

Abstract
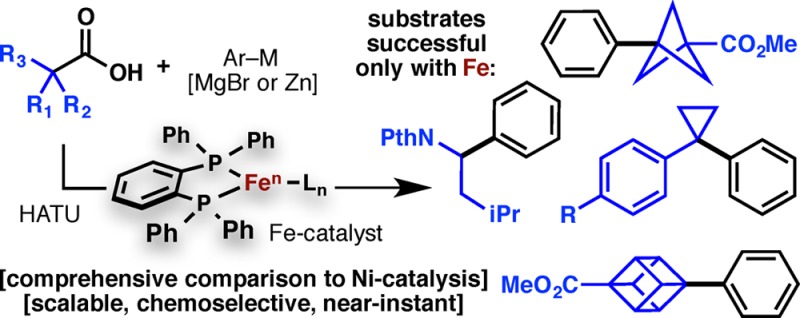
Cross-couplings of alkyl halides and organometallic species based on single electron transfer using Ni and Fe catalyst systems have been studied extensively, and separately, for decades. Here we demonstrate the first couplings of redox-active esters (both isolated and derived in situ from carboxylic acids) with organozinc and organomagnesium species using an Fe-based catalyst system originally developed for alkyl halides. This work is placed in context by showing a direct comparison with a Ni catalyst for >40 examples spanning a range of primary, secondary, and tertiary substrates. This new C–C coupling is scalable and sustainable, and it exhibits a number of clear advantages in several cases over its Ni-based counterpart.
New tools to access carbon frameworks via C–C cross-coupling are in great demand. Our group recently described a method to transform ubiquitous alkyl carboxylic acids into alkyl halide surrogates for use in such transformations.1,2 Quite fortuitously, many of the activated esters used to activate acids for amide-bond formation (such as NHPI, HOAt, and HOBt)3 can function as “redox-active” esters (RAEs), enabling decarboxylative Negishi- and Suzuki-type cross-couplings with organozincs1 and organoboronic acids,2 respectively. Like their alkyl halide counterparts, these processes have relied upon an in situ generated low-valent Ni-species.4 As these processes are generally accepted to proceed through a stepwise oxidative addition (single electron transfer, SET), it is logical that other transition metals capable of SET, such as Fe, might also function.5 Intrigued by the advertised advantages of Fe-based catalysts in C–C cross-coupling with alkyl halides (cost, toxicity, residual metal removal), we explored its use in RAE cross-coupling. Documented herein are the benefits of using an Fe-based catalyst in multiple contexts to enable scalable, near-instantaneous aryl–alkyl C–C bond formations between primary, secondary, and tertiary alkyl carboxylic acids and both arylzinc and arylmagnesium coupling partners.
The Fe-based system presents a number of tangible advances that could not have been predicted (Figure 1A). Lower catalyst loadings are generally needed, a more user-friendly procedure for in situ RAE formation is possible, and hindered (sometimes exotic) aryl–alkyl linkages are now accessible. The rapid kinetics observed even allow for the use of highly nucleophilic aryl Grignard reagents. The surprising activity of Fe to mediate RAE cross-coupling is non-obvious and further supports the hypothesis that such species are a proxy for alkyl halides in SET-based couplings.1,2,4,6
Figure 1.
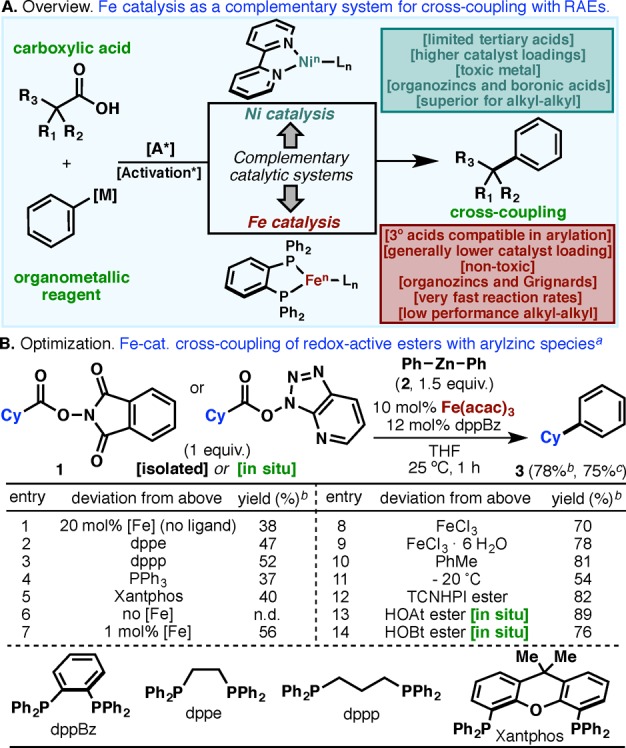
(A) Ni- and Fe-based complementary catalyst systems. (B) Invention and optimization of the Fe-catalyzed process. a0.1 mmol. bYield determined using cyclooctane as internal standard. cIsolated yield.
Figure 1B illustrates an abbreviated optimization table for the Fe-based coupling using the NHPI-based ester 1 derived from cyclohexanecarboxylic acid with Ph2Zn. The optimized conditions bear a striking resemblance to the protocols pioneered by Bedford7 and Nakamura8 for the analogous reaction of organozinc species with alkyl bromides. As with those findings, the dppBz diphosphine ligand proved to be ideal (Figure 1B, entries 1–5; see Supporting Information (SI) for an extensive list). It is striking to note that the reaction tolerates lower catalyst loadings (1 mol%, entry 7), extremely inexpensive sources of Fe (entry 8), and even hydrated forms of Fe (entry 9). As discussed below, the reaction is extremely rapid, and thus cryogenic temperatures can be employed (entry 11). Unlike other RAE-based cross-couplings, the Fe-based system operates equally well across a range of isolated or in situ-derived esters (entries 12–14). In practice, the coupling can proceed with the same simplicity (but even greater velocity) as amide bond formation using HATU or HBTU.
With a set of optimized conditions in hand, we explored the scope of this reaction with a series of primary, secondary, and tertiary carboxylic acids. In nearly all cases, direct comparisons can be made with the analogous Ni-catalyzed process. To the best of our knowledge, this represents the first instance where a C–C coupling has been benchmarked using the same substrates with Ni and Fe catalyst systems. It is anticipated that such a comparison will assist practitioners when selecting the best catalyst for a particular type of coupling. Further, the scope has been explored with both isolated procedures (NHPI or TCNHPI) and the in situ protocol (using HATU or HBTU) for RAE preparation. Since the latter provides similar yields in almost every case, we recommend the in situ protocol due to its procedural simplicity. In the case of simple secondary acid substrates (Table 1A), the yields observed were comparable to those obtained with the Ni-based system (3, 5–12, 14–20). One notable difference is the case of substituted cyclopentane 20, where an enhanced diastereoselectivity was observed under Fe catalysis (4:1 vs 1.3:1). Whereas similar trends can be seen with simple primary acids, including α-oxo acids (22, 29), a pyridine-containing substrate (23) was formed in higher yield when Fe was used (70% vs 33% for Ni).
Table 1. Scope of the Fe-Catalyzed Cross-Coupling of Redox-Active Esters with Arylzinc and Arylmagnesium Reagents.
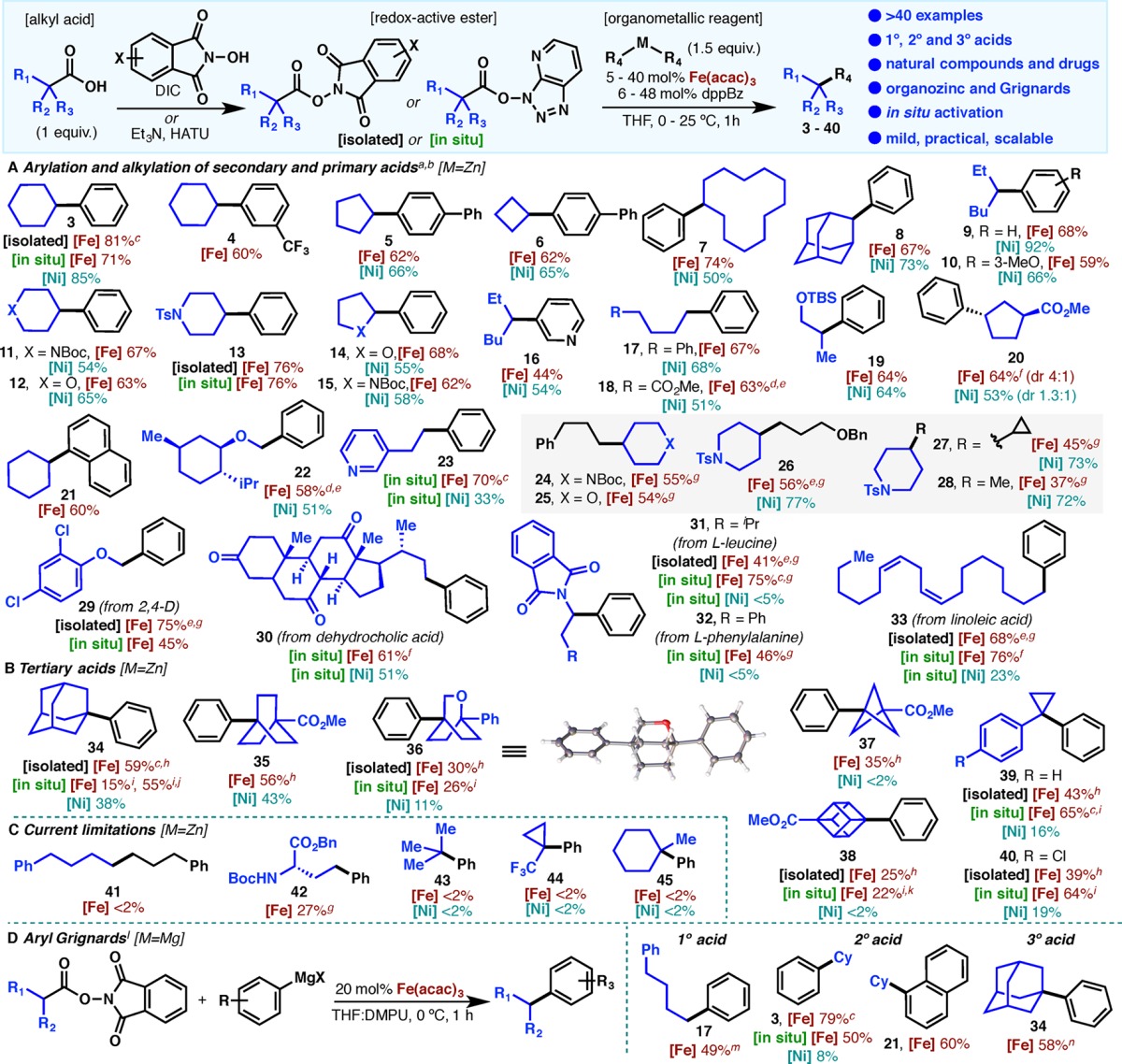
[Isolated] = isolated NHPI or TCNHPI ester (0.1 mmol, 1 equiv), Fe(acac)3 or FeCl3 (10 mol%), dppBz (12 mol%), and Ar2Zn or dialkylzinc (1.5 equiv) in THF at 25 °C for 1 h.
[In situ] = alkyl carboxylic acid (0.1 mmol, 1 equiv), Et3N (1 equiv), and HATU (1 equiv) in THF for 2 h at 25 °C, then addition of Fe(acac)3 (10 mol%), dppBz (20 mol%), and diarylzinc (2.5 equiv) at 25 °C for 1 h.
At 1.0 mmol scale.
Using FeCl3 (5 mol%) and dppBz (6 mol%).
Reaction at 0 °C.
At 0.5 mmol scale.
With Fe(acac)3 (20 mol%) and dppBz (24 mol%) in toluene.
With Fe(acac)3 (40 mol%), dppBz (48 mol%), and Ar2Zn (2.5 equiv) in PhMe.
Fe(acac)3 (20 mol%), dppBz (40 mol%), and Ar2Zn (2.5 equiv).
Using HBTU.
At 4.0 mmol scale.
Isolated NHPI ester (1 equiv), Fe(acac)3 (20 mol%), and ArMgBr (3 equiv) in DMPU:THF at 0 °C for 1 h.
Using 2 equiv of ArMgBr.
Using Fe(acac)3 (100 mol%) at 25 °C. Note: For all the comparative Ni-catalyzed cross-couplings, see SI.
Both Fe and Ni allow for chemoselective C–C couplings even in demanding natural product contexts (such as steroid 30), but the Fe-based system showed dramatically enhanced reactivity for the coupling of protected α-amino acids such as 31 and 32 (obtained as racemic mixtures). Cross-coupling between linoleic acid and diphenyl zinc also provided higher yield with the Fe-based system versus Ni (33, 76% vs 23% with the mass balance recovered starting material). For alkyl–alkyl cross-coupling (24–28), Ni catalysis provided superior results, although the yields obtained with Fe were still synthetically useful. As shown in Table 1C, Ni catalysis is the best option for primary–primary couplings such as that of 41 or glutamate systems such as 42.1b
On the other hand, striking differences were noted on tertiary carboxylic acid substrates (Table 1B). The yield of these extremely challenging coupling reactions was uniformly higher when Fe was used and, in some cases, entirely enabling. For example, bicyclo[1.1.1] (37) and cubane (38) frameworks have been outside of the realm of C–C coupling, even with alkyl halide starting materials (vide infra).9 Given the great importance of these motifs as phenyl bioisosteres in medicinal chemistry,10 the low but serviceable yields observed will be a welcome advance to access new areas of highly coveted chemical space. Of all the tertiary examples described herein, only adamantane (34) has been employed before in an analogous C–C coupling using the corresponding halide.11 It is worth noting that certain high-value tertiary acids are beyond the reach of the present Ni- or Fe-based systems such as 43–45.
Another instance where Fe catalysis differs from that with Ni is in Kumada-type C–C coupling (Table 1D). Thus, aryl Grignard reagents could be employed on primary (17), secondary (3, 21), and even tertiary substrates (34) in satisfactory yield. On the other hand, Ni catalysis provided only low conversion, with biphenyl being the predominant product.
Figure 2A highlights the scalability of this transformation in hypothetical medicinal and process scenarios, wherein the former employs chromatography and the latter uses solvent switches and crystallization as a means of purification. Starting with 1.4 g of carboxylic acid 46 in THF (10 mL) at room temperature, HATU (1.0 equiv) and Et3N (1.0 equiv) were added, followed by a THF solution of Fe/dppBz (15 mL). The reaction mixture was cooled to 0 °C, and a room-temperature diphenylzinc solution (2.5 equiv, 37 mL) was added. Standard workup (after 1 h) followed by chromatography furnished 69% of cross-coupled product 13. To demonstrate the potential of a process-scale application, the same reaction was carried out with 1 mol% Fe catalyst loading; 13 was purified by evaporation, dissolution with toluene, solvent partitioning, aqueous wash, and finally a single crystallization to deliver 61% yield of product (99% purity). Using a thermocouple, the temperature of the reaction was measured and an increase from 1.5 to 19 °C upon addition of the room-temperature Ph2Zn solution was observed. This suggests a manageable exotherm in the coupling step, supporting the scalability of the procedure. A stunning example of the potential of this coupling is depicted in Figure 2B with the stepwise cross-coupling of readily available cubane-dicarboxyl derivative 47. A series of decarboxylative couplings could be accomplished to furnish the differentially substituted triphenyl bioisosteres 49 and 50 in modest yield. It is worth mentioning that cubane structures are susceptible to decomposition in the presence of transition metals.12 The structures of these fascinating materials were verified by X-ray crystallography and represent a powerful entry into cubane functionalization with potential in both medicine and material science. To put this result in proper context, a recent publication demonstrated the enormous challenge of functionalizing cubane via cross-coupling: over 80 experiments were conducted using metalated cubanes (C-BPin, C-9BBN, C-Sn, C-Zn) or cubane-halide derivatives with Pd catalysis, and no cross-coupled products were detected.(13) Lastly, 6-bromohexanoic acid was enlisted in this coupling to discern the reactivity preference for a primary alkyl halide vs a primary carboxylic acid in C–C coupling under both Ni and Fe catalysis (Figure 2C). Surprisingly, RAEs react in preference to alkyl bromides under both Fe and Ni catalysis, with the former providing a higher yield and cleaner reaction profile than the latter (residual ZnCl2 and LiCl used in the Ni procedure results in −Br displacement; see SI for details).2
Figure 2.
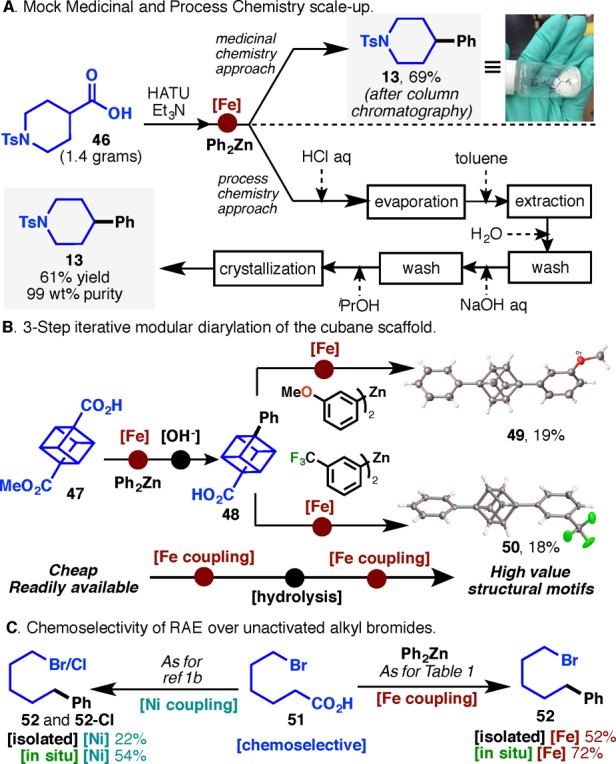
(A) Medicinal and process chemistry approaches for a hypothetical scale-up. (B) Three-step sequence for the bis-arylation of the cubane scaffold. (C) Example of chemoselectivity between RAE and alkyl bromides.
Although detailed mechanistic studies are beyond the scope of the current report, Figure 3 outlines a mechanistic picture modeled after the elegant studies of Bedford14 and others15 on analogous alkyl halide cross-couplings. Thus, initial reaction between the aryl nucleophile and the Fe(III) precatalyst would afford a reduced low-valent Fe–aryl complex (I), which could act as the catalytically active species. Such a highly reducing agent (I) would be capable of reducing the RAE via SET to its radical anion II. Fragmentation followed by recombination (supported by a radical clock experiment; see SI) with the Fe species III would afford IV, which upon reductive elimination delivers the final product and regenerates the low-valent catalyst V. Anion metathesis with another organometallic molecule would then restore the active catalyst I, thus closing the catalytic cycle. Finally, it is worth noting that the kinetics of the Fe-catalyzed process are extremely rapid, with complete conversion (ca. 75% yield) being observed within 1 min using substrate 1 (see SI). In comparison, Ni catalysis of the same transformation required >12 h to reach full conversion (85% yield).
Figure 3.
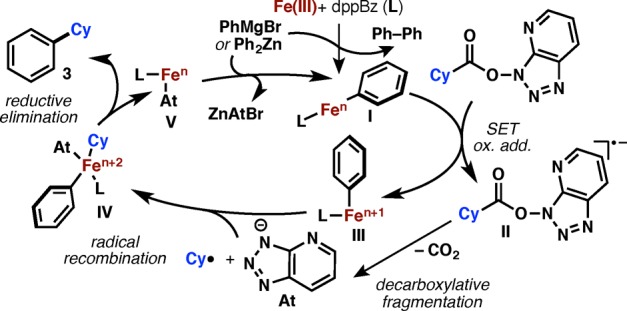
Mechanistic hypothesis for Fe-catalyzed cross-coupling between RAE and arylmetal species.
RAEs can now be employed in a manner similar to alkyl halides in Fe-catalyzed cross-coupling chemistry. To our knowledge, this is the first systematic side-by-side comparison of the differences between Ni and Fe catalysts across a range of substrates to reveal real-world practical advantages of each in certain contexts. The newly developed Fe-based system has obvious advantages in terms of ease of scale-up and sustainability but several unique and important differentiating features: near-instantaneous reaction rates, orthogonality of RAEs to alkyl bromides, applicability to tertiary systems including the venerable cubane coupling challenge, and superiority in the coupling of amino acid and unactivated primary systems.16
Acknowledgments
Financial support for this work was provided by NIH (GM-118176), Bristol-Myers Squibb, Daiichi Sankyo Co., Ltd. (research support to F.T.), postdoctoral fellowships from the Catalan Government (to J.C.), Austrian Science Fund (to L.W.), and Shenzhen HIWIN M&E Co. Ltd. (to T.-G.C.). We thank Ke Chen, Michael Schmidt, Frank Rinaldi, and Martin D. Eastgate for helpful discussions. We thank Dr. Craig M. Williams at Queensland University and Dr. John Tsanaktsidis at CSIRO Materials Science and Engineering for insightful discussions and a generous sample of cubane dimethyl ester. We are also grateful to Mr. Nathan Ferrandin for synthesis of starting materials, Mr. Jacob Edwards for initial investigations, and A. L. Rheingold and M. Gembicky for X-ray crystallographic analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b07172.
Author Contributions
§ F.T. and J.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Cornella J.; Edwards J. T.; Qin T.; Kawamura S.; Wang J.; Pan C.-M.; Gianatassio R.; Schmidt M.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2016, 138, 2174. 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Qin T.; Cornella J.; Li C.; Malins L. R.; Edwards J. T.; Kawamura S.; Maxwell B. D.; Eastgate M. D.; Baran P. S. Science 2016, 352, 801. 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Qin T.; Chen T.-G.; Wimmer L.; Edwards J. T.; Cornella J.; Vokits B.; Shaw S. A.; Baran P. S. Angew. Chem., Int. Ed. 2016, 55, 9676. 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefkens G. H. L.; Tesser G. I. J. Am. Chem. Soc. 1961, 83, 1263. 10.1021/ja01466a068. [DOI] [Google Scholar]
- Subsequent to our findings, Weix suggested that low-valent Ni species contribute to the activation of NHPI esters:Huihui K. M. M.; Caputo J. A.; Melchor Z.; Olivares A. M.; Spiewak A. M.; Johnson K. A.; DiBenedetto T. A.; Kim S.; Ackerman L. K. G.; Weix D. J. J. Am. Chem. Soc. 2016, 138, 5016. 10.1021/jacs.6b01533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of Fe-catalyzed cross-couplings of alkyl halides with arylmetal species, see:; a Tamura M.; Kochi J. K. J. J. Am. Chem. Soc. 1971, 93, 1487. 10.1021/ja00735a030. [DOI] [Google Scholar]; b Nakamura M.; Ito S.; Matsuo K.; Nakamura E. Synlett 2005, 11, 1794. 10.1055/s-2005-871541. [DOI] [Google Scholar]; c Nakamura M.; Matsuo K.; Ito S.; Nakamura E. J. Am. Chem. Soc. 2004, 126, 3686. 10.1021/ja049744t. [DOI] [PubMed] [Google Scholar]; d Przyojski J. A.; Veggeberg K. P.; Arman H. D.; Tonzetich Z. J. ACS Catal. 2015, 5, 5938. 10.1021/acscatal.5b01445. [DOI] [Google Scholar]; e Bedford R. B.; Bruce D. W.; Frost R. M.; Hird M. Chem. Commun. 2005, 4161. 10.1039/b507133j. [DOI] [PubMed] [Google Scholar]; f Bedford R. B.; Carter E.; Cogswell P. M.; Gower N. J.; Haddow M. F.; Harvey J. N.; Murphy D. M.; Neeve E. C.; Nunn J. Angew. Chem., Int. Ed. 2013, 52, 1285. 10.1002/anie.201207868. [DOI] [PubMed] [Google Scholar]; g Martin R.; Fürstner A. Angew. Chem., Int. Ed. 2004, 43, 3955. 10.1002/anie.200460504. [DOI] [PubMed] [Google Scholar]; h Nagano T.; Hayashi T. Org. Lett. 2004, 6, 1297. 10.1021/ol049779y. [DOI] [PubMed] [Google Scholar]; i Rao Volla C. M.; Vogel P. Angew. Chem., Int. Ed. 2008, 47, 1305. 10.1002/anie.200704858. [DOI] [PubMed] [Google Scholar]; j Noda D.; Sunada Y.; Hatakeyama T.; Nakamura M.; Nagashima H. J. Am. Chem. Soc. 2009, 131, 6078. 10.1021/ja901262g. [DOI] [PubMed] [Google Scholar]; k Hatakeyama T.; Hashimoto T.; Kondo Y.; Fujiwara Y.; Seike H.; Takaya H.; Tamada Y.; Ono T.; Nakamura M. J. Am. Chem. Soc. 2010, 132, 10674. 10.1021/ja103973a. [DOI] [PubMed] [Google Scholar]
- For NHPI esters utilized in PET events, see:; a Schnermann M. J.; Overman L. E. Angew. Chem., Int. Ed. 2012, 51, 9576. 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pratsch G.; Lackner G. L.; Overman L. E. J. Org. Chem. 2015, 80, 6025. 10.1021/acs.joc.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Okada K.; Okamoto K.; Oda M. J. Am. Chem. Soc. 1988, 110, 8736. 10.1021/ja00234a047. [DOI] [Google Scholar]; d Okada K.; Okamoto K.; Morita N.; Okubo K.; Oda M. J. Am. Chem. Soc. 1991, 113, 9401. 10.1021/ja00024a074. [DOI] [Google Scholar]
- Bedford R. B.; Huwe M.; Wilkinson M. C. Chem. Commun. 2009, 600. 10.1039/B818961G. [DOI] [PubMed] [Google Scholar]
- Hatakeyama T.; Kondo Y.; Fujiwara Y.; Takaya H.; Ito S.; Nakamura E.; Nakamura M. Chem. Commun. 2009, 1216. 10.1039/b820879d. [DOI] [PubMed] [Google Scholar]
- For Ph-substituted cubanes via PhLi radical addition, see:Eaton P. E. Angew. Chem., Int. Ed. Engl. 1992, 31, 1421. 10.1002/anie.199214211. [DOI] [Google Scholar]
- a Chalmers B. A.; et al. Angew. Chem., Int. Ed. 2016, 55, 3580. 10.1002/anie.201510675. [DOI] [PubMed] [Google Scholar]; b Stepan A. F.; et al. J. Med. Chem. 2012, 55, 3414. 10.1021/jm300094u. [DOI] [PubMed] [Google Scholar]; c Wlochal J.; Davies R. D. M.; Burton J. Org. Lett. 2014, 16, 4094. 10.1021/ol501750k. [DOI] [PubMed] [Google Scholar]
- For Fe- and Ni-catalyzed arylation of tertiary centers, see:; a Ghorai S. K.; Jin M.; Hatakeyama T.; Nakamura M. Org. Lett. 2012, 14, 1066. 10.1021/ol2031729. [DOI] [PubMed] [Google Scholar]; b Zultanski S. L.; Fu G. C. J. Am. Chem. Soc. 2013, 135, 624. 10.1021/ja311669p. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Joshi-Pangu A.; Wang C.-Y.; Biscoe M. R. J. Am. Chem. Soc. 2011, 133, 8478. 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bräse S.; Waegell B.; de Meijere A. Synthesis 1998, 1998, 148. 10.1055/s-1998-2013. [DOI] [Google Scholar]
- Chalmers B. A.; Chen A. P.-J.; Savage G. P.; Williams C. M. Aust. J. Chem. 2010, 63, 1108. 10.1071/CH10107. [DOI] [Google Scholar]
- Plunkett S.; Flanagan K. J.; Twamley B.; Senge M. O. Organometallics 2015, 34, 1408. 10.1021/acs.organomet.5b00151. [DOI] [Google Scholar]
- Adams C. J.; Bedford R. B.; Carter E.; Gower N. J.; Haddow M. F.; Harvey J. N.; Huwe M.; Cartes M. Á.; Mansell S. M.; Mendoza C.; Murphy D. M.; Neeve E. C.; Nunn J. J. Am. Chem. Soc. 2012, 134, 10333. 10.1021/ja303250t. [DOI] [PubMed] [Google Scholar]
- a Daifuku S. L.; Al-Afyouni M. H.; Snyder B. E. R.; Kneebone J. L.; Neidig M. L. J. Am. Chem. Soc. 2014, 136, 9132. 10.1021/ja503596m. [DOI] [PubMed] [Google Scholar]; b Daifuku S. L.; Kneebone J. L.; Snyder B. E. R.; Neidig M. L. J. Am. Chem. Soc. 2015, 137, 11432. 10.1021/jacs.5b06648. [DOI] [PMC free article] [PubMed] [Google Scholar]; For useful reviews addressing mechanistic aspects of Fe-catalyzed cross-couplings, see:; c Cassani C.; Bergonzini G.; Wallentin C.-J. ACS Catal. 2016, 6, 1640. 10.1021/acscatal.5b02441. [DOI] [Google Scholar]; d Bedford R. B. Acc. Chem. Res. 2015, 48, 1485. 10.1021/acs.accounts.5b00042. [DOI] [PubMed] [Google Scholar]
- This chemistry was field-tested in the context of both process (substrates 20 and 21) and medicinal (substrate 36, verified by X-ray crystallography) groups at Bristol-Myers Squibb.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.