

Abstract
Epacs (exchange proteins directly activated by cyclic AMP [cAMP]) act as downstream effectors of cAMP and play important roles in energy balance and glucose homeostasis. While global deletion of Epac1 in mice leads to heightened leptin sensitivity in the hypothalamus and partial protection against high-fat diet (HFD)-induced obesity, the physiological functions of Epac1 in white adipose tissue (WAT) has not been explored. Here, we report that adipose tissue-specific Epac1 knockout (AEKO) mice are more prone to HFD-induced obesity, with increased food intake, reduced energy expenditure, and impaired glucose tolerance. Despite the fact that AEKO mice on HFD display increased body weight, these mice have decreased circulating leptin levels compared to their wild-type littermates. In vivo and in vitro analyses further reveal that suppression of Epac1 in WAT decreases leptin mRNA expression and secretion by inhibiting cAMP response element binding (CREB) protein and AKT phosphorylation, respectively. Taken together, our results demonstrate that Epac1 plays an important role in regulating energy balance and glucose homeostasis by promoting leptin expression and secretion in WAT.
INTRODUCTION
Obesity has become a severe public health problem, and its prevalence has increased dramatically since the 1970s. In the United States, more than one-third of adults and approximately 17% of children are obese, defined as a body mass index (BMI) of 30 or above. It is projected that by 2030, more than 50% of the U.S. population will be obese (1). This dire outlook led the American Medical Association to officially recognize obesity as a disease. In addition to its direct health problems, obesity is also closely associated with other major human diseases, such as diabetes and cardiovascular diseases (2, 3). Despite its overwhelming prevalence and major health burdens, we know little about the molecular etiology of obesity and even less about its cures. At the physiology level, one major factor leading to the development of obesity is a chronic imbalance of energy homeostasis, which results in the accumulation of the excessive energy intake as fat in tissues, especially in adipocytes.
Adipose tissue not only is the main reservoir for fat storage but also is an important endocrine organ that secrets various adipose-derived hormones, called adipokines. As one of the most important and widely studied adipokines, leptin plays a critical role in energy balance, mainly by acting on receptors in the arcuate nucleus of the hypothalamus to suppress appetite and to increase energy expenditure (4, 5). While the circulating leptin level is proportional to the total body fat mass in rodents and human, in obese patients a reduced sensitivity to leptin leads to a failure in suppressing food intake despite increased fat stores and blood leptin levels. This apparent condition of leptin resistance is one major driver in developing obesity (6).
Recent studies have implicated the involvement of Epac1 (exchange protein directly activated by cyclic AMP [cAMP])-mediated signaling in leptin resistance (7, 8). As a major intracellular sensor of the second messenger cAMP, pharmacological activation of Epac proteins, but not protein kinase A (PKA), dampens leptin signaling in mice hypothalamus by reducing signal transducer and activator of transcription 3 (STAT3) phosphorylation levels (7). Consistent with the pharmacological studies, global Epac1 knockout mice exhibit increased leptin sensitivity, decreased food intake, and overall adiposity despite reduced serum leptin levels (9). These findings establish the importance of Epac1 in leptin signaling and energy homeostasis.
Epac1 is ubiquitously expressed (10), with abundant expression both in the adipose tissue and the central nervous system (CNS), which represents the main source of leptin production and the major target of leptin signaling, respectively. To date, it is not clear if the apparent phenotypes of Epac1 null mice are due to the loss of Epac1 functions specifically in peripheral adipose tissues, in CNS, or both. In this study, we investigate the adipose tissue-specific functions in energy balance and leptin signaling using adipose tissue Epac1-specific knockout mice.
MATERIALS AND METHODS
Reagents and cell lines.
EPAC selective agonists 8-(4-chlorophenylthio)-2-O-methyladenosine-3′,5′-cyclic monophosphate [007]) and its prodrug, acetoxymethyl ester (007-AM), with increased membrane permeability (11, 12), as well as 8-(4-methoxyphenylthio)-2′-O-methyladenosine-3′,5′-cyclic monophosphate (8-pMeOPT-2′-O-Me-cAMP) were purchased from Biolog Life Science Institute (Bremen, Germany). 3-(5-tert-butyl-isoxazol-3-yl)-2-[(3-chloro-phenyl)-hydrazono]-3-oxo-pro-pionitrile (ESI-09) is a selective EPAC inhibitor recently identified in our laboratory (13, 14). MK-2206, 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-2H-[1,2,4]triazolo[3,4-f][1,6]naphthyridin-3-one-di-hydrochloride, a selective AKT inhibitor, was from Selleck Chemicals, LLC. Antibodies specific against EPAC1 (4155), phosphorylated AKT serine 473 (9271), and phosphorylated cAMP response element binding protein (CREB) serine 133 (9198) were from Cell Signaling Technology. The 3T3-L1 mouse preadipocyte and human embryonic kidney 293 (HEK293) cell lines were obtained from the ATCC.
Mice.
Epac1+/LoxP neo mice with two LoxP sites flanking exons 3 and 5 of the Epac1 gene, generated as described previously (9), were crossed with FLPase deleter mice (15), which delete DNA sequences flanked by two FLP recombination target sites in all cell types. This resulted in the generation of Epac1+/LoxP mice with a floxed Epac1 allele no longer containing the neo cassette. Epac1+/LoxP mice were back crossed to the C57BL/6 background for more than 12 generations and then intercrossed to generate Epac1LoxP/LoxP homozygous mice. The transgenic Cre line, driven by the adipocyte-specific aP2/Fabp4 promoter [B6.Cg-Tg(Fabp4-cre)1Rev/J; JAX stock no. 005069], was obtained from Jackson Laboratory (16). Homozygous Epac1LoxP/LoxP mice were crossed with aP2-Cre mice to generate the adipose-specific Epac1LoxP/LoxP::αP2-Cre+/− progeny, referred to as AEKO. The Epac1LoxP/LoxP littermates without Cre gene expression were used as controls and are referred to as the wild type (WT). The mice were housed on a 12-h/12-h light-dark cycle in virus-free facilities with free access to food and water. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Texas Medical Branch or the University of Texas Health Science Center at Houston.
HFD studies.
Male AEKO and WT littermates were used. Mice were initially fed a chow diet (3.1 kcal/g, with 17% of calories from fat; Teklad 7912) and continued on the chow diet or switched to a high-fat diet (HFD; 5.24 kcal/g, with 60% of calories from fat; Research Diet D12492) on postnatal day 24. Food intake and body weight were examined daily and every 4 days, respectively.
Energy expenditure study using CLAMS.
This study was performed using a comprehensive laboratory animal monitoring system (CLAMS; Columbus Instruments) as described previously (17). Briefly, 5- to 6-week-old age- and sex-matched mice (n = 6 for each group) were housed individually in the chambers of CLAMS with free access to water and food at room temperature. The animals were allowed to become accustomed to their cages for 2 days, followed by 4 days of data collection. Mice were initially fed a chow diet and switched to HFD for the last 2 days. Food intake, O2 consumption, CO2 production, body weight, and beam break data were collected. Energy expenditure was calculated from oxygen consumption by indirect calorimetry.
Plasma leptin, insulin, and adiponectin measurements.
The plasma leptin concentration was determined by R&D Quantikine mouse leptin immunoassay kits (R&D Systems, MN). The plasma insulin concentration was measured by an ultrasensitive mouse insulin enzyme-linked immunosorbent assay (ELISA) kit (Crystal Chem Inc.). The plasma adiponectin concentration was monitored using the Quantikine mouse adiponectin/Acrp30 immunoassay kit supplied by R&D Systems.
Epac1 knockout in 3T3-L1 cells using clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 gene editing system.
An online design tool (http://crispr.mit.edu/) was used to identify the optimal single guide RNA (sgRNA) sequences. Two different sgRNA sequences with the highest scores were picked: Oligo 1, GTCATCTCCCTCGTGCAACGTGG, and Oligo 2, GCGGCTAGTTGGCCGATGGGTGG. These two oligonucleotides were cloned into the pLKO vector. The 3T3-L1 cells were transfected with pHAGE-EF1a-Cas9-IRES-BLAST plasmid with Lipofectamine 2000. Blasticidin was used to select cells stably expressing Cas9. Cas9-expressing 3T3-L1 cells then were transfected with Epac1-specific sgRNAs to knock out Epac1. Epac1 knockout cells were selected by puromycin and confirmed by quantitative real-time PCR (RT-qPCR) and Western blotting.
Rap1GAP transient transfection.
pFLAG-CMV2-Rap1GAP (18) or control vector was transfected into 3T3-L1 preadipocytes by electroporation according to the manufacturer's protocol established for 3T3-L1 cells using a Neon transfection system (Invitrogen). Thirty-six hours posttransfection, the cells were starved in serum-free Dulbecco's modified Eagle's medium (DMEM) for 4 h and treated with 007-AM at 10 μM or dimethyl sulfoxide (DMSO) vehicle for 25 min. After rinsing with phosphate-buffered saline (PBS), the cells were lysed in SDS sample buffer. Cell lysates with equal amounts of total proteins were load onto SDS-PAGE gels and subjected to immunoblotting analysis.
In vitro leptin secretion assay.
3T3-L1 cells were used for adipocyte differentiation as described previously (9). Leptin secretion assay was performed using differentiated 3T3-L1 adipocytes as described previously (19). Briefly, 3T3-L1 preadipocytes were maintained at 37°C with 5% CO2 in DMEM plus 10% fetal bovine serum (FBS). Adipocyte differentiation was induced 2 days after the cells became 100% confluent with a cocktail of 5 μg/ml insulin, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 1 μM dexamethasone (DEX), and 5 μM rosiglitazone. The cells were kept in the full cocktail for 3 days and then switched to the medium with 10% FBS plus insulin and rosiglitazone for 2 more days. The 3T3 adipocytes were then kept in DMEM–10% FBS medium for 1 more week. During this time, manipulations were performed to knock out Epac1 by CRISPR-Cas9 or inhibiting Epac1 activity by the specific inhibitor ESI-09, respectively. Cell culture supernatant was collected and concentrated by Amicon Ultra 0.5-ml centrifugal filters (Merck Millipore Ltd.) and used for measurement of leptin concentration. The leptin levels were normalized to the total protein in the culture supernatant.
Immunoblotting.
Tissues were homogenized in radioimmunoprecipitation assay (RIPA) buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.1% SDS, 0.5% Na-deoxycholate, 50 mM sodium fluoride plus 1% phosphatase inhibitor cocktail I, 0.5% phosphatase inhibitor cocktail II, and 1% protease inhibitor cocktail (Sigma-Aldrich, MO). The cells were directly collected in 1× SDS loading buffer to generate cell lysates. Equal amounts of protein were subjected to electrophoresis on polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated with primary antibodies overnight at 4°C, followed by secondary antibodies conjugated with horseradish peroxidase. Protein levels were visualized and quantified by ECL using a Bio-Rad ChemiDoc touch imaging system (Hercules, CA).
RT-qPCR analysis.
Total RNAs from white adipose tissue (WAT) and cells were extracted using TRIzol (Invitrogen) and further isolated using EconoSpin spin columns for RNA extraction (Epoch Life Science). RNA concentrations were determined by NanoDrop (Thermo Scientific). cDNAs were synthesized by reverse transcription from total RNA using iScript reverse transcription supermix for RT-qPCR (Bio-Rad). The iTaq universal SYBR green supermix kit (Bio-Rad) was used for quantitative PCR.
OGTT and ITT.
Oral glucose tolerance test (OGTT) and insulin tolerance test (ITT) were performed as described previously (9). For OGTT mice were fasted overnight, and for ITT mice were fasted for 4 h. In OGTT, mice were orally administered with d-glucose (ICN Biomedicals, Inc.) at a dose of 1 g/kg of body weight. In ITT, mice were administered intraperitoneally (i.p.) with human insulin (Eli Lilly) at a dose of 0.5 mU/g body weight. The tail-vein blood glucose concentration was determined using an OneTouch Ultra blood glucometer.
Statistical analysis.
Results are presented as means ± standard errors of the means (SEM). A P value of less than 0.05 was considered statistically significant. A Student t test was used to compare two groups. One-way or two-way analysis of variance (ANOVA) and the Bonferroni post hoc test were used to compare multiple groups.
RESULTS
AEKO mice display increased body weight and food intake on HFD.
To ascertain the functional role of Epac1 in adipose tissue, we generated adipose-specific Epac1 knockout mice (AEKO) using the Cre/LoxP site-specific recombination system. Disruption of Epac1 expression in WAT isolated from the epididymal fat pad (EWAT) was confirmed by Western blotting, whereas the expression of Epac1 in hypothalamus was not significantly affected. The levels of Epac1 in other fat depots, such as subcutaneous and brown fats, were also significantly reduced in the AEKO mice, although to a lesser extent than that of the gonadal fat pad (Fig. 1A and B). We therefore mainly focused on studies using the abdominal gonadal fat depot. We also examined the effect of αP2-Cre expression on Epac1 in macrophages, which express very low levels of Epac1 that could not be detected using immunoblot analysis. Real-time PCR confirmed that the mRNA expression level of Epac1 was not significantly altered in AEKO macrophages (Fig. 1C). On the other hand, the levels of Epac1 in isolated preadipocytes and cardiomyocytes were significantly reduced in AEKO mice (Fig. 1A and B).
FIG 1.

Tissue-specific disruption of Epac1 in WAT increases body weight gains and food intake on HFD. (A) Epac1 protein levels in epididymal white adipose tissue (EWAT), hypothalamus, subcutaneous white adipose tissue (SWAT), brown adipose tissue (BAT), preadipocytes, and cardiomyocytes of WT and AEKO mice. (B) Quantification of Epac1 protein expression in various tissues of WT and AEKO mice. (C) Quantification of Epac1 mRNA expression levels in EWAT and isolated macrophages. (D) Body weight changes of WT (n = 8) and AEKO (n = 7) mice on HFD. (E) Average daily food intake of WT (n = 8) and AEKO (n = 7) mice on HFD over 41 days. (F) Body weight changes of WT and AEKO mice on regular chow diet. Data are presented as means ± standard errors of the mean (SEM). *, P < 0.05; ***, P < 0.005; ****, P < 0.001.
The AEKO mice and WT controls had similar body weights from birth to postnatal day 24. After the mice were weaned and switched to an HFD, the AEKO mice gradually gained more body weight than WT controls and became significantly heavier than WT mice after 16 days on HFD (Fig. 1D). In agreement with the heavier body weight phenotype of AEKO mice on HFD, the average daily food intake of AEKO mice was significantly higher than that of the WT littermates (Fig. 1E). No significant difference in body weight between AEKO and WT groups over the same time period was observed when mice were fed on regular chow (Fig. 1F).
Deficiency of Epac1 in adipose tissue downregulates energy expenditure.
To determine the potential causes of the apparent difference in body weight, we examined the effects of adipose tissue-specific Epac1 deletion on energy homeostasis by monitoring energy expenditure using CLAMS. Age-matched and normal-chow-fed naive male AEKO and WT mice with similar body weights were used for the analyses. As shown in Fig. 2A and B, while both groups of mice had higher oxygen (O2) consumption rates at night, when the animals were more active regardless of the food sources, mice fed HFD had a higher O2 consumption rate than mice fed regular chow. Importantly, the AEKO mice showed significantly lower O2 consumptions than WT counterparts under HFD or chow diet. Similar observations were made for carbon dioxide (CO2) production. The AEKO mice had statistically lower CO2 production rates on both chow and HFD (Fig. 2C and D). The calculated respiratory exchange ratio, defined as the ratio between O2 consumption and CO2 production, was lower in HFD-fed mice for both groups, while there was no significant difference in respiratory exchange ratio between AEKO and WT mice under HFD or chow diet (Fig. 2E). Similarly, no statistically significant difference in food intake between AEKO and WT mice was observed, although AEKO mice had slightly higher food intake than WT mice when fed on HFD (Fig. 2F). This is consistent with the fact that mice on HFD burned more fatty acids, while mice on chow diet utilized carbohydrates as the predominant fuel source. Taken together, these data suggest that AEKO mice have an overall lower energy expenditure than the WT controls while maintaining a balanced carbohydrate/lipid oxidation ratio.
FIG 2.

Deletion of Epac1 in WAT downregulates energy expenditure. (A) Energy expenditure of WT and AEKO mice at the age of 5 to 6 weeks measured by O2 consumption using CLAMS. (B) Average O2 consumption of WT and AEKO mice. (C) Energy expenditure of WT and AEKO mice at the age of 5 weeks measured by CO2 production using CLAMS. (D) Average CO2 production of WT and AEKO mice. (E) The respiratory exchange ratio of WT and AEKO mice. (F) Daily food intake of WT and AEKO mice on regular chow diet and HFD. (G) Spontaneous activity profiles of WT and AEKO mice. (H) Average spontaneous locomotor activity of WT and AEKO mice. Data are means ± SEM. *, P < 0.05.
To determine if the apparent lower energy expenditure phenotype observed in AEKO mice was associated with a decrease in overall physical activities, we monitored and compared the spontaneous locomotor activity between AEKO and WT mice on HFD or chow diet. While there were no significant changes in the average total activity counts between AEKO and WT cohorts, AEKO mice showed a slight increase in voluntary locomotor activity consistently across all groups (Fig. 2G and H).
Loss of Epac1 in adipose tissue reduces plasma leptin levels.
To investigate if the aforementioned phenotypes of reduced energy expenditure and increased body weight and food intake were related to a change in leptin, one of the most important adipose-derived hormones for regulating energy homeostasis, we determined fasting plasma leptin levels. As shown in Fig. 3A, the measured fasting plasma leptin level (95.6 ± 3.4 ng/ml) of 15-week-old AEKO mice fed HFD for 16 weeks was significantly lower than that (109.5 ± 2.5 ng/ml) of the age-matched WT controls. Similar data of reduced leptin levels were obtained from AEKO mice fed ad libitum with water and food (Fig. 3B), whereas the levels of adiponectin in plasma were not significantly different between AEKO and WT mice (Fig. 3D). The reduced leptin levels in AEKO mice are particularly significant considering that these mice have increased body weights, which would normally be associated with an increase in leptin level. Therefore, these results suggest that Epac1 is directly involved in regulating the production and/or secretion of leptin in adipose tissue. This notion is further consistent with the finding that plasma leptin levels were significantly lower in newly weaned naive AEKO mice than those in age-matched WT mice on postnatal day 24, when there was no body weight difference between the two groups (Fig. 3C).
FIG 3.

Deletion of Epac1 in WAT reduces plasma leptin levels. (A) Fasting plasma leptin levels of AEKO mice and WT littermates after being on HFD for 16 weeks. (B) Plasma leptin levels of AEKO and WT mice fed on HFD ad libitum. (C) Plasma leptin levels of WT and AEKO mice at postnatal day 24 on chow diet. (D) Levels of plasma adiponectin from fasted AEKO and WT mice fed HFD. Data are means ± SEM. *, P < 0.05; ***, P < 0.005.
Disruption of Epac1 downregulates leptin mRNA expression.
To determine if Epac1 plays a role in controlling leptin expression at the transcriptional level, we measured the leptin mRNA expression levels in WAT of AEKO and WT mice using real-time PCR. Epac1 deficiency in WAT resulted in a marked reduction in leptin mRNA levels (Fig. 4A), while the levels of other adipokines, such as adiponectin, TNF, and resistin, were not significantly affected (Fig. 4D).
FIG 4.

Disruption of Epac1 in WAT downregulates leptin transcription. (A) Levels of leptin mRNA in WAT of WT and AEKO mice determined by RT-PCR. (B) Disruption of Epac1 in 3T3 preadipocytes by CRISPR-Cas9 gene editing system abolishes leptin mRNA level. (Inset) Expression levels of Epac1 in control and CRISPR-Cas9-targeted 3T3-L1 cells using Epac1-specific sgRNA. (C) Relative mRNA levels of leptin in 3T3-L1 adipocytes after 24 h of activation of Epac with an Epac-specific agonist (8-pMeOPT), inhibition with an Epac-specific antagonist (ESI09), or forskolin treatment. (D) Expression levels of adipokines in EWAT of AEKO and WT mice fed HFD. Ctl, control. Data are presented as means ± SEM. *, P < 0.05; ***, P < 0.005.
To determine if Epac1 plays a direct role in controlling leptin expression, we performed in vitro analyses using 3T3-L1, a model preadipocyte cell line widely used for studying adipocyte differentiation and functions, such as leptin secretion (19). We measured the expression of leptin in 3T3-L1 adipocytes in response to 8-pMeOPT-2′-O-Me-cAMP and forskolin, respectively. As shown in Fig. 4C, selective activation of Epac led to significant increases in leptin mRNA expression that could be negated by cotreatment with the Epac-specific antagonist ESI-09. In contrast, forskolin treatment led to reduced leptin mRNA expression, suggesting that PKA plays a negative and dominant role in leptin mRNA regulation while Epac exerts a positive effect. Consistent with the results obtained from pharmacological studies, deletion of Epac1 in 3T3-L1 cells by Epac1-specific single guide RNA (sgRNA) using the CRISPR-Cas9 gene editing system (Fig. 4B, inset) led to dramatic decreases in leptin mRNA levels (Fig. 4B).
Epac1 deletion or inhibition in 3T3-L1 adipocyte reduces leptin secretion.
To confirm that the apparent reduction in plasma leptin levels observed in AEKO mice was due to direct Epac1 deletion, not secondary effects such as in vivo compensation and/or adaptation, we monitored leptin secretion in differentiated 3T3-L1 adipocytes. As shown in Fig. 5A, suppressing Epac1 activity using an Epac-specific inhibitor, ESI-09 (13, 14, 20), led to dose-dependent inhibition of leptin secretion. Furthermore, consistent with the pharmacological data, knocking out Epac1 in 3T3 adipocytes using the CRISPR-Cas9 gene editing system significantly suppressed leptin secretion in differentiated 3T3-L1 adipocytes (Fig. 5B). To determine if activation of Epac is sufficient to induce leptin secretion and to examine the relative contributions of Epac and PKA, we stimulated 3T3-L1 adipocytes with an Epac selective agonist or a cAMP stimulator, forskolin. As shown in Fig. 5C, forskolin treatment inhibited leptin secretion, an observation consistent with previous publications. In contrast, activation of Epac by 007 led to a modest but significant increase in leptin secretion. The stimulatory effect of Epac activation on leptin secretion was largely suppressed by the cotreatment of an AKT inhibitor or ESI-09. Taken together, these data demonstrate a direct role of Epac1 in leptin secretion, supporting the in vivo observation of reduced plasma leptin levels in AEKO mice.
FIG 5.
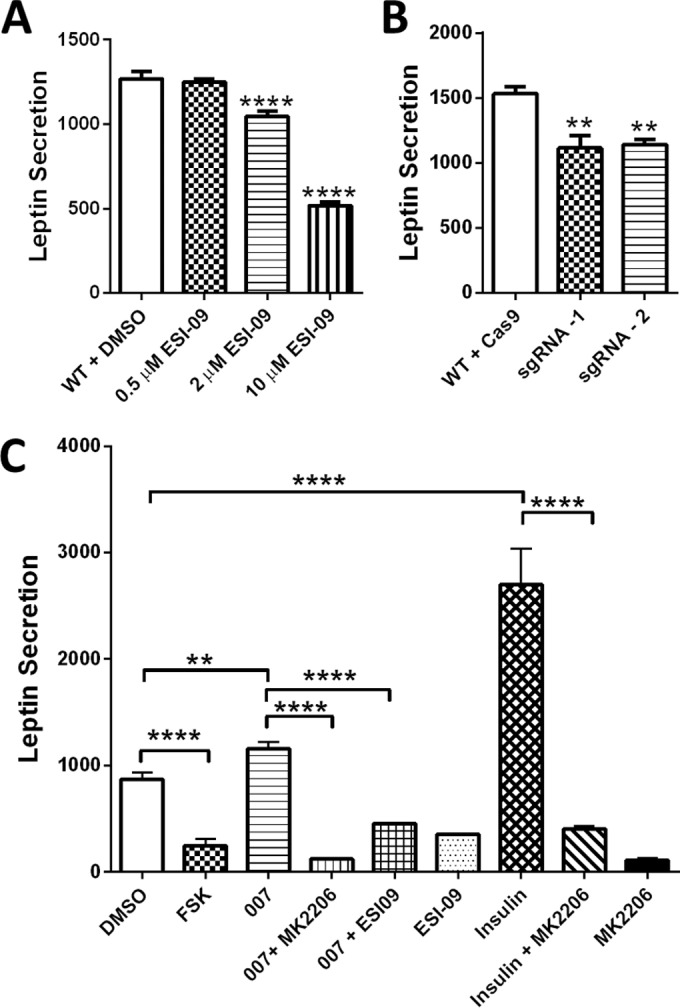
Regulation of leptin secretion in 3T3-L1 adipocytes by Epac1. (A) Inhibition of Epac1 by ESI-09 dose dependently suppresses leptin secretion by 3T3 adipocytes. (B) Gene editing of Epac1 by CRISPR-Cas9 system decreases leptin secretion by 3T3 adipocytes. (C) Regulation of leptin secretion by pharmacological modulation of Epac, PKA, and PKB. Data are means ± SEM. **, P < 0.01; ****, P < 0.001.
Deletion or inhibition of Epac1 in adipocytes suppresses AKT and CREB phosphorylation.
To determine the mechanisms that Epac1 may employ in regulating leptin expression and secretion, we investigated the activation status of signaling pathways downstream of cAMP/Epac/Rap1 in adipose tissues from AEKO and WT mice. Among multiple related signaling molecules probed, we consistently observed a reduced phosphorylated AKT/PKB level at serine 473 as well as a reduced phosphorylated CREB level at serine 133 in AEKO WAT compared to that of the WT control (Fig. 6A to C).
FIG 6.

Regulation of AKT and CREB phosphorylation by genetic and pharmacological modulation of Epac. (A) Representative levels of p-AKT serine 473 and p-CREB serine 133 in WAT of WT and AEKO mice after 16 weeks on HFD. (B) Quantitation of p-AKT serine 473 for 8 pairs of WT and AEKO mice. (C) Quantitation of p-CREB serine 133 for 5 pairs of WT and AEKO mice. (D) Regulation of p-AKT serine 473 and p-CREB serine 133 in 3T3-L1 adipocytes by 007-AM, a membrane-permeable Epac selective agonist. (E and F) Quantitation of p-AKT serine 473 (E) and p-CREB serine 133 (F) in response to 007-AM and MK2206 treatments. (G) Regulation of p-AKT serine 473 and p-CREB serine 133 in 3T3-L1 adipocytes by 007-AM in the presence or absence of Rap1GAP. (H and I) Quantitation of p-AKT serine 473 (H) and p-CREB serine 133 (I) in response to 007-AM treatments in the presence or absence of Rap1GAP. Data are presented as means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.005.
To determine if Epac1 activation in adipocytes is sufficient to activate AKT and CREB, we treated 3T3-L1 cells with a membrane-permeable Epac-specific activator, 007-AM. Activation of Epac by 007-AM led to a modest but reproducible increase in AKT and CREB phosphorylation. While Epac-mediated AKT phosphorylation could be reversed by MK-2206, a specific AKT inhibitor, the effects of MK-2206 on CREB phosphorylation were small and not statistically significantly (Fig. 6D to F). To further determine if Epac-mediated AKT and CREB phosphorylation is Rap1 dependent, we tested the effect of Rap1GAP, a Rap1-specific GTPase-activating protein that efficiently keeps Rap1 in its inactive GDP-bound state (21, 22), on Epac-mediated AKT and CREB phosphorylation. As shown in Fig. 6G and H, overexpression of Rap1GAP in 3T3-L1 cells blocked the ability of 007-AM to stimulate AKT phosphorylation, suggesting that Rap1 is required for Epac-induced AKT activation in 3T3-L1 cells. On the other hand, increases in CREB phosphorylation induced by 007-AM treatment were not affected by Rap1GAP expression (Fig. 6G and I).
AEKO mice have more severe HFD-induced glucose intolerance.
In light of our finding that deletion of Epac1 in adipose tissue reduces plasma leptin and disrupts energy balance, we investigated the effects of adipose-specific deletion of Epac1 on glucose homeostasis in HFD-fed mice by oral glucose tolerance test. After overnight fasting, AEKO mice had slightly higher basal blood glucose levels (171.8 ± 6.9 mg/dl) than WT controls (140.4 ± 7.7 mg/dl). Following a single dose (oral administration) of glucose, the blood glucose levels increased dramatically and peaked at 15 or 30 min in AEKO and WT mice, respectively (Fig. 7A). Starting from 30 min, the blood glucose levels were significantly elevated in AEKO mice compared to WT controls. Two hours after the glucose challenge, the blood glucose levels of WT mice returned to 175.3 ± 11.2 mg/dl, while AEKO mice had a significantly higher blood glucose level at 246.0 ± 16.6 mg/dl. In parallel to glucose levels, the plasma insulin levels were also monitored. No significant differences in insulin levels were observed between WT and AEKO mice at time 0 and 15 and 30 min after glucose administration (Fig. 7B). Furthermore, insulin tolerance test was performed to determine the insulin sensitivity, as shown in Fig. 7C, while the blood glucose levels were slightly elevated in AEKO mice starting from 15 min after insulin administration; these changes were not statistically significant. Taken together, these data suggest that AEKO mice have a compromised glucose clearance capability while maintaining insulin sensitivity similar to that of the WT controls.
FIG 7.
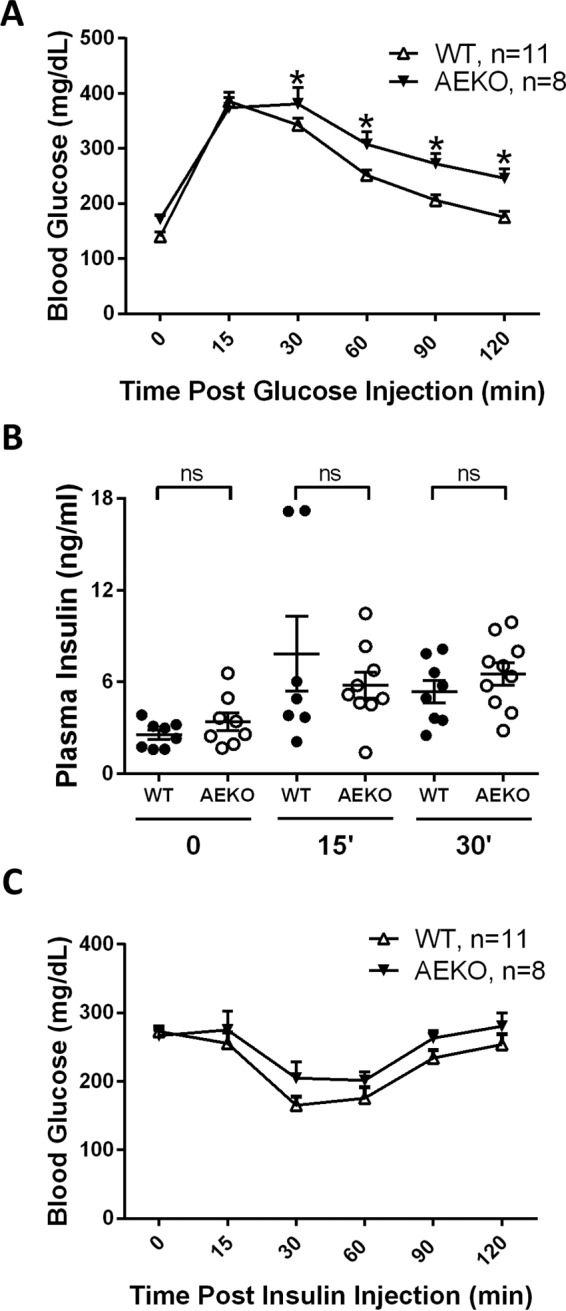
AEKO mice display more severe HFD-induced glucose intolerance. (A) Oral glucose tolerance test of 17-week-old HFD-fed mice (14 weeks on HFD). (B) Levels of insulin at basal fasting (time 0) and 15 and 30 min after oral glucose administration in AEKO and WT mice fed HFD during OGTT. (C) Insulin tolerance test of 19-week-old HFD-fed mice (16 weeks on HFD). Data are presented as means ± SEM. *, P < 0.05.
DISCUSSION
Leptin plays a critical role in maintaining energy balance and glucose homeostasis under normal physiological conditions. Dysregulation of leptin signaling under conditions such as leptin resistance disrupts energy homeostasis and leads to metabolic disorders, including obesity. Therefore, elucidating the signaling network important for the regulation of leptin functions is crucial for understanding the molecular etiology of obesity, as well as for developing new therapeutics for this growing health burden. Recent studies based on genetic and pharmacological manipulations have implicated Epac1 as an important signaling molecule for modulating leptin signaling in hypothalamus (7–9). However, the roles of Epac1 in peripheral tissues, particularly in WAT, the major tissue responsible for leptin production, remain unknown. To bridge this gap in our knowledge, we generated conditional adipose tissue-specific Epac1 knockout mice using the transgenic Cre line driven by the adipocyte protein 2 (αP2) gene promoter (16). Although αP2-Cre has been widely used for generating conditional adipose-specific knockout models, it has been reported that expression of αP2-driven Cre recombinase can be detected in macrophages (23) and brains (24) in certain instances. Therefore, in addition to confirming the knockdown of Epac1 in WAT, special attention was devoted to monitoring the expression levels of Epac1 in the hypothalamus and macrophages. We did not notice changes of Epac1 expression levels in either macrophages or hypothalamus in young AEKO mice. These results are consistent with a recent detailed study in which the tissue expression pattern of various adipose-specific Cre lines were systematically analyzed and compared to confirm the absence of recombination in the macrophages of αP2-Cre (25). Hence, the apparent phenotypes observed in AEKO mice should be due predominantly to the deficiency of Epac1 in peripheral tissues, in particular WAT.
Similar to Epac1 global knockout mice, AEKO mice showed decreased plasma leptin levels. However, when challenged by HFD, AEKO mice initially ate more and showed significantly higher body weight gains, which is the opposite of the phenotypes observed in mice with global Epac1 deletion. Conventional Epac1 KO mice have heightened leptin sensitivity and reduced food intake and are partially resistant to HFD-induced obesity (9). These unexpected opposing phenotypes offer important clues to the specific roles of Epac1 in various tissues. While phenotypes observed in the global Epac1 knockout mice reflect the integrated outcomes of Epac1 deletion from the whole body, including both CNS and peripheral effects, our AEKO mice exhibited only the specific consequences of Epac1 deletion in adipose tissues. It appears that Epac1 deletion in adipose tissues reduces circulating leptin levels, which explain the apparent phenotypes of AEKO mice, as these animals have normal Epac1 expression in hypothalamus and, thus, normal leptin sensitivity. Under such a condition, a reduced leptin level would lead to increased food intake and decreased energy expenditures. On the other hand, since CNS, especially hypothalamus, is the most important area for integrating metabolic signals and maintaining energy homeostasis, deletion of Epac1 in hypothalamus heightens leptin sensitivity and results in a more potent anorectic effect capable of overcoming the orexigenic effect of adipose tissue Epac1 deficiency in the global knockout mice (9). Taken together, our results further support a dominant role of Epac1 in CNS over peripheral tissues in regulating energy balance. In addition to energy balance, leptin has also been implicated in glucose homeostasis (26, 27). Consistent with this notion, AEKO mice fed on HFD are compromised in clearing glucose in OGTT while maintaining insulin sensitivity (Fig. 7). It is of particular interest that despite increased body weight, AEKO mice on HFD have decreased overall leptin levels, which can explain all observed phenotypic changes in energy balance and glucose homeostasis, as reduced leptin levels prompted the AEKO mice to be more prone to HFD-induced obesity by increasing food intake and decreasing energy expenditure.
Physiological levels of leptin usually correlate with adiposity and are regulated by food consumption and insulin, among other signals. As an important stress signal, the second messenger cAMP is known to regulate leptin production (28, 29). Increases in intracellular cAMP levels have been shown to downregulate leptin mRNA levels and to suppress leptin secretion in 3T3-L1 adipocytes and mesenchymal stem cells (30, 31). This inhibitory effect of cAMP on leptin production and secretion is PKA dependent, as inhibition of PKA is capable of rescuing cAMP-mediated downregulation of leptin (28–31). On the other hand, the role of Epac1, another important cAMP effector, in regulating adipocyte leptin production has not been explored. Our current studies based on in vivo tissue-specific Epac1 knockout and in vitro cellular analyses reveal that Epac1 plays a stimulatory role in adipose leptin production. Suppression of Epac1 by genetic manipulations and pharmacological inhibition in adipose tissue and adipocytes diminishes leptin mRNA expression levels and reduces leptin secretion, while activation of Epac by Epac-specific agonists increases leptin production. These observations suggest that cAMP-mediated leptin regulation is more complex and dynamic than was previously recognized, as Epac1 and PKA, the two distinct cAMP sensors, can act antagonistically in cAMP-mediated leptin regulation. Therefore, the net outputs of leptin production under the control of cAMP signaling are determined by the dynamic distribution/activation of PKA and Epac1 in adipose tissue to provide a more integrative and broad-range regulation.
Although the precise mechanisms of Epac1-mediated regulation of leptin production and secretion remain to be elucidated, our study suggests a potential role for AKT and CREB, respectively. The activation of AKT is well accepted to effectively promote leptin secretion in 3T3-L1 and primary adipocytes in response to insulin (32, 33). In addition, global Akt2−/− and Akt1+/− Akt2−/− mice exhibit type 2 diabetes concurrent with decreased levels of circulating leptin, supporting a major physiological role of AKT in leptin regulation (34). Similarly, in our model the levels of AKT phosphorylation in primary AEKO adipocytes and 3T3-L1 with Epac1 knocked out are significantly lower than those of the WT controls, consistent with the reduced leptin expression and secretion observed. Moreover, our studies reveal that Epac-specific agonists directly activate AKT phosphorylation in a Rap1-dependent manner, in agreement with our previous findings (35, 36). Interestingly, in macrophages, Epac1 is reported to form a complex between Rap1, AKT, and ILK, suggesting that Epac1 functions to translocate AKT to the plasma membrane, where this complex serves as a scaffold for phosphorylation of AKT by other kinases, such as phosphatidylinositol 3-kinase (PI3K) and PDPK1, shown to be pivotal in leptin signaling and AKT activation (37, 38).
The role of CREB as a transcriptional regulator of leptin has also been reported. Promoter analyses reveal the presence of an evolutionarily conserved cAMP response element (CRE) within the leptin promoter region of mouse, rat, and human (39–41). Super gel-shift assay using CREB antibody confirms direct physical interaction between CREB and CRE of the leptin promoter, which is required for Wnt3a-mediated leptin expression in primary and 3T3-L1 adipocytes (40). Furthermore, Soliman and colleagues showed that butyrate can stimulate leptin expression in fully differentiated human adipocytes at the transcriptional level by activating mitogen-activated protein kinase (MAPK) and phospho-CREB signaling in a time-dependent manner (42). While the significance of the Epac1/CREB pathway in the regulation of leptin expression within the context of adipose tissue has not been described, a recent study shows that 007, a cAMP analogue that specifically activates Epac, stimulates leptin expression by hCG, and that the overexpression of Epac and Rap1 proteins increases leptin promoter activity in placental cells (43), suggesting an alternative mechanism in addition to the well-established PKA/MAPK signaling pathways (44). Therefore, it is likely that a similar signaling network is in play for the regulation of leptin expression in adipocytes.
In summary, our study highlights an important and previously unknown role of Epac1 in regulation of leptin production in WAT. Disruption of Epac1 in adipose tissue suppresses leptin expression and secretion through downstream effectors CREB and AKT, respectively. Although CREB is a known regulatory target of AKT (45), results from our studies suggest that CREB acts independently from AKT, as inhibition of AKT has no effect on suppressing Epac1-mediated CREB phosphorylation. In addition, while Rap1 activation is required for Epac1-induced AKT phosphorylation, EPAC-mediated CREB phosphorylation is Rap1 independent. We are actively pursuing the mechanism of how CREB and AKT, acting downstream of Epac1 signaling, regulate leptin expression and secretion.
ACKNOWLEDGMENTS
We thank Lawrence Quilliam (Indiana University School of Medicine) for kindly providing the pFLAG-CMV2-Rap1GAP vector.
Funding Statement
This work was funded by a grant from the National Institute of General Medical Sciences (NIGMS) (R01GM066170) to Xiaodong Cheng and by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01DK09605) to Qingchun Tong.
REFERENCES
- 1.Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. 2011. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 378:815–825. doi: 10.1016/S0140-6736(11)60814-3. [DOI] [PubMed] [Google Scholar]
- 2.Kopelman PG. 2000. Obesity as a medical problem. Nature 404:635–643. [DOI] [PubMed] [Google Scholar]
- 3.Dixon JB. 2010. The effect of obesity on health outcomes. Mol Cell Endocrinol 316:104–108. doi: 10.1016/j.mce.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 5.Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr, Stoffel M, Friedman JM. 1996. Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 14:95–97. doi: 10.1038/ng0996-95. [DOI] [PubMed] [Google Scholar]
- 6.Myers MG, Cowley MA, Munzberg H. 2008. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol 70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- 7.Fukuda M, Williams KW, Gautron L, Elmquist JK. 2011. Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab 13:331–339. doi: 10.1016/j.cmet.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almahariq M, Mei FC, Cheng X. 2014. Cyclic AMP sensor EPAC proteins and energy homeostasis. Trends Endocrinol Metab 25:60–71. doi: 10.1016/j.tem.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yan J, Mei FC, Cheng H, Lao DH, Hu Y, Wei J, Patrikeev I, Hao D, Stutz SJ, Dineley KT, Motamedi M, Hommel JD, Cunningham KA, Chen J, Cheng X. 2013. Enhanced leptin sensitivity, reduced adiposity, and improved glucose homeostasis in mice lacking exchange protein directly activated by cyclic AMP isoform 1. Mol Cell Biol 33:918–926. doi: 10.1128/MCB.01227-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Banerjee U, Cheng X. 2015. Exchange protein directly activated by cAMP encoded by the mammalian rapgef3 gene: structure, function and therapeutics. Gene 570:157–167. doi: 10.1016/j.gene.2015.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chepurny OG, Leech CA, Kelley GG, Dzhura I, Dzhura E, Li X, Rindler MJ, Schwede F, Genieser HG, Holz GG. 2009. Enhanced Rap1 activation and insulin secretagogue properties of an acetoxymethyl ester of an Epac-selective cyclic AMP analog in rat INS-1 cells: studies with 8-pCPT-2′-O-Me-cAMP-AM. J Biol Chem 284:10728–10736. doi: 10.1074/jbc.M900166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vliem MJ, Ponsioen B, Schwede F, Pannekoek WJ, Riedl J, Kooistra MR, Jalink K, Genieser HG, Bos JL, Rehmann H. 2008. 8-pCPT-2′-O-Me-cAMP-AM: an improved Epac-selective cAMP analogue. Chembiochem 9:2052–2054. doi: 10.1002/cbic.200800216. [DOI] [PubMed] [Google Scholar]
- 13.Almahariq M, Tsalkova T, Mei FC, Chen H, Zhou J, Sastry SK, Schwede F, Cheng X. 2013. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol Pharmacol 83:122–128. doi: 10.1124/mol.112.080689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. 2015. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window.” Sci Rep 5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez CI, Buchholz F, Galloway J, Sequerra R, Kasper J, Ayala R, Stewart AF, Dymecki SM. 2000. High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat Genet 25:139–140. doi: 10.1038/75973. [DOI] [PubMed] [Google Scholar]
- 16.He W, Barak Y, Hevener A, Olson P, Liao D, Le J, Nelson M, Ong E, Olefsky JM, Evans RM. 2003. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 100:15712–15717. doi: 10.1073/pnas.2536828100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim ER, Wu Z, Sun H, Xu Y, Mangieri LR, Tong Q. 2015. Hypothalamic non-AgRP, non-POMC GABAergic neurons are required for postweaning feeding and NPY hyperphagia. J Neurosci 35:10440–10450. doi: 10.1523/JNEUROSCI.1110-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. 2003. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem 278:32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 19.Zeigerer A, Rodeheffer MS, McGraw TE, Friedman JM. 2008. Insulin regulates leptin secretion from 3T3-L1 adipocytes by a PI 3 kinase independent mechanism. Exp Cell Res 314:2249–2256. doi: 10.1016/j.yexcr.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsalkova T, Mei FC, Li S, Chepurny OG, Leech CA, Liu T, Holz GG, Woods VL Jr, Cheng X. 2012. Isoform-specific antagonists of exchange proteins directly activated by cAMP. Proc Natl Acad Sci U S A 109:18613–18618. doi: 10.1073/pnas.1210209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Polakis PG, Rubinfeld B, Evans T, McCormick F. 1991. Purification of a plasma membrane-associated GTPase-activating protein specific for rap1/Krev-1 from HL60 cells. Proc Natl Acad Sci U S A 88:239–243. doi: 10.1073/pnas.88.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubinfeld B, Munemitsu S, Clark R, Conroy L, Watt K, Crosier WJ, McCormick F, Polakis P. 1991. Molecular cloning of a GTPase activating protein specific for the Krev-1 protein p21rap1. Cell 65:1033–1042. doi: 10.1016/0092-8674(91)90555-D. [DOI] [PubMed] [Google Scholar]
- 23.Mao J, Yang T, Gu Z, Heird WC, Finegold MJ, Lee B, Wakil SJ. 2009. aP2-Cre-mediated inactivation of acetyl-CoA carboxylase 1 causes growth retardation and reduced lipid accumulation in adipose tissues. Proc Natl Acad Sci U S A 106:17576–17581. doi: 10.1073/pnas.0909055106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martens K, Bottelbergs A, Baes M. 2010. Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Lett 584:1054–1058. doi: 10.1016/j.febslet.2010.01.061. [DOI] [PubMed] [Google Scholar]
- 25.Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, Smyth G, Rourk M, Cederquist C, Rosen ED, Kahn BB, Kahn CR. 2013. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 62:864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, Lauzon D, Lee CE, Coppari R, Richardson JA, Zigman JM, Chua S, Scherer PE, Lowell BB, Bruning JC, Elmquist JK. 2010. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab 11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berglund ED, Vianna CR, Donato J Jr, Kim MH, Chuang JC, Lee CE, Lauzon DA, Lin P, Brule LJ, Scott MM, Coppari R, Elmquist JK. 2012. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Investig 122:1000–1009. doi: 10.1172/JCI59816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szkudelski T, Nowicka E, Szkudelska K. 2005. Leptin secretion and protein kinase A activity. Physiol Res 54:79–85. [DOI] [PubMed] [Google Scholar]
- 29.Szkudelski T. 2007. Intracellular mediators in regulation of leptin secretion from adipocytes. Physiol Res 56:503–512. doi: 10.1111/j.1439-0396.2006.00647.x. [DOI] [PubMed] [Google Scholar]
- 30.Yang DC, Tsay HJ, Lin SY, Chiou SH, Li MJ, Chang TJ, Hung SC. 2008. cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PLoS One 3:e1540. doi: 10.1371/journal.pone.0001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maeda T, Horiuchi N. 2009. Simvastatin suppresses leptin expression in 3T3-L1 adipocytes via activation of the cyclic AMP-PKA pathway induced by inhibition of protein prenylation. J Biochem 145:771–781. doi: 10.1093/jb/mvp035. [DOI] [PubMed] [Google Scholar]
- 32.Barthel A, Kohn AD, Luo Y, Roth RA. 1997. A constitutively active version of the Ser/Thr kinase Akt induces production of the ob gene product, leptin, in 3T3-L1 adipocytes. Endocrinology 138:3559–3562. doi: 10.1210/endo.138.8.5263. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Ali Y, Lim CY, Hong W, Pang ZP, Han W. 2014. Insulin-stimulated leptin secretion requires calcium and PI3K/Akt activation. Biochem J 458:491–498. doi: 10.1042/BJ20131176. [DOI] [PubMed] [Google Scholar]
- 34.Chen WS, Peng XD, Wang Y, Xu PZ, Chen ML, Luo Y, Jeon SM, Coleman K, Haschek WM, Bass J, Philipson LH, Hay N. 2009. Leptin deficiency and beta-cell dysfunction underlie type 2 diabetes in compound Akt knockout mice. Mol Cell Biol 29:3151–3162. doi: 10.1128/MCB.01792-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. 2002. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem 277:11497–11504. doi: 10.1074/jbc.M110856200. [DOI] [PubMed] [Google Scholar]
- 36.Cheng X, Ji Z, Tsalkova T, Mei F. 2008. Epac and PKA: a tale of two intracellular cAMP receptors. Acta Biochim Biophys Sin (Shanghai) 40:651–662. doi: 10.1111/j.1745-7270.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misra UK, Pizzo SV. 2005. Coordinate regulation of forskolin-induced cellular proliferation in macrophages by protein kinase A/cAMP-response element-binding protein (CREB) and Epac1-Rap1 signaling: effects of silencing CREB gene expression on Akt activation. J Biol Chem 280:38276–38289. doi: 10.1074/jbc.M507332200. [DOI] [PubMed] [Google Scholar]
- 38.Misra UK, Kaczowka S, Pizzo SV. 2008. The cAMP-activated GTP exchange factor, Epac1 upregulates plasma membrane and nuclear Akt kinase activities in 8-CPT-2-O-Me-cAMP-stimulated macrophages: gene silencing of the cAMP-activated GTP exchange Epac1 prevents 8-CPT-2-O-Me-cAMP activation of Akt activity in macrophages. Cell Signal 20:1459–1470. doi: 10.1016/j.cellsig.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao Y, Li Z, Gabrielsen JS, Simcox JA, Lee SH, Jones D, Cooksey B, Stoddard G, Cefalu WT, McClain DA. 2015. Adipocyte iron regulates leptin and food intake. J Clin Investig 125:3681–3691. doi: 10.1172/JCI81860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen ZL, Shao WJ, Xu F, Liu L, Lin BS, Wei XH, Song ZL, Lu HG, Fantus IG, Weng JP, Jin TR. 2015. Acute Wnt pathway activation positively regulates leptin gene expression in mature adipocytes. Cell Signal 27:587–597. doi: 10.1016/j.cellsig.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 41.Gong DW, Bi S, Pratley RE, Weintraub BD. 1996. Genomic structure and promoter analysis of the human obese gene. J Biol Chem 271:3971–3974. doi: 10.1074/jbc.271.8.3971. [DOI] [PubMed] [Google Scholar]
- 42.Soliman MM, Ahmed MM, Salah-Eldin AE, Abdel-Aal AA. 2011. Butyrate regulates leptin expression through different signaling pathways in adipocytes. J Vet Sci 12:319–323. doi: 10.4142/jvs.2011.12.4.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maymo JL, Perez Perez A, Maskin B, Duenas JL, Calvo JC, Sanchez Margalet V, Varone CL. 2012. The alternative Epac/cAMP pathway and the MAPK pathway mediate hCG induction of leptin in placental cells. PLoS One 7:e46216. doi: 10.1371/journal.pone.0046216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maymó JL, Pérez Pérez A, Dueñas JL, Calvo JC, Sánchez-Margalet V, Varone CL. 2010. Regulation of placental leptin expression by cyclic adenosine 5′-monophosphate involves cross talk between protein kinase A and mitogen-activated protein kinase signaling pathways. Endocrinology 151:3738–3751. doi: 10.1210/en.2010-0064. [DOI] [PubMed] [Google Scholar]
- 45.Du K, Montminy M. 1998. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem 273:32377–32379. doi: 10.1074/jbc.273.49.32377. [DOI] [PubMed] [Google Scholar]