

Abstract
Retinal degeneration 3 (RD3) protein, essential for normal expression of retinal membrane guanylyl cyclase (RetGC) in photoreceptor cells, blocks RetGC catalytic activity and stimulation by guanylyl cyclase-activating proteins (GCAPs). In a mouse retina, RD3 inhibited both RetGC1 and RetGC2 isozymes. Photoreceptors in the rd3/rd3 mouse retinas lacking functional RD3 degenerated more severely than in the retinas lacking both RetGC isozymes, consistent with a hypothesis that the inhibitory activity of RD3 has a functional role in photoreceptors. To map the potential target-binding site(s) on RD3, short evolutionary conserved regions of its primary structure were scrambled and the mutations were tested for the RD3 ability to inhibit RetGC1 and co-localize with the cyclase in co-transfected cells. Substitutions in 4 out of 22 tested regions, 87KIHP90, 93CGPAI97, 99RFRQ102, and 119RSVL122, reduced the RD3 apparent affinity for the cyclase 180–700-fold. Changes of amino acid sequences outside the Lys87–Leu122 central portion of the molecule either failed to prevent RD3 binding to the cyclase or had a much smaller effect. Mutations in the 93CGPAI97 portion of a predicted central α-helix most drastically suppressed the inhibitory activity of RD3 and disrupted RD3 co-localization with RetGC1 in HEK293 cells. Different side chains replacing Cys93 profoundly reduced RD3 affinity for the cyclase, irrespective of their relative helix propensities. We conclude that the main RetGC-binding interface on RD3 required for the negative regulation of the cyclase localizes to the Lys87–Leu122 region.
Keywords: guanylate cyclase (guanylyl cyclase), photoreceptor, phototransduction, retina, signal transduction, calcium-binding proteins, eye, cyclic GMP (cGMP), neurobiology
Introduction
Retinal membrane guanylyl cyclase (RetGC), 2 essential for the normal function of vertebrate photoreceptors, interacts with the following two types of regulatory proteins: (i) Ca2+ and Mg2+ sensors, guanylyl cyclase-activating proteins (GCAPs), which inhibit the cyclase at high Ca2+ levels in the dark and activate RetGC in response to illumination, when Ca2+ levels fall (reviewed in Ref. 1) and (ii) RD3 (retinal degeneration 3) protein (2), which inhibits RetGC and is required for normal accumulation of the cyclase in rod and cone outer segments (3–5). Although the lack of GCAPs alters the kinetics and light sensitivity of photoresponse without destroying photoreceptors (6, 7), the RD3 deficiency causes retinal degeneration in rd3/rd3 mouse strain and in human patients affected by congenital blindness (2). Photoreceptors lacking RD3 fail to produce normal levels of RetGC1 and RetGC2 isozymes and to accumulate them in the outer segment, indicating that binding of RD3 by the cyclase provides proper stability and/or trafficking of the cyclase (3, 5). We previously reported that RD3 is a very potent high-affinity inhibitor of the cyclase catalytic activity as well as its activation by GCAPs in vitro (4). Although the details of the mechanism through which GCAPs bind and regulate RetGC have gradually become understood, the function of RD3 as a negative regulator of the photoreceptor cyclase, as well as its physiological role, remains less clear. Adding even more importance to that subject, some mutations in RetGC1 that cause Leber congenital amaurosis, a severe from of an early-onset blindness, interfere with the cyclase binding of GCAPs and RD3 altogether (4, 5, 8, 9). We hypothesized that blocking the cyclase activity by high-affinity RD3 binding could prevent premature activation of RetGC in the inner segment of the photoreceptor, before it reaches its proper destination in the outer segment (4). Neither the structure of RD3 nor the molecular mechanisms of its binding to the target enzyme are presently understood. Therefore, the purpose of this study was to further elucidate the RD3 function as a negative regulator of the cyclase and to identify the parts of the RD3 molecule that form its RetGC-binding interface. We find that the main part of RetGC-binding interface occupies helical regions in the central portion of the RD3 molecule.
Results
RD3 Inhibits RetGC1 and RetGC2 Isozymes in Mouse Retinas
We previously demonstrated (4) that RD3 inhibited activity of recombinant RetGC1 in vitro and also strongly diminished the total RetGC activity in the retina. There are two isozymes of the cyclase, RetGC1 and RetGC2 (10–12), expressed in the retina, of which the latter accounts for one-fourth to one-third of the total GCAP-stimulated activity (13). Therefore, we tested whether or not both isozymes present in the native mouse photoreceptor membrane were sensitive to inhibition by RD3. To segregate the individual RetGC1 and RetGC2 isozyme activities, we used rod outer segment (ROS) fractions isolated from retinas lacking genes coding for GCAP1, GCAP2, and one of the RetGC isozymes: RetGC2−/− GCAPs−/− and RetGC1−/−GCAPs−/− (13), respectively. We found that mouse RD3 effectively suppressed basal catalytic activity of both native RetGC isozymes in the absence of GCAPs (Fig. 1, A and B). Human RD3 has been shown to prevent activation of a recombinant human RetGC1 by GCAP1 and GCAP2, by effectively competing with GCAPs (4). We found that mouse RD3 also suppressed activation of the native mouse RetGC isozymes by mouse GCAPs (Fig. 1, C and D) in GCAPs−/− ROS. What is more, RD3 effectively competed with the native mouse cyclase activation by GCAP1 or GCAP2 when its concentrations in the assay were ∼100-fold lower than GCAPs.
FIGURE 1.

RD3 binds both native isozymes of RetGC in mouse retinas and prevents mouse RetGC activation by GCAP1 and GCAP2. A and B, basal activities of RetGC1 (A) and RetGC2 (B) in ROS fractions isolated from their respective RetGC2−/−GCAPs−/− and RetGC1−/−GCAPs−/− retinas were assayed in the presence of recombinant mouse RD3 purified from E. coli; the data were fitted using the Synergy KaleidaGraph 4 software assuming the Hill function, a = a0/(1 + ([RD3]/IC50)h), where a is the activity of RetGC, [RD3] is the concentration of RD3, IC50 is the RD3 concentration that inhibited activation to half-maximal, and h is the Hill coefficient; the IC50 and the Hill coefficient values from the fit were 10 ± 2 nm and 0.78 ± 0.01 (n = 4) (RetGC2−/−GCAPs−/−), and 17 ± 3 and 0.8 ± 0.08 (n = 3) (RetGC1−/−GCAPs−/−). C and D, ROS fraction isolated from the GCAPs−/− knock-out mice was assayed in the presence of the indicated concentrations of recombinant mouse GCAP1 (C) or GCAP2 (D) in the absence (○,●) or in the presence of 15 nm (▵, ▴) or 45 nm (□, ■) mouse RD3; data are fitted assuming Michaelis-Menten hyperbolic function. Here and further, the data are mean average ± S.E. unless specified otherwise. See “Experimental Procedures” for other details.
Photoreceptor Degeneration in rd3/rd3 versus RetGC1−/− RetGC2−/− Mice
The lack of RD3, which diminishes the content of RetGCs in the outer segments (3, 5), strongly reduced the overall activity of the cyclase detectable in young rd3/rd3 mice (Fig. 2). Normalized per the photoreceptor content at 1 month of age (158 ± 15 nuclei count per 100 μm of the rd3/rd3 retina section, n = 8, versus 227 ± 14 in wild type, n = 6), the RetGC activity was 10-fold reduced in rd3/rd3 retinas at that age (0.07 ± 0.01 versus 0.68 ± 0.035 nmol of cGMP/min/retina in wild type, Fig. 2A). However, it was far from being completely absent. Moreover, consistent with the original observation by Friedman et al. (2), scotopic electroretinography (ERG) a- and b-wave amplitudes in young rd3/rd3 mice that we tested in this study were strongly reduced when compared with the wild type, but remained clearly detectable (Fig. 2B). In contrast, the double RetGC knock-out mice (indicated by GCdKO), which, despite only a minor loss of photoreceptor nuclei count at that age (202 ± 13 per 100 μm, n = 4), completely lacked detectable RetGC activity (Fig. 2A) and, as originally documented by Baehr et al. (14), were non-responsive even to the strongest light stimulus. In rd3/rd3, the average a-wave amplitude at 1 month of age was 105 ± 14 μV (n = 5), when compared with 454 ± 41 μV (n = 6) in wild type (p < 0.0001, unpaired t test), and the respective average b-wave amplitudes were 445 ± 59 and 886 ± 91 μV (p = 0.0037).
FIGURE 2.

Low RetGC content does not fully explain the severity of retinal degeneration in rd3/rd3 mice. A, RetGC activity is undetectable in RetGC1−/−RetGC2−/− double knock-out (indicated by GCdKO) and strongly reduced, but not completely eliminated in rd3/rd3 retinas (the activities are corrected for 30% photoreceptor loss at 1 month of age in rd3/rd3 and 12% loss in RetGC1−/−RetGC2−/−). Retinal homogenates from dark-adapted mice were assayed under infrared illumination as described under “Experimental Procedures.” B and C, scotopic ERG responses. B, representative individual traces for wild type, RetGC1−/−RetGC2−/− double knock-out (indicated by GCdKO), and rd3/rd3 mice; the arrow marks the delivery of the saturating 1-ms flash. C, the a-wave and b-wave variability at 1 month of age among wild type (open black circles), rd3/rd3 (closed red circles), and RetGC1−/−RetGC2−/− (closed black circles) mice. Horizontal bar, average; vertical bar, S.D. The average a-wave amplitude, mean ± S.E., was 454 ± 41 μV (n = 6) in wild type and 105 ± 14 μV (n = 5) in rd3/rd3 mice, and their respective average b-wave amplitudes were 886 ± 91 and 445 ± 59 μV. There were no detectable responses in five tested RetGC1−/−RetGC2−/− mice. D, photoreceptors degenerate more severely in rd3/rd3 than in RetGC1−/−RetGC2−/− mice. Top row, left to right, 5-μm sections of glutaraldehyde-fixed retinas from wild type, RetGC1−/−RetGC2−/−, and rd3/rd3 mice aged 5 months; note the difference in the thickness of the outer nuclear layer (red parenthesis) between the different lines. Bottom row, the same retina sections shown under higher magnification demonstrate that short outer segments were present in the remaining RetGC1−/−RetGC2−/− but not rd3/rd3 photoreceptors. Marked histological layers of the retina are: rpe, retinal pigment epithelium; ros, rod outer segments; onl, outer nuclear layer; opl, outer plexiform layer; inl, inner nuclear layer; ipl, inner plexiform layer; gcl, ganglion cell layer. Bar length, 50 μm. E, photoreceptor nuclei count in outer nuclear layer per 100-μm distance in a mid-portion, between the optic nerve and the periphery of the retina; error bar indicates S.D. Average photoreceptor nuclei counts in wild type (open black circles), RetGC1−/−RetGC2−/− (closed black circles), and rd3/rd3 (closed red circles) mice after 5 months of age were 237 ± 11 (n = 4), 113 ± 6 (n = 5), and 33 ± 6 (n = 5), respectively; the difference between all pairs was statistically significant (p < 0.0001 at 99.9% confidence level, analysis of variance/Bonferroni post hoc).
We hence reasoned that if the lower levels of RetGC in outer segments (3) were the sole cause for the rods and cones to degenerate in rd3/rd3 mice, then the deletion of both RetGC1 and RetGC2 genes, which completely eliminated the cyclase activity and the ERG light responses altogether (Fig. 2, A–C) (13, 14), would cause photoreceptors to degenerate even faster. However, a direct comparison of the retinal morphology between the rd3/rd3 and the RetGC1−/−RetGC2−/− mice at 5 months of age revealed just the opposite (Fig. 2D).
It is important to point out that genetic background can reportedly influence the rate of the rd3 retinal degeneration in different mouse strains (2). Therefore, to avoid ambiguity in making a direct comparison, in the present study, we first outcrossed rd3, RetGC1−/−, and RetGC2−/− lines to the C57B6J strain for 10 consecutive generations to make them conventionally congenic and only then bred them into their respective rd3/rd3 or RetGC1−/−RetGC2−/− homozygous states. The loss of photoreceptors in rd3/rd3 mice at 5 months was more pronounced than in the RetGC1−/−RetGC2−/− double knock-out (Fig. 2, D and E). The vast majority of the photoreceptor nuclei in the rd3/rd3 outer nuclear layer disappeared, leaving typically just one row of nuclei when compared with the 9–11 rows in the wild type (ERG also became undetectable in rd3/rd3 at that age (not shown)). In contrast, the retinas completely lacking the cyclase activity from birth retained as many as 4–6 nuclei rows, nearly half the normal photoreceptor layer thickness. The average number of photoreceptors in RetGC1−/−RetGC2−/− and rd3/rd3 retinas was respectively reduced to 48%, and 14% in normal retinas. Average photoreceptor nuclei counts in wild type, RetGC1−/−RetGC2−/−, and rd3/rd3 mice were 237 ± 11 (n = 4), 113 ± 6 (n = 5), and 33 ± 6 (n = 5), respectively; the difference between all pairs was significant (p < 0.0001 at 99.9% confidence level, analysis of variance/Bonferroni post hoc test).
The RetGC double knock-out retinas also had a thinner than normal yet clearly recognizable layer of outer segments, whereas none could be identified at that age in the rudiments of the rd3/rd3 photoreceptor cell layer (Fig. 2D, bottom row). The outer nuclear layer in several tested RetGC1−/−RetGC2−/− retinas still retained >40% normal nuclei count per 100 μm (110 ± 2, n = 4) even after 6 months of age, whereas only sporadic nuclei that possibly belonged to photoreceptors could be encountered in rd3/rd3 (7 ± 1, n = 3) (not shown). Evidently, these results argued that lower levels of RetGC expression could not fully explain the fast photoreceptor degeneration in rd3/rd3 mice and pointed at the lack of another RD3 function, its being a potent inhibitor of the cyclase (Fig. 1) (4), as a likely factor that contributed to the pace of degeneration.
Effect of RD3 Mutations on RetGC1 Inhibition
The structure of RD3 remains unknown, largely due to its previously noted poor solubility (4, 9), which hampers direct structural studies, and poor homology to proteins with known structures for three-dimensional modeling. Therefore, here we only refer to the predicted RD3 secondary structure (8) (Fig. 3).
FIGURE 3.

The primary structure of a human RD3 aligned with the mouse, bovine, and sheep protein sequences. Positions of the identical residues are marked in yellow, the H1–H4 regions are predicted α-helices, the boxed region is a predicted coiled-coil (after Molday et al. (8)), and the line marks regions lacking a defined secondary structure. The regions where scrambling of the peptide sequences reduced RetGC1 inhibition by RD3 by ≤50% (see Fig. 4) are highlighted in blue; those where the mutation reduced the extent of inhibition by ≥80% are highlighted in red.
We reasoned that the RetGC-binding interface on RD3 would likely include hydrophilic amino acid residues that are more likely to be exposed on the surface of the molecule and are most conserved among different animal species (Fig. 3). We hence selected 22 short peptide regions in a human RD3 primary structure containing hydrophilic residues, and then scrambled their sequences, either by simply changing the order of the same residues in the region or by additionally replacing some of the hydrophilic side chains in the sequence, such as positively charged to neutral or negatively charged or vice versa (Table 1). We chose to avoid changing strongly conserved extended hydrophobic regions to minimize the potential effects of changing parts of a hydrophobic core on the backbone fold. When we replaced hydrophobic residues in the mutated fragments, we used homologous replacements, such as Ile to Leu, Val or Ala. We then screened purified RD3 mutants for a drastic change in their ability to suppress RetGC1 activity at 0.1 μm RD3, concentration at which wild type RD3 suppressed GCAP1-activated RetGC1 10-fold (Fig. 4A). Among the 22 tested regions, mutations in four of them, 87KIHP90, 93CGPAI97, 99RFRQ102, and 119RSVL122, strongly negated the RD3 inhibitory effect: only ≤20% of the RetGC1 activity was lost when compared with the >90% loss in case of wild type RD3 (Table 1, Fig. 4B). One of these four regions, 93CGPAI97, was particularly sensitive to the change of its amino acid sequence.
TABLE 1.
Fractional RetGC1 activity in the presence of 0.1 μm RD3
The activity was assayed as described under “Experimental Procedures.” The fractional activity was defined as ARD3/A0, where A0 is the RetGC1 activity in the absence of RD3 in the standard assay and ARD3 is the RetGC1 activity in the presence of 0.1 μm RD3.
| Fragment | Mutation | Fractional RetGC1 activity |
|---|---|---|
| 21PAEM24 | APME | 0.21 ± 0.02 |
| 27ETLM30 | QMVT | 0.51 ± 0.02 |
| 35GQMR38 | ERLG | 0.19 ± 0.01 |
| 41ERQQ44 | KREQ | 0.22 ± 0.01 |
| 50AVRK53 | KAIE | 0.13 ± 0.01 |
| 55CTGVD59 | GNCDV | 0.34 ± 0.02 |
| 77ERLQ80 | QLRE | 0.12 ± 0.01 |
| 82EDVC85 | QVDS | 0.39 ± 0.01 |
| 87KIHP90 | PHRL | 0.86 ± 0.01a |
| 93CGPAI97 | PALSL | 0.93 ± 0.02a |
| 99RFRQ102 | FRQR | 0.87 ± 0.08a |
| 107QEPEV111 | PVQEE | 0.40 ± 0.01 |
| 112QEV114 | EVQ | 0.35 ± 0.01 |
| 119RSVL122 | LAER | 0.80 ± 0.01a |
| Met129 | D | 0.11 ± 0.00 |
| 132EEE134 | KKK | 0.12 ± 0.01 |
| 137KLTRQ141 | QRKTL | 0.09 ± 0.01 |
| 147RGSL150 | GRLS | 0.10 ± 0.01 |
| 154KTRAR158 | ENSES | 0.08 ± 0.01 |
| 164SDIRT168 | RSDTI | 0.10 ± 0.01 |
| 170EDVERD175 | KKAKQR | 0.05 ± 0.03 |
| 178PPP180 | AAA | 0.10 ± 0.03 |
| 186SMP188 | PRS | 0.13 ± 0.00 |
| Wild type | 0.08 ± 0.01 |
a RD3 mutants for which ARD3 ≥0.8 A0 in the standard assay conditions.
FIGURE 4.

Inhibition of a GCAP1-stimulated recombinant human RetGC1 by recombinant human RD3 and its mutants. A, membrane fraction from the RetGC1-expressing HEK293 cells was assayed in the presence of 1.5 μm GCAP1 and 1 mm EGTA as described under “Experimental Procedures.” The 0.1 μm concentration, at which wild type RD3 reduced RetGC1 activity in the standard assay nearly 10-fold, was used to compare the effect of RD3 mutants in Table 1. The data were fitted using the Synergy KaleidaGraph 4 software assuming the Hill function, a = 100 × (1 + ([RD3]/(K½)h)−1, where a is the activity of RetGC (% of maximal), [RD3] is the concentration of RD3, K½ is the RD3 concentration that inhibited activation to half-maximal, and h is the Hill coefficient; the K½ and the Hill coefficient from the fit were 3.3 nm and 0.66, respectively. The averaged data are mean average ± S.E. B, inhibition of RetGC1 by 0.1 μm RD3 mutants. The percentage of inhibition equals 100% × (1 − ARD3/A0), where ARD3/A0 is the fractional RetGC1 activity (see Table 1). The dashed line marks the level of inhibition by the wild type. Peptide sequences 87KIHP90, 93CGPAI97, 99RFRQ102, and 119RSVL122, where mutations reduced the inhibition by ≥80%, are marked in red.
The Affinity for RetGC1 Is Strongly Reduced by Mutations in the Central Region of RD3
The four most strongly affected RD3 mutants were tested for their dose dependence of RetGC1 inhibition (Fig. 5). The mutants were able to partially reduce RetGC1 activation at high concentrations, but when compared with the wild type, their apparent affinities became reduced 18–700-fold. Hence, the loss of the inhibitory activity in Fig. 4B and Table 1 was due to a dramatic reduction in the RD3 binding affinity for the target enzyme.
FIGURE 5.

Dose dependence of RetGC1 inhibition by wild type RD3 (●) and its mutants. RetGC1 in the assay was activated by 1.5 μm GCAP1 in the presence of 1 mm EGTA and assayed as described under “Experimental Procedures.” The data were fitted by Hill function using the Synergy KaleidaGraph 4 software as described in the legend for Fig. 4; the respective K½ values from the fit at R > 0.99 were 3.3 ± 0.3, 597 ± 44, 2440 ± 208, 1241 ± 105, and 918 ± 47 nm. The symbols are: ■, 87KIHP90 → PHRL; □, 93CGPAI97 → PALSL; ▴, 99RFRQ102 → FRQR; and (○), 119RSVL122 → LAER. The averaged data are mean average ± S.E.
To further verify that the binding to the target enzyme became affected, we tested the RD3 binding to RetGC1 using the in cyto method, in which GFP-tagged RD3 and mOrange-tagged RetGC1 were co-expressed in HEK293 cells (9, 15, 16). At high levels of expression, RD3 tends to precipitate inside the cells (9, 16), but when expressed at low levels in the absence of the target enzyme, it spreads mostly uniformly throughout the cytoplasm and the nucleus (9, 16) (Fig. 6A). In the presence of RetGC1, it becomes anchored to the endoplasmic reticulum and plasma membrane of HEK293 cells (Fig. 6B) via docking with the target enzyme (9, 16). Considering that the affinity of RD3 for the cyclase is 2–3 orders of magnitude greater than that of GCAPs (Fig. 1) (4), one could expect than even an ∼100-fold decrease in the affinity would not necessarily disrupt the co-localization pattern, because the affinity of the mutated RD3 could just be shifted to a range, where GCAPs, which bind the cyclase at the μm range (13), effectively co-localize with RetGC1 in cyto (9, 15, 16). However, more than 700-fold reduction in RD3 affinity for the cyclase after scrambling its short central 93CGPAI97 fragment was strong enough to completely prevent co-localization with the cyclase in cyto (Fig. 6D). The Pearson's correlation coefficient values averaged from multiple whole-cell images (Fig. 6, B–D) were 0.47 for the 93CGPAI97 → PALSL RD3, 0.94 for the wild type, and 0.92 for the 27ETLM30 → QMTV mutant (note that the Pearson's correlation coefficient values ≤0.5 indicate a complete lack of co-localization (17)).
FIGURE 6.
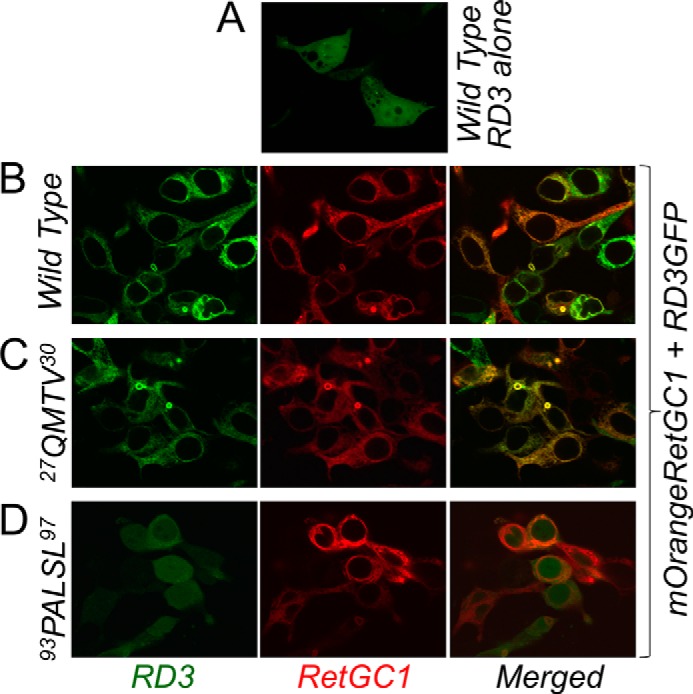
The effect of mutation on RD3 and RetGC1 co-localization in cyto. A, wild type RD3-GFP expressed in the presence of a plasmid not coding for RetGC1. B–D, mOrange-RetGC1 co-expressed with the wild type (B), 27ETLM30 → QMTV (C), or 93CGPAI97 → PALSL RD3. Note the lack of co-localization pattern in D. RD3-GFP and mOrange-RetGC1 were expressed in HEK293 cells as described under “Experimental Procedures.” Among the respective Pearson's correlation coefficient values for the wild type (0.94 ± 0.005, n = 47), 27ETLM30 → QMTV (0.92 ± 0.006, n = 40), and 93CGPAI97 → PALSL RD3 (0.47 ± 0.04, n = 26), the difference between the 93CGPAI97 → PALSL mutant and both the wild type and the 27ETLM30 → QMTV RD3 was statistically significant (p < 0.0001 at 99.9% confidence level).
Substitution of Cys93 Disrupts RetGC-binding Interface on RD3
The 93CGPAI97 sequence, which demonstrated the strongest reduction in cyclase binding activity among the mutated regions, is a part of a predicted α-helical H3 region (8) in RD3 (Fig. 3). The effects of the 93CGPAI97 → PALSL scrambling could reflect not only the change in a specific side chain composition in the RetGC-binding interface, but the disruption of the predicted H3 helix. Therefore, we further tested several individual amino acid substitutions in that region by taking into consideration their potential effects on the α-helical structure (18). Replacement of two residues that could have had a pronounced effect on the secondary structure, G94A and P95L, did not or only slightly affected the dose dependence of RetGC1 inhibition, whereas the C93P substitution reduced RD3 affinity for the cyclase almost 80-fold (Fig. 7A). Because Pro frequently disrupts protein secondary structure, we additionally substituted Cys93 of the wild type RD3 with those residues (Arg, Asp, Ser) that favor preservation of the α-helical structure better than Pro (18) (Fig. 7B). The apparent affinity in all those mutants was reduced 13–700-fold, but the extent of the affinity loss did not correlate with the relative helix propensity of the residues, Arg > Ser > Asp > Pro (18): the respective K½ values for the C93R, C93S, C93D, and C93P mutants were 399, 40, 2313, and 242 nm. Evidently, the absence of the sulfhydryl group per se was less critical than other properties of the side chain.
FIGURE 7.

Altering residues in the central H3 helix strongly weakens RD3 affinity for RetGC1. A and B, the effect of amino acid substitutions on dose dependence of RetGC1 inhibition. A, wild type RD3 (●), 93CGPAI97 → PALSL (○), C93P (▿), G94A (▾), and P95L (▴) B, effects of Cys93 substitution do not correlate with the helix propensity of the substituting residues: wild type RD3 (●), C93S (▾), C93R (♦), and C93D (♢). RetGC1 was assayed, and data were fitted as described in the legend for Fig. 2. The respective K½ values from the fit for the wild type, C93R, C93S, C93D, and C93P mutants were 3.3 ± 0.3, 399 ± 53, 40 ± 7, 2313 ± 83, and 242 ± 16 nm; the respective Hill coefficient values were 0.66 ± 0.03, 0.6 ± 0.02, 0.69 ± 0.02, and 0.5 ± 0.03. C, alkylation of Cys93 with NEM hampers RD3 binding to RetGC1. The symbols indicate dose dependence of RetGC1 inhibition in the presence of C55D/C85T RD3 mutant (□,▵) harboring a single Cys93, before (□) and after (▵) alkylation with NEM (the respective K½ values from the fit were 13.6 ± 2.3 and 298 ± 19 nm); ● indicates wild type. The averaged data are mean average ± S.E.
Elimination of Cys55 and Cys85 in C55D/C85T RD3 had little effect (K½ ∼14 nm), but alkylation of the remaining Cys93 in the C55D/C85T RD3 using N-ethylmaleimide (NEM) demonstrated nearly 100-fold reduced RD3 affinity for RetGC1 (K½ ∼0.3 μm) when compared with the wild type (Fig. 7C). The role of Cys93 was unlikely related to its potentially forming a disulfide bond, because (i) RetGC activity assays were conducted in the presence of dithiothreitol (DTT), (ii) substitution with Ser or Pro, although it profoundly hampered the inhibitory capacity of RD3, was less pronounced than replacement with Arg or Asp (Fig. 7, A and B); and iii) reduction of activity in C55D/C85T when compared with the wild type RD3 was minor (Fig. 7C).
Discussion
The important role of the GCAP-dependent calcium feedback regulation of RetGC in photoreceptor physiology (6–7, 10–12, 19–25) and abnormalities in the RetGC/GCAP regulation that cause multiple forms of congenital blindness (26–40) have been documented over the past two decades. At the same time, the role of RD3 (2), the protein implicated in trafficking (3, 5, 8) and more recently also identified as a strong negative regulator for the retinal guanylyl cyclase (4), remains much less understood.
Truncation of RD3 causes a recessive blindness accompanied by severe photoreceptor degeneration in rd3/rd3 mice and in human patients (2). One of the reasons why the lack of RD3 causes blindness is a strong reduction of RetGC1 and RetGC2 content in photoreceptor outer segments (3, 41), possibly as a result of RetGC trafficking being impaired in the absence of RD3 (5, 8). However, our present results argue that the severity of retinal degeneration associated with the lack of RD3 can hardly be explained by just the reduction in the cyclase content: even after a complete deletion of both RetGC1−/− and RetGC2−/−, photoreceptors degenerate less severely than in rd3/rd3 mice (Fig. 2). Interestingly, such a rudimentary rod ERG response as in rd3/rd3 is also typical for RetGC1−/− mice (42) and some human RetGC1 mutations causing Leber congenital amaurosis (28), but in neither of these cases do rods undergo massive degeneration.
The preceding study (4) gave rise to a hypothesis that blocking RetGC activity and/or shielding the cyclase from the GCAP-dependent activation occurs before the cyclase reaches its proper destination in the outer segment. The physiological data in Fig. 2 do not directly prove this hypothesis but further support such a possibility. We reason that the additional function of RD3 that improves the survival of the photoreceptors could be its ability to block RetGC activity/activation by GCAPs. That would presumably occur in the inner segment (4, 8), because RD3 does not effectively accumulate in the outer segments with the normal GCAPs content (5).
Indeed, RD3 strongly inhibits native RetGC1 and RetGC2 isozymes in the photoreceptor membranes at nm range (Fig. 1). This observation validates the results of our previous study utilizing a recombinant human RetGC1 (4) and confirms that RD3 is a potent inhibitor of RetGC whose affinity for the cyclase at least 100-fold exceeds that of GCAPs (4). Not only does RD3 directly bind to the cyclase (Fig. 7) (3, 15, 16), but its inhibitory action also results from the direct binding to the cyclase rather than GCAPs, because RD3 suppresses RetGC basal activity in the absence of GCAPs and also because it prevents activation of the cyclase in the presence of ∼100-fold molar excess of GCAPs (Fig. 1) (4).
To further understand the role of RD3/RetGC1 interactions in photoreceptor physiology, it is important to identify the part of the molecule that creates an interface for binding with RetGC and thus blocks the cyclase activity. The actual RD3 protein fold is presently unknown, first and foremost, because (i) RD3 content in the retina is very low (3) and (ii) when expressed as recombinant protein, it precipitates at the concentrations required for structural analysis (4, 16). However, functional assays, which use submicromolar concentrations of the soluble RD3 that are insufficient for structural studies, make it possible to test RD3 binding to RetGC based on the cyclase inhibition (Fig. 1) (4).
Dose dependence of RetGC inhibition by RD3 yields the Hill coefficients <1 (Figs. 1, 4, and 7). The negative cooperativity could indicate that more than one RD3 molecule binds to the catalytically active homodimer (43) of the cyclase and that the affinity of RD3 for RetGC1 decreases when another RD3 molecule is already bound to the cyclase. A more trivial explanation, however, is that the low Hill value reflects the tendency of recombinant RD3 to aggregate as the concentrations rise in the assay. This ambiguity can be resolved only when and if RD3 becomes available in a form that does not tend to aggregate at high concentrations.
Taken together, the results of this study indicate that the Lys87–Leu122 central part of the molecule (Fig. 3) plays the most critical role in forming the RetGC-binding interface (Figs. 5–7). This part includes the unstructured region 87KIHP90 between helixes H2 and H3, 93CGPAI97 and 99RFRQ102 in helix H3, and 119RSVL122 in the middle of the helix H4. Importantly, several different hydrophilic residues substituting a single Cys93 in the 93CGPAI97 fragment incapacitate RetGC1 binding irrespective of their relative helix-forming propensities (Fig. 7). Therefore, the role of the 93CGPAI97 is not limited to its forming the proper secondary structure, but rather involves either a direct contact with the cyclase or maintaining the proper conformation of an immediately adjacent region where such a contact is made. Which residues in the four central peptides directly contact RetGC and which of them are required for maintaining the proper conformation of the interface structure cannot be drawn from our results and will require further detailed structural studies.
In contrast to the central part of RD3, scrambling amino acid sequence in most of the N-terminal and C-terminal portions of the molecule caused surprisingly little effect. This includes the N-terminal helix H1, predicted to form a coiled-coil structure (8) (Fig. 3). A relatively modest 50% loss of the inhibitory effect at 0.1 μm RD3 occurred after scrambling the 27ETLM30 sequence located at the beginning of the coiled-coil motif (Fig. 4, Table 1). Other mutations in H1 helix had even less of an effect (Fig. 4, Table 1). Although those parts of the RD3 primary structure are less critical for the ability of RD3 to bind to the cyclase, we cannot exclude that the H1 helix and/or the long unstructured C-terminal tail instead contribute to the proposed RD3 function in RetGC1 trafficking (5, 8).
Recessive mutations in a mouse rd3 or a human RD3 gene that cause retinal degeneration and Leber congenital amaurosis truncate the protein in the middle of the putative RetGC1-binding interface (2). Mutations in other parts of RD3 were also found in patients with retinal dysfunction, although their role in the pathology was not defined (2). Some of those mutations also reduce the apparent affinity of RD3 for RetGC1 in vitro (4), although much less dramatically than mutations in the Lys87–Leu122 region. Hence, although the ability to bind the target is most critically defined by the central region of the molecule, other parts of RD3 can to some extent contribute to the binding, for example, by maintaining the proper fold of the protein.
Another issue that currently remains unresolved is how GCAPs and RD3 compete with each other over the same target enzyme. Evidently, the strong competition between GCAPs and RD3 for RetGC observed in vitro (Fig. 1) (4) also occurs in living photoreceptors in vivo. Although RD3 is nearly undetectable in rod outer segments of wild type mice, it accumulates in GCAPs−/− outer segments (5). In our opinion, this effect can more likely be explained by mutually exclusive binding of GCAPs and RD3 (4), rather than their synergy in forming complex with RetGC (5). We reason that the lack of GCAPs promotes accumulation of RD3 in the outer segment by eliminating that competition.
The RD3-binding part in RetGC1 also remains to be determined. It evidently involves the C-terminal portion of RetGC1 (3), a part of the enzyme that has little role in RetGC regulation by GCAPs (9, 16). Conversely, a part of GCAP-binding interface on the cyclase involves the RetGC dimerization domain, which is not critical for RD3 binding (9, 16). Thus, the binding interfaces for GCAPs and RD3 on the cyclase are non-identical. However, they likely overlap in a folded RetGC and can even share some common portions of the cyclase primary structure, because a number of disease-causing mutations in a kinase homology domain of RetGC1 disable binding of GCAPs and RD3 alike (5, 9, 16, 44). Site-directed mutagenesis complemented by molecular modeling could further help clarify the mechanisms of RetGC interaction with its regulatory proteins in photoreceptors.
Experimental Procedures
Animals
All animal work was conducted in accordance with the Public Health Service guidelines and approved by the Institutional Animal Care and Use Committee. The wild type C57B6J and rd3/rd3 mouse strains were purchased from JAX Research/The Jackson Laboratory, the RetGC1−/− line (42) was kindly provided by Dr. David Garbers (University of Texas), the RetGC2−/− line (14) was kindly provided by Dr. Wolfgang Baehr (University of Utah), and the GCAPs−/− line lacking both GCAP1 and GCAP2 (6) was kindly provided by Dr. Jeannie Chen (University of South California). The rd3, RetGC1−/−, and RetGC2−/− lines were maintained heterozygous by crossing to the C57B6 for 10 generations. After making all the lines congenic to the C57B6 background, the homozygous rd3/rd3 and RetGC1−/−RetGC2−/− lines were bred and verified by PCR and DNA sequencing.
Electroretinography
Mice were dark-adapted overnight, their pupils were dilated by 1% Tropicamide and 2.5% phenylephrine ophthalmic eye drops under dim red light, and mice were dark-adapted for another 15 min prior to the recordings. Retinal responses to light were recorded using a Phoenix Research Laboratories Ganzfeld ERG-2 setup from mice anesthetized by isoflurane inhalation (1.8–1.9% at the flow rate of 50 ml/min in a Kent Scientific SomnoSuite Small Animal Anesthesia System). The recordings were conducted in the dark, with the equipment controlling anesthesia and the ERG setup shielded by dark red acrylic plastic. During the recordings, the mice remained on a heated pad to maintain body temperature. To evoke maximal scotopic ERG response, 1-ms 504-nm light pulses of 1.2 × 106 photons μm−2 at the mouse retinal surface were delivered through the infrared camera-guided corneal electrode/LED light source in 3-min intervals, and the responses from 3–4 flashes were averaged.
Retinal Morphology
Mice were injected with a lethal dose of ketamine/xylazine anesthetic, perfused through the heart first with phosphate-buffered saline (PBS) and then with 2.5% glutaraldehyde in PBS. The eyes were surgically removed and fixed overnight in 2.5% glutaraldehyde, 2.5% formaldehyde, PBS solution at 4 °C. The fixed eyes were washed three times for 15 min each in PBS, soaked in PBS overnight, processed for paraffin embedding, sectioned, and stained with hematoxylin/eosin dye (AML Laboratories, Saint Augustine, FL). The retinas were photographed using an Olympus Magnafire camera mounted on an Olympus BX21 microscope. The photoreceptor nuclei in the outer nuclear layer of the retina were counted from a 425-μm fragment of the retina between the optic nerve and the periphery of the retina, and the densities of the nuclei per 100 μm were averaged from several frames for each retina. The average nuclei counts were then typically compared using 4–6 retinas of different genotypes.
Isolation of Mouse Retinas and ROS Membranes
ROS were isolated from dark-adapted RetGC2−/−GCAPs−/−, RetGC1−/−GCAPs−/− or GCAPs−/− mouse retinas under infrared illumination using a series of OptiPrep (Sigma-Aldrich) density gradient centrifugations, and rhodopsin concentrations in the isolated ROS fractions were determined as described previously in full detail (13). Aliquots of the mouse ROS were wrapped in aluminum foil, frozen in liquid N2, and stored at −70 °C. Whole mouse retinas for RetGC activity measurements were excised from the dark-adapted 4-week-old wild type, rd3/rd3, and RetGC1−/−RetGC2−/− eyes as described (13), wrapped in aluminum foil, frozen in liquid N2, and stored at −70 °C.
GCAP and RetGC Expression
N-Myristoylated GCAP1 and GCAP2 were expressed from pET11d vector in BLR(DE3) Escherichia coli strain harboring N-myristoyl transferase, extracted from inclusion bodies, and purified to ∼95% homogeneity using hydrophobic and size-exclusion chromatography as described previously in detail (15, 45). The recombinant mouse and human RD3 were expressed from pET11d vector in a BL21(DE3)CodonPlus E. coli strain (Agilent Technologies), extracted from inclusion bodies, and purified as described previously (4). RetGC1 was expressed in HEK293 cells from an Invitrogen pRCCMV vector using the calcium phosphate DNA precipitation transfection protocol, and the membrane fraction containing the recombinant RetGC1 was isolated as described previously in detail (9, 45).
Human RD3 Mutagenesis
Mutations were introduced in a human RD3 cDNA using a conventional “splicing by overlap extension” technique (46), using the cDNA's 5′-end-coding and the 3′-end-coding primers, 5′-AAGGACCATGGCTCTCATCTCATGGCTTCGGTGGAACGAGAAATT and 5′-GGATCCTCAGTCGGCTTTGGGCGCCCGGAAT, respectively, and primers that contained the designed nucleotide substitutions and at the same time generated overlapping regions for the subsequent splicing (46). The primary fragments were amplified in a PCR reaction using a human cDNA as a template and utilizing a Thermo Scientific Phusion Flash high-fidelity DNA polymerase. The fragments were then spliced in a second round of PCR using only the 5′-end and the 3′-end primers shown above. The spliced DNA constructs were purified using a Zymo Research DNA clean kit, digested with NcoI and BamHI endonucleases (New England Biolabs), inserted into the NcoI/BamHI sites of the pET11d vector (Novagen/Calbiochem), and verified by the automated DNA sequencing of the entire RD3 inserts in the resultant plasmids. To use the NcoI site for ligating the 5′-end of the constructs into the expression vector, the N-terminal Ser2 was replaced by Ala in all bacterially expressed variants of RD3, a modification that does not interfere with the ability of RD3 to bind and inhibit RetGC (4). For expression of a human RD3 fused at the C terminus with SuperGlo eGFP tag (Clontech) in HEK293 cells, the cDNA was amplified using a forward primer, 5′-TTTTTTAAGCTTGGGCCAGGGGCTATGTCTCTCATCTCA, and a reverse primer, 5′-AGCGGCAATTGTGAGTCGGCTTTGGGCGCCCGGAAT, digested with HindIII and MfeI endonucleases (New England Biolabs), and subcloned into the HindIII/EcoRI sites of a pQBIFn3 vector (Clontech). The original N-terminal residue, Ser2, remained preserved in these constructs.
RD3 Expression and Purification
The recombinant mouse and human RD3 were expressed from pET11d (Novagen/Calbiochem) vector in BL21(DE3)CodonPlus E. coli strain (Agilent Technologies), extracted from inclusion bodies, and purified as described previously (4) with the following modifications. Inoculated with a frozen stock, 5-ml bacterial cultures in a standard Luria broth were grown overnight in an incubator shaker at 37 °C, 200 rpm (Thermo Fisher Scientific) and then in 100-ml culture to reach A600 0.6–0.7. The protein expression was induced by 1 mm isopropyl-β-d-1-thiogalactopyranoside for 2 h. The bacterial cell pellet was harvested by centrifugation at 7000 rpm for 10 min in a Sorvall Fiberlite 14-6 × 250y rotor at 4 °C and frozen in −70 °C. The pellet was thawed, resuspended in 20 ml of 10 mm Tris-HCl (pH 7.5), 1 mm EDTA, 14 mm 2-mercaptoethanol buffer solution (TEM), and sonicated on ice for 2 min using 2-s ultrasonic pulses. After chilling on ice, the sonication step was repeated and the inclusion bodies from the disrupted cells were collected by centrifugation at 22,000 × g for 10 min, 4 °C, in a Sorvall Fiberlite F21-8 × 50y rotor. The pellet was resuspended in TEM, sonicated for 1 min on ice, and centrifuged using the same regime. The step was repeated one more time. White pellet containing inclusion bodies was dissolved in 2.5 ml of TEM buffer containing 2 mm EDTA and 8 m urea (BioXtra grade, Sigma-Aldrich) by gentle stirring for 1 h at 4 °C and then centrifuged at 22,000 × g for 10 min, 4 °C. The supernatant was dialyzed in a Pierce/Thermo Scientific 3-ml 10,000 MWCO Slide-A-Lyzer Cassette G2 at 4 °C against 1 liter of TEM buffer containing 0.1 mm EDTA for 3 h without stirring, and then overnight against 1 liter of TEM containing 0.1 mm EDTA and 7 mm 2-mercaptoethanol with gentle stirring, and finally for 4 h against 1 liter (fresh) of the same buffer. RD3 was precipitated from the dialyzed protein fraction by adding NaCl to 250 mm. Precipitated protein was collected by centrifugation at 22,000 × g, 4 °C, dissolved in TEM containing 8 m urea on ice, and then dialyzed as described above, except that EDTA concentration was 0.1 mm and 2-mercaptoethanol concentration was 14 mm at every step of the dialysis, and the last dialysis step continued for 24 h. The dialyzed protein solution was centrifuged at 5000 × g for 10 min at 4 °C in a standard 1.5-ml Eppendorf tube, the supernatant was collected, and the protein concentration was measured by absorbance at 280 nm in 50 mm Tris-HCl (pH 7.5) containing 7 m guanidine chloride, assuming 0.1 g/liter absorbance 1.41 (calculated using a ProtParam software available on-line from the ExPASy server). Protein solution was mixed with glycerol to final 35% v/v, aliquoted, frozen in liquid N2, and stored in −70 °C. Each aliquot was thawed only once, immediately before use in the RetGC assay. The final protein concentrations in the stock solutions typically were 10−6-10−5 m.
Guanylyl Cyclase Assays
RetGC activity was assayed as described previously (9, 45). Briefly, the assay mixture (25 μl) contained RetGC1 as a membrane fraction from HEK293 cells or retinal membranes, 30 mm MOPS-KOH (pH 7.2), 60 mm KCl, 4 mm NaCl, 1 mm DTT, 1 mm EGTA buffer at 10 mm MgCl2, 0.3 mm ATP, 4 mm cGMP, 1 mm GTP, 1 μCi of [α-32P]GTP, 0.1 μCi of [8-3H]cGMP (PerkinElmer), phosphodiesterase inhibitors zaprinast and dipyridamole, and variable concentrations of GCAPs and RD3. The [32P]cGMP product was quantified by a thin-layer chromatography on polyethylenimine cellulose plates (13, 16). RetGC activity in the whole-retina or isolated retinal ROS membranes was assayed under infrared illumination using infrared goggles, and the assay contained 10 mm creatine phosphate and 0.5 unit of creatine phosphokinase (13).
RD3/RetGC Co-localization in Cyto
For expression in HEK293 cells, RD3 was tagged at the 3′-end with a Clontech SuperGlo eGFP-coding cDNA and RetGC1 was tagged by the Clontech mOrange red fluorescent tag replacing a portion of the cyclase extracellular domain as described previously (9, 16). HEK293 cells were transfected in a Lab-Tek 4-well cover glass chamber using 0.01 μg of a RD3-GFP-coding pQBIFN3 plasmid, 1 μg of a mOrangeRetGC1-coding plasmid, and 3 μl/μg of DNA of the Promega FuGENE reagent as described previously (9, 16). Confocal images were taken after 24 h of incubation using an Olympus FV1000 spectral instrument with the respective 543- and 488-nm excitation wavelengths for the red and the green fluorochromes. No changes to the original images were made except for minor gamma corrections applied to the whole image for more clear presentation in the print. Quantitative analysis was performed using the original images, without gamma correction. Pearson's correlation coefficient for testing co-localization of RD3-GFP with mOrange-tagged RetGC1 variants was calculated using the Olympus FluoView FV10-ASW software as described previously (9, 15–16).
Alkylation of Cys93 in RD3 with NEM
RD3 mutant C55D/C85T harboring a single Cys93 was produced using the “splicing by overlap extension” method (46), isolated as described above, incubated for 1 h in 5 mm DTT on ice, and then separated from the reducing agent by gel filtration on a Sephadex G25 column in 5 mm argon-flushed sodium phosphate buffer (pH 7.2) and alkylated by incubating in the presence of 1 mm freshly prepared NEM (Sigma-Aldrich) for 1 h at room temperature, after which the reaction was stopped by adding 2 mm DTT. In a control sample, RD3 was treated under identical conditions, except that to prevent the Cys93 alkylation, NEM was preincubated with DTT prior to adding it into the reaction mixture. The cyclase activity assay contained 1 mm DTT in both cases.
Statistics
Unless specified otherwise, the averaged data are mean average ± S.E. Statistical significance for the difference between groups of data was evaluated by one-way analysis of variance followed by Bonferroni post hoc test or by unpaired Student t test using the Synergy KaleidaGraph 4 software.
Author Contributions
A. M. D. conceived and coordinated the study and wrote the paper, E. V. O. and A. M. D. bred the animal models and isolated photoreceptor membranes, A. M. D. recorded ERG, E. V. O. performed morphological analysis of mouse retinas, I. V. P. and A. M. D. constructed RD3 mutants, I. V. P. performed guanylyl cyclase assays, expression and purification of RD3 mutants, and transfection experiments, and I. V. P. and A. M. D. analyzed the data and prepared figures for publication.
Acknowledgments
We thank David Garbers, Wolfgang Baehr, and Jeannie Chen for providing mouse gene knock-out models and Charles Ternes for help with preparation of RD3 expression constructs.
This work was supported by National Institutes of Health Grant EY11522 from the NEI and a Pennsylvania Department of Health CURE Formula grant. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- RetGC
- retinal membrane guanylyl cyclase
- GCAP
- guanylyl cyclase-activating protein
- NEM
- N-ethylmaleimide
- RD3
- retinal degeneration 3 protein
- ROS
- rod outer segment(s)
- ERG
- electroretinography.
References
- 1. Dizhoor A. M., Olshevskaya E. V., and Peshenko I. V. (2010) Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (GCAPs): role in regulation of photoreceptor guanylyl cyclase. Mol. Cell. Biochem. 334, 117–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Friedman J. S., Chang B., Kannabiran C., Chakarova C., Singh H. P., Jalali S., Hawes N. L., Branham K., Othman M., Filippova E., Thompson D. A., Webster A. R., Andréasson S., Jacobson S. G., Bhattacharya S. S., et al. (2006) Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am. J. Hum. Genet. 79, 1059–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Azadi S., Molday L. L., and Molday R. S. (2010) RD3, the protein associated with Leber congenital amaurosis type 12, is required for guanylate cyclase trafficking in photoreceptor cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21158–21163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Peshenko I. V., Olshevskaya E. V., Azadi S., Molday L. L., Molday R. S., and Dizhoor A. M. (2011) Retinal degeneration 3 (RD3) protein inhibits catalytic activity of retinal membrane guanylyl cyclase (RetGC) and its stimulation by activating proteins. Biochemistry 50, 9511–9519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zulliger R., Naash M. I., Rajala R. V., Molday R. S., and Azadi S. (2015) Impaired association of retinal degeneration-3 with guanylate cyclase-1 and guanylate cyclase-activating protein-1 leads to Leber congenital amaurosis-1. J. Biol. Chem. 290, 3488–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mendez A., Burns M. E., Sokal I., Dizhoor A. M., Baehr W., Palczewski K., Baylor D. A., and Chen J. (2001) Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 98, 9948–9953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burns M. E., Mendez A., Chen J., and Baylor D. A. (2002) Dynamics of cyclic GMP synthesis in retinal rods. Neuron 36, 81–91 [DOI] [PubMed] [Google Scholar]
- 8. Molday L. L., Jefferies T., and Molday R. S. (2014) Insights into the role of RD3 in guanylate cyclase trafficking, photoreceptor degeneration, and Leber congenital amaurosis. Front. Mol. Neurosci. 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Evaluating the role of retinal membrane guanylyl cyclase 1 (RetGC1) domains in binding guanylyl cyclase-activating proteins (GCAPs). J. Biol. Chem. 290, 6913–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dizhoor A. M., Lowe D. G., Olshevskaya E. V., Laura R. P., and Hurley J. B. (1994) The human photoreceptor membrane guanylyl cyclase, RetGC, is present in outer segments and is regulated by calcium and a soluble activator. Neuron 12, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 11. Lowe D. G., Dizhoor A. M., Liu K., Gu Q., Spencer M., Laura R., Lu L., and Hurley J. B. (1995) Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc. Natl. Acad. Sci. U.S.A. 92, 5535–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang R. B., Foster D. C., Garbers D. L., and Fülle H. J. (1995) Two membrane forms of guanylyl cyclase found in the eye. Proc. Natl. Acad. Sci. U.S.A. 92, 602–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peshenko I. V., Olshevskaya E. V., Savchenko A. B., Karan S., Palczewski K., Baehr W., and Dizhoor A. M. (2011) Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (RetGC) from mouse photoreceptors. Biochemistry 50, 5590–5600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baehr W., Karan S., Maeda T., Luo D. G., Li S., Bronson J. D., Watt C. B., Yau K. W., Frederick J. M., and Palczewski K. (2007) The function of guanylate cyclase 1 and guanylate cyclase 2 in rod and cone photoreceptors. J. Biol. Chem. 282, 8837–8847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Peshenko I. V., Olshevskaya E. V., Lim S., Ames J. B., and Dizhoor A. M. (2014) Identification of target binding site in photoreceptor guanylyl cyclase-activating protein 1 (GCAP1). J. Biol. Chem. 289, 10140–10154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peshenko I. V., Olshevskaya E. V., and Dizhoor A. M. (2015) Dimerization domain of retinal membrane guanylyl cyclase 1 (RetGC1) is an essential part of guanylyl cyclase-activating protein (GCAP) binding interface. J. Biol. Chem. 290, 19584–19596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zinchuk V., and Zinchuk O. (2008) Quantitative colocalization analysis of confocal fluorescence microscopy images. Curr. Protoc. Cell Biol. 39, Chapter 4, Unit 4.19 [DOI] [PubMed] [Google Scholar]
- 18. Pace C. N., and Scholtz J. M. (1998) A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Koch K. W., and Stryer L. (1988) Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature 334, 64–66 [DOI] [PubMed] [Google Scholar]
- 20. Palczewski K., Subbaraya I., Gorczyca W. A., Helekar B. S., Ruiz C. C., Ohguro H., Huang J., Zhao X., Crabb J. W., Johnson R. S., et al. (1994) Molecular cloning and characterization of retinal photoreceptor guanylyl cyclase-activating protein. Neuron 13, 395–404 [DOI] [PubMed] [Google Scholar]
- 21. Dizhoor A. M., Olshevskaya E. V., Henzel W. J., Wong S. C., Stults J. T., Ankoudinova I., and Hurley J. B. (1995) Cloning, sequencing, and expression of a 24-kDa Ca2+-binding protein activating photoreceptor guanylyl cyclase. J. Biol. Chem. 270, 25200–25206 [DOI] [PubMed] [Google Scholar]
- 22. Imanishi Y., Yang L., Sokal I., Filipek S., Palczewski K., and Baehr W. (2004) Diversity of guanylate cyclase-activating proteins (GCAPs) in teleost fish: characterization of three novel GCAPs (GCAP4, GCAP5, GCAP7) from zebrafish (Danio rerio) and prediction of eight GCAPs (GCAP1–8) in pufferfish (Fugu rubripes). J. Mol. Evol. 59, 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scholten A., and Koch K.-W.(2011) Differential calcium signaling by cone specific guanylate cyclase-activating proteins from the zebrafish retina. PLoS ONE 6, e23117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sakurai K., Chen J., and Kefalov V. J. (2011) Role of guanylyl cyclase modulation in mouse cone phototransduction. J. Neurosci. 31, 7991–8000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Makino C. L., Peshenko I. V., Wen X. H., Olshevskaya E. V., Barrett R., and Dizhoor A. M. (2008) A role for GCAP2 in regulating the photoresponse: guanylyl cyclase activation and rod electrophysiology in GUCA1B knock-out mice. J. Biol. Chem. 283, 29135–29143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Makino C. L., Wen X. H., Olshevskaya E. V., Peshenko I. V., Savchenko A. B., and Dizhoor A. M. (2012) Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse: biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice. PLoS ONE 7, e47637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stone E. M. (2007) Leber congenital amaurosis: a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am. J. Ophthalmol. 144, 791–811 [DOI] [PubMed] [Google Scholar]
- 28. Jacobson S. G., Cideciyan A. V., Peshenko I. V., Sumaroka A., Olshevskaya E. V., Cao L., Schwartz S. B., Roman A. J., Olivares M. B., Sadigh S., Yau K. W., Heon E., Stone E. M., and Dizhoor A. M. (2013) Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum. Mol. Genet. 22, 168–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Perrault I., Rozet J. M., Calvas P., Gerber S., Camuzat A., Dollfus H., Châtelin S., Souied E., Ghazi I., Leowski C., Bonnemaison M., Le Paslier D., Frézal J., Dufier J. L., Pittler S., et al. (1996) Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat. Genet. 14, 461–464 [DOI] [PubMed] [Google Scholar]
- 30. Kelsell R. E., Gregory-Evans K., Payne A. M., Perrault I., Kaplan J., Yang R. B., Garbers D. L., Bird A. C., Moore A. T., and Hunt D. M. (1998) Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum. Mol. Genet. 7, 1179–1184 [DOI] [PubMed] [Google Scholar]
- 31. Tucker C. L., Woodcock S. C., Kelsell R. E., Ramamurthy V., Hunt D. M., and Hurley J. B. (1999) Biochemical analysis of a dimerization domain mutation in RetGC-1 associated with dominant cone-rod dystrophy. Proc. Natl. Acad. Sci. U.S.A. 96, 9039–9044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramamurthy V., Tucker C., Wilkie S. E., Daggett V., Hunt D. M., and Hurley J. B. (2001) Interactions within the coiled-coil domain of RetGC-1 guanylyl cyclase are optimized for regulation rather than for high affinity. J. Biol. Chem. 276, 26218–26229 [DOI] [PubMed] [Google Scholar]
- 33. Peshenko I. V., Moiseyev G. P., Olshevskaya E. V., and Dizhoor A. M. (2004) Factors that determine Ca2+ sensitivity of photoreceptor guanylyl cyclase. Kinetic analysis of the interaction between the Ca2+-bound and the Ca2+-free guanylyl cyclase activating proteins (GCAPs) and recombinant photoreceptor guanylyl cyclase 1 (RetGC-1). Biochemistry 43, 13796–13804 [DOI] [PubMed] [Google Scholar]
- 34. Payne A. M., Downes S. M., Bessant D. A., Taylor R., Holder G. E., Warren M. J., Bird A. C., and Bhattacharya S. S. (1998) A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum. Mol. Genet. 7, 273–277 [DOI] [PubMed] [Google Scholar]
- 35. Dizhoor A. M., Boikov S. G., and Olshevskaya E. V. (1998) Constitutive activation of photoreceptor guanylate cyclase by Y99C mutant of GCAP-1: possible role in causing human autosomal dominant cone degeneration. J. Biol. Chem. 273, 17311–17314 [DOI] [PubMed] [Google Scholar]
- 36. Sokal I., Dupps W. J., Grassi M. A., Brown J. Jr, Affatigato L. M., Roychowdhury N., Yang L., Filipek S., Palczewski K., Stone E. M., and Baehr W. (2005) A novel GCAP1 missense mutation (L151F) in a large family with autosomal dominant cone-rod dystrophy (adCORD). Invest. Ophthalmol. Vis. Sci. 46, 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Olshevskaya E. V., Calvert P. D., Woodruff M. L., Peshenko I. V., Savchenko A. B., Makino C. L., Ho Y. S., Fain G. L., and Dizhoor A. M. (2004) The Y99C mutation in guanylyl cyclase-activating protein 1 increases intracellular Ca2+ and causes photoreceptor degeneration in transgenic mice. J. Neurosci. 24, 6078–6085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Woodruff M. L., Olshevskaya E. V., Savchenko A. B., Peshenko I. V., Barrett R., Bush R. A., Sieving P. A., Fain G. L., and Dizhoor A. M. (2007) Constitutive excitation by Gly90Asp rhodopsin rescues rods from degeneration caused by elevated production of cGMP in the dark. J. Neurosci. 27, 8805–8815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kitiratschky V. B., Behnen P., Kellner U., Heckenlively J. R., Zrenner E., Jägle H., Kohl S., Wissinger B., and Koch K. W. (2009) Mutations in the GUCA1A gene involved in hereditary cone dystrophies impair calcium-mediated regulation of guanylate cyclase. Hum. Mutat. 30, E782–796 [DOI] [PubMed] [Google Scholar]
- 40. Jiang L., Zhang H., Dizhoor A. M., Boye S. E., Hauswirth W. W., Frederick J. M., and Baehr W. (2011) Long-term RNA interference gene therapy in a dominant retinitis pigmentosa mouse model. Proc. Natl. Acad. Sci. U.S.A. 108, 18476–18481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Molday L. L., Djajadi H., Yan P., Szczygiel L., Boye S. L., Chiodo V. A., Gregory-Evans K., Sarunic M. V., Hauswirth W. W., and Molday R. S. (2013) RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum. Mol. Genet. 22, 3894–3905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang R. B., Robinson S. W., Xiong W. H., Yau K. W., Birch D. G., and Garbers D. L. (1999) Disruption of a retinal guanylyl cyclase gene leads to cone-specific dystrophy and paradoxical rod behavior. J. Neurosci. 19, 5889–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu Y., Ruoho A. E., Rao V. D., and Hurley J. H. (1997) Catalytic mechanism of the adenylyl and guanylyl cyclases: modeling and mutational analysis. Proc. Natl. Acad. Sci. U.S.A. 94, 13414–13419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Peshenko I. V., Olshevskaya E. V., Yao S., Ezzeldin H. H., Pittler S. J., and Dizhoor A. M. (2010) Activation of retinal guanylyl cyclase RetGC1 by GCAP1: stoichiometry of binding and the effect of new LCA-related mutations. Biochemistry 49, 709–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Peshenko I. V., and Dizhoor A. M. (2007) Activation and inhibition of photoreceptor guanylyl cyclase by guanylyl cyclase activating protein 1 (GCAP-1): the functional role of Mg2+/Ca2+ exchange in EF-hand domains. J. Biol. Chem. 282, 21645–21652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Horton R. M., and Pease L. R. (1991) Recombination and mutagenesis of DNA sequences using PCR. in Directed Mutagenesis: A Practical Approach (McPherson M. J., ed), pp. 217–250, IRL Press at Oxford University Press, Oxford, UK [Google Scholar]