

Abstract
Objectives
This research study sought to improve the treatment of pancreatic cancer by improving the drug delivery of a promising AKT/PDK1 inhibitor, PHT-427, in poly(lactic-co-glycolic) acid (PLGA) nanoparticles.
Methods
PHT-427 was encapsulated in single-emulsion and double-emulsion PLGA nanoparticles (SE- and DE-PLGA-427). The drug release rate was evaluated to assess the effect of the second PLGA layer of DE-PLGA-427. Ex vivo cryo-imaging and drug extraction from ex vivo organs was used to assess the whole body biodistribution in an orthotopic model of MiaPaCa-2 pancreatic cancer. Anatomical MRI was used to noninvasively assess the effects of four weeks of nanoparticle-drug treatment on tumor size, and diffusion-weighted MRI longitudinally assessed changes in tumor cellularity.
Results
DE-PLGA-427 showed delayed drug release and longer drug retention in the pancreas relative to SE-PLGA-427. Diffusion-weighted MRI indicated a consistent decrease in cellularity during drug treatment with both types of drug-loaded nanoparticles. Both SE- and DE-PLGA-427 showed a 6-fold and 4-fold reduction in tumor volume relative to untreated tumors, and an elimination of primary pancreatic tumor in 68% of the mice.
Conclusions
These results indicated that the PLGA nanoparticles improved drug delivery of PHT-427 to pancreatic tumors, which improved the treatment of Mia PaCa-2 pancreatic cancer.
Keywords: pancreatic cancer, AKT/PDK1 inhibitor, Poly(lactic-co-glycolic) acid nanoparticles (PLGA), Magnetic Resonance Imaging (MRI), Apparent Diffusion Coefficient (ADC)
INTRODUCTION
Many chemotherapeutic drugs have poor cellular uptake, or have a short half-life of retention in vivo. These drugs can require a high dose to be administered to a patient, which can lead to unacceptable toxicity.1–4 These issues are especially problematic for the treatment of pancreatic carcinoma, because the normal pancreas tissue surrounding the pancreatic tumor can often have high fibrosity that inhibits drug delivery. This high fibrosity can also cause high interstitial pressure within the tumor, which can further inhibit drug delivery.5,6 Therefore, improved drug delivery to pancreatic cancer is especially needed for this cancer type.
Poly(lactic-co-glycolic acid) (PLGA) nanoparticles that encapsulate chemotherapies are shown to improve drug delivery to tumors, including pancreatic tumors. PLGA nanoparticles are biocompatible and biodegradable, and are approved by the FDA for clinical use.7–11 PLGA nanoparticles have passive drug targeting to tumors through the Enhanced Permeability and Retention (EPR) effect,12 and can protect pharmaceuticals against chemical and enzymatic degradation.
Drug-loaded PLGA nanoparticles often show a biphasic behavior when releasing the drug in solution. The PLGA nanoparticles have an initial “burst effect” that releases a fraction of drug.13–15 This initial drug release is assumed to primarily arise from the drug molecules that have loosely bound to the surface of the PLGA nanoparticle, or have been encapsulated near the surface of the nanoparticle. This initial drug release is followed by a slow and moderate degradation of the PLGA particles that releases the remaining encapsulated drug. However, this biphasic behavior may not be recapitulated within an in vivo environment, in which the degradation of the PLGA is affected by materials within the blood, fibrous tissue of the pancreas, the pancreatic tumor microenvironment, and the intracellular conditions within the tumor.16 A rapid degradation of the PLGA nanoparticles in vivo may reduce or eliminate the importance of the initial burst effect, which can change the design criteria for these drug delivery nanoparticles. To investigate the importance of the burst effect during in vivo drug delivery to a pancreatic tumor, we investigated the development of a PLGA nanoparticle with a second coating of PLGA, which cloaks the drugs that are loosely bound to the surface of the original PLGA nanoparticle. Double emulsion PLGA nanoparticles have been shown to encapsulate hydrophobic drugs with good efficiency and with good size characteristics under well-controlled conditions for formulation.10,17 We then investigated the therapeutic effects of the standard single-emulsion PLGA nanoparticle (SE-PLGA) and our new double emulsion PLGA nanoparticle (DE-PLGA) for delivering chemotherapy to a pancreatic tumor model.
Our study investigated the therapeutic effect of PHT-427 encapsulated in SE-PLGA or DE-PLGA. PHT-427 is a promising targeted chemotherapeutic agent that has shown strong effects against several cancers including pancreatic cancer.18 However, the hydrophobicity of PHT-427 limits its delivery.19 To investigate the merits of SE-PLGA-427 and DE-PLGA-427 in vitro, we synthesized and characterized the nanoparticles, particularly to study their drug encapsulation efficiency and drug release rates. We then investigated the pharmacokinetics of SE-PLGA-427 and DE-PLGA-427 through whole body ex vivo fluorescence imaging and ex vivo biodistribution studies of PHT-427 delivered to each organ. We also investigated the therapeutic effect of each drug-loaded nanoparticle by longitudinally assessing the cellularity of the pancreatic tumor with in vivo diffusion-weighted MRI.20 Together, these experiments were intended to investigate the relative importance of the burst effect of PLGA drug delivery when treating pancreatic tumors in vivo.
MATERIALS AND METHODS
Ester-terminated 50:50 Poly(DL-lactic-co-glycolidic acid) (PLGA, inherent viscosity range of 0.55–0.75 dL/g) was purchased from Lactel Absorbable Polymers (Durect Corp., Birmingham, AL; catalog number B6010-2P). The PHT-427 chemotherapeutic drug was synthesized following a previously reported protocol.18 Poly(vinyl alcohol) (PVA) that was 87–90% hydrolyzed and with average molecular weight 30,000–70,000 was purchased from Sigma-Aldrich (St. Louis, MO; catalog number P8136). Dichloromethane (DCM) that was 99.8% anhydrous was purchased from Sigma-Aldrich (St. Louis, MO). Paraformaldehyde was purchased from EMD Millipore (Merck KGaA, Darmstadt, Germany). Sesame seed oil was purchased from Sigma-Aldrich (St. Louis, MO). Heparin was purchased from APP Pharmaceuticals, LLC (Shaumburg, IL). Heparinized saline, 50 μl of 3000 U/L, was used for blood samples so they would not coagulate. Acetonitrile that was 99.9% anhydrous was purchased from Acros Organics (Thermo Fisher Scientific, Inc., Waltham, MA). A methyl ester version of tetramethylrhodamine (TMRM) was purchased from Life Technologies (Molecular Probes, Grand Island, NY). Trehalose was purchased from Sigma Aldrich (St. Louis, MO). Sodium Dodecyl Sulfate (SDS) was purchased from OmniPur (EMD Millipore, Darmstadt, Germany). Dulbecco’s Phosphate Buffered Saline (DPBS) 10x was purchased from Sigma Aldrich (St. Louis, MO).
Preparation of Drug-loaded Nanoparticles
SE-PLGA-427 was synthesized using an oil-water emulsion to entrap the hydrophobic PHT-427 in the PLGA.10,21 First, 100 mg of PLGA was dissolved overnight in 1 mL DCM. Then 40 mg of PHT-427 was also dissolved in the DCM. This PLGA solution was emulsified drop-wise in 2 ml of 0.5% PVA solution with rapid vortexing. The solution was sonicated with a Fisher Scientific Model 500 Sonic Dismembrator three times in 10 second bursts at approximately 200 watts of power. The nanoparticle mixture was then stirred in 100 ml of 0.3% PVA solution for three hours. The nanoparticle solution was centrifuged at 12,000 rpm for 10 minutes and washed with DI water. The nanoparticles were then sonicated for 10 minutes and vortexed to distribute the nanoparticles back into DI water. After three washes, trehalos was added at a 2:1 ratio of the expected yield of nanoparticles, then frozen in liquid nitrogen and lyophilized for 3 days until dry.
DE-PLGA-427 was synthesized using the same protocol with minor changes.10,22 A solution of 100 mg of PLGA and 40 mg of PHT-427 in 1 mL DCM was emulsified drop-wise in 2 ml of 0.5% PVA solution with rapid vortexing. The solution was then sonicated three times in 10 second bursts using a dismembrator. To create the second layer of PLGA on the nanoparticle, a second solution of 100 mg of PLGA in 1 mL DCM, but without additional drug, was emulsified drop-wise into the nanoparticle mixture with rapid vortexing. The combined nanoparticle mixture was then stirred in 100 ml solution of 0.3% PVA for three hours. The nanoparticle solution was centrifuged at 12,000 rpm for 10 minutes and washed with DI water. The nanoparticles were sonicated and vortexed to distribute into DI water. After three washes, trehalos was added at a 2:1 ratio of the expected yield of nanoparticles, then frozen in liquid nitrogen and lyophilized for 3 days until dry.
To create a calibration curve for average drug load within the nanoparticles, serial dilutions of free drug ranging from 10 to 500 μg/mL were created from a stock solution of 10 mg/mL PHT-427 in DMSO, and analyzed by HPLC.23 The samples were analyzed with a Surveyor high-performance liquid chromatography (HPLC) system (Thermo Finnigan, San Jose, CA) and a Luna C-18(2) particle size column with 3 μm particle size and a 100×4.6 mm column size. The chromatographic separation was achieved using a gradient mobile phase system consisting of acetonitrile with 0.05% formic acid (v/v) and water with 0.05% formic acid (v/v). The mobile phase flow rate was 0.3 mL/min and the acetonitrile in the mobile phase was increased in a linear fashion from 30% to 90% in 20 minutes, and held at 90% for 20 minutes, followed by 5 minutes of re-equilibration at 30% acetonitrile. Optical absorbance was collected at wavelengths ranging between 200–600 nm and chromatographs were extracted for the wavelengths ranging between 247–283 nm at approximately 34 minutes after injection. To test the average drug loading within the nanoparticles, serial dilutions were made from a stock solution of 450 μg/mL nanoparticles in DMSO. These serial dilutions were analyzed using same HPLC method described above. The loading of the PHT-427 drug in the nanoparticles was determined from the calibration curve.24
Characterization of Drug-loaded Nanoparticles
The average particle size and size distribution of the SE-PLGA-427 nanoparicles were analyzed with dynamic light scattering (DLS) using a nano Series Zetasizer (Malvern Instruments Ltd., Worcestershire, UK) with software version 6.20. The zeta potentials of these nanoparticles were also analyzed with the same instrument. SE-PLGA-427 were analyzed in DPBS 1x in a folded capillary cell. The refraction index used in the experiments was 1.59 for PLGA and 1.332 for DPBS.
Scanning electron microscopy (SEM) was performed to analyze the shape of the nanoparticles. The nanoparticles were gold-coated with a Hummer sputtering system (Anatech USA, Union City, CA). Nanoparticles were scraped onto a glass slide, mounted on the SEM stub, and placed in the gold-coat sputtering chamber.25 The SE-PLGA-427 and DE-PLGA-427 nanoparticles were each imaged with a S-4800 field emission scanning electron microscope (Hitachi Corp., Tokyo, Japan). Imaging was conducted in a conventional vacuum at 30 kV, 3.0 mm spot size, from 400x to 234,500x magnification, with an Everhart-Thornley Detector. Images were analyzed with Inspect S software version 3.1.4 (FEI Co., Hillsboro, OR).26 The DE-PLGA-427 particle size was calculated using SEM images with ImageJ, by calibrating the image size, Fast Fourier Transforming the image, thresholding the transformed image, and using an automated method to count the diameter of each particle in the “Analyze” tool of ImageJ.
Atomic force microscopy (AFM) was also used to analyze the morphology of the nanoparticles.27,28 The SE-PLGA-427 and DE-PLGA-427 nanoparticles were each scraped onto glass slides for imaging. Atomic force microscopy (AFM) images of the nanoparticles were captured using a Dimension 3100 atomic force microscope (Digital Instruments, Inc., Tonawanda, NY) with a 1 μm scan size, scan rate of 0.996 Hz, 1.302 V-2.5 V amplitude set point, integral gain 0.3250, proportional gain 0–.5131, drive amplitude 210.0 mV, a drive phase of −58.39, and sweep control frequency of 328.41 kHz.
A drug release study was performed with SE-PLGA-427 and DE-PLGA-427. Four round bottom flasks were suspended in oil baths maintained at 37°C. Then 2 mg of each nanoparticle to be analyzed was suspended in a dialysis membrane with 12,000–14,000 MWCO (Spectrum Laboratories, Inc., Rancho Dominguez, CA). A dialysis membrane containing a sample of SE-PLGA-427 was suspended in 50 mL of PBS, and a second dialysis membrane containing the same sample was suspended in 50 mL of PBS with 0.1% SDS. Likewise, dialysis membranes containing DE-PLGA-427 were suspended in 50 mL of PBS and in 50 mL of PBS with 0.1% SDS. Samples of 1 mL were periodically taken from each flask for analysis by HPLC to determine drug concentration, which was replaced with 1 mL of fresh solvent. The concentration was adjusted for the added 1 mL for an accurate analysis of each subsequently extracted sample. Samples were taken every 3 hours for the first few days, then every 12 hours for 13 days.
In vivo Studies
MiaPaCa-2 pancreatic tumor cells were authenticated and verified to be mycoplasma-free, and were transfected with the luciferase gene for investigations that were unrelated to this study.29 These cells were cultured in a 5% CO2 incubator and in complete RPMI-1640 media that was supplemented with 10% fetal bovine serum (FBS), and 100 U/mL penicillin and streptomycin. Cells were passaged using 0.25% trypsin.
All animal procedures were conducted with approval from the Institutional Animal Care and Use Committee of the University of Arizona. One million MiaPaCA-2 cells were subcutaneously implanted into eight female severe combined immunodeficient (SCID) donor mice by the Experimental Mouse Shared Service of the University of Arizona Cancer Center.30 Housing and care of the animals was provided by University Animal Care of the University of Arizona. Plugs from the MiaPaCa-2 flank tumors were harvested after 4 days of tumor growth. A second set of eight mice was implanted with a single plug. A single abdominal incision was made under sterile conditions, and the plug was inserted into the tail of the exposed pancreas. The average weight of the mice prior to imaging was 18.9 grams.
Each mouse was dosed with 100 mg of SE-PLGA-427, DE-PLGA-427, or PLGA nanparticles per kilogram of mouse body weight. Based on the drug loading measured during the characterization of the nanoparticles, this equated to 14 mg/kg and 19 mg/kg of encapsulated drug for the SE-PLGA-427 and DE-PLGA-427 nanoparticles, respectively. PHT-427 was administered at 10 mg/kg following a previous report.31 Each mouse was dosed once per week for four weeks. The i.p. administration of SE-PLGA-427 to each mouse was straightforward. Due to the larger sizes of DE-PLGA-427 nanoparticles, the solution of DE-PLGA-427 was constantly mixed prior to i.p. administration to avoid precipitation.
MRI studies were performed before the first treatment and 24 hours after each injection. Respiration gated, T2-weighted spin-echo MR images were acquired using a Biospec 7T MRI scanner (Bruker Biospin, Inc., Billerica, MA) and a spin-echo MRI acquisition sequence with 1297.6 ms TR, 10.6 – 66.8 ms TE, 6 echoes, 6×4 cm FOV, 234 × 312 μm in-plane resolution, 0.5 mm SL, 12 slices, and 2 averages. The total time to acquire the T2-weighted MR images was approximately 5 min.32 Respiration gated, diffusion weighted images were acquired with 3 orthogonal gradient directions, 501 ms TR, 27 ms TE, 6×4 cm FOV, 938×625 μm resolution, 0.5 mm SL, 2 slices, 2 averages, and b values of 100, 300, and 700 s/mm.20 The image slices were positioned in the center of the tumor or normal pancreas tissue. The total time to acquire the diffusion weighted MR images was approximately 10 min, and varied due to respiration gating. Respiration gating and the acquisition of two averages were key criteria of our protocol, which reduced the effect of motion artifacts. The parametric maps and average tissue values of the Apparent Diffusion Coefficient (ADC) were calculated using Paravision software v 5.1 (Bruker Biospin, Inc., Billerica, MA).33,34
Ex vivo Studies
Ex vivo fluorescence imaging was used to track the early distribution of nanoparticles in the mouse model. Both SE-PLGA-427 and DE-PLGA-427 nanoparticles were made with tetramethylrhodamine methyl ester (TMRM) by following the protocols described earlier. TMRM was added into the PLGA/DCM/PHT-427 tube during the synthesis of single emulsion nanoparticles, and TMRM was added to the second emulsion process during the synthesis of double emulsion nanoparticles.35 This procedure localized TMRM in an outer layer of the double emulsion nanoparticles to better match the localization of TMRM in the single emulsion nanoparticles.
Four mice with MiaPaCa-2 pancreatic tumors were placed on an alfalfa-free diet for 10 days (Teklad Global® 19% protein extruded rodent diet, Harlan Laboratories, Indianapolis, IN), then withdrawn from food for 24 hours. After completion of the diet, two mice were injected with the fluorescent TMRM-SE-PLGA-427 nanoparticles. One mouse was sacrificed at one hour post injection and the other mouse was sacrificed 3 hours post injection. Additionally, two mice were injected with the fluorescent TMRM-DE-PLGA-427 nanoparticles following the same timeline. Each mouse was placed in a 4.5×2×1 inch aluminum dish, which was filled with Frozen Section Compound 22 and frozen in liquid nitrogen.36
Ex vivo fluorescence imaging was performed with a CryoViz® imaging system (BioInVision, Cleveland, OH) by the Case Center for Imaging Research, Case Western Reserve University, Cleveland OH. Mice were scanned at 40 μm slice thickness with a 10.4 μm pixel size. There were 18 tiles of data collected for each slice. Images were collected with both bright-field imaging and fluorescence imaging. The fluorescence results were collected with an eGFP filter. Reconstructions were handled by the CryoViz® software and Amira imaging compilations were created for 3D visualization in Amira software (FEI Visualization Sciences Group, Burlington, MA).36
Quantifiable drug biodistribution was performed by extracting the PHT-427 from the collected organs. Three days after the last nanoparticle treatment, the mice were sacrificed and blood was obtained via cardiac draw in a needle with heparinized saline. Each organ was soaked in acetonitrile for 48 hours and placed on a shaker. The acetonitrile was removed and analyzed for HPLC for PHT-427 quantification.37,38
RESULTS
Synthesis of drug-loaded nanoparticles
Many factors can affect the design of the particles, including the type of PLGA solvent, emulsifier, emulsifier concentration, stir rate, sonication duration, porosity of the system, polymer molecular weight, monomer ratio, temperature of the bulk fluid, and type of apparatus used in preparation.10,21 Our synthesis method used established methods that accounted for some of these factors, and we also tested other factors to optimize our synthesis for PHT-427.
To develop our drug-loaded SE-PLGA-427 nanoparticle, we used PLGA with an inherent viscosity of 0.55–0.75. DCM was used as the solvent for our procedure, because the use of DCM produced nanoparticle spheroids with the most homogenous size distribution when compared to chloroform, acetone, and mixtures of these three solvents, as performed in this study. Furthermore, a comparison of methods with pure distilled water or PVA at 0.3%, 0.5%, 1%, and 3% concentrations of emulsifiers showed that 0.5% PVA yielded spherical nanoparticles with the most homogenous size distribution (Fig. 1A). Using these conditions, an average nanoparticle size of 491.7 ± 18.0 nm for SE-PLGA-427 was measured with DLS, which was significantly larger than SE-PLGA without drug (Fig. 1B,C).
Figure 1.

Characterization of drug-loaded PLGA nanoparticles. (A) 3D AFM of SE-PLGA-427 showed that these drug-loaded nanoparticles were spherical. DLS showed an average nanoparticle diameter of (B) 257.37 ± 2.04 nm for SE-PLGA and (C) 491.7 ± 17.58 nm for SE-PLGA-427. (D) SEM showed an average nanoparticle diameter of 19.23 ± 31.85 μm for DE-PLGA-427, (E) and showed that these DE nanoparticles were clumps of SE nanoparticles.
The SE-PLGA-427 nanoparticles had 14% drug loading by weight as determined via HPLC. Additionally, the SE-PLGA-427 nanoparticles had an electrostatic potential of -3.63 mV relative to a potential of −0.02 mV for SE-PLGA nanoparticles without drug. The average PLGA nanoparticle weight was 71% of the weight of the starting products.
The drop-wise addition of PLGA to the SE-PLGA-427 nanoparticles yielded DE-PLGA-427 particles with an average size of 19.23 +/− 31.85 μm (Fig. 1D). The total DE-PLGA-427 particle yield was 70% by weight, again demonstrating good yield. Double emulsion nanoparticles were verified as 19% drug loaded via HPLC. Additionally, the DE-PLGA-427 particles had an electrostatic potential of −3.56 mV. The greater encapsulation of PHT-427 by the double emulsification process was attributed to the protective outer layer of PLGA that reduced or prevented loss of PHT-427 on the surface of nanoparticles that were created by the first emulsification step.17 Therefore, this result showed that the DE-PLGA-427 nanoparticles had improved drug loading relative to the nanoparticles created by a single emulsion process.
Both AFM and SEM showed that multiple SE-PLGA-427 nanoparticles were clumped together by the second emulsifying step when creating the DE-PLGA-427 nanoparticles (Fig. 1D,E). From a technical perspective, SEM was preferred relative to AFM for imaging drug-loaded PLGA nanopaticles.39–42 SEM images for all particles were representative of the entire particle population. AFM images were more difficult to acquire due to the hydrophobicity of the encapsulated drug that caused the nanoparticles to adhere to the AFM cantilever, requiring large adjustments to acquire images.
We used HPLC to determine the drug release rates of each drug-loaded nanoparticle (Fig. 2). An initial drug release study was performed in PBS to be consistent with previous drug release studies. However, due to the poor solubility of PHT-427, sink conditions are not maintained in pure water, and therefore we also performed a drug release study in 0.1% SDS in PBS.13,16,43,44 After 12.5 days, PHT-427 was completely released from nanoparticle incubated in 0.1% SDS in PBS, while only 49% and 26% of the drug was released into PBS from the SE- and DE-PLGA-427 nanoparticles, respectively.
Figure 2.
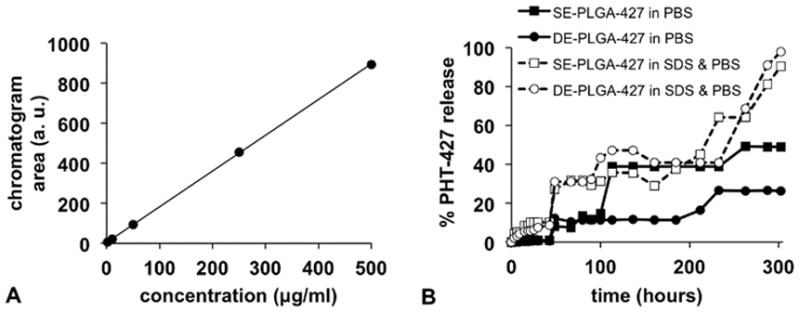
Evaluations of PHT-427 drug release from PLGA nanoparticles. (A) A calibration graph showed that HPLC analysis can precisely quantify drug concentration. (B) PHT-427 release from SE-PLGA and DE-PLGA nanoparticles was evaluated in PBS and SDS.
The DE-PLGA-427 nanoparticles in PBS had no release of PHT-427 until 14.5 hours into the study, whereas the SE-PLGA-427 nanoparticles released PHT-427 at 3 hours, which was the first measured time point. Similarly, the drug release from the DE-PLGA-427 nanoparticles was approximately half the rate of release from the SE-PLGA-427 nanoparticles during the first 6 hours in 0.1% SDS in PBS. These results indicate that the second layer of PLGA applied to the DE-PLGA-427 nanoparticles improved the retention of all loosely bound, surface-accessible PHT-427.
Drug biodistribution studies: 0–3 hours and 0–3 days
To determine the biodistributions of SE- and DE-PLGA-427 nanoparticles during the initial hours after i.p. administration, a version of each nanoparticle was synthesized to include the TMRM fluorophore. Mice were sacrificed one hour or three hours after administration of these fluorescent nanoparticles, and whole body cryo-imaging was used to image the biodistribution (Fig. 3). This unique cryo-imaging modality evaluated the entire mouse body, which was especially useful for assessing biodistribution in the i.p. cavity because invasive tissue harvesting within this cavity can strongly compromise the integrity of drug location.
Figure 3.
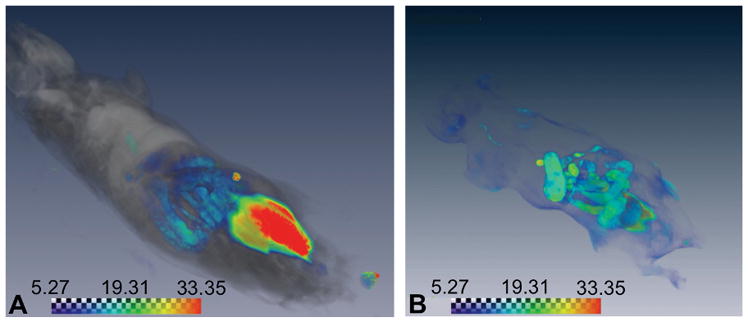
Initial biodistributions of drug-loaded nanoparticles. The whole body cryoimages of the biodistributions of DE-PLGA-427 (A) one hour and (B) three hours after i.p. administration. The scale bars represent relative signal amplitudes using results in an arbitrary unit recorded by the imaging system.
These results showed that the payload encapsulated by the SE- and DE-PLGA-427 had the same biodistribution one hour and three hours after administration. After one hour, most of the fluorescence signal arose from the injection site just inside the intraperitoneal cavity. Weak fluorescence was detected near the intestines and vasculature, indicating that the nanoparticles were distributing within the i.p. cavity but had not appreciably left this cavity. After three hours, stronger fluorescence signals were observed in the vasculature and gallbladder, showing the early events of biodistribution. This delayed entry into the vasculature demonstrated a need for the i.p.-administered nanoparticles to postpone drug release for at least a few hours to allow the particles to arrive at the cancer location and deliver the chemotherapy treatment. Therefore, the delayed drug release from the DE-PLGA-427 nanoparticles may have advantages relative to SE-PLGA-427 nanoparticles when the nanoparticles are delivered i.p.
A drug biodistribution study was performed three days after administering PHT-427 or the drug-loaded PLGA nanoparticles. Upon sacrifice, organs were harvested and shaken in acetonitrile to extract the drug, and the extract was analyzed with HPLC (Fig. 4). Each treatment resulted in high drug uptake in liver, especially when a PLGA nanoparticle was used for drug delivery. Notably, the DE-PLGA-427 nanoparticles clearly maintained a longer retention of the drug in the tumor and pancreas relative to SE-PLGA-427 or treatment with PHT-427. Furthermore, treatment with DE-PLGA-427 resulted in detectable drug distribution in lung, spleen and intestines.
Figure 4.
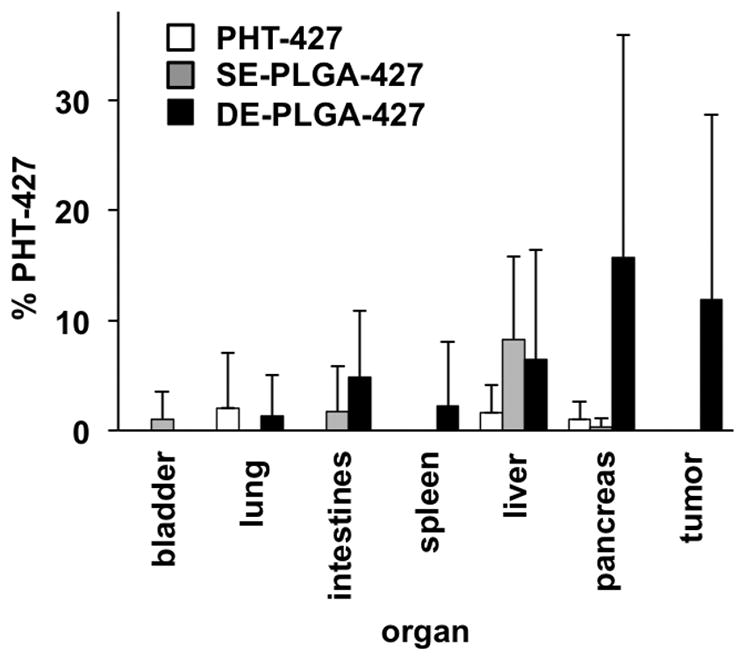
Ex vivo analyses of PHT-427 drug biodistribution in a MiaPaCa-2 pancreatic tumor model. Tissues were excised three days after treatment, and PHT-427 was extracted from each tissue and analyzed with HPLC. These results were quantified using the calibration graph shown in Figure 2A, with six samples per analysis.
Longitudinal In vivo studies: 0–4 weeks
Anatomical MR images were acquired to locate primary tumors within the pancreas (Fig. 5A). Diffusion-weighted MR images verified the location of each tumor (Fig. 5B), and were used to quantitatively measure the Apparent Diffusion Coefficient (ADC), which is an indication of cellularity in normal tissues and tumors (Fig. 5C–G).33 Normal tissues of the pancreas were measured to have an average ADC of 1.2×10−3 mm2/sec, which agreed with previously reported results.45,46 For comparison, the pancreatic tumors showed ADC values as low as 0.6×10−3 mm2/sec, which was attributed to the increased density and fibrosity in the pancreatic tumor tissue.32,47
Figure 5.
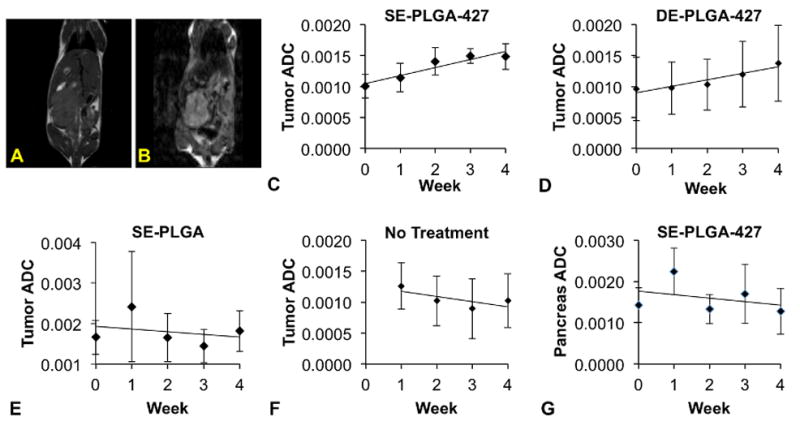
MRI assessments of the drug-treated MiaPaCa-2 tumor model. (A) T2-weighted MRI was used to measure tumor volume. (B) Diffusion-weighted MRI was used to evaluate cellular changes in tumors and normal tissues. (C) SE-PLGA-427 (n=7) and (D) DE-PLGA-427 (n=8) caused a monotonic increase in the Apparent Diffusion Coefficient (ADC) of water in the pancreatic tumor. (E) SE-PLGA without drug (n=7) and (F) untreated mice (n=8) showed a decrease in the Apparent Diffusion Coefficient (ADC) of water in the pancreatic tumor. (G) The ADC decreased in normal pancreatic tissue (n=8).
Mice treated with SE- and DE-PLGA-427 nanoparticles showed an average pre-treatment ADC value of 1.0×10−3 mm2/sec. The ADC values of the two cohorts for testing with SE- and DE-PLGA-427 nanoparticles were statistically indistinguishable (p > 0.05). After 4 weeks of treatment, ADC values of the tumor rose linearly to a value of 1.5×10−3 mm2/sec and 1.4×10−3 mm2/sec, respectively, which were not statistically different from each other (p > 0.5), but were significantly different than the ADC value before initiating treatment (p < 0.05). The rate of ADC increase over the 4 weeks was 1.0×10−4 mm2/sec per week for both nanoparticle treatments. This increase in water diffusion rates indicated that the cell density and/or fibrosity of the pancreatic tumor had decreased with either nanoparticle treatment, thereby allowing water to diffuse more freely within the tumor tissue. More importantly, these results showed that both nanoparticle treatments had the same effect on cell density and/or fibrosity in the pancreatic tumor.
We performed control studies to ensure that our diffusion-weighted MRI results with SE- and DE-PLGA-427 were due to a treatment effect. Prior to treatment, the average ADC values of pancreatic tumors of all cohorts of mice were statistically indistinguishable (p> 0.35 for all comparisons of groups eventually treated with SE-PLGA-427, DE-PLGA-427, PLGA nanoparticles, and the group of mice that was not treated). Mice with pancreatic tumors receiving no treatment had an initial average ADC value of 1.3 x10−3 mm2/sec, which dropped to 1.0 ×10−3 mm2/sec after four weeks, for a rate of -0.8 ×10−4 mm2/sec per week. This result suggested that the pancreatic tumors increased in cell density and/or fibrosity without treatment. Second, the MiaPACa-2 tumors treated with PLGA nanoparticle that did not include PHT-427 had an initial ADC value of 1.9×10−3 mm2/sec, and decreased to 1.7 ×10−3 mm2/sec after four weeks of treatment, at a rate of −0.7×10−4 mm2/sec. Finally, mice that had no tumors and normal pancreas tissues that had an initial average ADC value of 1.9×10−3 mm2/sec, which decreased at a rate of −0.9×10−4 mm2/sec per week to reach a final ADC value of 1.3×10−3 mm2/sec. Therefore, the increase in water diffusion rate observed during treatment with SE- and DE-PLGA-427 nanoparticles must be due to the delivered PHT-427 drug.
Tumor volume monitoring with MRI compared to ex vivo analysis
The tumor volume was longitudinally monitored with T2-weighted MRI to evaluate effectiveness of the SE- and DE-PLGA-427 nanoparticle treatment, relative to treatment with PLGA nanoparticle or with no treatment (Fig. 6A). Prior to initiating treatments, the average tumor volumes of each cohort of mice were statistically indistinguishable (p > 0.05). The pancreatic tumors increased 18-fold and 6-fold in volume with 4 weeks of PLGA treatment or with no treatment, respectively (this tumor growth with PLGA treatment vs. no treatment was not statistically different). For comparison, four weeks of treatment with SE- and DE-PLGA-427 nanoparticles caused the tumors to have an average volume that was 6-fold and 4-fold lower in average volume of the pancreatic tumors, respectively, relative to the average volume of untreated tumors. Notably, the average size of the pancreatic tumors after four weeks of treatment with SE- and DE-PLGA-427 nanoparticles were statistically indistinguishable from each other (p > 0.5). Furthermore, treatment with SE- and DE-PLGA-427 nanoparticles caused 67% and 62% of the pancreatic tumors to completely disappear, respectively (Fig. 6B). These in vivo results were verified with ex vivo gross pathological analyses. All mice that were untreated had pancreatic tumors after 4 weeks. Similarly, all mice that were treated with PLGA nanoparticles without drug also had pancreatic tumors after 4 weeks. These results showed that the SE- and DE-PLGA-427 nanoparticles had a good treatment effect against pancreatic cancer.
Figure 6.

Effects of drug-loaded nanoparticles on tumor burden. (A) T2-weighted MRI showed no tumor growth after four weeks of treatment with SE- and DE-PLGA-427, while PLGA-treated and untreated tumors continued to grow. (B) Only 33% and 38% of mice had a pancreatic tumor after 4 weeks of treatment as shown by T2-weighted MRI, which was confirmed with gross pathological analyses.
DISCUSSION
Previous studies with a mouse model of pancreatic cancer demonstrated that SE-PLGA-427 provided more therapeutic effect relative to treatment with PHT-427 alone.31 In particular, the hydrophobicity of this drug prevented delivery to the tumor without encapsulation in a nanoparticle. Our study provides additional evidence for the need of PLGA nanoparticles to deliver PHT-427 to the pancreatic tumor. Our study showed that SE- and DE-PLGA-427 is transported from the site of administration in the i.p. cavity to the vasculature in 1–3 hours after injection; these drug-loaded nanoparticles deliver PHT-427 to the pancreas and pancreatic tumor within three days; the delivered drug causes consistent changes in cellular density and/or fibrosity as monitored with diffusion-weighted MRI; and the delivered PHT-427 consistently shrinks or eliminates the pancreatic tumor. Therefore, this study shows the merits for using PLGA nanoparticles for delivering hydrophobic drugs to pancreatic tumors.
Our in vitro study showed that the SE-PLGA-427 nanoparticles had a biphasic release of PHT-427 drug, while the DE-PLGA-427 nanoparticles had a delayed release of the drug. This biphasic release of the smaller nanoparticle has been described as a potential advantage, because an initially strong exposure to the drug, followed by longer-term exposure, may have greater therapeutic effect. However, our in vivo studies showed that both SE- and DE-PLGA-427 had the same therapeutic effects. Therefore, the potential advantages of SE-PLGA-427 are inconsequential to treating pancreatic cancer. This conclusion aligns with the idea that total drug exposure governs therapeutic efficacy more than the peak dose of the drug. Moreover, our study demonstrates that efforts to optimize the size and drug release rates from nanoparticles during in vitro studies may not be relevant to in vivo studies, and therefore in vivo studies with drug delivery nanoparticles should be more rapidly pursued. For example, 19 mg/kg of PHT-427 delivered with DE-PLGA-427 was 36% greater than the 14 mg/kg of PHT-427 delivered with SE-PLGA-427 in this study. The effects of this difference in drug delivery could be investigated in future studies, and these future studies should be performed in vivo.
Intraperitoneal injection of nanoparticle drug delivery vehicles is a common route of administration, which can avoid harm to the capillaries of the respiratory system encountered with intravenous injection. The delayed release of drug from DE-PLGA-427 via intraperitoneal delivery may have some advantages. Our whole body cryo-imaging study showed that both drug delivery nanoparticles had similar delivery and uptake into the vasculature during the first three hours after i.p. administration. Therefore, the delayed release of drug from the DE-PLGA-427, while this nanoparticle still resides in the i.p cavity, may be an advantage for delivering drug to the pancreatic tumor. Furthermore, the biodistribution study of drug three days after administration of DE-PLGA-427 showed that PHT-427 still remained in the tumor, while no drug was detected in the tumor three days after administration of SE-PLGA-427. This result suggests that the drug was more quickly released from the SE-PLGA-427 nanoparticle and was metabolized or cleared by the tumor and/or liver, while the drug was retained in the DE-PLGA-427 nanoparticle for at least three days, and was prevented from being cleared or metabolized. This longer drug retention and slower release may have advantages for treating pancreatic cancer with PHT-427 and other similar hydrophobic chemotherapies.
Non-invasive imaging studies can provide advantages when evaluating longitudinal changes in tumor morphology within mouse models of orthotopic tumors.48–50 For comparison, serial sacrifice for ex vivo analyses may not necessarily capture longitudinal changes. Furthermore, MRI provides the opportunity to assess physicochemical properties of tumors and normal tissues, such as the cellular density and/or fibrosity as monitored with diffusion-weighted MRI. The ADC values calculated from diffusion-weighted MRI quantitatively demonstrated the treatment of the tumor.48
The ex vivo analysis of harvested organs cannot adequately evaluate drug biodistribution in the i.p. cavity because drug-loaded nanoparticles can distribute within organs and spaces between organs. Therefore, our study used whole body cryo-imaging for this analysis. This new modality can determine drug distribution by freezing the whole mouse, slicing the carcass at 40 μm thickness, analyzing tissue en facia with fluorescence imaging, and reconstructing a three-dimensional image of the distribution of the fluorescent material throughout the whole mouse. Whole body cryo-imaging provides rapid analyses relative to traditional and laborious histopathology of the whole mouse, and provides excellent 10.4 μm spatial resolution of relative to traditional in vivo fluorescence imaging that has ~3 mm spatial resolution.
Although mouse models of pancreatic tumors have improved the development of chemotherapies and drug delivery for treating this cancer type, these models do not sufficiently mimic the carcinogenic and tumor microenvironment of human pancreatic cancer. In particular, an intact immune system is needed to stimulate fibrous growth in normal pancreas tissues that surround a pancreatic tumor, and yet orthotopic tumor models require the use of an immunocompromised mouse model to avoid rejection of the implanted tumor cells.51–54 A transgenic pancreatic cancer model is more appropriate for studying drug delivery to this tumor type, because a transgenic model can retain an intact immune system. Future studies of drug delivery with PLGA nanoparticles should consider the use of these transgenic models.
As a summary, though in vitro drug release results from the SE-PLGA-427 and DE-PLGA-427 demonstrate a delay in drug release from the DE-PLGA-427 compared to the immediate release in SE-PLGA-427 particles, the longitudinal monitoring of tumor volume and ex vivo organ analysis showed no difference in the overall treatment of the pancreatic tumor model. Therefore, the so-called “burst effect” of PLGA nanoparticles had little or no effect on the overall treatment of the pancreatic cancer. The longer retention and drug release from DE-PLGA-427 particles may provide advantages for treating pancreatic cancer relative to the SE-PLGA-427 nanoparticles. Non-invasive imaging is an outstanding tool for evaluating nanoparticle biodistributions and treatment effects at the tissue and cellular levels.
Acknowledgments
The authors thank Douglas Cromey and the Experimental Mouse Shared Resource of the University of Arizona Cancer Center, Tucson, AZ. The authors also thank Joseph Molter and Dr. Chris Flask of the Case Center for Imaging Research for studies with the CryoViz™ imaging system at Case Western Reserve University.
Source of Funding: This research is supported by National Institute of Health (NIH) grants R01CA167183-01 and P50 CA95060. JEK acknowledges support from the United States Army Advanced Civil Schooling Program.
Footnotes
Conflicts of Interest
EJM has financial interests in PHusis Therapeutics, Inc (San Diego, CA), and the other authors have no conflicts of interest to declare.
Contributor Information
Joseph E. Kobes, Biomedical Engineering Graduate Interdisciplinary Program, University of Arizona, Tucson, AZ. Chemistry/Life Science Department, United States Military Academy, West Point, NY
Iman Daryaei, Department of Chemistry and Biochemistry, University of Arizona, Tucson, AZ
Christine M. Howison, Department of Biomedical Engineering, University of Arizona, Tucson, AZ
Jordan G. Bontrager, Biomedical Engineering Graduate Interdisciplinary Program, University of Arizona, Tucson, AZ
Rachael W. Sirianni, Barrow Brain Tumor Research Center, Barrow Neurological Institute, Phoenix, AZ
Emmanuelle J. Meuillet, University of Arizona Cancer Center, University of Arizona, Tucson, AZ
Mark D. Pagel, Department of Medical Imaging, University of Arizona, Tucson, AZ. University of Arizona Cancer Center, University of Arizona, Tucson, AZ
References
- 1.Yeo CJ, Cameron JL. Prognostic factors in ductal pancreatic cancer. Langenbecks Arch Surg. 1998;383:129–133. doi: 10.1007/s004230050104. [DOI] [PubMed] [Google Scholar]
- 2.Kindler HL. Front-line therapy of advanced pancreatic cancer. Semin Oncol. 2005;32:S33–36. doi: 10.1053/j.seminoncol.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Chang DT, Schellenberg D, Shen J, et al. Stereotactic radiotherapy for unresectable adenocarcinoma of the pancreas. Cancer. 2009;115:665–672. doi: 10.1002/cncr.24059. [DOI] [PubMed] [Google Scholar]
- 4.Xu Q, Zhang TP, Zhao YP. Advances in early diagnosis and therapy of pancreatic cancer. Hepatobiliary Pancreat Dis Int. 2011;10:128–135. doi: 10.1016/s1499-3872(11)60021-0. [DOI] [PubMed] [Google Scholar]
- 5.Di Costanzo F, Sdrobolini A, Gasperoni S. Possibilities of palliation in pancreatic cancer. Tumori. 1999;85:S47–53. [PubMed] [Google Scholar]
- 6.Sato N, Yamabuki T, Takano A, et al. Wnt inhibitor Dickkopf-1 as a target for passive cancer immunotherapy. Cancer Res. 2010;70:5326–5336. doi: 10.1158/0008-5472.CAN-09-3879. [DOI] [PubMed] [Google Scholar]
- 7.Yang Q, Williams D, Owusu-Ababio G, et al. Controlled release tacrine delivery system for the treatment of Alzheimer’s disease. Drug Deliv. 2011;8:93–98. doi: 10.1080/107175401750177061. [DOI] [PubMed] [Google Scholar]
- 8.Vasir JK, Labhasetwar V. Biodegradable nanoparticles. In: Friedman T, Rossi J, editors. Gene Transfer: Delivery and Expression of DNA and RNA. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2007. pp. 561–566. [Google Scholar]
- 9.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011;3:1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCall RL, Sirianni RW. PLGA nanoparticles formed by single- or double-emulsion with vitamin E-TPGS. J Vis Exp. 2013;82:51015. doi: 10.3791/51015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadat Tabatabaei Mirakabad F, Nejati-Koshki K, Akbarzadeh A, et al. PLGA-based nanoparticles as cancer drug delivery systems. Asian Pac J Cancer Prev. 2014;15:517–535. doi: 10.7314/apjcp.2014.15.2.517. [DOI] [PubMed] [Google Scholar]
- 12.Householder KT, DiPerna DM, Chiung EP, et al. Intravenous delivery of camtothecin-loaded PLGA nanoparticles for the treatment of intracranial glioma. Int J Pharmaceutics. 2015;479:374–380. doi: 10.1016/j.ijpharm.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez E, Baro M, Soriano I, et al. In vivo-in vitro study of biodegradable and osteointegrable gentamicin bone implants. Eur J Pharm Biopharm. 2011;52:151–158. doi: 10.1016/s0939-6411(01)00169-2. [DOI] [PubMed] [Google Scholar]
- 14.Jiang G, Qiu W, DeLuca PP. Preparation and in vitro/in vivo evaluation of insulin-loaded poly(acryloyl-hydroxyethyl starch)-PLGA composite microspheres. Pharm Res. 2003;20:452–459. doi: 10.1023/a:1022668507748. [DOI] [PubMed] [Google Scholar]
- 15.Onoue S, Kuriyama K, Uchida A, et al. Inhalable sustained-release formulation of glucagon: in vitro amyloidogenic and inhalation properties, and in vivo absorption and bioactivity. Pharm Res. 2011;28:1157–1166. doi: 10.1007/s11095-011-0379-8. [DOI] [PubMed] [Google Scholar]
- 16.Lam XM, Duenas ET, Daugherty AL, et al. Sustained release of recombinant human insulin-like growth factor-I for treatment of diabetes. J Control Release. 2000;67:281–292. doi: 10.1016/s0168-3659(00)00224-8. [DOI] [PubMed] [Google Scholar]
- 17.Cohen-Sela E, Chorny M, Koroukhov N, et al. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. J Control Release. 2009;133:90–95. doi: 10.1016/j.jconrel.2008.09.073. [DOI] [PubMed] [Google Scholar]
- 18.Moses SA, Ali MA, Zuohe S, et al. In vitro and in vivo activity of novel small-molecule inhibitors targeting the pleckstrin homology domain of protein kinase B/AKT. Cancer Res. 2009;69:5073–5081. doi: 10.1158/0008-5472.CAN-08-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meuillet EJ, Zuohe S, Lemos R, et al. Molecular pharmacology and antitumor activity of PHT-427, a novel Akt/phosphatidylinositide-dependent protein kinase 1 pleckstrin homology domain inhibitor. Mol Cancer Ther. 2010;9:706–717. doi: 10.1158/1535-7163.MCT-09-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Morgan DE, Buchsbaum DJ, et al. Early therapy evaluation of combined anti-death receptor 5 antibody and gemcitabine in orthotopic pancreatic tumor xenografts by diffusion-weighted magnetic resonance imaging. Cancer Res. 2008;68:8369–8376. doi: 10.1158/0008-5472.CAN-08-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Awotwe-Otoo D, Zidan AS, Rahman Z, et al. Evaluation of anticancer drug-loaded nanoparticle characteristics by nondestructive methodologies. AAPS PharmSciTech. 2012;13:611–622. doi: 10.1208/s12249-012-9782-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tewes F, Munnier E, Antoon B, et al. Comparative study of doxorubicin-loaded poly(lactide-co-glycolide) nanoparticles prepared by single and double emulsion methods. Eur J Pharm Biopharm. 2007;66:488–492. doi: 10.1016/j.ejpb.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Abdelghany SM, Schmid D, Deacon J, et al. Enhanced antitumor activity of the photosensitizer meso-Tetra(N-methyl-4-pyridyl) porphine tetra tosylate through encapsulation in antibody-targeted chitosan/alginate nanoparticles. Biomacromolecules. 2013;14:302–310. doi: 10.1021/bm301858a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarron PA, Marouf WM, Quinn DJ, et al. Antibody targeting of camptothecin-loaded PLGA nanoparticles to tumor cells. Bioconjug Chem. 2008;19:1561–1569. doi: 10.1021/bc800057g. [DOI] [PubMed] [Google Scholar]
- 25.Meuillet EJ, Mania-Farnell B, George D, et al. Modulation of EGF receptor activity by changes in the GM3 content in a human epidermoid carcinoma cell line, A431. Exp Cell Res. 2000;256:74–82. doi: 10.1006/excr.1999.4509. [DOI] [PubMed] [Google Scholar]
- 26.Kranz H, Bodmeier R. Structure formation and characterization of injectable drug loaded biodegradable devices: in situ implants versus in situ microparticles. Eur J Pharm Sci. 2008;34:164–172. doi: 10.1016/j.ejps.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Vasir JK, Labhasetwar V. Quantification of the force of nanoparticle-cell membrane interactions and its influence on intracellular trafficking of nanoparticles. Biomaterials. 2008;29:4244–4252. doi: 10.1016/j.biomaterials.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nahar M, Jain NK. Preparation, characterization and evaluation of targeting potential of amphotericin B-loaded engineered PLGA nanoparticles. Pharm Res. 2009;26:2588–2598. doi: 10.1007/s11095-009-9973-4. [DOI] [PubMed] [Google Scholar]
- 29.Wenot MK, Molter J, Flask CA, et al. In vivo dual substrate bioluminescence imaging. J Vis Exp. 2011;56:3245. doi: 10.3791/3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Damaraju VL, Bouffard DY, Wong CK, et al. Synergistic activity of troxacitabine (Troxatyl) and gemcitabine in pancreatic cancer. BMC Cancer. 2007;7:121. doi: 10.1186/1471-2407-7-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lucero-Acuña A, Jeffery JJ, Abril ER, et al. Nanoparticle Delivery of a AKT/PDK1 Inhibitor Improves the Therapeutic Effect Against Pancreatic Cancer. Int J Nanotech. 2014;9:5653–5665. doi: 10.2147/IJN.S68511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W, Zhang Z, Nicolai J, et al. Magnetization transfer MRI in pancreatic cancer xenograft models. Magn Reson Med. 2012;68:1291–1297. doi: 10.1002/mrm.24127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagmann P, Jonasson L, Maeder P, et al. Understanding diffusion MR imaging techniques: from scalar diffusion-weighted imaging to diffusion tensor imaging and beyond. Radiographics. 2006;26:S205–223. doi: 10.1148/rg.26si065510. [DOI] [PubMed] [Google Scholar]
- 34.O’Donnell LJ, Westin CF. An introduction to diffusion tensor image analysis. Neurosurg Clin N Am. 2011;22:185–196. doi: 10.1016/j.nec.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaumet M, Gurny R, Delie F. Localization and quantification of biodegradable particles in an intestinal cell model: the influence of particle size. Eur J Pharm Sci. 2009;36:465–473. doi: 10.1016/j.ejps.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Fei B, Wang H, Muzic RF, et al. Deformable and rigid registration of MRI and microPET images for photodynamic therapy of cancer in mice. Med Phys. 2006;33:753–760. doi: 10.1118/1.2163831. [DOI] [PubMed] [Google Scholar]
- 37.Bocker R, Estler CJ. A high pressure liquid chromatographic method for the determination of tetracyclines in blood and organs of experimental animals. Arzneimittelforschung. 1979;29:1690–1693. [PubMed] [Google Scholar]
- 38.Leitner A, Zollner P, Lindner W. Determination of the metabolites of nitrofuran antibiotics in animal tissue by high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr A. 2001;939:49–58. doi: 10.1016/s0021-9673(01)01331-0. [DOI] [PubMed] [Google Scholar]
- 39.Mu L, Teo MM, Ning HZ, et al. Novel powder formulations for controlled delivery of poorly soluble anticancer drug: application and investigation of TPGS and PEG in spray-dried particulate system. J Control Release. 2005;103:565–575. doi: 10.1016/j.jconrel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 40.Li D, Yamamoto H, Takeuchi H, et al. A novel method for modifying AFM probe to investigate the interaction between biomaterial polymers (chitosan-coated PLGA) and mucin film. Eur J Pharm Biopharm. 2010;75:277–283. doi: 10.1016/j.ejpb.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Luo R, Venkatraman SS, Neu B. Layer-by-layer polyelectrolyte-polyester hybrid microcapsules for encapsulation and delivery of hydrophobic drugs. Biomacromolecules. 2013;14:2262–2271. doi: 10.1021/bm4003915. [DOI] [PubMed] [Google Scholar]
- 42.Fortunati E, Mattioli S, Visai L, et al. Combined effects of Ag nanoparticles and oxygen plasma treatment on PLGA morphological, chemical, and antibacterial properties. Biomacromolecules. 2013;14:626–636. doi: 10.1021/bm301524e. [DOI] [PubMed] [Google Scholar]
- 43.Fotaki N, Vertzoni M. Biorelevant dissolution methods and their applications in in vitro-in vivo correlations for oral formulations. Open Drug Delivery J. 2010;4:2–13. [Google Scholar]
- 44.Allison SD. Analysis of initial burst in PLGA microparticles. Expert Opin Drug Delivery. 2008;5:615–628. doi: 10.1517/17425247.5.6.615. [DOI] [PubMed] [Google Scholar]
- 45.Schoennagel BP, Habermann CR, Roesch M, et al. Diffusion-weighted imaging of the healthy pancreas: apparent diffusion coefficient values of the normal head, body, and tail calculated from different sets of b-values. J Magn Reson Imaging. 2011;34:861–865. doi: 10.1002/jmri.22743. [DOI] [PubMed] [Google Scholar]
- 46.Herrmann J, Schoennagel BP, Roesch M, et al. Diffusion-weighted imaging of the healthy pancreas: ADC values are age and gender dependent. J Magn Reson Imaging. 2013;37:886–891. doi: 10.1002/jmri.23871. [DOI] [PubMed] [Google Scholar]
- 47.Wang DQ, Zeng MS, Shi X, et al. Comparison of MRI manifestations and histopathologic findings in pancreatic head carcinoma in vivo ex vivo. Zhonghua Zhong Liu Za Zhi. 2008;30:347–351. [PubMed] [Google Scholar]
- 48.Kim S, Pickup S, Hsu O, et al. Diffusion tensor MRI in rat models of invasive and well-demarcated brain tumors. NMR Biomed. 2008;21:208–216. doi: 10.1002/nbm.1183. [DOI] [PubMed] [Google Scholar]
- 49.Jansen SA, Conzen SD, Fan X, et al. In vivo MRI of early stage mammary cancers and the normal mouse mammary gland. NMR Biomed. 2011;24:880–887. doi: 10.1002/nbm.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montelius M, Ljungberg M, Horn M, et al. Tumour size measurement in a mouse model using high resolution MRI. BMC Med Imaging. 2012;12:1–7. doi: 10.1186/1471-2342-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Higuchi Y, Herrera P, Muniesa P, et al. Expression of a tumor necrosis factor alpha transgene in murine pancreatic beta cells results in severe and permanent insulitis without evolution towards diabetes. J Exp Med. 1992;176:1719–1731. doi: 10.1084/jem.176.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawakami Y, Inoue K, Hayashi H, et al. Subcutaneous xenotransplantation of hybrid artificial pancreas encapsulating pancreatic B cell line (MIN6): functional and histological study. Cell Transplant. 1997;6:541–545. doi: 10.1177/096368979700600519. [DOI] [PubMed] [Google Scholar]
- 53.Casanova ML, Bravo A, Ramirez A, et al. Exocrine pancreatic disorders in transsgenic mice expressing human keratin 8. J Clin Invest. 1999;103:1587–1595. doi: 10.1172/JCI5343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spector I, Zilberstein Y, Lavy A, et al. Involvement of host stroma cells and tissue fibrosis in pancreatic tumor development in transgenic mice. PLoS One. 2012;7:e41833. doi: 10.1371/journal.pone.0041833. [DOI] [PMC free article] [PubMed] [Google Scholar]