

Abstract
Galectin-3 is an adhesion/growth-regulatory protein with a modular design comprising an N-terminal tail (NT, residues 1–111) and the conserved carbohydrate recognition domain (CRD, residues 112–250). The chimera-type galectin interacts with both glycan and peptide motifs. Complete 13C/15N-assignment of the human protein makes NMR-based analysis of its structure beyond the CRD possible. Using two synthetic NT polypeptides covering residues 1–50 and 51–107, evidence for transient secondary structure was found with helical conformation from residues 5 to 15 as well as proline-mediated, multi-turn structure from residues 18 to 32 and around PGAYP repeats. Intramolecular interactions occur between the CRD F-face (the 5-stranded β-sheet behind the canonical carbohydrate-binding 6-stranded β-sheet of the S-face) and NT in full-length galectin-3, with the sequence P23GAW26…P37GASYPGAY45 defining the primary binding epitope within the NT. Work with designed peptides indicates that the PGAX motif is crucial for self-interactions between NT/CRD. Phosphorylation at position Ser6 (and Ser12) (a physiological modification) and the influence of ligand binding have minimal effect on this interaction. Finally, galectin-3 molecules can interact weakly with each other via the F-faces of their CRDs, an interaction that appears to be assisted by their NTs. Overall, our results add insight to defining binding sites on galectin-3 beyond the canonical contact area for β-galactosides.
Keywords: adhesion, apoptosis, lectin, phosphorylation, self-association
Introduction
A key question in order to understand aspects of cell sociology is to define the structural basis for the flow of biological information. Oligosaccharides are an ideal platform to store such messages (Laine 1997; Spiro 2002; Zuber and Roth 2009; Corfield and Berry 2015; Gabius 2015; Hennet and Cabalzar 2015; Ledeen and Wu 2015; Schengrund 2015). Guiding to the topic of our report, endogenous sugar receptors (lectins) serve as a link to translate this type of biochemical “wording” into cellular activities (for a recent review, see Solís et al. 2015). Besides the direct contact to the glycan, the modular structure of a lectin is then crucial for triggering post-binding effects (Gabius et al. 2011, 2015). The topological display of the carbohydrate recognition domains (CRDs) is known to differ within lectin families, a challenge for structure–function analysis.
Among the members of the galectin protein family, galectin-3 (Gal-3) is unique, due to its phylogenetically strictly conserved trimodular design (Hughes 1994; Lobsanov and Rini 1997; Cooper 2002; Houzelstein et al. 2004; Leffler et al. 2004; Kaltner and Gabius 2012). This lectin (present in the nucleus, cytoplasm and extracellular matrix) usually binds to glycans containing β-galactosides, as well as to certain proteins and lipids (Woo et al. 1990; Mey et al. 1996; Hughes 1999; Haudek et al. 2010; Dawson et al. 2013; Funasaka et al. 2014). Each section of its structure shown in Figure 1, i.e. the N-terminal peptide, the non-triple helical collagen-like repeats (8–13 in vertebrates) and the CRD, is essential for its distinct functions. However, a complete structure-based rationalization for its activities, which appear to be of clinical relevance, e.g. in cardiac dysfunction and (osteo)arthritis (Liu et al. 2009; Hrynchyshyn et al. 2013; Li et al. 2013; Toegel et al. 2014), has yet to be attained. Whereas crystallographic analysis of the CRD proved feasible (Seetharaman et al. 1998), attempts to crystallize the full-length protein have been unsuccessful. Therefore, the only option to proceed on this level of Gal-3 structural analysis is to engineer variants with a shortened N-terminal tail (NT) and to systematically test their tendency to crystallize (Kopitz et al. 2014; Flores-Ibarra et al. 2015). In order to define the structure of the full-length protein, including sites for intra- and intermolecular self-binding, work in solution is thus necessary. In fact, sequential assignment of 1H, 13C and 15N resonances of the CRD of human Gal-3 had opened the door for NMR studies (Umemoto and Leffler 2001; Umemoto et al. 2003; Diehl et al. 2010). Moving on from this basis, assignment work was recently completed for the full-length lectin (Ippel et al. 2015). This information now provides the opportunity to address numerous structure-based questions. The satisfying correlation between crystallographic and NMR data are highlighted by the detection of the Pro117 cis-conformation in solution (Ippel et al. 2015), previously observed in crystals (Saraboji et al. 2012).
Fig. 1.

(A) Schematic overview of the trimodular design of human Gal-3. The NT is composed of the peptide with two sites for Ser phosphorylation (N) and the nine non-triple helical collagen-like repeat units (I–IX), these being followed by the CRD. Cleavage sites for MMPs, PSA and bacterial collagenase are indicated by arrows (adapted from Kopitz et al. 2014). (B) Amino acid sequence of the NT. Repeats I–IX and 7-fold occurrence of the pentapeptide PGAYP are indicated by boxes and shaded boxes, respectively. (C) Crystal structure of the CRD (PDB code 3ZSJ; Seetharaman et al. 1998). The question marks highlight the current uncertainty about the conformation of the NT, and the arrow identifies the contact site for glycan binding. This figure is available in black and white in print and in color at Glycobiology online.
Looking at the full-length protein, differential scanning calorimetry and nano-electrospray ionization mass spectrometry had suggested the presence of secondary structure within the NT and the occurrence of two conformers, one likely compact and the other relatively unfolded, as indicated by a bimodal charge distribution (Agrwal et al. 1993; Kopitz et al. 2003). Interestingly, modeling and NMR analysis of a slightly extended hamster Gal-3 CRD (Barboni et al. 2000; Birdsall et al. 2001) and initial comparison of 1H,15N chemical shifts from human Gal-3 CRD (Umemoto and Leffler 2001) and full-length human Gal-3 (Ippel et al. 2015) had shaped the idea for intramolecular interactions that would underlie the generation of a folded form. Given the physiological release of peptides from the NT by proteolysis up to its entire length, as listed in Figure 1, such self-binding may thus be impaired by proteolytic processing, with functional consequences. The present NMR study starts with assessing whether the NT sequence itself had any elements of secondary structure. Whether an interaction between Gal-3's CRD and its tail (as free 15N-labeled polypeptide or as an integral part of the full-length lectin) can take place is then answered. In this context, preparative production of Gal-3 with serine phosphorylation within the N-terminal peptide (see Figure 1) facilitates study of this physiological post-translational modification, whereas synthesis of the DiLacNAc tetrasaccharide enables us to monitor its influence on ligand binding. Finally, we employed NMR spectroscopy to investigate concentration-dependent changes in the quaternary structure of Gal-3.
Results and discussion
Search for secondary structure in the NT
Previous NMR studies with hamster Gal-3 NT detected no “residual signals for NH protons showing that these had been completely exchanged for deuterium, thus indicating the absence of secondary structure containing strong hydrogen bonds” (Birdsall et al. 2001). Because 1H–15N HSQC resonances from the NT in Gal-3 exhibit relatively narrow line widths and are minimally dispersed falling between 8.5 and 7.5 ppm, Ippel et al. (2015) concluded that the NT was highly dynamic with random coil structure. Analysis of nuclear Overhauser effect spectroscopy (NOESY) results obtained with full-length Gal-3 failed to detect long-range NOEs from within the NT segment. However, very weak NOEs may be obscured by resonance overlap and differential relaxation rates between the CRD and NT. To circumvent these shortcomings, we divided the NT into two halves and used NMR spectroscopy to investigate the respective synthetic peptides, namely residues 1–50 and 51–107 from the NT sequence, as illustrated in Figure 1. 1H NOESY spectra of peptide 1–50 showed, e.g. the presence of i to (i − 4) and i to (i − 3) αβ NOEs between His8 and Ser12 (Figure 2A) and Ser6 and Asp9 (Figure 2B). All of the observed long-range NOEs are summarized in Figure 2C. These data demonstrate the presence of transient helical conformation between Phe5 and Gly15, as well as Pro-mediated, multiple turns between residues 18 and 32, in particular around Trp22/Trp26 whose aromatic rings may stack. Upon reducing the peptide's length to residues 2–21, no secondary structure NOEs could be detected (data not shown). This result suggests that the length of the NT sequence in this region is crucial for formation/stabilization of the observed secondary structure. In contrast to the NT's first half, the second peptide 51–107 yielded only a few, rather weak long-range NOEs around or within Pro-induced turns.
Fig. 2.

(A and B) Two expansions (αβ region) from a 1H NOESY spectrum (1H frequency of 700 MHz NMR, mixing time of 350 ms) of the NT polypeptide 1–50 (∼1 mM) are shown. Several (i,i+ 1), (i,i+ 3) and (i,i+ 4) NOEs are labeled as discussed in the text. (C) A summary of these NOEs observed in NT peptide 1–50 is shown. Resonance assignments for NT peptide 1–50 were made by using standard 1H homonuclear protocols, primarily based on DIPSI and NOESY data recorded at pH 3.3 and 7. NT polypeptides were dissolved as TFA salts and buffer exchanged. Solution conditions are described in Materials and methods. This figure is available in black and white in print and in colour at Glycobiology online.
Overall, transient folding interactions within the NT and between the NT and the CRD provide a structural basis to help explain the increase in susceptibility of the NT to collagenase digestion upon denaturation and the detection of a thermal denaturation transition of ∼48°C for NT residues 1–137 of murine Gal-3 at pH 10 (Agrwal et al. 1993). That the Tm with full-length Gal-3 is 39°C (Agrwal et al. 1993) suggests the influence of structural context on maintaining the protein fold by intra- and/or intermolecular interactions.
Intramolecular interactions between NT and CRD
Because our goal here was to better define NT interactions with the CRD, we have further assigned multiple resonances arising from Pro cis–trans isomerization (exemplified in Supplementary data, Figure S1A and B) and thereby have been able to better interpret our NMR spectra of Gal-3. In addition, we have been able to work with the natural material, free of any engineering, so that a His-tag was not present in the peptides or the protein, as studied by Halimi et al. (2014). We assessed interactions between the NT and CRD in a number of ways. First, we compared chemical shifts from 15N-labeled full-length Gal-3 with those from the 15N-labeled Gal-3 CRD. HSQC spectral overlays are shown in Figure 3A. Using our complete resonance assignments for Gal-3, 1H, 15N-weighted chemical shift differences (Δδ) are plotted as a function of the amino acid sequence in Figure 3B, and the largest Δδ-values are identified by coloring in the structural model given in Figure 3C. Contact sites for the NT in full-length Gal-3 comprise β-strands 7, 8 and 9 on the F-face. This finding is in general agreement with a recent analysis using His-tagged Gal-3 (Halimi et al. 2014). Here, we studied His-tag-free material to exclude the possibility that the His-tag itself could affect interactions between the NT and CRD. Although we observed some changes with residues from the S-face (sugar binding face), these are relatively minor and could be explained by allosteric effects induced by interactions of the NT with the F-face. Interestingly, we previously observed binding of polysaccharides (galactomannans) to the F-face of Gal-1 and Gal-3 (Miller et al. 2009, 2012, 2016), and in these instances, ligand binding at the F-face allosterically attenuated affinity for lactose at the S-face and vice versa. Thus, the F-face these galectins appears to possess functional features for binding to both peptides and polysaccharides with binding to one face influencing affinity of interactions at the other.
Fig. 3.

(A) 15N–1H HSQC spectra (1H frequency of 700 MHz) of 20 µM 15N-labeled full-length Gal-3 (blue) and 20 µM 15N-labeled Gal-3 CRD (red) are overlaid. Some highly shifted backbone and side chain NH resonances are labeled using assignments previously reported (Ippel et al. 2015). (B) Chemical shift differences (Δδ) of CRD resonances in full-length Gal-3 and Gal-3 CRD are plotted vs. the amino acid sequence of Gal-3 CRD. (C) Δδ values are highlighted in the X-ray crystal structure of Gal-3 CRD. Residues that are most highly shifted are highlighted in red (2 SD above average), followed by orange (1 SD above average), gray (above the average) and green (below the average). The average value was 0.05 ± 0.04 ppm (SD). A molecule of lactose is shown in space-filling format for orientation. Solution conditions are described in Materials and methods. This figure is available in black and white in print and in color at Glycobiology online.
Next, we used two synthetic NT polypeptides (i.e. residues 1–50 and 51–107) in titrations with 15N-labeled Gal-3 CRD to provide further evidence of these interactions. Peptide-induced shifting of CRD resonances in HSQC spectra are shown in Figure 4A with the overlay of several HSQC spectra from the titration with NT peptide 1–50. HSQC titration results with peptide 51–107 were essentially the same (Supplementary data, Figure S2). The binding sites for these polypeptides were delineated from plots of chemical shift changes (Δδ) for peptides 1–50 and 51–107 vs. the Gal-3 CRD residue number (Figure 4B and C). Of note, we found that both peptides, regardless of their having dissimilar sequences, perturb the same set of resonances within the CRD, an observation that was confirmed by a regression coefficient of 0.98 from a plot of Δδ values for one peptide vs. the other (insert to Figure 4C). This indicates that both NT peptides interact with the same CRD site which is located within the 5-stranded β-sheet at the backside of the CRD (F-face, opposite the 6-stranded β-sheet S-face with the canonical carbohydrate-binding site). CRD residues showing the largest shift changes with NT peptide 1–50 are marked on the Gal-3 CRD structure in Figure 4D. As noted previously (Berbís et al. 2014), shorter NT-derived peptides representing proline-rich repeats III and IV also interact with residues at the CRD F-face, albeit differently from these two larger NT peptides, suggesting that a longer sequence better captures structural elements (e.g. folding) characteristic for the NT and for optimal interactions with the CRD.
Fig. 4.

(A) 15N–1H HSQC spectra (1H frequency of 700 MHz) for the 15N-labeled Gal-3 CRD (20 µM) in the absence (blue contours) and in the presence of NT peptide 1–50 at concentrations of 20 µM (orange), 60 µM (yellow), 160 µM (purple), 400 µM (green) and 1.11 mM (light blue/gray) are superimposed. For comparison, the spectrum of 20 µM 15N-labeled full-length Gal-3 (red contours) is superimposed on the Gal-3 CRD spectra, showing that shift changes of the most perturbed peaks in the titration with Gal-3 CRD track directly to the corresponding peak positions in full-length Gal-3 (black lines). Solution conditions are described in Materials and methods. (B) Chemical shift differences (Δδ) of Gal-3 CRD resonances ± NT peptide 1–50 (1.1 mM) are shown vs. the amino acid sequence. (C) Net Δδ values of Gal-3 CRD resonances ± NT polypeptide 51–107 (1.27 mM) are shown vs. the amino acid sequence. The insert presents a plot of Δδ values observed with peptide 1–50 vs. Δδ values observed with peptide 51–107. The regression coefficient (R) of 0.98 indicates that both peptides interact with the same residues on the F-face of the CRD. (D) Chemical shift differences from NT peptide 1–50 are color highlighted on the X-ray crystal structure of Gal-3 CRD (PDB code 3ZSJ; Seetharaman et al. 1998). Residues that are highly shifted are highlighted in red (2 SD above average), followed by orange (1 SD above average), gray (above the average) and green (below the average). The average value was 0.04 ± 0.04 ppm (SD). For orientation, a molecule of lactose is shown at its binding site. (E) Δδ values of the most shifted resonances (192, 198, 199, 202–204, 210–213 and 215–220) with peptide 1–50 are plotted vs. the peptide concentration. (F) Δδ values of the most shifted resonances (192, 198, 199, 202–204, 210–213 and 215–220) with peptide 51–107 are plotted vs. the peptide concentration. Lines connecting data points in both (E) and (F) are shown simply as visual aids. (G) Expansions of HSQC data are shown for three of the most highly shifted resonances (V202, K210, A216) during the titrations with NT 1–50 and NT 51–107 as labeled. The blue contours are Gal-3 CRD resonances in the absence of either NT peptide, and the red contours are for the same CRD resonances from full-length Gal-3. This figure is available in black and white in print and in color at Glycobiology online.
Our HSQC titrations provide additional information about the binding of these NT polypeptides to the Gal-3 CRD. Figure 4E and F plot Δδ values for the most shifted resonances (residues 192, 198, 199, 202–204, 210–213 and 215–220) vs. the concentration of each NT peptide as labeled. Although saturation was not reached with either peptide, Δδ values tracked along a straight line upon increasing the NT peptide concentration (Figure 4A; Supplementary data, Figure S2) and moved directly towards Δδ values observed for the same CRD resonances in full-length Gal-3. This is more clearly seen in Figure 4G which shows HSQC expansions for three of the most highly shifted resonances (V202, K210 and A216) with NT 1–50 and NT 51–107 as labeled. The blue contours are Gal-3 CRD resonances in the absence of either NT peptide, and the red contours are for the same CRD resonances in full-length Gal-3. NT peptide-induced chemical shifts track along a straight line; therefore, NT peptide binding can be explained by a two-state model in which the NT merely associates and dissociates from the F-face of the CRD. Because the titration was limited by NT peptide solubility and increasing viscosity, we estimated equilibrium dissociation constants (Kd) by using the chemical shifts of the same 16 most shifted resonances in full-length Gal-3 to represent 100% Gal-3 bound and taking the peptide concentration at 50% Gal-3 bound. The average of these 16 values yielded KD values of 917 and 1058 µM for the binding of NT peptides 1–50 and 51–107, respectively. This relatively weak binding is consistent with our HSQC data, where resonances are smoothly shifted during the titrations, indicating that interactions fall within the fast exchange regime on the chemical shift time scale. Overall, our data indicate that NT-binding interactions with the CRD are the same for free NT-derived peptides and for the NT tethered to the CRD in intact Gal-3.
To define the binding epitope within the NT, we recorded 15N HSQC spectra of this complete portion of Gal-3, i.e. the 15N-labeled NT (residues 1–108), shown in Supplementary data, Figure S3. Resonances were assigned using procedures as described previously (Ippel et al. 2015), and their chemical shifts were compared with those from the NT sequence in 15N-labeled full-length Gal-3 (Figure 5). Even though the largest Δδ values are observed within the N-terminal part of the NT (residues 23–26, 37–40 and 42–45), other significant shifts are noted at residues 3–5, 52–56, 62, 66, 77–81, 93–96 and 101–108. Whereas changes at the C-terminal part of the NT (residues 101–108) could be explained simply by changes to the covalent structure of the peptide (i.e. covalently bonded to the CRD in one case but not the other), most other changes were found to occur within or around PGAX sequences, especially those within the first 50 N-terminal residues (i.e. P23GAW26, P37GAS40, P42GAY45, P51GAY54 and P76GAY79). The Gal-3 NT contains multiple PGAX repeats, in particular PGAY repeats (see Figure 1). Thus, it appears as though the PGAX motif is a defining element for the CRD F-face binding epitope on the NT tail.
Fig. 5.

Chemical shift differences (Δδ) between HSQC resonances from the 15N-labeled NT (1–111) obtained by recombinant production and those NT resonances from 15N-labeled full-length Gal-3. Δδ values are shown vs. the amino acid sequence of Gal-3 NT.
To support this proposal, we investigated effects on 15N-labeled Gal-3 CRD induced by the presence of a synthetic tetrapeptide, i.e. PGAY. Addition of PGAY at the same concentration as the two long NT-derived peptides 1–50 or 51–107 had very little, if any, effect on Gal-3 CRD resonances. However, when the PGAY concentration was increased 4-fold, responses became significant, although less than with either of these NT peptides (Figure 6A). Furthermore, because PGAX sequences are often proximal to each other within the NT (e.g. P37GASYPGAY45), we performed the HSQC study using a 16mer that repeated the PGAY sequence four times (i.e. PGAYPGAYPGAYPGAY). HSQC spectra of 15N-labeled Gal-3 CRD in the absence and presence of this 16mer are shown in Supplementary data, Figure S4. Compared with effects from tetrapeptide PGAY, the 16mer peptide produced far greater Δδ changes (Figure 6B). Overall, the trend in Δδ values is the same as that observed with either NT peptide 1–50 or 51–107, as demonstrated by correlation plots shown in Figure 6C and D. In aggregate, these data provide clear evidence that the PGAX sequence is indeed the primary determinant for NT binding to the F-face of the CRD. Moreover, because the largest Δδ values are found within the N-terminal part of the NT (P23GAW26 and P37GASYPGAY45), it is this PGAX-containing NT segment (vis-à-vis that from the C-terminal part) that best promotes interactions with the F-face of the CRD.
Fig. 6.

Effects of PGAY and (PGAY)4 peptides on Gal-3 CRD. Chemical shifts of HSQC resonances were determined for 15N-labeled Gal-3 CRD in the absence and presence of 4.45 mM PGAY tetrapeptide or 1.1 mM 16mer peptide (PGAY)4, and their Δδ values are shown vs. the amino acid sequence of Gal-3 CRD in (A) and (B), respectively. Correlation plots show Δδ values for the 16mer (PGAY)4 peptide vs. Δδ values for NT peptides 1–50 (C) and 51–107 (D). NMR data were acquired at a 1H frequency of 700 MHz.
To investigate whether NT interactions with the CRD F-face perturb, the interior of the lectin's β-sandwich structure and thereby possibly mediate allosteric effects at sites on the S-face, we recorded 1H–13C HSQC spectra in the absence and presence of NT peptides 1–50 and 51–107, focusing on the well-resolved 13C-methyl region and therefore on various aliphatic side chains within the interior. These results are shown in HSQC spectral overlays in Figure 7A, and the most shifted resonances are highlighted on the Gal-3 CRD structure (Figure 7B and C). Most of these methyl groups are located within the CRD β-sandwich. One of the key NT-interacting residues on the CRD, i.e. Gln201 (shown in stick structure in Figure 7B and C), falls centrally among these methyls. The binding of NT-derived polypeptides to the surface of the F-face therefore can induce conformational and/or dynamical alterations within the interior of the lectin. This in turn can explain the bimodal charge distribution observed by mass spectrometry, which suggested that the protein can adopt extended and compact conformations (Kopitz et al. 2003). In addition, the absence of spectral differences with a co-mixture of both peptides (Figure 7A) supports the conclusion that the two peptides compete for the same site on the CRD F-face and exert the same influence on residues within the CRD β-sandwich.
Fig. 7.

(A) The methyl region from constant time 1H–13C HSQC spectra (1H frequency of 700 MHz) of [13C,15N]-labeled Gal-3 CRD (20 µM) is shown as acquired during titrations with NT polypeptides 1–50 and 51–107. Overlays of four HSQC spectra are documented for Gal-3 CRD in the absence of peptides (dark blue contours) and in the presence of 320 µM NT peptide 1–50 (orange), 1.11 mM NT polypeptide 1–50 (cyan) or 1.11 mM NT polypeptide plus 1 mM NT polypeptide 51–107 (red). Chemical shift differences were observed to be very similar with either NT polypeptide. Addition of an extra 200 µM [13C,15N]-labeled Gal-3 CRD to the mixture containing CRD/NT(1–50)/NT(51–107) partially reverses the direction of observed chemical shift changes in the Gal-3 CRD spectrum, in line with competitive binding of NT polypeptides to the CRD of Gal-3. Bleed-through of 13C-methyl resonances (natural abundance) from both peptides (Val methyls from NT peptide 51–107; Leu methyls from NT peptide 1–50, and Ala methyls from both peptides) are shown in light blue. (B and C) The positions of the branched amino acids are marked in two orientations of the structure of the Gal-3 CRD highlighting the largest chemical shift changes in green (space-filling format). For this experimental set-up, Gal-3 CRD and NT polypeptides were dissolved in an aqueous (99.9% D2O) solution of 20 mM potassium phosphate buffer (pH 6.9) and 8 mM DTT, at 30°C. This figure is available in black and white in print and in color at Glycobiology online.
Effects from phosphorylation of Gal-3
At this point, we have established that the N-terminal part of the NT is crucial for interactions with the F-face of the CRD and that it also displays some propensity to form α-helix or multiple-turn conformations (residues 5 to ∼32). This is interesting, because there are two sites within this NT region (Ser6 and Ser12) that are known to be phosphorylated (Huflejt et al. 1993) with functional significance, i.e. in nuclear export (Tsay et al. 1999; Takenaka et al. 2004). Therefore, we also investigated phosphorylated Gal-3 (Ser6 and Ser12) prepared by using casein kinase 1 for large-scale production (Kübler et al. 2008). Incorporation yields were ∼70% at Ser6 and 20% at Ser12. HSQC chemical shift differences observed between resonances from 15N-labeled Gal-3 and its phosphorylated form are shown in Figure 8A, with an expansion given in Figure 8B. The highly downfield shifted NH resonance of pSer6 is consistent with introduction of a phosphate group on the Ser6 side chain (Selenko et al. 2008), as also noted with a pSer6-containing N-terminal 20mer peptide (Pep-2, residues 2–21, in Berbís et al. 2014). Amino acids in the vicinity of pSer6 are also significantly perturbed as expected (Figure 8B). Perturbations at NH resonances of residues 3 and 8–10 (and beyond) induced by this modification are consistent with helical structure within this region as discussed above.
Fig. 8.

(A) The 15N–1H HSQC spectra (1H frequency of 700 MHz) obtained with 15N-labeled Gal-3 (200 µM) (red) and phosphorylated 15N-labeled Gal-3 (pGal-3, 200 µM) (blue) are superimposed. Assignments of pSer6 and other perturbed amide resonances around this site in pGal-3 were confirmed by using 3D HNHA (heteronuclear 1H–15N–13Cα–1H through-bond correlation) and 3D-edited NOESY experiments. (B) An expansion of the region in these 15N–1H HSQC spectra where most NT resonances are found is shown. (C) Chemical shift differences (Δδ) between pGal-3 and Gal-3 CRD (as done for Gal-3 and Gal-3 CRD in Figure 5B) are plotted vs. the amino acid sequence of Gal-3. (D) Chemical shift differences (Δδ) between pGal-3 and unmodified Gal-3 from HSQC data shown in (A) are plotted vs. the amino acid sequence of Gal-3. (E) Expansion of the Δδ plot in (D) is shown to better visualize changes in residues within the CRD due to the presence of NT pSer6. The 11 β-strands (β1 to β11) in the protein are identified in (C) and (E) with blue-filled rectangles above each plot. Solution conditions are described in Materials and methods. This figure is available in black and white in print and in color at Glycobiology online.
Within the CRD of full-length Gal-3, significant shifts are also noted. Upon detailed inspection, comparison of pGal-3 and Gal-3 CRD shows that the Δδ distribution (Figure 8C) is essentially the same as that observed between Gal-3 and Gal-3 CRD (Figure 7B). This indicates that the presence of the phosphate group(s) apparently does not affect NT interactions with the F-face of the CRD to any great extent, and if anything, it may actually weaken them somewhat based on smaller Δδ values observed with pGal-3 (Figure 8C vis-à-vis Figure 7B). This contrasts with results on a shorter NT-derived 20mer peptide (Pep-1 and -2 in Berbís et al. 2014), where interactions were only observed with the pSer6 peptide and not at all with the non-phosphorylated 20mer peptide. This difference could be explained by use of higher concentrations of the two mentioned peptides (2 mM) that could accentuate effects from weak binding interactions and/or by more preferred interactions between the CRD and other segments in the longer, more folded NT in full-length Gal-3. Further analysis comparing HSQC data from pGal-3 and Gal-3 reveals that, while Δδ values are largest for residues around pSer6 at the N-terminus (Figure 8D) as discussed above, the Δδ distribution within the CRD (Figure 8E, expanded from Figure 8D) shows some significant differences, albeit small, within the canonical carbohydrate-binding site on the S-face. This suggests that the presence of the phosphate group at Ser6 may have some influence on ligand binding to the lectin.
Intermolecular interactions between Gal-3 molecules
In addition to providing insight into intramolecular reactivity profiles, HSQC spectral analysis can also help elucidate an intriguing property of Gal-3, namely reversible formation of complexes ranging from dimer to higher-order homo-oligomers. It is generally accepted that at low concentration Gal-3 is a monomer which at higher concentrations can aggregate. This has been measured for example by chemical cross-linking, dynamic light scattering, electron microscopy, fluorescence correlation spectroscopy (FCS) and ultracentrifugation (Hsu et al. 1992; Ochieng et al. 1993; Mehul et al. 1994; Birdsall et al. 2001; Morris et al. 2004; Göhler et al. 2010; Lepur et al. 2012; Halimi et al. 2014). Here, we used pulsed field gradient (PFG) NMR self-diffusion experiments to measure Gal-3 diffusion coefficients at 30°C over a concentration range of 42–420 µM. At lower concentrations, the diffusion coefficients for ligand-free Gal-3 and lactose-loaded Gal-3 are 1.1 × 10−6 and 1.17 × 10−6 cm2/s, respectively. Similar values have been reported for an even lower concentration of Gal-3 of ∼1 µM by FCS (Göhler et al. 2010). A gradual decrease in these diffusion coefficients was measured by PFG NMR at Gal-3 concentrations above ∼100 µM. At 420 µM Gal-3, diffusion coefficients were 0.88 × 10−6 and 0.89 × 10−6 cm2/s for ligand-free and ligand-loaded lectin, respectively. Assuming a diffusion coefficient of ∼0.67 × 10−6 cm2/s for dimeric Gal-3 based on its molecular weight and the Stokes–Einstein spherical model, the weight-averaged quaternary structure falls between that of monomer and dimer. At the lower concentrations of Gal-3 (20–50 µM), we also found that diffusion coefficients for Gal-3 or Gal-3 CRD are not significantly altered by the presence of NT peptides 1–50 or 51–107. This indicates that the free NT does not interact to any great extent with Gal-3 and that Gal-3 self-association is not significantly influenced by their presence.
This tendency for weak association is also observed in HSQC spectra, as shown at 20 µM and 420 µM (Figure 9A). Of particular interest, these data enable us to infer sites of contact between Gal-3 molecules, and thus contribute to our understanding of the relative involvement of NT/CRD in Gal-3 self-aggregation. In this regard, note that, at the higher concentration, resonances from the CRD (and not the NT) are by far the most decreased in intensity or broadened (positive ΔIntensity values). Resonances of the NT are generally increased in intensity or narrowed (negative ΔIntensity values, Figure 9B), consistent with mere displacement of the NT from the F-face of the CRD. This is not observed at the lower Gal-3 concentration, where aggregation is absent and signals from both CRD and NT display similar signal to noise ratios (Figure 9A). These data tell us that intermolecular Gal-3 interactions occur between their CRDs and apparently not between their NTs under conditions of this experiment.
Fig. 9.

(A) 15N–1H HSQC spectra (1H frequency of 700 MHz) obtained with 15N-labeled Gal-3 at 20 µM (blue) and 15N-labeled Gal-3 at 420 µM (red) are superimposed. As discussed in the text, Gal-3 is essentially monomeric at 20 µM, and oligomerizes at increased concentrations. At 20 µM, line widths are rather uniform and narrow for both CRD and NT, unlike those at 420 µM that are differentially broadened. In these presentations, HSQC contour levels were adjusted to accentuate the resonance broadening observed at 420 µM (blue) and to view contours at 20 µM (red). Overall, all resonances are observed at both concentrations. The insert shows an expansion of a region from these overlays of HSQC spectra. (B) Intensity changes (ΔI) of CRD resonances in full-length Gal-3 at 20 and 420 µM shown in (A) are plotted vs. the amino acid sequence of Gal-3. Intensity changes (ΔIntensity) were calculated as (1 − Inti/Into), where Inti is the resonance intensity at 420 µM Gal-3 and Into is the resonance intensity at 20 µM Gal-3. For this comparison, intensities in both spectra were first normalized to the intensities of NH resonances from the first 10 NT residues. (C) Chemical shift differences of CRD resonances in full-length Gal-3 at 20 and 420 µM shown in (A) are plotted vs. the amino acid sequence of Gal-3. Residues showing the largest chemical shift differences (>0.011 ppm) are highlighted in orange on the structure of Gal-3 CRD (insert, PDB code 3ZSJ; Seetharaman et al. 1998). Solution conditions are described in Materials and methods. In addition, 25 mM lactose was added to each sample to neutralize effects from even a small amount of lactose remaining in the higher concentration sample following buffer exchange. This figure is available in black and white in print and in color at Glycobiology online.
1H–15N HSQC chemical shift mapping reveals that shift differences are largest for residues within the F-face of the CRD (Figure 9C, marked in orange on the structure of Gal-3 CRD in the insert). This suggests that the F-face mediates interactions between two CRDs. Furthermore, we observed a decrease in the diffusion coefficient (i.e. greater aggregation) upon raising the temperature from 15 to 40°C (data not shown). This suggests that hydrophobic forces mediate interactions between molecules of Gal-3. Normally, one associates greater intermolecular dissociation with an increase in temperature; however, when hydrophobic forces dominate inter-molecular interactions, association is enhanced as the temperature is increased. In addition, the observation of sharper NT resonances upon Gal-3 aggregation (Figure 9B) is most likely explained by dissociation of the NT into solution when two CRDs associate via their F-faces. Nevertheless, because diffusion coefficients for truncated Gal-3 CRDs (having no NT tails) show no concentration dependence up to ∼500 µM (i.e. no association), it appears that the NT likely does play some role in mediating interactions between CRDs in full-length Gal-3.
Given the complexity of available data on ligand-induced Gal-3 aggregation, e.g. describing different effects between lactose and lacto-N-neo-tetraose (DiLacNAc), oligovalent glycoclusters and the nonavalent glycoprotein asialofetuin (Yang et al. 1998; Ahmad et al. 2004; Lepur et al. 2012; Halimi et al. 2014), we also investigated the effect of DiLacNAc binding on Gal-3 self-association. For this experiment, we used a spacered derivative of this natural tetrasaccharide, in which the CH3 group of the N-acetyl group at the C2 ring position of the reducing end Glc residue was replaced with a trifluoro-methyl group (a potential sensor for binding to galectins using 19F-based NMR spectroscopy (Diercks et al. 2009; Matei et al. 2013) with very minor, if any, influence on affinity) and the reducing end of the sugar is extended by an O1-linked ethyl azide group. HSQC spectra of 15N-labeled Gal-3 in the absence (black peaks) and presence of this ligand (red peaks) are shown overlaid in Figure 10A.
Fig. 10.

(A) Overlays of 15N–1H HSQC spectra (1H frequency of 850 MHz) of 15N-labeled Gal-3 CRD (20 µM) are shown in the absence (black contours) and in the presence of 40 µM DiLacNAc derivative (red contours). Solution conditions are described in Materials and methods. (B) Chemical shift differences (Δδ) of CRD resonances from 15N-labeled full-length Gal-3 (20 µM) in the absence and presence of 40 µM tetrasaccharide are shown vs. the amino acid sequence of Gal-3. (C) Residues showing the largest chemical shift differences are highlighted in red (>1 SD above the average) and orange (between the average and 1 SD) on the structure of the Gal-3 CRD (PDB code 3ZSJ; Seetharaman et al. 1998). Solution conditions are described in Materials and methods. (D) Intensity changes of CRD resonances from 15N-labeled full-length Gal-3 in the absence and presence of tetrasaccharide are shown vs. the amino acid sequence of Gal-3. This figure is available in black and white in print and in color at Glycobiology online.
Whereas some Gal-3 resonances are highly chemically shifted by the presence of the tetrasaccharide, others are not. Spectral effects are indicative of slow chemical exchange on the chemical shift time scale. During the titration, new resonances associated with the ligand-loaded state appear and increase in intensity, as resonances of ligand-free Gal-3 decrease in intensity. Thus, the Kd for this ligand should be in the low micromolar range, consistent with literature data on DiLacNAc and related glycans (Leffler and Barondes 1986; Sato and Hughes 1992; Knibbs et al. 1993; Ahmad et al. 2002; Hirabayashi et al. 2002; Halimi et al. 2014). Chemical shift differences (Δδ) of resonances between ligand-loaded and -free Gal-3 are plotted vs. the amino acid sequence of the lectin in Figure 10B, with residues showing the largest Δδ values highlighted in red (>1 SD above the average) and orange (between the average and 1 SD) on the Gal-3 CRD structure in Figure 10C. Note that the largest Δδ values are observed at residues within the canonical carbohydrate-binding S-face, indicating that DiLacNAc binds to the canonical site for a β-galactoside.
We also observed changes in intensities between unligated and ligated Gal-3 resonances, as shown in Figure 10D. These generally follow trends observed with Δδ values (Figure 10B) and are consistent with slow exchange binding of DiLacNAc to the S-face and consequent effects at the F-face. Because equal populations of ligand-free and -loaded Gal-3 at 20 µM occur at 10 µM DiLacNAc, binding stoichiometry is 1:1 as expected. More importantly, while chemical shift and intensity changes of some resonances from residues within the F-face are relatively large (indicated by F-face-labeled bars in Figure 10B and D), effects at residues within the NT are all very small, if present at all. If the NT were involved in intermolecular Gal-3 interactions, then Δδ and ΔIntensity values would have been much greater for residues within the NT, and they are not. Any such small changes of resonances within the NT may be explained simply by release of the NT from the CRD upon association of Gal-3 CRDs. Thus, our data indicate that ligand-induced Gal-3 self-association occurs weakly and via interactions between CRD F-faces of Gal-3 molecules, consistent with an earlier report by Lepur et al. (2012). This finding is supported by the concentration dependence of HSQC spectra for ligand-free Gal-3, as well as by changes in diffusion coefficients that suggest formation of transient Gal-3 aggregates no larger than dimers. As also mentioned above, however, this does not exclude the possibility that the NT is somehow involved in CRD–CRD interactions.
Conclusions
The primary interactions between the Gal-3 NT and CRD occur between the N-terminal segment of the NT and the F-face of the CRD, with the sequence P23GAW26…P37GASYPGAY45 defining the primary binding epitope within the NT. Along with the remainder of the N-terminal part of the NT (residues 5–18), this segment has the propensity to form α-helix and/or multiple-turn structures, which could help promote binding, albeit relatively weak, to the CRD. Even though phosphorylation of Ser6/12 has little effect on interactions between the NT and CRD F-face, it may allosterically influence carbohydrate binding to the S-face. In addition, although Gal-3 self-association is enhanced by S-face binding of DiLacNAc, the Gal-3 aggregation process occurs primarily via interactions between CRD F-faces, with the NT apparently playing some as yet undefined role in promoting this aggregation event. Overall, this study has advanced our knowledge of Gal-3 structure and will spur the study of interactions of multi-functional Gal-3 with its many in situ binding partners, especially with protein counter-receptors such as Bcl-2, as well as with complex polysaccharides.
Materials and methods
Galectin-3 expression and purification
Full-length human Gal-3 (residues 1–250) and human Gal-3 CRD (residues 108–250) were recombinantly produced with [15N]NH4Cl and/or U-[13C]glucose as medium additive(s) in Escherichia coli BL21 (DE3)-pLysS cells (Promega, Mannheim, Germany) using the cDNA inserted into the pET12a vector platform (Novagen, Darmstadt, Germany) at 30°C with 400 µM IPTG induction for 16 h. The protein was purified by affinity chromatography on lactosylated Sepharose 4B as a crucial step; lactose was removed by several rounds of ultrafiltration and gel filtration (PC-10), and the protein was checked for purity and activity as previously described (Kübler et al. 2008; Sanchez-Ruderisch et al. 2010; Berbís et al. 2014).
Preparation of 15N-enriched NT of Galectin-3
The 15N-enriched N-terminal tail (15N-NT) of Gal-3 was prepared from 15N-enriched His-tagged NT (15N-His-NT) by thrombin digestion. Briefly, cDNA for His-NT was inserted into the pET28a vector between BamHI and EcoRI sites. Escherichia coli BL21 (DE3) cells were transformed with this construct and induced to express protein in minimal medium with 15NH4Cl as the nitrogen source. Bacteria were grown at 37°C for 12 h and induced to express 15N-His-NT by 0.2 mmol/L IPTG at 25°C for 16 h. After centrifugation, bacteria were resuspended in phosphate buffer and lysed by ultrasonication. The cell lysate was incubated with Ni-NTA agrose beads. After extensive washing, the beads with bound 15N His-NT were digested by 2 U/mg thrombin. His-tag-free 15N-NT was then released into the buffer and recovered following dialysis.
Galectin-3 phosphorylation
The 15N-labeled, full-length protein was phosphorylated with constitutively active casein kinase 1 on a preparative scale, and then subjected to mass spectrometric phospho-peptide analysis after enrichment by titanium dioxide nano-column chromatography, as previously described (Kübler et al. 2008; Kaltner et al. 2011).
Peptide synthesis
Peptides used in this study were manually synthesized using tert-butyloxycarbonyl (tBoc)-based solid phase peptide synthesis and then purified by using HPLC as previously described (Schnölzer et al. 1992). Four peptides were produced: (i) NT 1–50, comprising NT amino acid residues 1–50: MADNFSLHDA-LSGSGNPNPQ-GWPGAWGNQP-AGAGGYPGAS-YPGAYPGQAP, (ii) NT 51–107, comprising NT residues 51–107: PGAYPGQAPP-GAYHGAPGAY-PGAPAPGVYP-GPPSGPGAYP-SSGQPSAPGA-YPATGPY, (iii) tetrapeptide PGAY, and (iv) (PGAY)4: PGAYPGAYPGAYPGAY. In short, these peptides were prepared on a 0.2 mmol scale using the in situ neutralization/activation procedure for butyloxycarbonyl-/benzyl- (Boc-/Bzl)-based peptide synthesis using HCTU (N-[1H-6-chlorobenzotriazol-1-yl)-(dimethylamino)methylene] N-methylmethanaminium hexafluorophosphate N-oxide) instead of HBTU (2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) as a coupling reagent. MBHA (p-methylbenzhydrylamine) resin (1 meq/g) was used as the solid support. In the case of NT 1–50, glycines were replaced by 15N-glycines (Sigma).
After synthesis, the resin-bound protected peptide was treated with 4 mL mercaptoethanol/2 mL DIPEA (diisopropylethyl-amine) in 14 mL DMF (dimethlformamide) (2 × 30 min) to remove the Dnp protecting group from histidine. Next the resin-bound peptide was treated with TFA (trifluoroacetic acid) (2 × 1 min) to remove the N-terminal Boc group, then washed sequentially with DMF, DCM and 1:1 (v/v) DCM/MeOH and dried. Thereafter, the peptides were deprotected and cleaved from the resin by treatment with anhydrous HF for 1 h at 0°C, using 4 v% p-cresol as a scavenger. Following cleavage, the peptides were precipitated with ice-cold diethylether, dissolved in 0.1 M sodium acetate buffer (pH 4) containing 6 M Gn.HCl, and purified by preparative HPLC using a Vydac C-18 column (250 × 10 mm, 10 µm) and a linear gradient of acetonitrile in water/0.1%TFA (flow rate of 5 mL/min; 0.2% B/min). Fractions containing the product were identified by ESI-MS, pooled and lyophilized. ESI-MS was performed on an Applied Biosystems SCIEX API 150 EX electrospray ionization quadrupole (ESI-Q) mass spectrometer. Peptide masses were calculated from the experimental mass-to-charge (m/z) ratios of all the protonation states observed in the ESI-MS spectrum of a peptide using Analyst 1.4.2 software (SCIEX).
Chemical synthesis of the DiLacNAc derivative
General procedures
Unless noted, chemical reagents and solvents were used without further purification from commercial sources. Dry solvents as CH2Cl2, Et2O and THF were obtained from a PureSolv-EN™ solvent purification system (Innovation Technology Inc.). Concentration in vacuo was performed using a Buchi rotary evaporator. The 1H/13C/19F NMR spectra (δ in ppm, relative to TMS in CDCl3) were recorded using Varian spectrometers (Varian, Palo Alto, CA) (400/101 or 500/125 MHz) at 25°C. Assignments were aided by 1H–1H and 1H–13C correlation experiments. HRMS spectra were recorded on a micromass LCT instrument from Waters. LRMS spectra were recorded on a Waters micromass Quattro Micro LC–MS/MS instrument using electrospray ionization (ESI) in either positive or negative mode. Optical rotations were recorded on a Perkin-Elmer polarimeter (Model 343) at the sodium D-line (589 nm) at 20°C using a 1 dm cell and are not corrected. Silica gel chromatography was carried out using Davisil LC60A (Grace Tech., Columbia, MD) SiO2 (40–63 µm) silica gel. All reactions were monitored by thin-layer chromatography (TLC). TLC was performed on Merck DC-Alufolien plates precoated with silica gel 60 F254. They were visualized with UV-light (254 nm) fluorescence quenching, and/or by charring with an 8% H2SO4 dip and/or ninhydrin dip. Deprotected sugars were lyophilized using a freeze-dryer Alpha 1–2 Ldplus (Christ Ltd.), with a pressure of 0.035 mbar and ice condenser temperature −55°C.
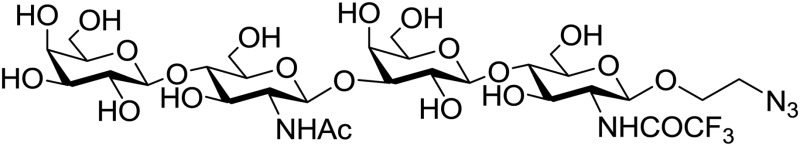
Complete experimental details and reaction schemes for the synthesis of the spacered tetrasaccharide are provided in Supplementary Material. Its chemical structure is shown below.
NMR spectroscopy
NMR experiments were performed at 30°C on Bruker Ascend 700 MHz or Bruker Avance III 700 or 850 NMR spectrometer, each equipped with a cryogenically cooled z-gradient TXI probe. Samples for 1H–15N HSQC experiments contained 15N-labeled human Gal-3 CRD or 15N-labeled full-length human Gal-3 at certain concentrations between 20 and 420 µM, and were prepared in 20 mM potassium phosphate buffer (pH 6.9) made out of 96% H2O plus 4% D2O for the field-frequency lock. Lactose, still present from affinity elution during purification, was removed by extensive filtration over Amicon (EMD Millipore, Darmstadt, Germany) Ultra-4 ultracentrifugation filters with a molecular cut off of 3 kDa (applying five to six subsequent buffer subsequent exchange steps starting from 4.75 mL volume and concentrating down to 250 µL). Resonance perturbations (chemical shift and resonance broadening differences) were monitored using sequence-specific heteronuclear (1H, 15N, 13C) assignments for full-length human Gal-3 (Ippel et al. 2015) and 1H and 15N assignments for Gal-3 CRD (Umemoto and Leffler 2001).
Pulsed field gradient (PFG) measurements on Gal-3 were carried out at 30°C and 700 MHz using the Bruker pulse sequence stebpg1s19 with stimulated echo, bipolar gradients and Watergate solvent suppression.
Processing of NMR data was performed using the Bruker Topspin 3.2 program (Rheinstetten, Germany), and Sparky 3.114 software (Goddard and Kneller 1995) was used to analyze 2D and 3D spectra. Peak integration of 1D spectra was carried out using Topspin, whereas PFG data were fitted and analyzed with Word Excel macros.
Chemical shifts were internally referenced to DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid), and chemical shift differences (Δδ) were calculated as [(Δ1H)2 + (0.25Δ15N)2]1/2. Intensity changes (ΔIntensity) were calculated as (1 − Inti/Into), where Inti is the resonance intensity at some new condition (addition of peptide or more Gal-3) and Into is the resonance intensity initially. A value of 0 indicates the absence of resonance broadening, and a value of 1 indicates that the resonance is no longer observable. A negative value indicates that resonances have increased in intensity, i.e. become more narrow. When conditions between samples (e.g. concentrations) differed, intensities in both spectra were first normalized to intensities of resonances from the first 10 N-terminal residues.
Supplementary data
Supplementary data for this article are available online at http://glycob.oxfordjournals.org/.
Funding
Funding for NMR instrumentation at the University of Minnesota was provided by the Office of the Vice President for Research, the Medical School, the College of Biological Sciences, NIH, NSF and the Minnesota Medical Foundation. H.L. was funded in part by the Kunst and Alice Wallenberg Foundation, Sweden, and H.-J.G.by the Verein zur Förderung des biologisch-technologischen Fortschritts in der Medizin e.V. (Heidelberg, Germany). Generous financial support by the ITN network GLYCOPHARM (contract no. 317297; to J.K., J.J.-B., S.O. and H.-J.G.) is gratefully acknowledged, as are inspiring discussions with Drs. B. Friday, M. Ilsüm and A. Leddoz as well as the valuable recommendations of both reviewers.
Conflict of interest statement
None declared.
Abbreviations
CRD, carbohydrate recognition domain; FCS, fluorescence correlation spectroscopy; Gal-1, galectin-1; Gal-3, galectin-3; pGal-3, phosphorylated Gal-3; Gal-7, galectin-7; HSQC, heteronuclear single quantum coherence; NMR, nuclear magnetic resonance; NOE(SY), nuclear Overhauser effect spectroscopy; NT, N-terminal tail; PFG, pulsed field gradient.
Supplementary Material
References
- Agrwal N, Sun Q, Wang SY, Wang JL. 1993. Carbohydrate-binding protein 35. I. Properties of the recombinant polypeptide and the individuality of the domains. J Biol Chem. 268:14932–14939. [PubMed] [Google Scholar]
- Ahmad N, Gabius H-J, André S, Kaltner H, Sabesan S, Roy R, Liu B, Macaluso F, Brewer CF. 2004. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 279:10841–10847. [DOI] [PubMed] [Google Scholar]
- Ahmad N, Gabius H-J, Kaltner H, André S, Kuwabara I, Liu F-T, Oscarson S, Norberg T, Brewer CF. 2002. Thermodynamic binding studies of cell surface carbohyrate epitopes to galectins-1, -3, and -7: Evidence for differential binding specificities. Can J Chem. 80:1096–1104. [Google Scholar]
- Barboni EA, Bawumia S, Henrick K, Hughes RC. 2000. Molecular modeling and mutagenesis studies of the N-terminal domains of galectin-3: Evidence for participation with the C-terminal carbohydrate recognition domain in oligosaccharide binding. Glycobiology. 10:1201–1208. [DOI] [PubMed] [Google Scholar]
- Berbís MA, André S, Cañada FJ, Pipkorn R, Ippel H, Mayo KH, Kübler D, Gabius H-J, Jiménez-Barbero J. 2014. Peptides derived from human galectin-3 N-terminal tail interact with its carbohydrate recognition domain in a phosphorylation-dependent manner. Biochem Biophys Res Commun. 443:126–131. [DOI] [PubMed] [Google Scholar]
- Birdsall B, Feeney J, Burdett ID, Bawumia S, Barboni EA, Hughes RC. 2001. NMR solution studies of hamster galectin-3 and electron microscopic visualization of surface-adsorbed complexes: Evidence for interactions between the N- and C-terminal domains. Biochemistry. 40:4859–4866. [DOI] [PubMed] [Google Scholar]
- Cooper DNW. 2002. Galectinomics: Finding themes in complexity. Biochim Biophys Acta. 1572:209–231. [DOI] [PubMed] [Google Scholar]
- Corfield AP, Berry M. 2015. Glycan variation and evolution in the eukaryotes. Trends Biochem Sci. 40:351–359. [DOI] [PubMed] [Google Scholar]
- Dawson H, André S, Karamitopoulou E, Zlobec I, Gabius H-J. 2013. The growing galectin network in colon cancer and clinical relevance of cytoplasmic galectin-3 reactivity. Anticancer Res. 33:3053–3059. [PubMed] [Google Scholar]
- Diehl C, Engstrom O, Delaine T, Hakansson M, Genheden S, Modig K, Leffler H, Ryde U, Nilsson UJ, Akke M. 2010. Protein flexibility and conformational entropy in ligand design targeting the carbohydrate recognition domain of galectin-3. J Am Chem Soc. 132:14577–14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diercks T, Ribeiro JP, Canada FJ, André S, Jiménez-Barbero J, Gabius H-J. 2009. Fluorinated carbohydrates as lectin ligands: Versatile sensors in 19F-detected saturation transfer difference NMR spectroscopy. Chem Eur J. 15:5666–5668. [DOI] [PubMed] [Google Scholar]
- Flores-Ibarra A, Ruiz FM, Vértesy S, André S, Gabius H-J, Romero A. 2015. Preliminary X-ray crystallographic analysis of an engineered variant of human chimera-type galectin-3 with a shortened N-terminal domain. Acta Crystallogr F. 71:184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funasaka T, Raz A, Nangia-Makker P. 2014. Nuclear transport of galectin-3 and its therapeutic implications. Semin Cancer Biol. 27:30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabius H-J. 2015. The magic of the sugar code. Trends Biochem Sci. 40:341. [DOI] [PubMed] [Google Scholar]
- Gabius H-J, André S, Jiménez-Barbero J, Romero A, Solís D. 2011. From lectin structure to functional glycomics: Principles of the sugar code. Trends Biochem Sci. 36:298–313. [DOI] [PubMed] [Google Scholar]
- Gabius H-J, Kaltner H, Kopitz J, André S. 2015. The glycobiology of the CD system: A dictionary for translating marker designations into glycan/lectin structure and function. Trends Biochem Sci. 40:360–376. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. 1995. Sparky 3. San Francisco: University of California. [Google Scholar]
- Göhler A, André S, Kaltner H, Sauer M, Gabius H-J, Doose S. 2010. Hydrodynamic properties of human adhesion/growth-regulatory galectins studied by fluorescence correlation spectroscopy. Biophys J. 98:3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halimi H, Rigato A, Byrne D, Ferracci G, Sebban-Kreuzer C, ElAntak L, Guerlesquin F. 2014. Glycan dependence of galectin-3 self-association properties. PLoS ONE. 9:e111836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haudek KC, Spronk KJ, Voss PG, Patterson RJ, Wang JL, Arnoys EJ. 2010. Dynamics of galectin-3 in the nucleus and cytoplasm. Biochim Biophys Acta. 1800:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennet T, Cabalzar J. 2015. Congenital disorders of glycosylation: A concise chart of glycocalyx dysfunction. Trends Biochem Sci. 40:377–384. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J, Hashidate T, Arata Y, Nishi N, Nakamura T, Hirashima M, Urashima T, Oka T, Futai M, Müller WEG et al. 2002. Oligosaccharide specificity of galectins: A search by frontal affinity chromatography. Biochim Biophys Acta. 1572:232–254. [DOI] [PubMed] [Google Scholar]
- Houzelstein D, Gonçalves IR, Fadden AJ, Sidhu SS, Cooper DNW, Drickamer K, Leffler H, Poirier F. 2004. Phylogenetic analysis of the vertebrate galectin family. Mol Biol Evol. 21:1177–1187. [DOI] [PubMed] [Google Scholar]
- Hrynchyshyn N, Jourdain P, Desnos M, Diebold B, Funck F. 2013. Galectin-3: A new biomarker for the diagnosis, analysis and prognosis of acute and chronic heart failure. Arch Cardiovasc Dis. 106:541–546. [DOI] [PubMed] [Google Scholar]
- Hsu DK, Zuberi RI, Liu F-T. 1992. Biochemical and biophysical characterization of human recombinant IgE-binding protein, an S-type animal lectin. J Biol Chem. 267:14167–14174. [PubMed] [Google Scholar]
- Huflejt ME, Turck CW, Lindstedt R, Barondes SH, Leffler H. 1993. L-29, a soluble lactose-binding lectin, is phosphorylated on serine 6 and serine 12 in vivo and by casein kinase I. J Biol Chem. 268:26712–26718. [PubMed] [Google Scholar]
- Hughes RC. 1994. Mac-2: A versatile galactose-binding protein of mammalian tissues. Glycobiology. 4:5–12. [DOI] [PubMed] [Google Scholar]
- Hughes RC. 1999. Secretion of the galectin family of mammalian carbohydrate-binding proteins. Biochim Biophys Acta. 1473:172–185. [DOI] [PubMed] [Google Scholar]
- Ippel H, Miller MC, Berbís MA, Suylen D, André S, Hackeng TM, Canada FJ, Weber C, Gabius H-J, Jiménez-Barbero J et al. 2015. 1H, 13C, and 15N backbone and side-chain chemical shift assignments for the 36 proline-containing, full length 29 kDa human chimera-type galectin-3. Biomol NMR Assign. 9:59–63. [DOI] [PubMed] [Google Scholar]
- Kaltner H, Gabius H-J. 2012. A toolbox of lectins for translating the sugar code: The galectin network in phylogenesis and tumors. Histol Histopathol. 27:397–416. [DOI] [PubMed] [Google Scholar]
- Kaltner H, Kübler D, López-Merino L, Lohr M, Manning JC, Lensch M, Seidler J, Lehmann WD, André S, Solís D et al. 2011. Toward comprehensive analysis of the galectin network in chicken: Unique diversity of galectin-3 and comparison of its localization profile in organs of adult animals to the other four members of this lectin family. Anat Rec. 294:427–444. [DOI] [PubMed] [Google Scholar]
- Knibbs RN, Agrwal N, Wang JL, Goldstein IJ. 1993. Carbohydrate-binding protein 35. II. Analysis of the interaction of the recombinant polypeptide with saccharides. J Biol Chem. 268:14940–14947. [PubMed] [Google Scholar]
- Kopitz J, André S, von Reitzenstein C, Versluis K, Kaltner H, Pieters RJ, Wasano K, Kuwabara I, Liu F-T, Cantz M et al. 2003. Homodimeric galectin-7 (p53-induced gene 1) is a negative growth regulator for human neuroblastoma cells. Oncogene. 22:6277–6288. [DOI] [PubMed] [Google Scholar]
- Kopitz J, Vértesy S, André S, Fiedler S, Schnölzer M, Gabius H-J. 2014. Human chimera-type galectin-3: Defining the critical tail length for high-affinity glycoprotein/cell surface binding and functional competition with galectin-1 in neuroblastoma cell growth regulation. Biochimie. 104:90–99. [DOI] [PubMed] [Google Scholar]
- Kübler D, Hung C-W, Dam TK. 2008. Phosphorylated human galectin-3: Facile large-scale preparation of active lectin and detection of structural changes by CD spectroscopy. Biochim Biophys Acta. 1780:716–722. [DOI] [PubMed] [Google Scholar]
- Laine RA. 1997. The information-storing potential of the sugar code. In: Gabius H-J, Gabius S, editors. Glycosciences: Status and Perspectives. London-Weinheim: Chapman & Hall; p. 1–14. [Google Scholar]
- Ledeen RW, Wu G. 2015. The multi-tasked life of GM1 ganglioside, a true factotum of life. Trends Biochem Sci. 40:407–418. [DOI] [PubMed] [Google Scholar]
- Leffler H, Barondes SH. 1986. Specificity of binding of three soluble rat lung lectins to substituted and unsubstituted mammalian beta-galactosides. J Biol Chem. 261:10119–10126. [PubMed] [Google Scholar]
- Leffler H, Carlsson S, Hedlund M, Qian Y, Poirier F. 2004. Introduction to galectins. Glycoconj J. 19:433–440. [DOI] [PubMed] [Google Scholar]
- Lepur A, Carlsson MC, Novak R, Dumic J, Nilsson UJ, Leffler H. 2012. Galectin-3 endocytosis by carbohydrate independent and dependent pathways in different macrophage like cell types. Biochim Biophys Acta. 1820:804–818. [DOI] [PubMed] [Google Scholar]
- Li S, Yu Y, Koehn CD, Zhang Z, Su K. 2013. Galectins in the pathogenesis of rheumatoid arthritis. J Clin Cell Immunol. 4:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, D'Ambrosio M, Liao Td, Peng H, Rhaleb NE, Sharma UC, André S, Gabius H-J, Carrataro OA. 2009. N-Acetyl-seryl-aspartyl-lysyl-proline prevents cardiac remodeling and dysfunction induced by galectin-3, a mammalian adhesion/growth-regulatory lectin. Am J Physiol Heart Circ Physiol. 296:H404–H412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsanov YD, Rini JM. 1997. Galectin structure. Trends Glycosci Glycotechnol. 9:145–154. [Google Scholar]
- Matei E, André S, Glinschert A, Infantino AS, Oscarson S, Gabius H-J, Gronenborn AM. 2013. Fluorinated carbohydrates as lectin ligands: Dissecting glycan-cyanovirin interactions by using 19F-NMR spectroscopy. Chem Eur J. 19:5364–5374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehul B, Bawumia S, Martin SR, Hughes RC. 1994. Structure of baby hamster kidney carbohydrate-binding protein CBP30, an S-type animal lectin. J Biol Chem. 269:18250–18258. [PubMed] [Google Scholar]
- Mey A, Leffler H, Hmama Z, Normier G, Revillard JP. 1996. The animal lectin galectin-3 interacts with bacterial lipopolysaccharides via two independent sites. J Immunol. 156:1572–1577. [PubMed] [Google Scholar]
- Miller MC, Ippel H, Suylen D, Klyosov AA, Traber PG, Hackeng T, Mayo KH. 2016. Binding of polysaccharides to human galectin-3 at a non-canonical site in its carbohydrate recognition domain. Glycobiology. 26:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MC, Klyosov A, Mayo KH. 2009. The α-galactomannan Davanat binds galectin-1 at a site different from the conventional galectin carbohydrate binding site. Glycobiology. 19:1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MC, Klyosov A, Mayo KH. 2012. Structural features for α-galactomannan binding to galectin-1. Glycobiology. 22:543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S, Ahmad N, André S, Kaltner H, Gabius H-J, Brenowitz M, Brewer CF. 2004. Quaternary solution structures of galectins-1, -3 and -7. Glycobiology. 14:293–300. [DOI] [PubMed] [Google Scholar]
- Ochieng J, Platt D, Tait L, Hogan V, Raz T, Carmi P, Raz A. 1993. Structure-function relationship of recombinant human galactoside-binding protein. Biochemistry. 32:4455–4460. [DOI] [PubMed] [Google Scholar]
- Sanchez-Ruderisch H, Fischer C, Detjen KM, Welzel M, Wimmel A, Manning JC, André S, Gabius H-J. 2010. Tumor suppressor p16INK4a: Downregulation of galectin-3, an endogenous competitor of the pro-anoikis effector galectin-1, in a pancreatic carcinoma model. FEBS J. 277:3552–3563. [DOI] [PubMed] [Google Scholar]
- Saraboji K, Hakansson M, Genheden S, Diehl C, Qvist J, Weininger U, Nilsson UJ, Leffler H, Ryde U, Akke M et al. 2012. The carbohydrate-binding site in galectin-3 is preorganized to recognize a sugar-like framework of oxygens: Ultra-high-resolution structures and water dynamics. Biochemistry. 51:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Hughes RC. 1992. Binding specificity of a baby hamster kindey lectin for H type I and II chains, polylactosamine glycans, and appropriately glycosylated forms of laminin and fibronectin. J Biol Chem. 267:6983–6990. [PubMed] [Google Scholar]
- Schengrund C-L. 2015. Gangliosides: Glycosphingolipids essential for normal neural development and function. Trends Biochem Sci. 40:397–406. [DOI] [PubMed] [Google Scholar]
- Schnölzer M, Alewood P, Jones A, Alewood D, Kent SBH. 1992. In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high-yield assembly of difficulty sequences. Int J Pept Protein Res. 40:180–193. [DOI] [PubMed] [Google Scholar]
- Seetharaman J, Kanigsberg A, Slaaby R, Leffler H, Barondes SH, Rini JM. 1998. X-ray crystal structure of the human galectin-3 carbohydrate recognition domain at 2.1-Å resolution. J Biol Chem. 273:13047–13052. [DOI] [PubMed] [Google Scholar]
- Selenko P, Frueh DP, Elsaesser SJ, Haas W, Gygi SP, Wagner G. 2008. In situ observation of protein phosphorylation by high-resolution NMR spectroscopy. Nat Struct Mol Biol. 15:321–329. [DOI] [PubMed] [Google Scholar]
- Solís D, Bovin NV, Davis AP, Jiménez-Barbero J, Romero A, Roy R, Smetana K Jr, Gabius H-J. 2015. A guide into glycosciences: How chemistry, biochemistry and biology cooperate to crack the sugar code. Biochim Biophys Acta. 1850:186–235. [DOI] [PubMed] [Google Scholar]
- Spiro RG. 2002. Protein glycosylation: Nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 12:43R–56R. [DOI] [PubMed] [Google Scholar]
- Takenaka Y, Fukumori T, Yoshii T, Oka N, Inohara H, Kim H-RC, Bresalier RS, Raz A. 2004. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol. 24:4395–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toegel S, Bieder D, André S, Kayser K, Walzer SM, Hobusch G, Windhager R, Gabius H-J. 2014. Human osteoarthritic knee cartilage: Fingerprinting of adhesion/growth-regulatory galectins in vitro and in situ indicates differential upregulation in severe degeneration. Histochem Cell Biol. 142:373–388. [DOI] [PubMed] [Google Scholar]
- Tsay YG, Lin NY, Voss PG, Patterson RJ, Wang JL. 1999. Export of galectin-3 from nuclei of digitonin-permeabilized mouse 3T3 fibroblasts. Exp Cell Res. 252:250–261. [DOI] [PubMed] [Google Scholar]
- Umemoto K, Leffler H. 2001. Assignment of 1H, 15N and 13C resonances of the carbohydrate recognition domain of human galectin-3. J Biomol NMR. 20:91–92. [DOI] [PubMed] [Google Scholar]
- Umemoto K, Leffler H, Venot A, Valafar H, Prestegard JH. 2003. Conformational differences in liganded and unliganded states of galectin-3. Biochemistry. 42:3688–3695. [DOI] [PubMed] [Google Scholar]
- Woo H-J, Shaw LM, Messier JM, Mercurio AM. 1990. The major non-integrin laminin-binding protein of macrophages is identical to carbohydrate-binding protein 35 (Mac-2). J Biol Chem. 265:7097–7099. [PubMed] [Google Scholar]
- Yang R-Y, Hill PN, Hsu DK, Liu F-T. 1998. Role of the carboxyl-terminal lectin domain in self-association of galectin-3. Biochemistry. 37:4086–4092. [DOI] [PubMed] [Google Scholar]
- Zuber C, Roth J. 2009. N-Glycosylation. In: Gabius H-J, editor. The Sugar Code. Fundamentals of Glycosciences. Weinheim, Germany: Wiley-VCH; p. 87–110. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.