

Abstract
Tumor-associated alterations in RNA splicing result either from mutations in splicing-regulatory elements or changes in components of the splicing machinery. This review summarizes our current understanding of the role of splicing-factor alterations in human cancers. We describe splicing-factor alterations detected in human tumors and the resulting changes in splicing, highlighting cell-type-specific similarities and differences. We review the mechanisms of splicing-factor regulation in normal and cancer cells. Finally, we summarize recent efforts to develop novel cancer therapies, based on targeting either the oncogenic splicing events or their upstream splicing regulators.
INTRODUCTION
We have known for many years that tumors exhibit abnormal splicing patterns, compared to normal tissues. Changes in splicing occur for genes involved in every step of the transformation process (for review, see David et al. 2010). For example, the increased expression of anti-apoptotic isoforms of genes such as BCL2L1, CASP2, or FAS, has been extensively documented in various tumors types; alternative splicing of CD44, FGFR2, RAC1, or MST1R has been linked with the acquisition of invasive properties; and VEGFA splice variants are involved in angiogenesis regulation (Fig. 1). However, in the past few years we have started to appreciate that many of these tumor-associated splicing changes reflect alterations in particular components of the splicing machinery (Fig. 1). The core spliceosome plus associated regulatory factors comprise more than 300 proteins and five small nuclear RNAs (snRNAs), and catalyzes both constitutive and regulated alternative splicing (Hegele et al. 2012). The U1, U2, U4, U5, and U6 snRNAs participate in several key RNA–RNA and RNA–protein interactions during spliceosome assembly and splicing catalysis. These snRNAs associate with seven “Sm” core proteins and additional proteins to form small nuclear ribonucleoprotein particles (snRNPs). Other protein subcomplexes also play key roles, such as the SF3A and B complexes, and the PRP19-associated complexes dubbed NTC and NTR. The architecture of the spliceosome undergoes extensive remodeling in preparation for, during, and after splicing. In addition to the core spliceosome, regulatory proteins are involved in modulating the splicing reaction. These include RNA-binding proteins that function as activators or repressors of splicing by binding specifically to exonic or intronic enhancer or silencer elements, respectively, and they are involved in both constitutive and alternative splicing (for review, see Biamonti et al. 2014). In this review, we discuss the various splicing-factor alterations detected in human tumors, their cell-type specificity, as well as their specific roles in tumor development and progression.
FIGURE 1.
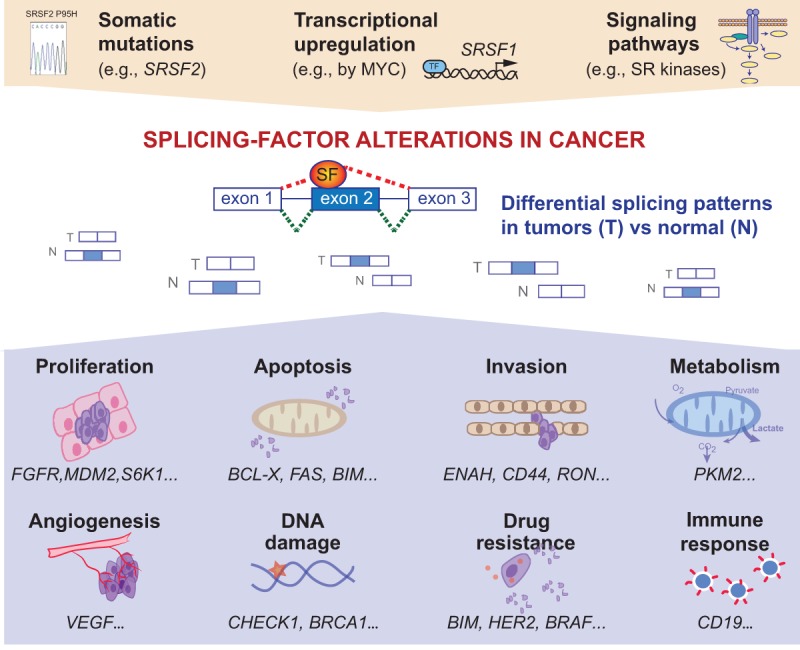
Splicing-factor alterations in human tumors. Human tumors exhibit somatic mutations in splicing regulators, or changes in splicing-factor levels in response to cell signaling or transcriptional regulation. These alterations in splicing factors promote differential splicing patterns in tumors compared to normal tissues. Alterations in alternative splicing events lead to the production of pro-tumorigenic isoforms that have been linked to various steps of tumorigenesis, including proliferation, apoptosis, invasion, metabolism, angiogenesis, DNA damage, or even drug resistance and immune response.
RECURRENT SOMATIC MUTATIONS OF CORE SPLICEOSOME COMPONENTS IN HEMATOLOGICAL MALIGNANCIES
Recently, large-scale sequencing projects identified recurrent somatic mutations in certain components of the spliceosome in several types of hematological malignancies, including myelodysplastic syndromes (MDS), other myeloid neoplasms, and chronic lymphocytic leukemia (CLL) (Table 1; Yoshida et al. 2011; Bejar et al. 2012; Papaemmanuil et al. 2013). These mutations occur most commonly in four genes: SF3B1 (splicing factor 3b subunit 1), SRSF2 (serine/arginine-rich splicing factor 2), U2AF1 (U2 small nuclear RNA auxiliary factor 1), and ZRSR2 (zinc finger RNA binding motif and serine/arginine rich 2), and almost always as somatic heterozygous missense mutations that are mutually exclusive (Papaemmanuil et al. 2011; Wang et al. 2011; Yoshida et al. 2011). In a very detailed review, Yoshida and Ogawa (2014) discussed the discovery of splicing-factor mutations and their correlation with tumor classification. Here we will focus on the functional differences and similarities between mutant splicing factors in hematological malignancies.
TABLE 1.
Recurrent splicing-factor mutations in human malignancies

SFB3B1—splicing factor 3b subunit 1
SF3B1, the most frequently mutated component of the spliceosome in cancer, is involved in the recognition of the intronic branch point sequence (BPS) during selection of the 3′ splice site (3′SS) (Fig. 2). SF3B1 is a component of the SF3B complex, which associates with the SF3A complex and U2 snRNP to form the 17S U2 complex. U2 snRNP binds to BPSs via SF3B14, and to U2AF2 via SF3B1 to stabilize the base-pairing interaction between U2 snRNA and the BPS, leading to the formation of the spliceosomal A complex. SF3B1 mutations are found in a variety of myeloid malignancies, with extremely high recurrence (48%–57%) in MDS subtypes that show increased ring sideroblasts (RARS/RCMD-RS) (Malcovati et al. 2011; Yoshida et al. 2011; Damm et al. 2012; Patnaik et al. 2012; Visconte et al. 2012), as well in 6%–26% of CLLs (Table 1). SF3B1 mutations are clustered in several hot spots, including K700, E622, R625, H662, and K666, all of which are located within HEAT (Huntingtin, Elongation factor 3, protein phosphatase 2A, Targets of rapamycin 1) repeats that extend from exon 12 to exon 15 (Fig. 2). In addition, SF3B1 mutations of residues K700 and K666 have been reported in 1.8% of unselected breast tumors and 4% of luminal breast tumors (Ellis et al. 2012; Maguire et al. 2015), as well as in 3% of pancreatic ductal adenocarcinomas (Biankin et al. 2012), whereas mutations of residues R625 and K666 are found in 15%–29% of uveal melanomas (Furney et al. 2013; Harbour et al. 2013; Martin et al. 2013) and 1% of cutaneous melanomas (Table 1; Kong et al. 2014).
FIGURE 2.
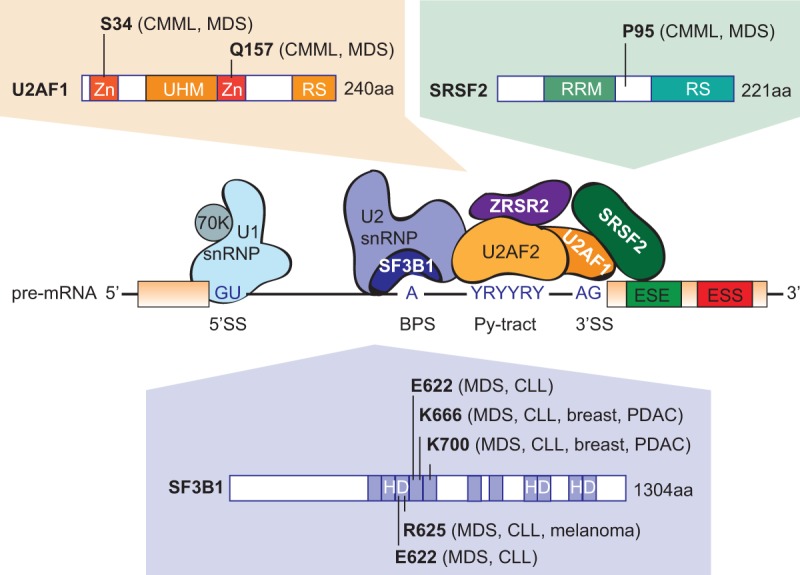
Recurrent splicing-factor mutations in human malignancies. Hotspot mutations in the genes coding for splicing factors U2AF1, SRSF2, and SF3B1 detected in myelodysplasia, as well as other tumor types, are indicated. In contrast, ZRSR2 mutations are distributed evenly across the gene (not shown). Domain abbreviations: (Zn) zinc finger domain, (UHM) U2AF homology motif domain, (RS) arginine/serine-rich domain, (RRM) RNA-recognition motif, (HD) heat domain.
SF3B1 mutations correlate with good prognosis in MDS and melanoma, but with poor prognosis in CLL. The SF3B1 K700 mutant promotes utilization of a different BPS compared to wild-type SF3B1, and recognizes a cryptic 3′SS with a short and weak polypyrimidine tract, located upstream of the canonical 3′SS (Darman et al. 2015). Similarly, SF3B1 R625 and K666 mutations result in deregulated splicing at a subset of splice sites, mostly involving the use of alternative or cryptic 3′SS (DeBoever et al. 2015; Alsafadi et al. 2016).
SRSF2—serine/arginine-rich splicing factor 2
The second most frequently mutated splicing regulator in MDS is SRSF2, a member of the serine–arginine (SR) protein family, which is involved in both constitutive and alternative splicing. SRSF2 bound to an exon plays a role in recruiting U2AF to the upstream 3′SS and U1 snRNP to the downstream 5′SS (Fig. 2). SRSF2 mutations and insertions/deletions map to a unique hotspot centered on residue P95, within the region linking the N-terminal RNA recognition motif (RRM) and the C-terminal RS domain (Fig. 2). Recurrent SRSF2 mutations have been found in various hematological malignancies (Table 1), with higher frequencies in patients with MDS (20%–30%) and chronic myelomonocytic leukemia (CMML) (50%) (Yoshida et al. 2011; Papaemmanuil et al. 2013). SRSF2 mutations correlate with poor prognosis, thus suggesting a major functional difference compared to SF3B1 mutations. In addition, no recurrent SRSF2 mutations have been reported to date in solid tumors.
Conditional expression from the endogenous murine locus of the Srsf2P95H allele in the bone marrow leads to impaired hematopoietic differentiation in heterozygous animals; in addition, mice heterozygous for the Srsf2P95H allele have distinct hematopoietic defects, compared to animals with heterozygous or homozygous deletion of Srsf2 (Kim et al. 2015). SRSF2 mutations are associated with changes in mRNA splicing patterns of key hematopoietic regulators, both in primary murine and patient samples, such as EZH2 and BCOR (Kim et al. 2015). The mutant SRSF2 protein exhibits altered RNA-binding specificity, rather than a loss of RNA-binding activity (Kim et al. 2015). Wild-type SRSF2 recognizes the consensus binding motifs CCNG and GGNG with similar affinity, whereas SRSF2P95H preferentially binds CCNG, which is present within exons that are differentially spliced. The altered interaction of mutant SRSF2 with RNA is due to the mutations affecting the conformations of the termini of SRSF2′s RRM domain, as deduced from NMR experiments (Kim et al. 2015).
U2AF1—U2 small nuclear RNA auxiliary factor
U2AF1 mutations are detected in 5%–15% of MDS and 5%–17% of CMML, as well as at lower rates in other hematological malignancies (Table 1). They are also found in 3% of lung tumors (Imielinski et al. 2012). The mutations are clustered in two hotspots centered on residues S34 and Q157, located within the two conserved zinc-finger domains (Fig. 2; Yoshida et al. 2011). U2AF is a heterodimer involved in 3′SS selection, with the small subunit, U2AF1 interacting with the AG dinucleotide, and the large subunit, U2AF2 interacting with the polypyrimidine tract. The MDS mutations in U2AF1 alter RNA splicing and promote mis-splicing of genes in ways that presumably contribute to abnormal hematopoiesis (Graubert et al. 2012; Quesada et al. 2012; Przychodzen et al. 2013; Brooks et al. 2014; Ilagan et al. 2015; Okeyo-Owuor et al. 2015; Shirai et al. 2015).
ZRSR2—zinc finger RNA binding motif and serine/arginine rich 2
In contrast to the above mutations, ZRSR2 mutations are distributed evenly across the gene sequence, and most of them disrupt the translational reading frame. This mutation pattern suggests loss of function, rather than gain/change of function, for the ZRSR2 mutations in MDS. ZRSR2 mutations are less frequent in MDS or in CMML than SF3B1, U2AF1, or SRSF2 mutations (Table 1). ZRSR2 associates with the U2AF heterodimer and plays a role in 3′SS recognition by participating in the formation of the spliceosomal A complex (Fig. 2). shRNA-mediated knockdown (KD) of ZRSR2 does not affect splicing of U2-type introns, but leads to the retention of U12-type introns (Madan et al. 2015), an atypical minor class of introns which are distinct from the canonical U2-dependent introns and represent only ∼0.5% of all human introns (for review, see Turunen et al. 2013).
Other RNA-binding proteins
Finally, alterations in other RNA-binding proteins have been reported at very low frequencies (0.5–1.5%) in hematological malignancies, such as de novo AML, CLL, or MDS, including mutations in PRPF8, SF3A1, LUCL7L2, SF1, U2AF2, HNRNPK, SRSF6, SRSF1, SRSF7, TRA2β, SRRM2, DDX1, DDX23, CELF4 (Makishima et al. 2012; Quesada et al. 2012; The Cancer Genome Atlas Research Network 2013; Walter et al. 2013). The existence of such mutations suggests that alterations in multiple steps of spliceosome assembly and splicing regulation can contribute to hematological malignancies.
ALTERATIONS IN SPLICING-FACTOR LEVELS IN SOLID TUMORS
Interestingly, very few recurrent mutations in splicing regulators have been detected to date in solid tumors, suggesting a fundamental difference in splicing targets and/or regulation in hematological malignancies compared to solid tumors. Mutations in SF3B1 (located on Chr. 2q33) have been reported in 1.8% of breast tumors, and more specifically in 4% of a subset of luminal breast tumors (Ellis et al. 2012; Maguire et al. 2015), and are significantly associated with the presence of estrogen-receptor (ER)-positive tumor cells, AKT1 mutations, and distinct copy-number alterations, including frequent chromosomal-segment gains on 16q12–q13 and 16q21–q22, and losses on 1p36–p35, 16q11–q13, and 16q21–q23 (Ellis et al. 2012; Maguire et al. 2015). SF3B1 mutations are also found at low frequency in pancreatic tumors (Biankin et al. 2012), as well as melanoma (Furney et al. 2013; Harbour et al. 2013; Martin et al. 2013; Kong et al. 2014). U2AF1 mutations have been detected in 3% of lung tumors (Imielinski et al. 2012). Interestingly, other splicing-factor mutations recurrent in blood malignancies, i.e., in SRSF2 or ZRSR2, have not yet been detected in solid tumors. However, solid tumors do exhibit frequent alterations in splicing factors; these are mostly changes in levels, occurring through changes in gene copy number and/or changes in gene expression (Fig. 1). Several regulatory splicing factors, such as SRSF1 (Karni et al. 2007; Anczukow et al. 2012), SRSF3 (Jia et al. 2010), SRSF6 (Karni et al. 2007; Cohen-Eliav et al. 2013), HNRNPA2/B1 (Golan-Gerstl et al. 2011), or HNRNPH (Lefave et al. 2011), have oncogenic properties, whereas other factors, including QKI (Zong et al. 2014), RBM5, RBM6, and RBM10 (Bechara et al. 2013), act as tumor suppressors (Table 2). Additional splicing regulators are also involved in tumorigenesis, although their function as oncogenes or tumor suppressors seems less clearly defined and can be different across tissue types (see below) (Table 2).
TABLE 2.
Splicing-factor expression changes in human malignancies
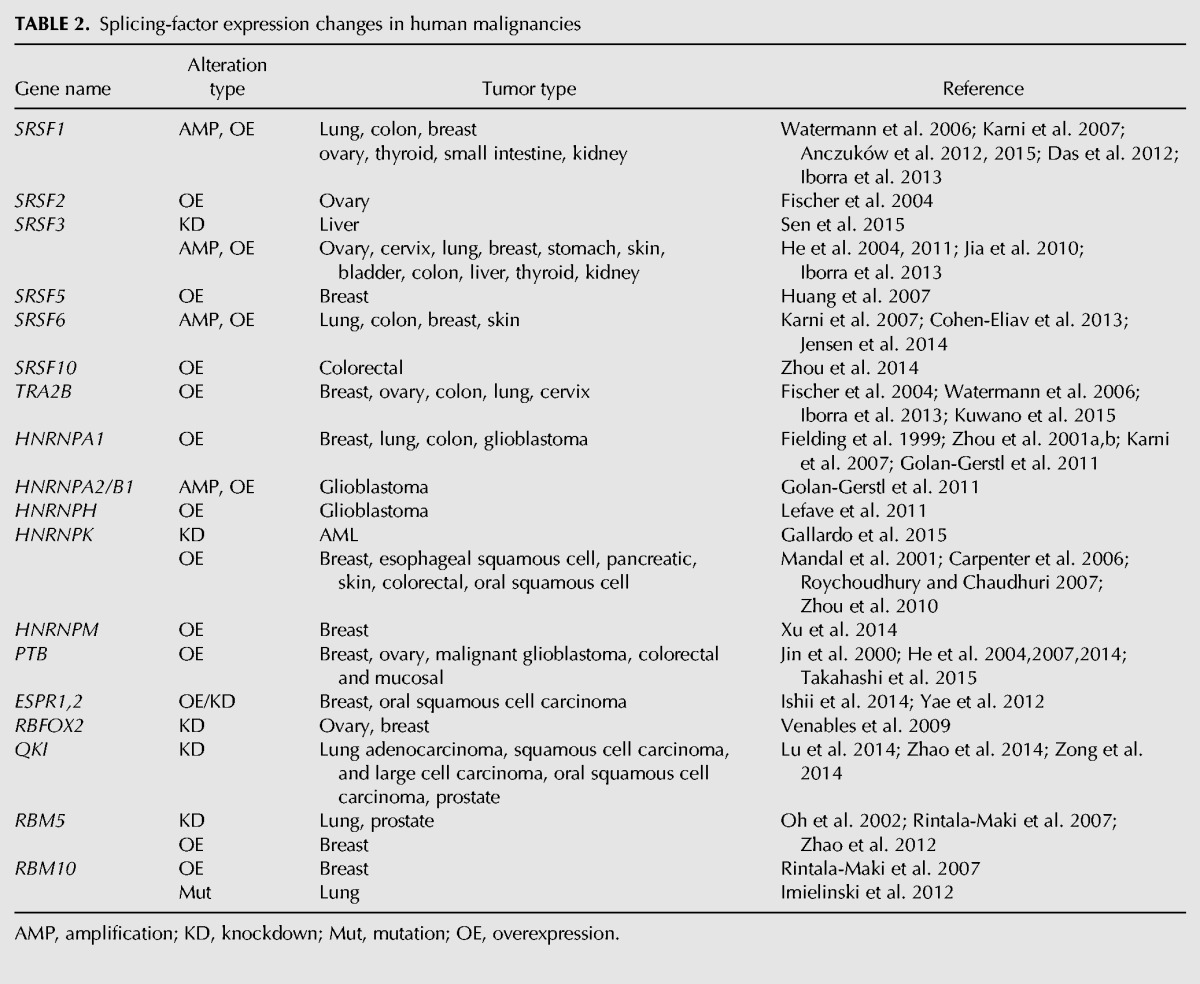
These RNA-binding proteins elicit changes in alternative splicing in a concentration-dependent manner, and, thus, changes in their levels can alter pre-mRNA splicing of many genes related to cancer, even in the absence of mutations. Alternative splicing changes have been linked to cancer through post-transcriptional regulation of components of many of the cellular processes considered as “hallmarks” of cancer, including cell proliferation, apoptosis, metabolism, invasion, and angiogenesis. However, the biological consequences of these global changes in alternative splicing are only beginning to be unraveled. We will briefly discuss selected examples of oncogenic or tumor suppressor splicing factors that have been found to contribute to cancer. For a detailed review of oncogenic spliced isoforms, see David and Manley (2010).
SRSF1—serine/arginine-rich splicing factor 1
The splicing factor SRSF1 (previously known as SF2/ASF) is a prototypical SR protein involved in both constitutive and alternative splicing, but it has also been implicated in additional functions, such as regulating mRNA transcription, stability and nuclear export, nonsense-mediated mRNA decay (NMD), translation, and protein sumoylation (for review, see Das and Krainer 2014). SRSF1 is an essential gene and Srsf1-null mice are embryonic lethal (Xu et al. 2005). Tissue-specific deletion of Srsf1 in mouse heart leads to lethality about 6–8 wk after birth, due to heart failure (Xu et al. 2005). These mice have defective Ca2+ metabolism, attributed to mis-splicing of the Ca2+/calmodulin-dependent kinase IIδ (CAMKIIδ), which leads to a defective contractile apparatus and cardiomyopathy. SRSF1 was also the first member of the SR protein family to be identified as a proto-oncogene, highlighting the important role of alternative splicing in tumorigenesis (Karni et al. 2007).
SRSF1 is frequently overexpressed in many solid tumors, relative to their respective normal controls, including tumors of the breast (13%), colon (25%), lung (25%), as well as thyroid, small intestine, kidney, and ovarian tumors (Watermann et al. 2006; Karni et al. 2007; Anczukow et al. 2012, 2015; Das et al. 2012; Iborra et al. 2013). Modest SRSF1 overexpression is sufficient to promote transformation of immortal rodent fibroblasts (Karni et al. 2007), as well as human mammary epithelial cells in vitro and in vivo (Fig. 3; Anczukow et al. 2012). SRSF1 up-regulation in some breast cancers is attributable to amplification of its locus as part of the Chr. 17q23 amplicon, which is associated with breast malignancies with poor prognosis (Karni et al. 2007). In addition, SRSF1 is a direct transcriptional target of the MYC oncoprotein, and high levels of SRSF1 are detected in MYC-expressing tumors (Anczukow et al. 2012; Das et al. 2012). SRSF1 expression is also regulated at the level of splicing, in response to the splicing factor Sam 68 (Valacca et al. 2010). Sam68 activity switches alternative splicing of the SRSF1 transcript from an NMD-targeted isoform to the major, translatable isoform, thus resulting in an increase in SRSF1 protein levels.
FIGURE 3.
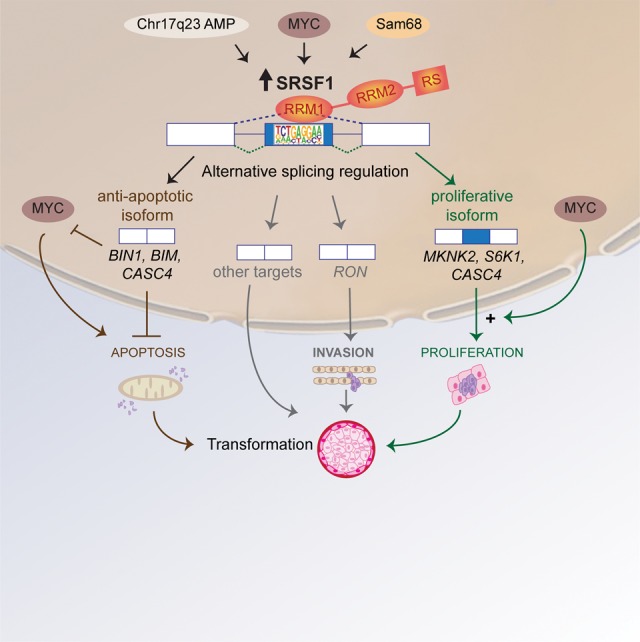
Model for SRSF1's roles in transformation in breast cancer. Increased expression of SRFS1 in human tumors results from several distinct types of alterations, such as amplification of the Chr. 17q23 amplicon, transcriptional regulation of SRFS1 downstream from MYC, or splicing regulation of SRSF1 pre-mRNA by Sam68. Up-regulation of SRSF1 promotes splicing changes in target genes involved in apoptosis, cell motility, proliferation, and other cellular functions. SRSF1 activates splicing by binding to an exonic motif recognized by its RRM1. SRSF1 overexpression promotes expression of anti-apoptotic isoforms unable to interact with pro-apoptotic factors, or that inhibit the action of pro-apoptotic factors, such as MYC or members of the Bcl-2 family. In parallel, SRSF1 overexpression promotes expression of isoforms that stimulate translation and cell proliferation by increasing phosphorylation of translation activators, such as S6 or eIF4E, or by inhibiting translational repressors, such as 4EBP1. In addition, MYC can cooperate with SRSF1 in transformation, and has a synergistic effect in the activation of the eIF4E pathway. By increasing proliferation and decreasing apoptosis, SRSF1 promotes cellular transformation in mammary epithelial cells.
SRSF1 comprises two RRMs and a C-terminal RS domain (Fig. 4). A structure–function dissection of the modular protein domains of SRSF1 showed that SRSF1's role in regulating proliferation and apoptosis during transformation involves splicing targets recognized primarily by RRM1, and does not require SRFS1's activity in NMD or physical interaction with mTOR (Anczukow et al. 2012). SRSF1 overexpression affects specific pre-mRNA splicing events in the MST1R (Ron) proto-oncogene and the kinases MKNK2 and S6K1, promoting the expression of pro-oncogenic isoforms (Ghigna et al. 2005; Karni et al. 2007; Anczukow et al. 2012). In addition, SRSF1 controls splicing of the tumor suppressor BIN1 and the apoptotic factor BCL2L11 (BIM), promoting the expression of isoforms that lack pro-apoptotic activity and contribute at least in part to SRSF1's activity in transformation (Fig. 3; Anczukow et al. 2012). In addition, SRSF1 regulates splicing of BCL2L1, MCL1, and CASP2 and 9, and its KD induces G2 cell-cycle arrest and apoptosis (Li et al. 2005; Moore et al. 2010). Finally, SRSF1-regulated splicing targets were identified by RNA-seq profiling of an organotypic three-dimensional cell culture model that mimics a context relevant to breast cancer, as well as of human breast tumors with SRSF1 overexpression (Anczukow et al. 2015). Overexpression of the SRSF1-regulated, exon-9-included CASC4 isoform—a target consistently observed in the different data sets—increased proliferation and decreased apoptosis, partially recapitulating SRSF1's oncogenic effects (Anczukow et al. 2015).
FIGURE 4.

Domain structure of splicing factors altered in solid tumors. The chromosomal locus of each splicing-factor-encoding human gene is shown on the left. For each RNA-binding protein (RBP) representative of the indicated families, the annotated protein domains or regions are shown in the diagrams (see legend for details), along with the size (in amino acids) of the human protein.
Interestingly, SRSF1-mediated oncogenesis appears to be dependent on inactivation of the p53 tumor suppressor pathway, a finding that could potentially be exploited when developing therapies against SRSF1-driven or -dependent tumors. p53 induction is one of the primary responses to SRSF1 overexpression in primary human and murine fibroblasts, and results in cells entering a state of premature cellular senescence (Fregoso et al. 2013). Briefly, SRSF1 plays a role in the nucleolar stress pathway, by stabilizing the interaction between ribosomal protein RPL5 and the E3 ubiquitin ligase MDM2 (Fregoso et al. 2013). Sequestration of MDM2 in this complex results in stabilization of the p53 tumor suppressor, which then mediates the stress response. Consistent with the stabilization of p53, increased SRSF1 expression in primary human fibroblasts decreases cellular proliferation and eventually triggers oncogene-induced senescence.
SRSF3—serine/arginine-rich splicing factor 3
Among the other SR proteins, SRSF3 (previously known as SRp20) is also frequently up-regulated in human tumors (Fig. 4). SRSF3 is an essential gene: Srsf3 knockout (KO) in mice results in an early lethal phenotype, suggesting a fundamental and nonredundant function for each SR protein in embryonic development (Jumaa et al. 1999). In human tumors, SRSF3 is overexpressed in ovarian, cervical, lung, colon, breast, stomach, skin, bladder, thyroid, liver, and kidney cancer (He et al. 2004, 2011; Jia et al. 2010; Iborra et al. 2013), at least in part due to copy-number changes in Chr. 6p21. Paradoxically, decreased expression of SRSF3 in human hepatocellular carcinoma has also been observed (Sen et al. 2015). Overexpression of SRSF3 promotes transformation on immortal rodent fibroblasts, and KD reduces tumor growth of HeLa cells (Jia et al. 2010). SRSF3 has been shown to regulate splicing of HIPK2, a homeodomain-interacting protein, controlling the switch between the full-length isoform and the Δ8 isoform, which induces cell death (Kurokawa et al. 2014). SRSF3 also plays a role in regulating the splicing switch between the PKM2 and PKM1 isoforms of pyruvate kinase, the key metabolic enzyme underlying the Warburg effect on cancer cells (Wang et al. 2012a,b). Finally, SRSF3 has been shown to be essential for hepatocyte differentiation and metabolic function in mice, by regulating splicing of target genes involved in glucose and lipid metabolism (Sen et al. 2013). Interestingly, genetic deletion of SRSF3 in hepatocytes causes fibrosis and the development of metastatic hepatocellular carcinoma with aging (Sen et al. 2015).
SRSF6—serine/arginine-rich splicing factor 6
SRSF6 (formerly SRp55) is overexpressed in 50% of lung and colon tumors, as well as in breast tumors, in part due to an amplification at the Chr. 20q12 locus (Fig. 4; Karni et al. 2007; Cohen-Eliav et al. 2013). SRSF6 protein levels are also frequently elevated in basal-cell carcinomas, squamous-cell carcinomas, and malignant melanomas (Jensen et al. 2014). SRSF6 overexpression enhances proliferation and inhibits cell death of mouse lung epithelial cells, whereas SRSF6 KD does not inhibit proliferation, suggesting that SRSF6 is not required for proper growth or survival of these cells, but overexpression may enhance proliferation by gain of function (Cohen-Eliav et al. 2013). SRSF6 is required for both tumor initiation and maintenance of lung cancer and colon cancer cells. In lung and colon, SRSF6 regulates splicing of INSR and leads to the production of the more mitogenic isoform of the insulin receptor (Cohen-Eliav et al. 2013). In human fibroblasts, SRSF6 controls the splicing switch of the kinase MKNK2 and reduces the tumor suppressive isoform Mnk2a (Karni et al. 2007). Finally, SRSF6 has a role in normal wound healing that is potentially related to its role in cancer (Jensen et al. 2014). Overexpression of SRSF6 causes skin hyperplasia in mice by deregulating tissue homeostasis, and results in strong up-regulation of Krt6, Krt16, Il1b, and many other wound-healing markers. The effects of SRSF6 on wound healing assayed in vitro depend on controlling splicing of the Tnc tenascin-C isoforms (Jensen et al. 2014).
TRA2β—transformer 2β homolog (Drosophila)
The SR-like TRA2 proteins are conserved across the animal kingdom, but separate gene paralogs encoding TRA2α and TRA2β proteins evolved early in vertebrate evolution (for review, see Best et al. 2014). KO experiments in mice showed that Tra2β is essential for embryonic and brain development (Mende et al. 2010; Roberts et al. 2014). In humans, TRA2β expression changes in several cancers, and this factor has been implicated in the pathology of other diseases, including spinal muscular atrophy, Alzheimer's disease, and frontotemporal dementia with Parkinsonism linked to Chr. 17 (Fig. 4; for review, see Long and Caceres 2009). TRA2β is up-regulated in tumors of the lung, ovary, cervix, colon, and breast (Fischer et al. 2004; Watermann et al. 2006; Iborra et al. 2013). TRA2β up-regulation is associated with invasive breast cancer, and medium to high TRA2β expression is associated with poor prognosis in cervical cancer (Fischer et al. 2004; Watermann et al. 2006; Iborra et al. 2013). Although TRA2β expression is associated with cancer cell survival, its contribution to disease progression is still poorly understood. Up-regulation of TRA2β promotes several splicing switches, including in CD44, which were previously associated with tumor progression and metastasis (Watermann et al. 2006). Another example of a TRA2β splicing target is the nuclear autoantigenic sperm protein NASP, which is important for cell survival (Grellscheid et al. 2011). TRA2β also regulates splicing of the estrogen receptor α, decreasing the expression of the ERaΔ7 isoform, which correlates with a better outcome in endometrial cancer (Hirschfeld et al. 2015). Finally, TRA2β regulates turnover of BCL2 mRNA, in a splicing-independent fashion, by competing with miR-204 for binding to the 3′UTR (Kuwano et al. 2015).
hnRNPA1 and hnRNPA2/B1—heterogeneous nuclear ribonucleoprotein A1 and A2
hnRNPA1 and A2/B1 are structurally related members of the hnRNP protein family, and are likely to play roles in cancer. hnRNPA2/B1 are two spliced isoforms encoded by the same gene, and B1 comprises 12 additional amino acids near the N-terminus (Fig. 4). hnRNPs are a diverse superfamily of abundant RNA-binding proteins expressed in most human tissues (for review, see Chaudhury et al. 2010). The hnRNP A/B family is a subset of hnRNP proteins, with closely related sequences and a conserved modular structure. They regulate alternative splicing, frequently by antagonizing SR proteins, in part through the recognition of exonic splicing silencer elements (Chaudhury et al. 2010). Additional functions of these proteins in mRNA trafficking, and in replication and transcription of cytoplasmic RNA viruses, have also been described. Several studies have reported overexpression of hnRNPA1 or A2/B1 in human tumors, including breast, lung, colon, and glioblastoma (Fielding et al. 1999; Zhou et al. 2001a,b; Karni et al. 2007; Golan-Gerstl et al. 2011). hnRNPA2 is an oncogenic driver in glioblastoma and other brain tumors, and its expression level is correlated with poor prognosis (Golan-Gerstl et al. 2011). In the brain, hnRNPA2 modulates alternative splicing and induces the expression of oncogenic isoforms of the tumor suppressors BIN1 and WWOX, the anti-apoptotic proteins encoded by CFLAR and CASP9, the insulin receptor gene IR, and the MST1R proto-oncogene (Golan-Gerstl et al. 2011). Furthermore, hnRNPA1 and A2 modulate alternative splicing of the glycolytic enzyme PKM2 in cancer cells, suggesting a role in the regulation of tumor metabolism (Clower et al. 2010; David et al. 2010).
hnRNPK—heterogeneous nuclear ribonucleoprotein K
In a subset of AML patients, the Chr. 9q21.32 genomic segment comprising the HNRNPK gene is specifically lost, suggesting that a tumor suppressor may reside at this locus (Fig. 4; Kronke et al. 2013). Additionally, AML patients harboring a partial deletion of Chromosome 9 exhibit a significant decrease in hnRNPK protein expression (Gallardo et al. 2015). Hnrnpk KO is embryonic lethal in mice, whereas Hnrnpk haploinsufficiency results in reduced survival, increased tumor formation, genomic instability, and the development of transplantable hematopoietic neoplasms with myeloproliferation (Gallardo et al. 2015). Reduced hnRNPK expression attenuates p21 activation, down-regulates C/EBP levels, and activates STAT3 signaling (Gallardo et al. 2015). Together, these data suggest that hnRNPK can act as a tumor suppressor in hematological disorders. Conversely, hnRNPK was shown to be overexpressed in a variety of cancers, including breast, esophageal squamous cell, pancreatic, skin, colorectal, and oral squamous cell carcinoma (Mandal et al. 2001; Carpenter et al. 2006; Roychoudhury and Chaudhuri 2007; Zhou et al. 2010). hnRNPK overexpression results in enhanced malignancy and metastasis in vitro, at least in part through alternative splicing of target genes that regulate the extracellular matrix, cell motility, and angiogenesis (Gao et al. 2013). hnRNPK also regulates splicing of the anti-apoptotic CFLAR inhibitory protein and the vascular endothelial growth factor VEGF in cancer cells (31, 32). Several studies suggested that increased hnRNPK expression results in a potential oncogenic effect through regulation of c-Myc in solid tumors (Notari et al. 2006; Roychoudhury and Chaudhuri 2007), thus suggesting a fundamental difference between hnRNPK's activity in solid tumors versus hematological malignancies.
PTB—polypyrimidine tract-binding protein
PTB (also known as hnRNPI), is an RNA-binding protein that binds preferentially to pyrimidine-rich sequences (Fig. 4). PTB expression levels are significantly up-regulated in breast, ovarian, glioblastoma, colorectal, and mucosal cancerous tissues compared with the corresponding normal tissues (Jin et al. 2000; He et al. 2004, 2007, 2014; Takahashi et al. 2015). PTB controls alternative splicing of target genes, such as pyruvate kinase PKM2 (Clower et al. 2010), fibroblast growth factor receptor-1α-exon FGFR-1 (Jin et al. 2000), and multidrug resistance protein 1 (MRP1), which contributes to the drug-resistance phenotype associated with many cancers (He et al. 2004). PTB levels are elevated in cancer cell lines, as well as endometrial tumor tissues, compared to normal tissues, although no correlation was observed between PTB expression and tumor grading (Wang et al. 2008). PTB is also overexpressed in most ovarian tumors, compared with matched normal tissue, and its overexpression is associated with an increased number of expressed MRP1 spliced isoforms (He et al. 2004, 2007). Furthermore, PTB KD decreases breast and ovarian tumor cell proliferation, anchorage-independent growth, and invasiveness in vitro (He et al. 2007, 2014), and is accompanied by an increased ratio of PKM1 versus PKM2 isoform and increased oxygen consumption (He et al. 2014). PTB overexpression also enhances anchorage-dependent growth of immortalized human mammary epithelial cells (HMECs) (He et al. 2014); however, overexpression of PTB alone is not sufficient to transform murine fibroblasts (Wang et al. 2008). Together, these results suggest that PTB plays a role in breast tumorigenesis.
Other hnRNPs
hnRNPM expression is associated with aggressive breast cancers and correlates with increased expression of the CD44 standard (CD44s) splice isoform, which comprises exons 1–5 and 16–20, and with the mesenchymal status of breast tumors (Fig. 4; Xu et al. 2014). hnRNPM plays a role in both EMT induction and maintenance, at least in part by potentiating TGFβ signaling and controlling CD44 isoform switching from the CD44 variable (CD44v) exon-8-containing splice isoform to the CD44s isoform (Xu et al. 2014).
Splicing factor hnRNPH, which is up-regulated in glioblastoma multiforme, controls splicing of MADD, a death-domain adaptor protein, to generate an antagonistic anti-apoptotic isoform that promotes survival and proliferation, and similarly mediates the splicing switch to a ligand-independent, constitutively active variant of the tyrosine kinase receptor MST1R (Ron), which promotes migration and invasion (Fig. 4; Lefave et al. 2011).
ESRP1 and 2—epithelial splicing regulator protein 1 and 2
ESRP1 (epithelial splicing regulatory protein 1, also known as RBM35) and ESRP2 (epithelial splicing regulatory protein 2, also known as RBM35B) are two structurally related splicing factors, encoded by genes on Chr. 8 and Chr. 16, respectively, and expressed specifically in epithelial cells (Fig. 4; Warzecha et al. 2009a). The mammalian ESRPs and their orthologs in chicken, D. melanogaster, and C. elegans comprise three RRMs and display significant phylogenetic sequence conservation within these domains, particularly within the first RRM (Warzecha et al. 2009a). Mice with ablation of Esrp1 develop cleft lip and palate, whereas tissue-specific ablation in the epidermis revealed ESRP1's requirement for establishing a proper skin barrier (Bebee et al. 2015). Loss of both Esrp1 and Esrp2 results in widespread developmental defects (Bebee et al. 2015). ESRPs regulate alternative splicing events associated with epithelial cell phenotypes, and both proteins are down-regulated during the epithelial-mesenchymal transition (EMT) (Warzecha et al. 2009a). ESRP splicing targets are enriched in genes involved in cytoskeletal dynamics, cell motility, cell–cell junctions, and pathways involved in EMT, including CD44, ENAH, and FGFR2 isoforms (Warzecha et al. 2009a,b; Shapiro et al. 2011; Dittmar et al. 2012). During oral squamous cell carcinogenesis, ESRPs are up-regulated relative to their levels in normal epithelium, but down-regulated in invasive fronts (Ishii et al. 2014). Importantly, ESRPs are expressed in the lymph nodes, where carcinoma cells metastasize and colonize (Ishii et al. 2014). In head and neck carcinoma cell lines, ESRP1 and ESRP2 suppress cancer cell motility through distinct mechanisms: ESRP1 KD affects the dynamics of the actin cytoskeleton through induction of RAC1 splicing isoforms, whereas ESRP2 KD attenuates cell-cell adhesion through increased expression of EMT-associated transcription factors (Ishii et al. 2014). In addition, ectopic expression of ESRP1 suppresses the malignant phenotypes of colon and breast cancer cells, suggesting that ESRP1 is a tumor suppressor (Leontieva and Ionov 2009; Horiguchi et al. 2012). In contrast, high levels of ESRP1 expression are associated with lower survival rate in breast cancer patients (Yae et al. 2012). These paradoxical findings may reflect yet unappreciated differences between the tumor subtypes or in the experimental models, which should be further characterized in detail. Thus, ESRP1 is likely to play a dual role during tumor progression, with the potential to act either as a positive or a negative regulator.
RBFOX2—RNA binding protein, Fox-2 homolog
The RBFOX family of splicing regulators includes three mammalian paralogs, RBFOX1 (A2BP1), RBFOX 2 (RBM9), and RBFOX3 (NeuN), which control tissue-specific alternative splicing of exons in brain, muscle, epithelial, and mesenchymal cells, as well as in embryonic stem cells (Kuroyanagi 2009). RBFOX proteins have a single RRM, highly homologous among the three paralogs, and their binding sites are exceptionally highly conserved in sequence (UGCAUG) and position across vertebrate evolution (Sun et al. 2012). Mutations or deletions in RBFOX1 and RBFOX3 are found in epilepsy patients (Bhalla et al. 2004; Lal et al. 2013a,b), whereas copy-number variations in RBFOX1 are associated with autism spectrum disorders and spinocerebellar ataxias (Bill et al. 2013; Weyn-Vanhentenryck et al. 2014). Mouse models with Rbfox KO or KD show extensive defects in neuronal and muscle physiology, suggesting that these proteins play key roles in normal development (Gehman et al. 2011, 2012). A role for RBFOX2 in cancer has also been proposed. Its levels are significantly lower in epithelial ovarian cancer than in normal tissue (Venables et al. 2009). In breast cancer, alternative splicing events occurring in cell lines from the claudin-low subtype are enriched in the RBFOX2 binding motif (Fig. 4; Lapuk et al. 2010). RBFOX2-regulated splicing events were also identified in multiple solid tumor types, including breast, colon, and pancreatic tumors (Danan-Gotthold et al. 2015). RBFOX2 was identified as one of the potential drivers of the mesenchymal splicing signature in a mammary cell line, following induction of Twist expression (Shapiro et al. 2011). In this context, the loss of RBFOX2 in mesenchymal cells leads to a partial reversion of the epithelial phenotype, with reduced levels of vimentin, changes in morphology, and restricted migration (Shapiro et al. 2011). Furthermore RBFOX2 regulates events that differ between epithelial and mesenchymal cancer cell lines (Venables et al. 2013). Thus, RBFOX2 may be important to specify the mesenchymal tissue-specific splicing profiles both in normal and in cancer tissues.
RBM5, 6, and 10—RNA binding motif protein 5, 6, and 10
RBM5, RBM6, and RBM10 are highly homologous RNA-binding proteins that share 30–50% amino-acid sequence identity (Fig. 4; Bechara et al. 2013). They comprise two RRMs and an RS-domain, as well as a zing-finger domain, a G-patch domain, and a nuclear localization signal (Bechara et al. 2013). The genes encoding RBM5 and RBM6 map to the Chr. 3p21.3 region, which is frequently deleted in heavy smokers, in lung cancer, and in other tissue carcinomas (Angeloni 2007). RBM5 protein levels are down-regulated in 75% of primary lung cancers (Oh et al. 2002), as well as in prostate and breast cancer samples (Zhao et al. 2012). Down-regulation of RBM5 is considered to be one molecular signature associated with metastasis of various solid tumors (Ramaswamy et al. 2003). On the other hand, overexpression of RBM5 in breast cancer samples was also reported (Rintala-Maki et al. 2007), paradoxically suggesting that both up- and down-regulation of RBM5 can play a role in tumor progression. RBM6 and 10 also show altered expression in breast cancer (Rintala-Maki et al. 2007). Overexpression of RBM5 in various cell lines leads to growth arrest, apoptosis, and delayed tumor growth when cells are injected into nude mice (Mourtada-Maarabouni and Williams 2002; Oh et al. 2002, 2006; Mourtada-Maarabouni et al. 2003). RBM5-mediated apoptosis is associated with up-regulation of the pro-apoptotic protein BAX and down-regulation of the anti-apoptotic proteins BCL-2 and BCL-XL (Mourtada-Maarabouni and Williams 2002; Oh et al. 2006; Sutherland et al. 2010). RBM5/6 and RBM10 antagonistically regulate proliferation of breast and lung cancer cells, and display distinct positional effects on alternative splicing regulation (Bechara et al. 2013). The Notch pathway regulator NUMB is a key splicing target of RBM5, 6, and 10, which contributes to their role in colony formation in vitro and in tumor xenograft growth in vivo (Bechara et al. 2013). Finally, RBM10 is also among the most frequently mutated genes in lung adenocarcinoma (Imielinski et al. 2012). The RBM10 mutations identified in patients disrupt NUMB splicing regulation and promote proliferation of cancer cell lines (Bechara et al. 2013; Hernandez et al. 2016).
QKI—quaking KH domain containing RNA binding
Quaking KH domain containing RNA binding (QKI) protein belongs to the STAR (signal transduction and activation of RNA) family of RNA-binding proteins (Fig. 4; for review, see Darbelli and Richard 2016). QKI has been implicated in the regulation of myocyte and oligodendrocyte differentiation, as well as in endothelial-cell function (Darbelli and Richard 2016). QKI haploinsufficiency is associated with the 6q terminal deletion syndrome, which is characterized by intellectual disabilities (Darbelli and Richard 2016). QKI is also frequently down-regulated in lung adenocarcinoma, squamous cell carcinoma, and large cell carcinoma, compared with normal lung, and its down-regulation is significantly associated with poor prognosis (Zong et al. 2014). QKI expression is also reduced in oral squamous cell carcinoma, compared to normal mucosa (Lu et al. 2014), as well as in prostate cancer samples, at least in part due to the high levels of promoter methylation (Zhao et al. 2014). QKI is highly expressed in benign prostatic hyperplasia, but not in carcinomatous tissue (Zhao et al. 2014). In addition, the decrease in QKI expression closely correlates with the Gleason score, poor differentiation, degree of invasion, lymph-node metastasis, distant metastasis, tumor grading, and poor survival (Zhao et al. 2014). QKI overexpression impairs proliferation and transformation of lung and prostate cancer cells (Zhao et al. 2014; Zong et al. 2014), as well as reduces cancer stem cell sphere formation and stem-cell-associated gene expression in oral cancer cells (Lu et al. 2014). In a tumor-implantation nude-mouse model, QKI expression significantly reduces the tumor-initiation rate, tumor size, and lung-metastasis rate, whereas QKI KD has the opposite effects (Lu et al. 2014; Zong et al. 2014). QKI expression is also down-regulated in lung, colon, and breast tumors, but is up-regulated in kidney cancer, and QKI-regulated splicing events are altered in the corresponding tumor types (Danan-Gotthold et al. 2015). QKI regulates splicing of NUMB, which in turn suppresses cell proliferation and prevents the activation of the Notch signaling pathway (Zong et al. 2014). QKI may also directly regulate SOX2 expression via specific binding to its mRNA 3′UTR in a cis-element-dependent way (Lu et al. 2014). Recently, MYB-QKI fusions were identified as a specific and single candidate-driver event in angiocentric gliomas (Bandopadhayay et al. 2016). In vitro and in vivo functional studies demonstrated that MYB-QKI rearrangements promote tumorigenesis through three mechanisms: MYB activation by truncation, enhancer translocation driving aberrant MYB-QKI expression, and hemizygous loss of the QKI tumor suppressor (Bandopadhayay et al. 2016).
REGULATION OF THE SPLICING MACHINERY IN TUMORS
How changes in splicing-factor levels occur in human tumors remains poorly understood. In the absence of genomic alterations at loci encoding splicing regulators, alterations in splicing-factor expression likely result from changes in transcriptional or post-transcriptional regulation, as well as in signaling pathways.
Splicing-factor regulation by the transcription factor MYC
A link between the transcription factor oncoprotein MYC and the splicing machinery has emerged in the past few years. Genes encoding several splicing factors, such as SRSF1, HNRNPA1, HNRNPA2, or PTB, were shown to be direct transcriptional targets of MYC (Figs. 1, 3; David et al. 2010; Anczukow et al. 2012; Das et al. 2012). Furthermore, SRSF1 not only contributes to MYC's oncogenic activity (Das et al. 2012), but also cooperates with MYC in transformation, promoting the formation of more aggressive breast tumors (Anczukow et al. 2012). In addition, two recent studies revealed that components of the spliceosome, such as PRMT5 and BUD31, are essential for MYC to function as an oncogene in lymphoma and breast cancer, respectively (Hsu et al. 2015; Koh et al. 2015). As MYC is the most frequently amplified oncogene in human cancers and plays a crucial role in transformation, therapies that exploit the spliceosome would be very attractive. Both studies uncovered an essential role of the splicing machinery in MYC-driven transformation, and identified multiple associated abnormal splicing events, including intron retention (Hsu et al. 2015; Koh et al. 2015). Interestingly, MYC appears to alter splicing by somewhat different mechanisms in lymphomagenesis versus breast cancer. In the former context, MYC hyperactivation affects the levels of specific splicing regulators (Koh et al. 2015), whereas in the latter context, it promotes a global increase in pre-mRNA levels (Hsu et al. 2015), although up-regulation of particular splicing regulators was also previously reported (David et al. 2010; Anczukow et al. 2012; Das et al. 2012). These ostensibly different findings suggest that many of the splicing changes associated with cancer are context-dependent.
Post-transcriptional regulation of SRSFs
SRSFs are essential genes whose expression is tightly regulated at the post-transcriptional level. For example, SRSF1 or SRFS2 regulate splicing of introns in the 3′UTR of their own pre-mRNA; splicing out of these introns introduces an exon–exon junction downstream from the normal stop codon, which targets the mRNA to NMD (Sureau et al. 2001; Lareau et al. 2007a,b). This mechanism is highly conserved and shared by other SR proteins (Sureau et al. 2001; Lareau et al. 2007a,b; Lareau and Brenner 2015). SRSF1 autoregulation can also occur via nuclear retention of alternative SRSF1 transcript variants or regulation of the translational efficiency of other variants (Sun et al. 2010). Autoregulation of SR proteins serves as a negative feedback loop, in which increased SR protein levels promote an increase in unproductive spliced variants of their own transcripts. Furthermore, several miRNAs targeting SRSF1 mRNA have been identified, including mir-7, miR-28, miR-505, miR-10a, and miR-10b (Verduci et al. 2010; Wu et al. 2010; Meseguer et al. 2011). Finally, SRSF1 expression is also regulated at the level of splicing by the splicing factor Sam68 (Valacca et al. 2010). In response to ERK1/2 signaling, Sam68 switches splicing of the SRSF1 transcript from the NMD-targeted isoforms to the major, translatable isoform, thus resulting in an increase in SRSF1 protein levels. Sam68-mediated SRSF1 overexpression is associated with oncogenic phenotypes, such as increased cell-proliferation, anchorage-independent growth, cell motility and invasion, and epithelial–mesenchymal transition in colon cancer cell lines (Valacca et al. 2010). Thus, SRSF1 transcript levels are fine-tuned by various post-transcriptional mechanisms, though the precise contributions of each step in response to various stimuli remain to be determined.
SR protein regulation by phosphorylation
SR protein localization and activity are regulated by a dynamic cycle of phosphorylation/dephosphorylation, mostly at serine residues within the RS-domain (for a detailed review, see Long and Caceres 2009). For example, phosphorylation of SRSF1's RS domain enhances protein–protein interactions with other RS-domain-containing splicing factors, such as U1–70K (Xiao and Manley 1997), whereas dephosphorylation of SR and SR-related proteins is required for splicing catalysis (Tazi et al. 1993; Cao et al. 1997). The SR protein kinase family (SRPK) (Gui et al. 1994; Wang et al. 1998), Clk/Sty kinase family (Colwill et al. 1996) and topoisomerase I (Rossi et al. 1996) have been identified as the main regulators of SR protein phosphorylation. A hypo-phosphorylated RS domain is required for the interaction of shuttling SR proteins with the TAP/NFX1 nuclear export receptor (Huang et al. 2004), whereas the RS domain needs to be rephosphorylated before SR proteins can return to the nucleus (Ding et al. 2006). Dephosphorylation also plays an important role in sorting SR proteins in the nucleus, where shuttling SR proteins and nonshuttling SR proteins are recycled via different recycling pathways (Lin et al. 2005). In the cytoplasm, dephosphorylation of the RS domain enhances mRNA binding by SRSF1 and contributes to its stimulatory role in translation (Sanford et al. 2005). The regulation of SR protein phosphorylation has been implicated in the control of cancer-associated splicing events. For example, growth-factor-induced alternative splicing of the fibronectin EDA exon involves phosphorylation of SRSF1 and SRSF7 by AKT kinase (Blaustein et al. 2004). This effect is most likely indirect and occurs via SRPKs. Indeed, AKT was shown to activate SRPKs in response to EGF-signaling, thus leading to enhanced SRPK nuclear translocation and SR protein phosphorylation (Zhou et al. 2012). Multiple components of the AKT pathway function as oncogenes or tumor suppressors, and overexpression of SRPK1 is seen in multiple tumor types (Hayes et al. 2006, 2007; Plasencia et al. 2006; Krishnakumar et al. 2008; Amin et al. 2011); however, how perturbations in phosphorylation processes lead to changes in splicing-factor activity in human tumors remains to be defined.
TARGETING ALTERNATIVE-SPLICING ALTERATIONS FOR CANCER THERAPY
The idea of targeting the spliceosome is not new, and the first general splicing inhibitors were initially identified in the late 1990s, during the course of characterizing certain anti-tumor drugs (for review, see Bonnal et al. 2012). However, improvements in RNA-targeting therapeutics and in particular in antisense-oligonucleotide pharmacology, as well as a better understanding of the mode of action of these molecules, have created novel therapeutic opportunities. Two conceptually different approaches to splicing inhibition are currently being tested.
Blocking splicing-factor activity with small-molecule inhibitors
The first approach is to target general components of the splicing machinery and inhibit splicing at a global level, e.g., using small-molecule inhibitors of the SF3B complex, or of kinases that phosphorylate SR proteins (Bonnal et al. 2012). These drugs inhibit very basic steps in splice-site recognition, lack splicing-target specificity, and can potentially have broad cytotoxic effects. However, several studies have reported that cancer cells are more sensitive to these drugs, compared to normal cells (Bonnal et al. 2012), suggesting that general inhibition of splicing could be a viable anti-tumor strategy. For example, SF3B1 inhibitors, such as spliceostatin A, pladienolide-B, GEX1A, and E1707, partially inhibit splicing in cultured cells and animal models, and show selective action, i.e., greater potency, on tumor cells (for review, see Ohe and Hagiwara 2015). Thus SF3B1 inhibitors are currently being evaluated in the context of splicing-factor mutations in preclinical models of MDS (Lee et al. 2016).
Another way to block splicing-factor activity is to inhibit the regulatory kinases that phosphorylate the SR proteins (Ohe and Hagiwara 2015). For example, inhibition of Cdc2-like kinase 1 (CLK1), dual-specificity-tyrosine-phosphorylation-regulated kinase 1A (DYRK1A), or SR protein-specific kinase (SRPK) affects alternative splicing and induces exon skipping of SR-regulated exons in cell cultures (Ohe and Hagiwara 2015).
Exon-specific splicing modulation
An alternative approach to target splicing alterations is to directly target a tumor-specific splicing event, e.g., by using antisense oligonucleotides that bind to a transcript in a sequence-specific manner to redirect splicing (for review, see Kole et al. 2012). Briefly, antisense oligonucleotides are short single-stranded nucleic-acid molecules with various chemical modifications—compared to RNA or DNA—that bind to complementary sequences on the target pre-mRNA in the nucleus. The target sequences can be, e.g., splice sites, splicing enhancers, or splicing silencers, which are normally recognized by the core splicing machinery or by a particular regulatory splicing factor (activator or repressor). Thus, such antisense compounds can be used either to promote or inhibit a splicing event in a target-specific manner. This approach is expected to have few off-targets effects—provided that unique complementary sequences in the transcriptome are targeted—and with appropriate target selection, it might be more tumor-specific, compared to general splicing inhibitors. However, identifying a key splicing event, or more likely a set of splicing events, required for transformation and tumor maintenance, will require a concerted effort.
Finally, small-molecules that can specifically modulate a single alternative splicing event are currently being investigated as an alternative to antisense-oligonucleotide therapies. For example, several groups identified compounds that enhance SMN2 exon 7 splicing to promote the production of a full-length SMN protein (Hastings et al. 2009; Naryshkin et al. 2014; Palacino et al. 2015), an approach with therapeutic potential for spinal muscular atrophy. Likewise, small molecules that promote the inclusion of exon 20 in the IKBKAP pre-mRNA have been reported, and may have therapeutic utility for familial dysautonomia (Yoshida et al. 2015). However, it is still unknown to what extent this type of strategy may be broadly applicable to other splicing events. These molecules were initially identified using cell-based reporter screens, and while potential mechanisms of action have been proposed, additional work will be required to understand what degree of target specificity is achievable, and to what extent these approaches are generalizable.
In summary, splicing-factor alterations detected in human tumors are a growing area of interest in cancer research, and represent potential targets for personalized cancer therapies. However, the precise roles of splicing factors in normal tissue physiology and the consequences of their dysregulation in disease are still underexplored. A better understanding of the cell-type specificity and functions of cancer-associated splicing factors will be crucial for the development of new therapeutic strategies.
ACKNOWLEDGMENTS
O.A. and A.R.K. acknowledge support from the National Cancer Institute (CA13106 and K99CA178206).
Author contributions: Both authors wrote, discussed, revised, and approved the final manuscript.
Footnotes
Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.057919.116.
REFERENCES
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, et al. 2011. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20: 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. 2012. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Akerman M, Clery A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. 2015. SRSF1-regulated alternative splicing in breast cancer. Mol Cell 60: 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeloni D. 2007. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief Funct Genomic Proteomic 6: 19–39. [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O'Rourke R, Gibson WJ, et al. 2016. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet 48: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebee TW, Park JW, Sheridan KI, Warzecha CC, Cieply BW, Rohacek AM, Xing Y, Carstens RP. 2015. The splicing regulators Esrp1 and Esrp2 direct an epithelial splicing program essential for mammalian development. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara EG, Sebestyen E, Bernardis I, Eyras E, Valcarcel J. 2013. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell 52: 720–733. [DOI] [PubMed] [Google Scholar]
- Bejar R, Stevenson KE, Caughey BA, Abdel-Wahab O, Steensma DP, Galili N, Raza A, Kantarjian H, Levine RL, Neuberg D, et al. 2012. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 30: 3376–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best A, Dalgliesh C, Kheirollahi-Kouhestani M, Danilenko M, Ehrmann I, Tyson-Capper A, Elliott DJ. 2014. Tra2 protein biology and mechanisms of splicing control. Biochem Soc Trans 42: 1152–1158. [DOI] [PubMed] [Google Scholar]
- Bhalla K, Phillips HA, Crawford J, McKenzie OL, Mulley JC, Eyre H, Gardner AE, Kremmidiotis G, Callen DF. 2004. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J Hum Genet 49: 308–311. [DOI] [PubMed] [Google Scholar]
- Biamonti G, Catillo M, Pignataro D, Montecucco A, Ghigna C. 2014. The alternative splicing side of cancer. Semin Cell Dev Biol 32: 30–36. [DOI] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. 2012. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 491: 399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill BR, Lowe JK, Dybuncio CT, Fogel BL. 2013. Orchestration of neurodevelopmental programs by RBFOX1: implications for autism spectrum disorder. Int Rev Neurobiol 113: 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Coso OA, Bissell MJ, Kornblihtt AR, Srebrow A. 2004. Mammary epithelial-mesenchymal interaction regulates fibronectin alternative splicing via phosphatidylinositol 3-kinase. J Biol Chem 279: 21029–21037. [DOI] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, Valcarcel J. 2012. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 11: 847–859. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. 2014. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9: e87361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bueno R, Stawiski EW, Goldstein LD, Durinck S, De Rienzo A, Modrusan Z, Gnad F, Nguyen TT, Jaiswal BS, Chirieac LR, et al. 2016. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat Genet 48: 407–416. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network. 2013. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, Jamison SF, Garcia-Blanco MA. 1997. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA 3: 1456–1467. [PMC free article] [PubMed] [Google Scholar]
- Carpenter B, McKay M, Dundas SR, Lawrie LC, Telfer C, Murray GI. 2006. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br J Cancer 95: 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury A, Chander P, Howe PH. 2010. Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: Focus on hnRNP E1’s multifunctional regulatory roles. RNA 16: 1449–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clower CV, Chatterjee D, Wang Z, Cantley LC, Vander Heiden MG, Krainer AR. 2010. The alternative splicing repressors hnRNP A1/A2 and PTB influence pyruvate kinase isoform expression and cell metabolism. Proc Natl Acad Sci 107: 1894–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Eliav M, Golan-Gerstl R, Siegfried Z, Andersen CL, Thorsen K, Orntoft TF, Mu D, Karni R. 2013. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J Pathol 229: 630–639. [DOI] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. 1996. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J 15: 265–275. [PMC free article] [PubMed] [Google Scholar]
- Damm F, Thol F, Kosmider O, Kade S, Loffeld P, Dreyfus F, Stamatoullas-Bastard A, Tanguy-Schmidt A, Beyne-Rauzy O, de Botton S, et al. 2012. SF3B1 mutations in myelodysplastic syndromes: clinical associations and prognostic implications. Leukemia 26: 1137–1140. [DOI] [PubMed] [Google Scholar]
- Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. 2015. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 43: 5130–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbelli L, Richard S. 2016. Emerging functions of the Quaking RNA-binding proteins and link to human diseases. Wiley Interdiscip Rev RNA 7: 399–412. [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- Das S, Krainer AR. 2014. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol Cancer Res 12: 1195–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Anczukow O, Akerman M, Krainer AR. 2012. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1: 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL. 2010. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24: 2343–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. 2010. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463: 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, Adams JA, Fu XD. 2006. Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell 17: 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP. 2012. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol 32: 1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding P, Turnbull L, Prime W, Walshaw M, Field JK. 1999. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: a clinical marker of early lung cancer detection. Clin Cancer Res 5: 4048–4052. [PubMed] [Google Scholar]
- Fischer DC, Noack K, Runnebaum IB, Watermann DO, Kieback DG, Stamm S, Stickeler E. 2004. Expression of splicing factors in human ovarian cancer. Oncol Rep 11: 1085–1090. [PubMed] [Google Scholar]
- Fregoso OI, Das S, Akerman M, Krainer AR. 2013. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol Cell 50: 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. 2013. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov 3: 1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo M, Lee HJ, Zhang X, Bueso-Ramos C, Pageon LR, McArthur M, Multani A, Nazha A, Manshouri T, Parker-Thornburg J, et al. 2015. hnRNP K is a haploinsufficient tumor suppressor that regulates proliferation and differentiation programs in hematologic malignancies. Cancer Cell 28: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao R, Yu Y, Inoue A, Widodo N, Kaul SC, Wadhwa R. 2013. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J Biol Chem 288: 15046–15056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M Jr, Mody I, Black DL. 2011. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet 43: 706–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M Jr, Otis TS, Black DL. 2012. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G. 2005. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 20: 881–890. [DOI] [PubMed] [Google Scholar]
- Golan-Gerstl R, Cohen M, Shilo A, Suh SS, Bakacs A, Coppola L, Karni R. 2011. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res 71: 4464–4472. [DOI] [PubMed] [Google Scholar]
- Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, et al. 2012. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet 44: 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grellscheid S, Dalgliesh C, Storbeck M, Best A, Liu Y, Jakubik M, Mende Y, Ehrmann I, Curk T, Rossbach K, et al. 2011. Identification of evolutionarily conserved exons as regulated targets for the splicing activator tra2β in development. PLoS Genet 7: e1002390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui JF, Lane WS, Fu XD. 1994. A serine kinase regulates intracellular localization of splicing factors in the cell cycle. Nature 369: 678–682. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. 2013. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45: 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings ML, Berniac J, Liu YH, Abato P, Jodelka FM, Barthel L, Kumar S, Dudley C, Nelson M, Larson K, et al. 2009. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci Transl Med 1: 5ra12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes GM, Carrigan PE, Beck AM, Miller LJ. 2006. Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res 66: 3819–3827. [DOI] [PubMed] [Google Scholar]
- Hayes GM, Carrigan PE, Miller LJ. 2007. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res 67: 2072–2080. [DOI] [PubMed] [Google Scholar]
- He X, Ee PL, Coon JS, Beck WT. 2004. Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin Cancer Res 10: 4652–4660. [DOI] [PubMed] [Google Scholar]
- He X, Pool M, Darcy KM, Lim SB, Auersperg N, Coon JS, Beck WT. 2007. Knockdown of polypyrimidine tract-binding protein suppresses ovarian tumor cell growth and invasiveness in vitro. Oncogene 26: 4961–4968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Arslan AD, Pool MD, Ho TT, Darcy KM, Coon JS, Beck WT. 2011. Knockdown of splicing factor SRp20 causes apoptosis in ovarian cancer cells and its expression is associated with malignancy of epithelial ovarian cancer. Oncogene 30: 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Arslan AD, Ho TT, Yuan C, Stampfer MR, Beck WT. 2014. Involvement of polypyrimidine tract-binding protein (PTBP1) in maintaining breast cancer cell growth and malignant properties. Oncogenesis 3: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Luhrmann R, Stelzl U. 2012. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell 45: 567–580. [DOI] [PubMed] [Google Scholar]
- Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. 2016. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol 13: 466–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschfeld M, Ouyang YQ, Jaeger M, Erbes T, Orlowska-Volk M, Zur Hausen A, Stickeler E. 2015. HNRNP G and HTRA2-BETA1 regulate estrogen receptor alpha expression with potential impact on endometrial cancer. BMC Cancer 15: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, Fujii H, Yamaguchi A, Miyazawa K, Miyazono K, et al. 2012. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene 31: 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. 2015. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 525: 384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Yario TA, Steitz JA. 2004. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci 101: 9666–9670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CS, Shen CY, Wang HW, Wu PE, Cheng CW. 2007. Increased expression of SRp40 affecting CD44 splicing is associated with the clinical outcome of lymph node metastasis in human breast cancer. Clin Chim Acta 384: 69–74. [DOI] [PubMed] [Google Scholar]
- Iborra S, Hirschfeld M, Jaeger M, Zur Hausen A, Braicu I, Sehouli J, Gitsch G, Stickeler E. 2013. Alterations in expression pattern of splicing factors in epithelial ovarian cancer and its clinical impact. Int J Gynecol Cancer 23: 990–996. [DOI] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. 2012. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Saitoh M, Sakamoto K, Kondo T, Katoh R, Tanaka S, Motizuki M, Masuyama K, Miyazawa K. 2014. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem 289: 27386–27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MA, Wilkinson JE, Krainer AR. 2014. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat Struct Mol Biol 21: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia R, Li C, McCoy JP, Deng CX, Zheng ZM. 2010. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int J Biol Sci 6: 806–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, McCutcheon IE, Fuller GN, Huang ES, Cote GJ. 2000. Fibroblast growth factor receptor-1 α-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res 60: 1221–1224. [PubMed] [Google Scholar]
- Jumaa H, Wei G, Nielsen PJ. 1999. Blastocyst formation is blocked in mouse embryos lacking the splicing factor SRp20. Curr Biol 9: 899–902. [DOI] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. 2007. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14: 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, Al-Haddawi M, Tan SY, Osato M, Sabo A, et al. 2015. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 523: 96–100. [DOI] [PubMed] [Google Scholar]
- Kole R, Krainer AR, Altman S. 2012. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nat Rev Drug Discov 11: 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Krauthammer M, Halaban R. 2014. Rare SF3B1 R625 mutations in cutaneous melanoma. Melanoma Res 24: 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar S, Mohan A, Kandalam M, Ramkumar HL, Venkatesan N, Das RR. 2008. SRPK1: a cisplatin sensitive protein expressed in retinoblastoma. Pediatr Blood Cancer 50: 402–406. [DOI] [PubMed] [Google Scholar]
- Kronke J, Bullinger L, Teleanu V, Tschurtz F, Gaidzik VI, Kuhn MW, Rucker FG, Holzmann K, Paschka P, Kapp-Schworer S, et al. 2013. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 122: 100–108. [DOI] [PubMed] [Google Scholar]
- Kurokawa K, Akaike Y, Masuda K, Kuwano Y, Nishida K, Yamagishi N, Kajita K, Tanahashi T, Rokutan K. 2014. Downregulation of serine/arginine-rich splicing factor 3 induces G1 cell cycle arrest and apoptosis in colon cancer cells. Oncogene 33: 1407–1417. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H. 2009. Fox-1 family of RNA-binding proteins. Cell Mol Life Sci 66: 3895–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwano Y, Nishida K, Kajita K, Satake Y, Akaike Y, Fujita K, Kano S, Masuda K, Rokutan K. 2015. Transformer 2β and miR-204 regulate apoptosis through competitive binding to 3′ UTR of BCL2 mRNA. Cell Death Differ 22: 815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D, Reinthaler EM, Altmuller J, Toliat MR, Thiele H, Nurnberg P, Lerche H, Hahn A, Moller RS, Muhle H, et al. 2013a. RBFOX1 and RBFOX3 mutations in rolandic epilepsy. PLoS One 8: e73323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal D, Trucks H, Moller RS, Hjalgrim H, Koeleman BP, de Kovel CG, Visscher F, Weber YG, Lerche H, Becker F, et al. 2013b. Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia 54: 265–271. [DOI] [PubMed] [Google Scholar]
- Lapuk A, Marr H, Jakkula L, Pedro H, Bhattacharya S, Purdom E, Hu Z, Simpson K, Pachter L, Durinck S, et al. 2010. Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res 8: 961–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Brenner SE. 2015. Regulation of splicing factors by alternative splicing and NMD is conserved between kingdoms yet evolutionarily flexible. Mol Biol Evol 32: 1072–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Brooks AN, Soergel DA, Meng Q, Brenner SE. 2007a. The coupling of alternative splicing and nonsense-mediated mRNA decay. Adv Exp Med Biol 623: 190–211. [DOI] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. 2007b. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446: 926–929. [DOI] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. 2016. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22: 672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, Cartegni L. 2011. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J 30: 4084–4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontieva OV, Ionov Y. 2009. RNA-binding motif protein 35A is a novel tumor suppressor for colorectal cancer. Cell Cycle 8: 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Wang J, Manley JL. 2005. Loss of splicing factor ASF/SF2 induces G2 cell cycle arrest and apoptosis, but inhibits internucleosomal DNA fragmentation. Genes Dev 19: 2705–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Xiao R, Sun P, Xu X, Fu XD. 2005. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell 20: 413–425. [DOI] [PubMed] [Google Scholar]
- Long JC, Caceres JF. 2009. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 417: 15–27. [DOI] [PubMed] [Google Scholar]
- Lu W, Feng F, Xu J, Lu X, Wang S, Wang L, Lu H, Wei M, Yang G, Wang L, et al. 2014. QKI impairs self-renewal and tumorigenicity of oral cancer cells via repression of SOX2. Cancer Biol Ther 15: 1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho JS, Weigelt B, Natrajan RC. 2015. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol 235: 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. 2012. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 119: 3203–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, et al. 2011. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood 118: 6239–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jadersten M, Jansson M, Elena C, Galli A, Walldin G, Della Porta MG, et al. 2015. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood 126: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Vadlamudi R, Nguyen D, Wang RA, Costa L, Bagheri-Yarmand R, Mendelsohn J, Kumar R. 2001. Growth factors regulate heterogeneous nuclear ribonucleoprotein K expression and function. J Biol Chem 276: 9699–9704. [DOI] [PubMed] [Google Scholar]
- Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. 2013. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet 45: 933–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mende Y, Jakubik M, Riessland M, Schoenen F, Rossbach K, Kleinridders A, Kohler C, Buch T, Wirth B. 2010. Deficiency of the splicing factor Sfrs10 results in early embryonic lethality in mice and has no impact on full-length SMN/Smn splicing. Hum Mol Genet 19: 2154–2167. [DOI] [PubMed] [Google Scholar]
- Meseguer S, Mudduluru G, Escamilla JM, Allgayer H, Barettino D. 2011. MicroRNAs-10a and -10b contribute to retinoic acid-induced differentiation of neuroblastoma cells and target the alternative splicing regulatory factor SFRS1 (SF2/ASF). J Biol Chem 286: 4150–4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian SA, Smith AE, Kulasekararaj AG, Kizilors A, Mohamedali AM, Lea NC, Mitsopoulos K, Ford K, Nasser E, Seidl T, et al. 2013. Spliceosome mutations exhibit specific associations with epigenetic modifiers and proto-oncogenes mutated in myelodysplastic syndrome. Haematologica 98: 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Wang Q, Kennedy CJ, Silver PA. 2010. An alternative splicing network links cell-cycle control to apoptosis. Cell 142: 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourtada-Maarabouni M, Williams GT. 2002. RBM5/LUCA-15—tumour suppression by control of apoptosis and the cell cycle? Sci World J 2: 1885–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourtada-Maarabouni M, Sutherland LC, Meredith JM, Williams GT. 2003. Simultaneous acceleration of the cell cycle and suppression of apoptosis by splice variant delta-6 of the candidate tumour suppressor LUCA-15/RBM5. Genes Cells 8: 109–119. [DOI] [PubMed] [Google Scholar]
- Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG, et al. 2014. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 345: 688–693. [DOI] [PubMed] [Google Scholar]
- Notari M, Neviani P, Santhanam R, Blaser BW, Chang JS, Galietta A, Willis AE, Roy DC, Caligiuri MA, Marcucci G, et al. 2006. A MAPK/HNRPK pathway controls BCR/ABL oncogenic potential by regulating MYC mRNA translation. Blood 107: 2507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh JJ, West AR, Fishbein MC, Slamon DJ. 2002. A candidate tumor suppressor gene, H37, from the human lung cancer tumor suppressor locus 3p21.3. Cancer Res 62: 3207–3213. [PubMed] [Google Scholar]
- Oh JJ, Razfar A, Delgado I, Reed RA, Malkina A, Boctor B, Slamon DJ. 2006. 3p21.3 tumor suppressor gene H37/Luca15/RBM5 inhibits growth of human lung cancer cells through cell cycle arrest and apoptosis. Cancer Res 66: 3419–3427. [DOI] [PubMed] [Google Scholar]
- Ohe K, Hagiwara M. 2015. Modulation of alternative splicing with chemical compounds in new therapeutics for human diseases. ACS Chem Biol 10: 914–924. [DOI] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. 2015. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29: 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, et al. 2015. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol 11: 511–517. [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Cazzola M, Boultwood J, Malcovati L, Vyas P, Bowen D, Pellagatti A, Wainscoat JS, Hellstrom-Lindberg E, Gambacorti-Passerini C, et al. 2011. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med 365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. 2013. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 122: 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patnaik MM, Lasho TL, Hodnefield JM, Knudson RA, Ketterling RP, Garcia-Manero G, Steensma DP, Pardanani A, Hanson CA, Tefferi A. 2012. SF3B1 mutations are prevalent in myelodysplastic syndromes with ring sideroblasts but do not hold independent prognostic value. Blood 119: 569–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasencia C, Martinez-Balibrea E, Martinez-Cardus A, Quinn DI, Abad A, Neamati N. 2006. Expression analysis of genes involved in oxaliplatin response and development of oxaliplatin-resistant HT29 colon cancer cells. Int J Oncol 29: 225–235. [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. 2013. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 122: 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Conde L, Villamor N, Ordonez GR, Jares P, Bassaganyas L, Ramsay AJ, Bea S, Pinyol M, Martinez-Trillos A, et al. 2012. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet 44: 47–52. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. 2003. A molecular signature of metastasis in primary solid tumors. Nat Genet 33: 49–54. [DOI] [PubMed] [Google Scholar]
- Rintala-Maki ND, Goard CA, Langdon CE, Wall VE, Traulsen KE, Morin CD, Bonin M, Sutherland LC. 2007. Expression of RBM5-related factors in primary breast tissue. J Cell Biochem 100: 1440–1458. [DOI] [PubMed] [Google Scholar]
- Roberts JM, Ennajdaoui H, Edmondson C, Wirth B, Sanford JR, Chen B. 2014. Splicing factor TRA2B is required for neural progenitor survival. J Comp Neurol 522: 372–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi F, Labourier E, Forne T, Divita G, Derancourt J, Riou JF, Antoine E, Cathala G, Brunel C, Tazi J. 1996. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 381: 80–82. [DOI] [PubMed] [Google Scholar]
- Roychoudhury P, Chaudhuri K. 2007. Evidence for heterogeneous nuclear ribonucleoprotein K overexpression in oral squamous cell carcinoma. Br J Cancer 97: 574–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford JR, Ellis JD, Cazalla D, Caceres JF. 2005. Reversible phosphorylation differentially affects nuclear and cytoplasmic functions of splicing factor 2/alternative splicing factor. Proc Natl Acad Sci 102: 15042–15047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Jumaa H, Webster NJ. 2013. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat Commun 4: 1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S, Langiewicz M, Jumaa H, Webster NJ. 2015. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 61: 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. 2011. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS Genet 7: e1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. 2015. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 27: 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, Esquivel E, Yu A, Seepo S, Olsen SR, et al. 2015. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47: 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Sinha R, Karni R, Krainer AR. 2010. SF2/ASF autoregulation involves multiple layers of post-transcriptional and translational control. Nat Struct Mol Biol 17: 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Zhang Z, Fregoso O, Krainer AR. 2012. Mechanisms of activation and repression by the alternative splicing factors RBFOX1/2. RNA 18: 274–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J. 2001. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20: 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland LC, Wang K, Robinson AG. 2010. RBM5 as a putative tumor suppressor gene for lung cancer. J Thorac Oncol 5: 294–298. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Nishimura J, Kagawa Y, Kano Y, Takahashi Y, Wu X, Hiraki M, Hamabe A, Konno M, Haraguchi N, et al. 2015. Significance of polypyrimidine tract-binding protein 1 expression in colorectal cancer. Mol Cancer Ther 14: 1705–1716. [DOI] [PubMed] [Google Scholar]
- Takita J, Yoshida K, Sanada M, Nishimura R, Okubo J, Motomura A, Hiwatari M, Oki K, Igarashi T, Hayashi Y, et al. 2012. Novel splicing-factor mutations in juvenile myelomonocytic leukemia. Leukemia 26: 1879–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tazi J, Kornstadt U, Rossi F, Jeanteur P, Cathala G, Brunel C, Luhrmann R. 1993. Thiophosphorylation of U1-70K protein inhibits pre-mRNA splicing. Nature 363: 283–286. [DOI] [PubMed] [Google Scholar]
- Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. 2012. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 119: 3578–3584. [DOI] [PubMed] [Google Scholar]
- Turunen JJ, Niemela EH, Verma B, Frilander MJ. 2013. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 4: 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, Ghigna C, Biamonti G. 2010. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 191: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, Durand M, Couture S, Froehlich U, Lapointe E, et al. 2009. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16: 670–676. [DOI] [PubMed] [Google Scholar]
- Venables JP, Brosseau JP, Gadea G, Klinck R, Prinos P, Beaulieu JF, Lapointe E, Durand M, Thibault P, Tremblay K, et al. 2013. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol Cell Biol 33: 396–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verduci L, Simili M, Rizzo M, Mercatanti A, Evangelista M, Mariani L, Rainaldi G, Pitto L. 2010. MicroRNA (miRNA)-mediated interaction between leukemia/lymphoma-related factor (LRF) and alternative splicing factor/splicing factor 2 (ASF/SF2) affects mouse embryonic fibroblast senescence and apoptosis. J Biol Chem 285: 39551–39563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconte V, Makishima H, Jankowska A, Szpurka H, Traina F, Jerez A, O'Keefe C, Rogers HJ, Sekeres MA, Maciejewski JP, et al. 2012. SF3B1, a splicing factor is frequently mutated in refractory anemia with ring sideroblasts. Leukemia 26: 542–545. [DOI] [PubMed] [Google Scholar]
- Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, et al. 2013. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 27: 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Lin W, Dyck JA, Yeakley JM, Songyang Z, Cantley LC, Fu XD. 1998. SRPK2: a differentially expressed SR protein-specific kinase involved in mediating the interaction and localization of pre-mRNA splicing factors in mammalian cells. J Cell Biol 140: 737–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Norton JT, Ghosh S, Kim J, Fushimi K, Wu JY, Stack MS, Huang S. 2008. Polypyrimidine tract-binding protein (PTB) differentially affects malignancy in a cell line-dependent manner. J Biol Chem 283: 20277–20287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. 2011. SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N Engl J Med 365: 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Chatterjee D, Jeon HY, Akerman M, Vander Heiden MG, Cantley LC, Krainer AR. 2012a. Exon-centric regulation of pyruvate kinase M alternative splicing via mutually exclusive exons. J Mol Cell Biol 4: 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Jeon HY, Rigo F, Bennett CF, Krainer AR. 2012b. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol 2: 120133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. 2009a. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Shen S, Xing Y, Carstens RP. 2009b. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6: 546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watermann DO, Tang Y, Zur Hausen A, Jager M, Stamm S, Stickeler E. 2006. Splicing factor Tra2-β1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res 66: 4774–4780. [DOI] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Mele A, Yan Q, Sun S, Farny N, Zhang Z, Xue C, Herre M, Silver PA, Zhang MQ, et al. 2014. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep 6: 1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Sun S, Tu K, Gao Y, Xie B, Krainer AR, Zhu J. 2010. A splicing-independent function of SF2/ASF in microRNA processing. Mol Cell 38: 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao SH, Manley JL. 1997. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev 11: 334–344. [DOI] [PubMed] [Google Scholar]
- Xu X, Yang D, Ding JH, Wang W, Chu PH, Dalton ND, Wang HY, Bermingham JR Jr, Ye Z, Liu F, et al. 2005. ASF/SF2-regulated CaMKIIδ alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 120: 59–72. [DOI] [PubMed] [Google Scholar]
- Xu Y, Gao XD, Lee JH, Huang H, Tan H, Ahn J, Reinke LM, Peter ME, Feng Y, Gius D, et al. 2014. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev 28: 1191–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H, et al. 2012. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 3: 883. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Ogawa S. 2014. Splicing factor mutations and cancer. Wiley Interdiscip Rev RNA 5: 445–459. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kataoka N, Miyauchi K, Ohe K, Iida K, Yoshida S, Nojima T, Okuno Y, Onogi H, Usui T, et al. 2015. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc Natl Acad Sci 112: 2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Li R, Shao C, Li P, Liu J, Wang K. 2012. 3p21.3 tumor suppressor gene RBM5 inhibits growth of human prostate cancer PC-3 cells through apoptosis. World J Surg Oncol 10: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhang G, Wei M, Lu X, Fu H, Feng F, Wang S, Lu W, Wu N, Lu Z, et al. 2014. The tumor suppressing effects of QKI-5 in prostate cancer: a novel diagnostic and prognostic protein. Cancer Biol Ther 15: 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Allred DC, Avis I, Martinez A, Vos MD, Smith L, Treston AM, Mulshine JL. 2001a. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res Treat 66: 217–224. [DOI] [PubMed] [Google Scholar]
- Zhou J, Nong L, Wloch M, Cantor A, Mulshine JL, Tockman MS. 2001b. Expression of early lung cancer detection marker: hnRNP-A2/B1 and its relation to microsatellite alteration in non-small cell lung cancer. Lung Cancer 34: 341–350. [DOI] [PubMed] [Google Scholar]
- Zhou R, Shanas R, Nelson MA, Bhattacharyya A, Shi J. 2010. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int J Cancer 126: 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Li X, Cheng Y, Wu W, Xie Z, Xi Q, Han J, Wu G, Fang J, Feng Y. 2014. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat Commun 5: 4581. [DOI] [PubMed] [Google Scholar]
- Zong FY, Fu X, Wei WJ, Luo YG, Heiner M, Cao LJ, Fang Z, Fang R, Lu D, Ji H, et al. 2014. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet 10: e1004289. [DOI] [PMC free article] [PubMed] [Google Scholar]