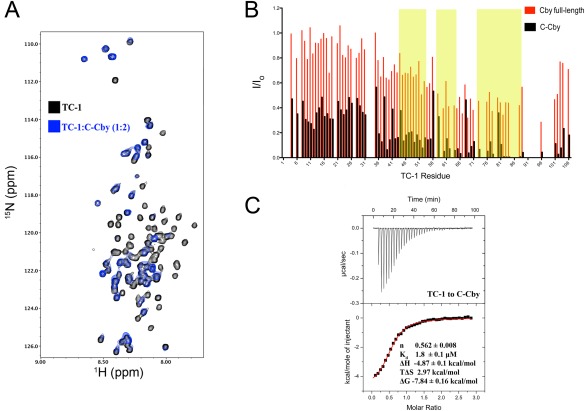
(A) Overlay of 1H‐15N HSQC spectrum of TC‐1 in the absence (black) and presence (blue) of full‐length C‐Cby at a 1:2 ratio (TC‐1 50 μM:C‐Cby 100 μM). (B) Intensity ratios (I/Io) for assigned TC‐1 amide resonances in the presence (I) or absence (Io) of either full‐length Cby (as shown in Fig. 5) or C‐Cby. Yellow shaded areas correspond to the three regions on TC‐1 with high‐helical propensity as determined by chemical shift analysis.8 (C) An ITC thermal profile of TC‐1 binding to C‐Cby. Thermodynamic experimental values are displayed. Kd is the dissociation constant ΔH, ΔS, and ΔG are the change in enthalpy, entropy, and Gibbs free energy upon binding at T = 298 K, respectively.
The greater intensity losses observed in the TC‐1 spectrum upon titration with C‐Cby suggest that C‐Cby binds TC‐1 with a higher affinity than the full‐length protein. This was confirmed by ITC experiments, which revealed that C‐Cby binds to TC‐1 with a Kd of ∼ 2 μM [Fig. 7(C), Supporting Information Fig. 3(A)]. On the other hand, the interactions between TC‐1/full‐length Cby and TC‐1/CbyΔC20 are too weak to produce discernible enthalpy changes with the protein concentrations (which are limited by the proteins’ solubilities) used in the ITC experiments. [Supporting Information Fig. 3(B,C)].
Intriguingly, when Coil‐Cby was added to TC‐1 in a 2:1 ratio, no changes in the HSQC spectrum were observed [Supporting Information Fig. 2(C)]. The lack of observed binding may be due to its potential aggregation at the concentration (∼100 μM) used for these NMR experiments. Attempts to concentrate the protein to > 200 μM led to the observation of a gel‐like precipitate, again highlighting the importance of disordered regions for the solubility of Cby.
Discussion
The mapping of coiled‐coil domains using NMR spectroscopy has proven to be a challenge for not only Cby, but also for other partially unstructured proteins such as CHOP, Par‐4, and NA14.31, 32, 33 For those three proteins, the only observable HSQC peaks correspond to residues located in disordered regions. As demonstrated in this study, HDX‐MS is a valuable alternative technique for deciphering which residues make up these coiled‐coil domains.
Cby first exhibits HDX protection in large fragments comprised of residues 60‐84 and 60‐90. Our ability to set a defined boundary for the N‐terminus of the coiled‐coil is limited by the size of the peptic fragments produced during online digestion. Meanwhile, a peptic fragment comprising residues 85‐90 displays further reduced HDX rates while residues 91‐98 display virtually no deuterium uptake over the course of the experiment. These results align well with the predicted coiled‐coil region, residues 68‐102.7
The HDX data revealed a previously unrecognized structural feature, that is, considerable disorder in the C‐terminal extension of Cby (residues 102‐126), where deuterium levels around 90% were observed for the earliest time point of t = 1 min. This behavior does not necessarily imply that the C‐terminal extension is completely unfolded. Instead, it is more likely that this region exhibits rapid coil/helix fluctuations. Following the standard HDX model42 one can estimate the average deuteration rate constant as kHDX = Kop × kch, where Kop is the coil/helix equilibrium constant, and kch ≈ 7 min−1 is the “chemical” exchange rate constant at pH 5. An assumed value of Kop = 0.3 corresponds to a deuteration level of 90% for t = 1 min. Thus, our HDX data suggest that the residual helicity of the C‐terminal extension could be as high as 77% (considering that a coil/helix equilibrium constant of 23/77 yields Kop = 0.3). Although the C‐terminal extension is not part of the coiled‐coil domain, it is not visible in the HSQC spectra of full‐length Cby or its C‐terminal half (C‐Cby). It is likely that the proximity of the C‐terminal extension to the coiled‐coil and the suggested coil/helix fluctuations collectively contribute to signal broadening of these residues.
Our NMR and CD spectroscopy experiments demonstrate that Cby's C‐terminal extension is expendable for coiled‐coil formation, in good agreement with a yeast two‐hybrid assay that demonstrated full‐length Cby could bind to a large Cby fragment (residues 60‐112), where the C‐terminal extension's last 14 residues are not present.16 However, the absence of the C‐terminal extension results in a reduction in Cby's solubility. Importantly, this extension plays a major functional role as it contains Cby's nuclear localization signal, residues 123‐126, which binds to importin‐α.5
From our NMR experiments, Cby's coiled‐coil domain mediates the interaction with TC‐1. For the Cby/TC‐1 interaction, no binding was observed between TC‐1 and the CbyC25 peptide, while the CbyΔC20 construct was observed to bind TC‐1 by NMR spectroscopy. Notably, while the coiled‐coil is required for the Cby/TC‐1 complex formation, we failed to detect interactions between Coil‐Cby and TC‐1 by NMR, possibly due to the aggregation of the former at high concentration. Despite remaining α‐helical as shown by CD spectroscopy, the absence of both disordered ends on Coil‐Cby drastically reduces the solubility of this domain.
The C‐Cby construct bound to TC‐1 with much greater affinity compared to full‐length Cby. Our ITC data, based on the calculated molar ratio, suggests that C‐Cby binds to TC‐1 as a dimer. We speculate that the bound C‐Cby dimer likely lies along the helical region of TC‐1's C‐terminal half. The difference in affinity for TC‐1 between full‐length and C‐Cby may be due to a higher entropic cost of binding for full‐length Cby compared to C‐Cby. Alternatively, the disordered N‐terminus of Cby may inhibit the interaction by acting as an entropic bristle, that is, an unstructured region that impedes surrounding macromolecules via random, thermally driven motions.43
In summary, our HDX‐MS data help characterize the structure and dynamics of the NMR‐invisible C‐terminal half of Cby. The data reveal that Cby is comprised of a disordered N‐terminus, a short coiled‐coil, and an unstructured C‐terminal extension. These three structural elements were assessed for their importance to Cby's interaction with the Wnt signaling enhancer TC‐1. Our results also provide insight into how Cby's disordered regions and coiled‐coil work in concert to assemble higher order protein complexes, such as the trimolecular Cby/14‐3‐3/β‐catenin6 or Cby/clusterin/NBPF1 complexes.15 Cby's N‐terminus comprises the binding motifs for a number of binding targets, including 14‐3‐3 protein and clusterin. In the free state, Cby's N‐terminus may reduce the affinity for some binding targets that bind to its C‐terminal half, as demonstrated in this work with TC‐1. However, this inhibition may be abrogated if the N‐terminus is present in a bound state. Such a mechanism may allow for cooperative, sequential formation of multi‐molecular Cby complexes.
Materials and Methods
Protein expression and purification
The expression and purification protocols for full‐length Cby, CbyΔC20 (deletion of the last 20 residues of Cby), N‐Cby (residues 1‐63), and TC‐1 were carried out as described previously.8, 10, 17 The pET‐15b plasmid containing human TC‐1 with an N‐terminal His6‐tag was a gift from Dr. E. Chua (University of Sydney, Australia).10 The His‐tags remained uncleaved from the recombinant Cby constructs to increase their solubility.
C‐Cby (residues 67‐126) and Coil‐Cby (residues 67‐104) were cloned into the Gateway destination vector pDEST‐HisMBP. The His‐MBP fusion constructs were expressed in Esherichia Coli (E. coli) BL21(DE3) pLysS in M9 minimal media and induced with 0.75 mM isopropyl‐β‐D‐thiogalactopyranoside (IPTG; Bioshop). For pull‐down assays the His‐tagged proteins were purified from crude lysate by affinity chromatography using Ni‐Sepharose 6 Fast Flow beads (Amersham Biosciences). To purify C‐Cby and Coil‐Cby, the His‐MBP tag were cleaved by overnight incubation with tobacco etch virus (TEV) protease at room temperature in 20 mM Tris‐HCl, 50 mM NaCl, pH 7.5. The tag‐less Cby constructs precipitated out of solution during the cleavage and were subsequently re‐solubilized in 20 mM Tris‐HCl, 100 mM NaCl, 6 M Guanidine, pH 7.5. The protein was passed through a Superdex 200 column (GE Healthcare) in the same guanidine‐containing buffer. For refolding, C‐Cby was dialyzed immediately into 10 mM ammonium acetate, pH 5.0 while Coil‐Cby was first dialyzed into 10 mM sodium acetate, 8 M urea, pH 5.0, and later into 10 mM ammonium acetate, pH 5.0. All experiments of this work were performed at pH 5.0 as both Cby and TC‐1 are soluble at low pH.
Peptide synthesis
The Cby peptide comprising its C‐terminal 25 residues (CbyC25) was purchased from the TUFTS University Core facility. The peptide was dissolved and dialyzed in 10 mM ammonium acetate, pH 5.0 for NMR experiments.
Hydrogen/deuterium exchange – mass spectrometry
HDX‐MS of Cby was performed by mixing protein stock solution in a 1:9 ratio with D2O‐based labelling buffer at pH‐meter reading of 5.0. The final protein concentration was 2 μM. Aliquots were removed at selected time points ranging between 1 min and 2 h. These aliquots were quenched at pH 2.4 by addition of HCl on ice, followed by flash freezing in liquid nitrogen. Prior to analysis the aliquots were rapidly thawed to ∼0°C and injected into a nanoACQUITY HDX/UPLC (Waters, Milford, MA)40 for peptic digestion, trapping, and peptide separation. A peptic cleavage map of Cby is provided in Supporting Information Fig. S1. Deuteration percentages were calculated as 100% × (m – m 0)/(m 100 – m 0) where m is the centroid m/z for the partially deuterated peptide of interest, and where m 0 and m 100 correspond to minimally and fully deuterated controls, respectively.40
Circular dichroism spectropolarimetry
CD spectra and thermal melting curves were collected on a Jasco J‐810 spectropolarimeter (Easton, MD). Spectra of the various Cby constructs (0.15 mg/mL to 0.30 mg/mL) were collected at 25°C in10 mM ammonium acetate, pH 5.0, with 5 accumulated scans for each construct. For the melting curves, the temperature was scanned from 15°C to 95°C, with the ellipticity monitored at 222 nm. The analysis program CDSSTR included in the DichroWeb online analysis software was used to deconvolute the CD spectra.44 Deconvolution was performed using the SMP180 (optimized for 190‐240 nm) reference set.45
NMR spectroscopy
NMR experiments were carried out on a Varian Inova 600‐MHz spectrometer equipped with xyz‐gradient triple resonance probe at 25°C in 10 mM ammonium acetate buffer, pH 5.0. Samples contained 10% D2O and 1 mM 2,2‐dimethyl‐2‐sila‐pentane‐5‐sulfonic acid (DSS) as an internal standard for chemical shift referencing. Data were processed using NMRPipe46 and analyzed using NMRView.47 The backbone assignment of TC‐1 was previously completed by our lab (BMRB accession code: 15141).8
Isothermal titration calorimetry
ITC experiments were performed on a VP‐ITC instrument (MicroCal) at 25°C in 10 mM ammonium acetate, pH 5.0. 8 μL aliquots of 230 μM TC‐1 were titrated stepwise into the 1.4 mL sample cell containing 20 μM C‐Cby. Titrations of TC‐1 to buffer and buffer to C‐Cby revealed negligible heat changes. The dissociation constant (Kd), molar binding stoichiometry (n), and the binding enthalpy (ΔH), entropy (ΔS), and Gibbs free energy (ΔG) were determined by fitting the binding isotherm to a one‐site model with MicroCal Origin7 software. The duplicate set of thermodynamic derived binding parameters are found in Supporting Information Figure 3. Protein concentrations were determined from amino acid analysis (Amino Acid Analysis Facility, SickKids, Toronto, ON).
Supporting information
Supporting Information
Supporting Information
Supporting Information
Significance: The protein Chibby plays a key regulatory role in Wnt signaling by antagonizing the transcriptional activity of β‐catenin. By using an array of biophysical techniques, we have dissected the structural properties of this intrinsically disordered protein and the way it interacts with TC‐1, another key regulator in the signaling pathway. The results provide insights into the interplay between disordered regions and coiled‐coil motifs, two commonly found structural elements in hub proteins, involved in protein‐protein interactions.
REFERENCES
- 1. Schepers A, Clevers H (2012) Wnt signaling, stem cells, and cancer of the gastrointestinal tract. Cold Spring Harb Perspect Biol 4:a007989. [DOI] [PMC free article] [PubMed]
- 2. Polakis P (2012) Drugging Wnt signalling in cancer. Embo J 31:2737–2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Clevers H, Nusse R (2012) Wnt/beta‐catenin signaling and disease. Cell 149:1192–1205. [DOI] [PubMed] [Google Scholar]
- 4. Takemaru KI, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT (2003) Chibby, a nuclear beta‐catenin‐associated antagonist of the Wnt/Wingless pathway. Nature 422:905–909. [DOI] [PubMed] [Google Scholar]
- 5. Li FQ, Mofunanya A, Fischer V, Hall J, Takemaru KI (2010) Nuclear‐cytoplasmic shuttling of Chibby controls beta‐catenin signaling. Mol Biol Cell 21:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li FQ, Mofunanya A, Harris K, Takemaru KI (2008) Chibby cooperates with 14‐3‐3 to regulate beta‐catenin subcellular distribution and signaling activity. J Cell Biol 181:1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mofunanya A, Li FQ, Hsieh JC, Takemaru KI (2009) Chibby forms a homodimer through a heptad repeat of leucine residues in its C‐terminal coiled‐coil motif. BMC Mol Biol 10:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gall C, Xu HY, Brickenden A, Ai XJ, Choy WY (2007) The intrinsically disordered TC‐1 interacts with Chibby via regions with high helical propensity. Protein Sci 16:2510–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jung YS, Bang S, Choi K, Kim E, Kim Y, Kim J, Park J, Koo H, Moon RT, Song K, Lee I (2006) TC1 (C8orf4) enhances the Wnt/beta‐catenin pathway by relieving antagonistic activity of Chibby. Cancer Res 66:723–728. [DOI] [PubMed] [Google Scholar]
- 10. Chua EL, Young L, Wu WM, Turtle JR, Dong QH (2000) Cloning of TC‐1 (C8orf4), a novel gene found to be overexpressed in thyroid cancer. Genomics 69:342–347. [DOI] [PubMed] [Google Scholar]
- 11. Xu HT, Liu Y, Liu SL, Miao Y, Li QC, Wang EH (2013) TC‐1 (C8orf4) expression is correlated with differentiation in ovarian carcinomas and might distinguish metastatic ovarian from metastatic colorectal carcinomas. Virchows Arch 462:281–287. [DOI] [PubMed] [Google Scholar]
- 12. Zhang P, Cao HY, Bai LL, Li WN, Wang Y, Chen SY, Zhang L, Yang LH, Xu HT, Wang EH (2015) The high expression of TC1 (C8orf4) was correlated with the expression of beta‐catenin and cyclin D1 and the progression of squamous cell carcinomas of the tongue. Tumor Biol 36:7061–7067. [DOI] [PubMed] [Google Scholar]
- 13. Su K, Huang LJ, Li WH, Yan XL, Li XF, Zhang ZP, Jin FG, Lei J, Ba GZ, Liu BY, Wang XP, Wang YJ (2013) TC‐1 (c8orf4) enhances aggressive biologic behavior in lung cancer through the Wnt/beta‐catenin pathway. J Surg Res 185:255–263. [DOI] [PubMed] [Google Scholar]
- 14. Burke MC, Li FQ, Cyge B, Arashiro T, Brechbuhl HM, Chen XW, Siller SS, Weiss MA, O'Connell CB, Love D, Westlake CJ, Reynolds SD, Kuriyama R, Takemaru KI (2014) Chibby promotes ciliary vesicle formation and basal body docking during airway cell differentiation. J Cell Biol 207:123–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vandepoele K, Staes K, Andries V, Van Roy F (2010) Chibby interacts with NBPF1 and clusterin, two candidate tumor suppressors linked to neuroblastoma. Exp Cell Res 316:1225–1233. [DOI] [PubMed] [Google Scholar]
- 16. Hidaka S, Konecke V, Osten L, Witzgall R (2004) PIGEA‐14, a novel coiled‐coil protein affecting the intracellular distribution of polycystin‐2. J Biol Chem 279:35009–35016. [DOI] [PubMed] [Google Scholar]
- 17. Mokhtarzada S, Yu C, Brickenden A, Choy WY (2011) Structural characterization of partially disordered human Chibby: Insights into its function in the Wnt‐signaling pathway. Biochemistry 50:715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Killoran RC, Fan JS, Yang DW, Shilton BH, Choy WY (2015) Structural analysis of the 14‐3‐3 zeta/Chibby interaction involved in Wnt/beta‐catenin signaling. Plos One 10:e0123934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dos Santos HG, Abia D, Janowski R, Mortuza G, Bertero MG, Boutin M, Guarin N, Mendez‐Giraldez R, Nunez A, Pedrero JG, Redondo P, Sanz M, Speroni S, Teichert F, Bruix M, Carazo JM, Gonzalez C, Reina J, Valpuesta JM, Vernos I, Zabala JC, Montoya G, Coll M, Bastolla U, Serrano L (2013) Structure and non‐structure of centrosomal proteins. Plos One 8:e62633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nido GS, Mendez R, Pascual‐Garcia A, Abia D, Bastolla U (2012) Protein disorder in the centrosome correlates with complexity in cell types number. Mol Biosyst 8:353–367. [DOI] [PubMed] [Google Scholar]
- 21. Anurag M, Singh GP, Dash D (2012) Location of disorder in coiled coil proteins is influenced by its biological role and subcellular localization: a GO‐based study on human proteome. Mol Biosyst 8:346–352. [DOI] [PubMed] [Google Scholar]
- 22. Galea CA, High AA, Obenauer JC, Mishra A, Park CG, Punta M, Schllessinger A, Ma J, Rost B, Slaughter CA, Kriwacki RW (2009) Large‐scale analysis of thermostable, mammalian proteins provides insights into the intrinsically disordered proteome. J Proteome Res 8:211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Flock T, Weatheritt RJ, Latysheva NS, Babu MM (2014) Controlling entropy to tune the functions of intrinsically disordered regions. Curr Opin Struc Biol 26:62–72. [DOI] [PubMed] [Google Scholar]
- 24. Uversky VN (2013) The most important thing is the tail: Multitudinous functionalities of intrinsically disordered protein termini. FEBS Lett 587:1891–1901. [DOI] [PubMed] [Google Scholar]
- 25. Hegde ML, Tsutakawa SE, Hegde PM, Holthauzen LMF, Li J, Oezguen N, Hilser VJ, Tainer JA, Mitra S (2013) The disordered C‐terminal domain of human DNA glycosylase NEIL1 contributes to its stability via intramolecular interactions. J Mol Biol 425:2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vuzman D, Levy Y (2012) Intrinsically disordered regions as affinity tuners in protein‐DNA interactions. Mol Biosyst 8:47–57. [DOI] [PubMed] [Google Scholar]
- 27. Trudeau T, Nassar R, Cumberworth A, Wong ETC, Woollard G, Gsponer J (2013) Structure and intrinsic disorder in protein autoinhibition. Structure 21:332–341. [DOI] [PubMed] [Google Scholar]
- 28. Konrat R (2014) NMR contributions to structural dynamics studies of intrinsically disordered proteins. J Magn Reson 241:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kosol S, Contreras‐Martos S, Cedeno C, Tompa P (2013) Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 18:10802–10828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jensen MR, Ruigrok RWH, Blackledge M (2013) Describing intrinsically disordered proteins at atomic resolution by NMR. Curr Opin Struc Biol 23:426–435. [DOI] [PubMed] [Google Scholar]
- 31. Rodriguez‐Rodriguez M, Trevino MA, Laurents DV, Arranz R, Valpuesta JM, Rico M, Bruix M, Jimenez MA (2011) Characterization of the structure and self‐recognition of the human centrosomal protein NA14: implications for stability and function. Protein Eng Des Sel 24:883–892. [DOI] [PubMed] [Google Scholar]
- 32. Libich DS, Schwalbe M, Kate S, Venugopal H, Claridge JK, Edwards PJB, Dutta K, Pascal SM (2009) Intrinsic disorder and coiled‐coil formation in prostate apoptosis response factor 4. Febs J 276:4134–4152. [DOI] [PubMed] [Google Scholar]
- 33. Singh VK, Pacheco I, Uversky VN, Smith SP, MacLeod RJ, Jia ZC (2008) Intrinsically disordered human C/EBP homologous protein regulates biological activity of colon cancer cells during calcium stress. J Mol Biol 380:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Konermann L, Rodriguez AD, Sowole MA (2014) Type 1 and Type 2 scenarios in hydrogen exchange mass spectrometry studies on protein‐ligand complexes. Analyst 139:6078–6087. [DOI] [PubMed] [Google Scholar]
- 35. Keppel TR, Howard BA, Weis DD (2011) Mapping unstructured regions and synergistic folding in intrinsically disordered proteins with amide H/D exchange mass spectrometry. Biochemistry 50:8722–8732. [DOI] [PubMed] [Google Scholar]
- 36. Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25:158–170. [DOI] [PubMed] [Google Scholar]
- 37. Englander SW (2006) Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectr 17:1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhu MM, Rempel DL, Du ZH, Gross ML (2003) Quantification of protein‐ligand interactions by mass spectrometry, titration, and H/D exchange: PLIMSTEX. J Am Chem Soc 125:5252–5253. [DOI] [PubMed] [Google Scholar]
- 39. Lupas A (1996) Prediction and analysis of coiled‐coil structures. Method Enzymol 266:513–525. [DOI] [PubMed] [Google Scholar]
- 40. Wales TE, Fadgen KE, Gerhardt GC, Engen JR (2008) High‐speed and high‐resolution UPLC separation at zero degrees Celsius. Anal Chem 80:6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lau SYM, Taneja AK, Hodges RS (1984) Synthesis of a model protein of defined secondary and quaternary structure ‐ effect of chain‐length on the stabilization and formation of 2‐stranded alpha‐helical coiled‐coils. J Biol Chem 259:3253–3261. [PubMed] [Google Scholar]
- 42. Bai Y, Milne JS, Mayne L, Englander SW (1993) Primary structure effects on peptide group hydrogen exchange. Proteins 17:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoh JH (1998) Functional protein domains from the thermally driven motion of polypeptide chains: A proposal. Proteins 32:223–228. [PubMed] [Google Scholar]
- 44. Whitmore L, Wallace BA (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res 32:W668–W673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Abdul‐Gader A, Miles AJ, Wallace BA (2011) A reference dataset for the analyses of membrane protein secondary structures and transmembrane residues using circular dichroism spectroscopy. Bioinformatics 27:1630–1636. [DOI] [PubMed] [Google Scholar]
- 46. Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRpipe ‐ a multidimensional spectral processing system based on Unix pipes. J Biomol NMR 6:277–293. [DOI] [PubMed] [Google Scholar]
- 47. Johnson BA (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol 278:313–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information