

Abstract
Trypanosoma cruzi is the etiological agent of Chagas disease, affecting approximately 10 million people in the Americas and with some 40 million people at risk. The objective of this study was to evaluate the anti-T. cruzi activity of three new diamidines that have a 3,4-ethylenedioxy extension of the thiophene core, designated MB17, MB19, and MB38. All three diamidines exhibited dose-dependent inhibition of epimastigote replication. The mechanisms of action of these diamidines were investigated. Unlike MB17 and MB19, MB38 exhibited a significant increase in the number of annexin-propidium iodide double-labeled cells compared to levels in control parasites. As MB17 had shown a lower 50% inhibitory concentration (IC50) against epimastigote growth, the mechanism of action of this drug was studied in more detail. MB17 triggered a decrease in the intracellular ATP levels. As a consequence, MB17 affected the genomic DNA and kinetoplast DNA (kDNA) and impaired the parasite cell cycle. Moreover, MB17 caused DNA fragmentation, with a more severe effect on kDNA than on nuclear DNA, resulting in dyskinetoplastic cells. MB17 was tested for toxicity and effectiveness for the treatment of infected CHO-K1 cells, exhibiting a 50% cytotoxic concentration (CC50) of 13.47 ± 0.37 μM and an IC50 of 0.14 ± 0.12 μM against trypomastigote release. MB17 also diminished the infection index by 60% at 0.5 μM. In conclusion, despite belonging to the same family, these diamidines have different efficiencies. To summarize, MB17 was the most potent of these diamidines against epimastigotes, producing DNA damage preferentially in kDNA, impairing the parasite cell cycle, and decreasing the infection index and trypomastigote release from infected mammalian host cells, with a high selectivity index (SI) (<90). These data suggest that MB17 could be an interesting lead compound against T. cruzi.
INTRODUCTION
American trypanosomiasis, or Chagas disease, affects approximately 10 million people, with 40 million people at risk of acquiring the infection. Chagas disease is endemic to South and Central America and the southern states of the United States. Currently, it is also spreading in Europe and the rest of North America due to the immigration of infected people, as well as through transmission of the disease by transfusions, transplants, and congenital mechanisms (1, 2). The disease is caused by the protozoan Trypanosoma cruzi, which shares some distinctive features with other trypanosomes, such as the presence of a flagellum and of a kinetoplast, a complex structure bearing the mitochondrial genome. In this case, the mitochondrial genomic DNA is referred to as kinetoplast DNA, or kDNA. It consists of a great number of relaxed circular DNA molecules interlocked with each other to form a catenated DNA network (3). T. cruzi has a complex life cycle, which occurs within invertebrate and vertebrate hosts (4–6). Chagas disease presents two clinical phases, the acute phase, which appears shortly after infection and is characterized by an evident parasitemia and lack of IgG antibodies, and a chronic phase, which is characterized by the absence of evident parasitemia and a robust humoral immune response. The symptoms of the chronic phase, which affects approximately 30% of the infected population, include cardiomyopathy, heart failure, and digestive tract abnormalities, such as megacolon and megaesophagus. The severity of these symptoms generally determines the morbidity of the disease (2, 7, 8). Currently, the only two clinically available drugs for treating the infection are benznidazole and nifurtimox. Both drugs, despite various side effects, are effective during the acute phase, but their efficiencies during the chronic phase, which is when most cases are diagnosed, are controversial due to the low compliance of patients to the long-term dosing that is required for the successful treatment of infection (1, 3, 7). Therefore, it is important to identify new drugs against T. cruzi. DNA-binding compounds are one of the most widely studied classes of agents characterized by biological effects, such as antiprotozoal, antiviral, antibacterial, and antitumoral activity. Beginning in the 1970s, a large number of diamidine derivatives were synthesized to identify more effective drugs. As a result, several compounds, such as 4′,6′-diamidino-2-phenylindole (DAPI), diminazene (Berenil), furamidine, and pentamidine, were shown to be therapeutically useful agents, particularly against several protozoan diseases (9–11).
A well-known example is pentamidine, which has been used clinically against trypanosomiasis, leishmaniasis, and Pneumocystis carnii pneumonia for over 70 years (3, 11). Although the complete mechanism of action of aromatic diamidines is not fully understood, several studies suggest that some of their activities are related to DNA binding and the subsequent inhibition of DNA-dependent enzymes, such as topoisomerases, polymerases, nucleases, and helicases. For trypanosomatids, it was proposed that the parasite dies as a consequence of interference with the complex structure and regulation of the unique kDNA (12–14), making this structure a differential drug target (10, 11, 15). Therefore, diamidines constitute promising templates for the design and development of new drugs against trypanosomatids (10, 16–18). Our group has shown an antiparasitic effect in a new diamidine characterized by a 3,4-ethylenedioxy extension of the thiophene core in the central unit (18, 19). In the present study, we evaluated the anti-T. cruzi effects of three recently described aromatic diamidines with 3,4-ethylenedioxy extensions of the thiophene core, designated MB17, MB19, and MB38 (Fig. 1).
FIG 1.
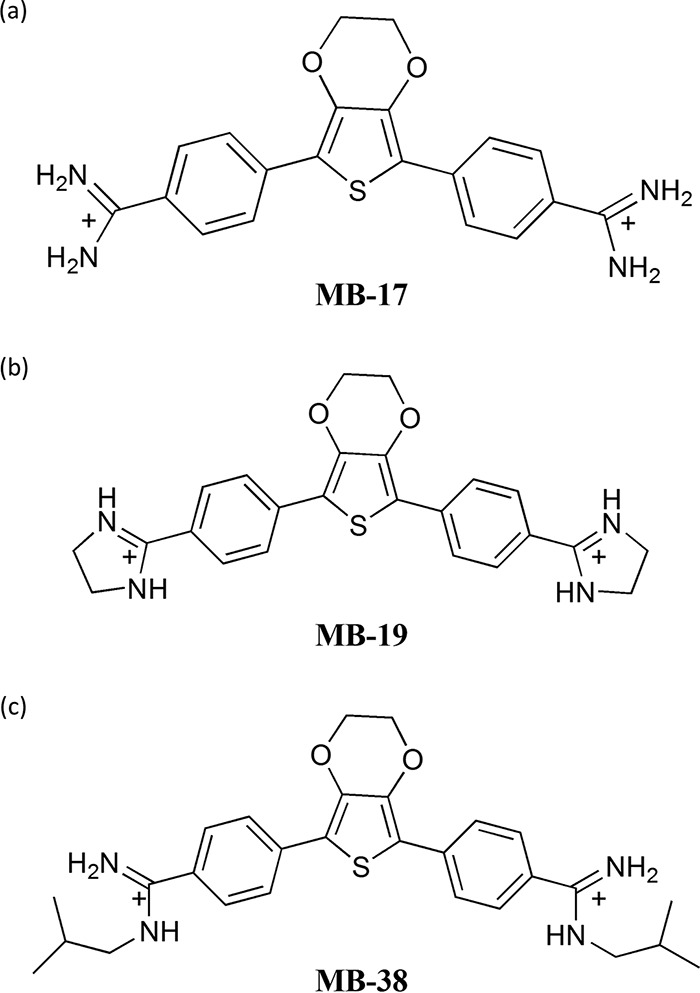
Chemical composition of the three tested compounds MB17, MB19, and MB38.
MATERIALS AND METHODS
Chemicals and reagents.
MB17, MB19, and MB38 were synthesized as previously reported (19). An MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltretazolium bromide] assay and a kit for bioluminescence assays in somatic cells were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fluo-4 acetoxymethyl ester (AM) was purchased from Invitrogen (Eugene, Oregon, USA). Annexin-Alexa 647 was provided by Gustavo P. Amarante-Mendes's Laboratory (Institute of Biomedical Sciences University of São Paulo). Culture medium and fetal calf serum (FCS) were purchased from Cultilab (Campinas, SP, Brazil).
Cells and parasites.
T Chinese hamster ovary cell line CHO-K1 was cultivated in RPMI medium supplemented with 10% heat-inactivated FCS, 0.15% (wt/vol) NaCO3, 100 units/ml penicillin, and 100 mg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. T. cruzi CL strain clone 14 epimastigotes (20) were maintained in exponential growth phase by subculturing every 48 h in liver infusion tryptose (LIT) medium supplemented with 10% FCS at 28°C. Trypomastigotes were obtained by infection of CHO-K1 cells with cultured trypomastigotes, as described previously (21). Trypomastigotes were collected from the extracellular medium 5 or 6 days after infection.
Growth inhibition assays.
T. cruzi epimastigotes in the exponential growth phase (5.0 × 107 cells/ml) were cultured in fresh LIT medium. The cells were treated with different concentrations of drugs or not treated (negative control). A combination of 60 μM rotenone–0.5 μM antimycin (RA) was used as a positive control for inhibition, as previously described (22). The cells (2.5 × 106 cell/ml) were transferred to 96-well culture plates and incubated at 28°C. Cell proliferation was quantified by reading the optical density (OD) at 620 nm for 9 days. The OD values were converted to cell density values (cells per milliliter) using a linear regression equation previously obtained under the same conditions. The concentration of compound that inhibited 50% of parasite proliferation (50% inhibitory concentration, or IC50) was determined during the exponential growth phase (fifth day) by fitting the data to a typical sigmoidal dose-response curve using GraphPad Prism, version 5. The compounds were evaluated in quadruplicate in each experiment, and the results correspond to three independent experiments.
Growth inhibition recovery assays.
T. cruzi epimastigotes in the exponential growth phase (5.0 × 107 cells/ml) were treated for 48 h with either 0.5 or 1.5 μM MB17 or with a combination of 60 μM rotenone and 0.5 μM antimycin (RA) or were left untreated (negative control). Then, parasites were washed and resuspended in fresh LIT medium with or without 0.5 or 1.5 μM MB17 or RA, such that half of the treated parasites remained under treatment, and half of the treated parasites were incubated under treatment-free conditions. Cell proliferation was assessed for 11 days by the absorbance reading (wavelength [λ] of 620 nm) as described above. The compounds were evaluated in triplicate in each experiment, and the results correspond to three independent experiments.
Flow cytometry analysis.
To discriminate between apoptotic, late apoptotic, and necrotic death, the treated or untreated epimastigotes were harvested and stained with propidium iodide (PI) and annexin V-Alexa 647 (Molecular Probes) according to the manufacturer's instructions. The fluorescence was measured using a FACSCalibur (BD Biosciences), with 10,000 events collected and analyzed using Flowing Software, version 2.
Determination of T. cruzi intracellular ATP levels.
Intracellular ATP levels were measured in treated and control epimastigote forms. To assess the effect of a given compound on the levels of intracellular ATP, a kit for monitoring bioluminescence in somatic cells was purchased from Sigma-Aldrich and used according to the manufacturer's instructions. Briefly, 50 μl of phosphate-buffered saline (PBS) was added to 100 μl of cellular ATP-releasing reagent, and the mixture was added to a 50-μl suspension of 5.0 × 106 parasites, either treated or untreated (controls). Light emission levels were measured on a Spectra Max I3 fluorometer at 570 nm (23).
Analysis of DNA damage by TUNEL assay.
To detect in situ DNA fragmentation and compare parasite groups treated with MB17 at the IC50 and IC80 [MB17 (IC50) and MB17 (IC80), respectively] and control parasites, we employed a terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) technique, using a fluorescein-based apoptosis detection system (fluorescein kit; Promega). Samples of each group, containing ∼1 × 107 parasites/ml in exponential phase, were collected by centrifugation in 4% paraformaldehyde for 10 min. Parasites were washed with PBS and permeabilized by the addition of 0.1% Triton X-100 for 10 min at 26°C; 0.1 M glycine was added for 5 min to neutralize remaining aldehyde groups. The TUNEL assay was performed according to the manufacturer's protocol. Vectashield mounting medium with DAPI (Vector Laboratories) was added as an antifade mounting solution and to stain nuclear and kinetoplast DNA. Cells were analyzed using a fluorescence microscope (Olympus BX51), and images were captured using Olympus Cell F software.
DNA content and cell cycle analysis.
In the three analyzed groups [control untreated, MB17 (IC50)-treated, and MB17 (IC80)-treated cells], cells were collected by centrifugation (2,500 × g for 5 min), washed in 1× PBS, fixed in 70% ethanol for 12 h, and incubated with 10 μg/ml RNase A (Thermo Scientific) for 10 min at 26°C. To measure the DNA content, parasite cells were stained with 40 μg/ml propidium iodide (Molecular Probes/Invitrogen) and analyzed with an acoustic focusing cytometer (Attune; Applied Biosystems). Histograms (number of counts by BL2 area), scatter plots (side scatter [SSC] area by forward scatter [FSC] area) (data not shown), and gates for each cell cycle phase were performed using Attune cytometric software, version 1.2.5.
Effect of MB17 on mammalian cell viability.
CHO-K1 cells (5.0 × 105 cells/ml) were seeded in 24-well plates in RPMI medium supplemented with FCS (10%), with or without (control) different concentrations (ranging between 0.5 and 10 μM) of MB17. Cell viability was evaluated 48 h after the initiation of treatment using the MTT assay (24).
Effect of MB17 on amastigote replication and trypomastigote release.
CHO-K1 cells (5.0 × 104 per well) were maintained in 24-well plates in RPMI medium supplemented with 10% FCS at 37°C. To perform the infections, the cells were incubated with trypomastigote forms (2.5 × 106 per well) for 4 h. After this period, free parasites were removed by washing the plates twice with PBS, and the cells were incubated overnight in RPMI medium supplemented with 10% FCS at 37°C in the presence of different concentrations of MB17 or left untreated (control). The plates were then incubated at 33°C to allow the parasite to complete its infection cycle, as previously reported (21). To measure the effect on amastigote replication, after 48 h the CHO-K1 cells and parasites were fixed with 4% paraformaldehyde and stained with Hoechst. Images were taken by microscopy. Cells, parasites, and infected cells were counted using ImageJ software. The infection index (percentage of infected cells × the number of parasites per cell) was calculated. To measure the effect of MB17 on the infection of CHO-K1 cells, trypomastigotes were collected from the extracellular medium on the fifth day and counted in a Neubauer chamber (23). All experiments were performed in technical and biological triplicates.
Fluorescence microscopy analysis.
T. cruzi epimastigotes and intracellular forms were incubated with MB17 (4 μM) for 5 days. The cells were then washed with PBS and fixed with 4% paraformaldehyde for fluorescence microscopy. MB17 fluorescence was observed at an excitation wavelength (λexcitation) of 488 nm and emission wavelength (λemission) of 535 nm. The data were analyzed using a fluorescence microscope (Axiophot; Carl Zeiss Jena GmbH, Jena, Germany) with a Zeiss LP-520 filter and a magnification of ×1,000.
MB17 uptake assay.
T. cruzi epimastigotes in the exponential growth phase (5.0 × 105 cells) were incubated in the presence of MB17 (1.5 μM) for different durations. Then the parasites were washed and resuspended in PBS. The uptake of MB17 was measured using the fluorescence of this compound at λexcitation of 488 nm and λemission of 515 nm on a Spectra Max M3 fluorometer (Molecular Devices). The data were fit to a typical one-phase decay curve using GraphPad Prism, version 5. The compound was evaluated in triplicate.
Statistical analysis.
One-way analysis of variance (ANOVA) followed by a Tukey posttest was used for statistical analysis. Student's t test was used to analyze differences between groups, and P values of less than 0.05 were considered statistically significant.
RESULTS
MB17, MB19, and MB38 affect the growth of epimastigote forms.
To elucidate the effect of the three new DNA-binding diamidines against T. cruzi, we first determined whether these compounds inhibit epimastigote growth. A negative control (cells cultured in LIT medium without drugs) and a positive control (cells cultured in LIT medium supplemented with RA) were also performed. MB17, MB19, and MB38 exhibited dose-dependent inhibition of epimastigote growth at 28°C and pH 7.5, the optimal growth conditions for these cells (Fig. 2). We observed significant growth inhibition between the treated cells and the control, with IC50s of epimastigote growth of 0.5 ± 0.13 μM for MB17, 3.7 ± 0.29 μM for MB19, and 8.3 ± 1.49 μM for MB38. The MB17, MB19, and MB38 IC80 values were also computed, giving 1.5 ± 0.51, 8.0 ± 0.94, and 16 ± 3.2 μM, respectively. Of the three diamidine-derived compounds evaluated, MB17 showed the best growth-inhibitory effect against T. cruzi epimastigotes.
FIG 2.

Growth curve of epimastigote forms of T. cruzi. Growth curves in the presence of different concentrations of MB17 (a), MB19 (b), or MB38 (c) and dose-response curves (d, e, and f) in the presence of different concentrations of compounds. Treatments were performed with MB17 (a and d), MB19 (b and e), and MB38 (c and f). ●, 0 μM; ◽, inhibition control (0.5 μM antimycin and 60 μM rotenone); ▲, 0.005 μM (MB17), 2 μM (MB19), 4 μM (MB38); ▼, 0.025 μM (MB17), 4 μM (MB19), and 6 μM (MB38); △, 0.05 μM (MB17), 6 μM (MB19), and 8 μM (MB38); ▽, 0.1 μM (MB17), 7 μM (MB19), and 10 μM (MB38); ◆, 0.25 μM (MB17), 8 μM (MB19), and 12 μM (MB38); ♢, 0.5 μM (MB17), 9 μM (MB19), and 16 μM (MB38); ■, 1 μM (MB17), 10 μM (MB19), and 18 μM (MB38); ○, 2 μM (MB17), 12 μM (MB19), and 20 μM (MB38); X, 5 μM (MB17), 20 μM (MB19), and 25 μM (MB38); ⭑, 10 μM (MB17), 25 μM (MB19), and 30 μM (MB38).
Cell death mechanisms.
To identify the cell death mechanisms triggered by MB17, MB19, and MB38, we first investigated the morphological and biochemical changes of epimastigote cells. Programmed cell death (PCD) is characterized by the exposure of phosphatidylserine on the extracellular face of the cytoplasmic membrane, whereas classical necrosis is characterized by plasma membrane permeabilization (25–27). Parasites treated with various concentrations of MB17, MB19, and MB38 (corresponding to the IC50 and IC80 values) were incubated with annexin V-Alexa 647 to assess the external exposure of phosphatidylserine (a feature of PCD) and with propidium iodide to assess plasma membrane permeabilization (a feature of classical necrosis). The cells were then subjected to analysis by flow cytometry (Fig. 3). Interestingly, treatment with MB38, but not MB17 or MB19, produced a significant difference in the percentage of annexin-propidium iodide double-labeled cells compared to that in untreated parasites. With MB38 at the IC50 dose, 24.4% of double-labeled cells were positive, whereas with MB38 at the IC80 dose 55% of double-labeled cells and 27% of PI-single-labeled cells were positive. MB17 and MB19 did not produce a significant difference in the percentages of positive cells compared to levels in the control.
FIG 3.

Analysis of extracellular exposure of phosphatidylserine by annexin V/propidium iodide labeling. The parasites were treated with MB17, MB19, and MB38 at concentrations corresponding to the IC50s (0.5 ± 0.13, 3.7 ± 0.29, and 8.3 ± 1.49 μM, respectively) and IC80s (1.5 ± 0.5, 8.0 ± 0.94, and 16 ± 3.2 μM, respectively) for 5 days. After this period, the parasites were labeled with annexin V and propidium iodide and analyzed by flow cytometry as follows: untreated epimastigotes (a), parasites treated with MB17 at concentrations corresponding to the IC50 (b) and IC80 (e), parasites treated with MB19 at concentrations corresponding to the IC50 (c) and IC80, (f), and parasites treated with MB38 at concentrations corresponding to the IC50 (d) and IC80 (g). Quantitative analysis of the parasites treated with MB17, MB19, and MB38, as indicated, are also shown. One-way ANOVA followed by a Tukey posttest was used for statistical analysis to compare the treatments to values in the respective control. ***, P < 0.001; **, P < 0.01 (Tukey posttest).
MB17 affects parasite replication.
As MB17 was the most promising drug based on the IC50, we focused on its mechanism of action. To investigate the action of MB17 in growth impairment, we first analyzed its subcellular fate, taking advantage of its fluorescence properties, which allowed us to visualize it by fluorescence microscopy. As expected for a DNA binder, MB17 and Hoechst colocalized in T. cruzi epimastigotes, trypomastigotes, amastigotes, and intracellular epimastigote-like forms (Fig. 4), showing that MB17 binds to both genomic DNA and kDNA, which is consistent with its previously described DNA-binding activity (18, 19). Interestingly, treated epimastigotes exhibited an increased number of double-flagellated trypanosomes (15% at IC50 and 30% at IC80) compared to control levels (see Fig. S1 in the supplemental material). These double-flagellated parasites frequently presented two nuclei and one kinetoplast. Taken together, these results suggest that MB17 interferes with the epimastigote cell cycle. Therefore, to check if the cell cycle could be impaired by MB17, parasites were either left untreated (control) or were treated with concentrations corresponding to the IC50 and IC80 and submitted to cell cycle analysis by flow cytometry. Cells treated with MB17 presented an accumulation of cells with a 2N amount of DNA compared to that of the control cells (Fig. 5).
FIG 4.

Fluorescence microscopy analysis of the intracellular distribution of MB17. Parasites were treated with 4 μM MB17, fixed with 4% paraformaldehyde, incubated with Hoechst, and analyzed by light microscopy (a, f, k, and p) and by fluorescence for Hoechst staining (b, g, l, and q) and MB17 staining (c, h, m, and r). Merged images for MB17 and Hoechst (d, i, n, and s) and for light microscopy, MB17, and Hoechst (e, j, o, and t) were obtained. Magnifications: ×1,000 (a to o) and ×600 (p to t).
FIG 5.

Effect of MB17 on epimastigote cell cycle using fluorescence-activated cell sorting. Control cells and cells treated with MB17 (IC50) or MB17 (IC80) for 5 days were treated with RNase A and stained with propidium iodide, and their DNA content was analyzed by fluorescence-activated cell sorting. Histograms (a) and quantifications of the numbers of cells in each stage of the cell cycle (b) are shown. Representative histograms of three independent experiments are shown, and results are compared between control and MB17 (IC50)-treated G1-phase cells and between control and MB17 (IC80)-treated G1-phase cells. **, P < 0.01; *, P < 0.05 (Student's t test).
MB17 damages both genomic and kinetoplastic DNA.
As MB17 colocalized with DNA markers, we analyzed its effect on both genomic DNA (gDNA) and kDNA. To evaluate DNA fragmentation, a TUNEL assay was performed on epimastigotes that had been cultured for 5 days at concentrations corresponding to the IC50 and IC80. At both concentrations, gDNA and kDNA were intensively labeled, while untreated parasites remained unlabeled, indicating that MB17 produces DNA fragmentation (Fig. 6a). Controls were performed by submitting the cells to a TUNEL assay without TdT enzyme (negative control) or by submitting the cells to the TUNEL assay after a treatment with DNase I (positive control). Additional positive and negative controls were unlabeled cells and cells with nearly 100% labeling of both gDNA and kDNA, respectively (see Fig. S2 in the supplemental material). Interestingly, at both concentrations, the percentage of cells with labeled kDNA was higher than the percentage of cells with labeled gDNA, which shows that kDNA was more sensitive to the treatment (Fig. 6b). Consequently, MB17 preferentially targets the kinetoplast rather than the nucleus. By DAPI analysis of MB17 (IC80)-treated cells, we noted a high number of cells with no kinetoplast. Therefore, cells were treated with IC50 or IC80 MB17 and subjected to DAPI staining, and then the numbers of cells with 1 or 2 nuclei and 0, 1, or 2 kinetoplasts were counted (Fig. 7). At the concentration corresponding to the IC50, no differences were detected with respect to the control level. However, at the concentration corresponding to the IC80, over 70% of the cells were dyskinetoplastic. As MB17 seemed to affect preferentially the kDNA, we were interested in evaluating the possible variation of the level of the main mitochondrial metabolism product, ATP. Epimastigotes were incubated with or without (control) MB17 at concentrations corresponding to the IC50 for 24 h or 120 h (5 days), and then ATP levels were measured using a bioluminescence assay. Both treatments led to decreased intracellular ATP levels (Fig. 8). The results from ATP quantification indicate that MB17 affected the mitochondrial functions, particularly the energy metabolism of the parasite.
FIG 6.
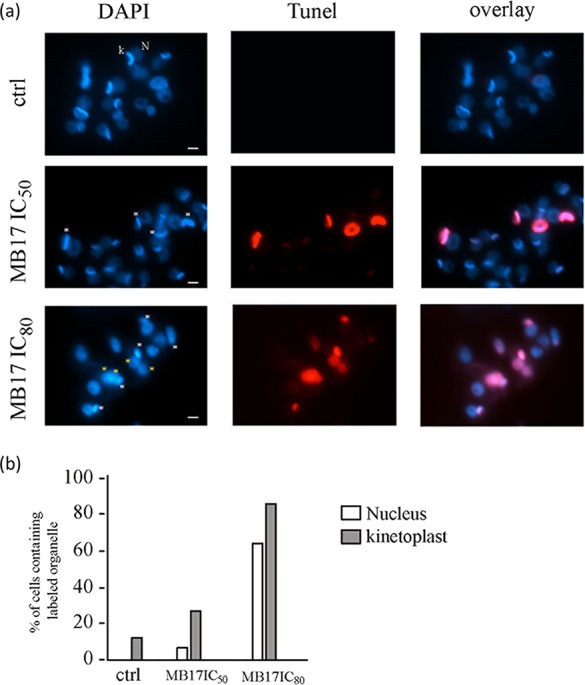
MB17 induces DNA fragmentation. (a) Control cells and cells treated with MB17 (IC50) or MB17 (IC80) were subjected to a TUNEL assay (red) and stained with DAPI (blue). White asterisks indicate TUNEL-positive kinetoplasts, and yellow asterisks indicate TUNEL-positive nuclei. N, nucleus; k, kinetoplast. Bar, 2 μm. (b) The percentage of TUNEL-positive nuclei or kinetoplasts was plotted after analysis of 100 cells from each treatment.
FIG 7.
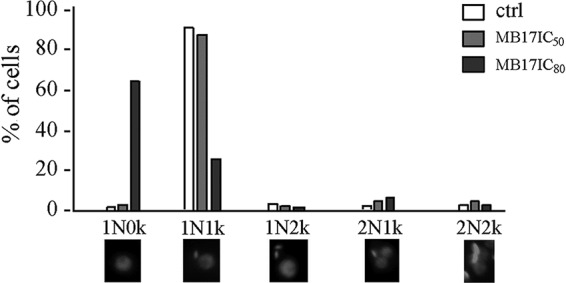
Analysis of MB17 effect on T. cruzi DNA pattern. Control cells and cells treated with MB17 (IC50) or MB17 (IC80) were stained with DAPI. Patterns were observed and quantified. The graph shows the numbers obtained from 150 analyzed cells. N, nucleus; k, kinetoplast.
FIG 8.

Quantification of ATP levels in T. cruzi. ATP levels of treated or untreated (control) epimastigotes were measured after 24 h (a) or 5 days (b). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (in comparison to the control; Tukey posttest). AU, arbitrary units.
Further experiments were performed to determine whether MB17 interferes with parasite proliferation in a reversible or irreversible manner. Epimastigotes were treated or not treated (control) with MB17 at a concentration corresponding to the IC50 or the IC80. In addition, a control treatment was also performed with RA. At the second day after treatment, treated and control parasites were washed and resuspended in fresh medium with or without MB17 or with or without RA. Parasite growth was monitored for the following 9 days. All MB17-treated cells showed the same growth inhibition, independent of whether treatment was continuous for 11 days or for 2 days, and this inhibition persisted in both cases after washout (Fig. 9a). The parasites treated with RA showed growth inhibition during the treatment, but they were able to recover when the drugs were washed out. These results indicate that MB17 irreversibly inhibited epimastigote proliferation. To evaluate the rate of MB17 uptake, its incorporation was measured as a function of time (Fig. 9b). As reported for T. brucei, a fast uptake of the drug was detected, showing saturation at approximately 100 s and accumulation of fluorescence associated with both gDNA and kDNA (28).
FIG 9.

Reversibility of the effect of MB17 on epimastigotes. Epimastigotes were either left untreated or treated with MB17 or a combination of 60 μM rotenone–0.5 μM antimycin (RA) for 48 h, and then MB17 or RA was either removed or not from the culture medium. The culture growth was followed up until the stationary phase (11 days). ●, control (no treatment); ○, control, washed; ■, treatment with RA; ◽, treatment with RA, washed; ▲, treatment with 0.5 μM MB17; △, treatment with 0.5 μM MB17, washed; ▼, treatment with 1.5 μM MB17; ▽, treatment with 1.5 μM MB17, washed. The arrow indicates when MB17 or RA was removed.
MB17 selectively inhibits the intracellular cycle of T. cruzi.
To evaluate the effect of MB17, we first evaluated the toxicity of the compound in the mammalian host cells. For this purpose, CHO-K1 cells were incubated in RPMI medium supplemented with MB17 at different concentrations, ranging from 1 to 100 μM, and cytotoxicity was evaluated by MTT assay. A value for the 50% cytotoxic concentration (CC50) of 13.47 ± 0.37 μM was obtained (Fig. 10a and b). According to this result, a range of MB17 doses from 0.1 to 5 μM was selected to evaluate the IC50 for trypomastigote release after an entire infection cycle on CHO-K1 cells. To measure the effect of MB17 on the intracellular cycle of T. cruzi, CHO-K1 cells were infected with trypomastigotes for 4 h. The cells were washed twice with PBS to eliminate the noninternalized parasites, and then they were incubated with culture medium that was either untreated (control) or supplemented with different MB17 concentrations. To calculate the effect of MB17 on the infection index, the cells were infected, and at 2 days postinfection the cultures were fixed and stained. The number of trypomastigotes released by infected cells after an infection cycle was also determined. We observed a dose-dependent decrease of trypomastigote release, which allowed us to measure an MB17 IC50 for trypomastigote bursting [IC50(Tryp)] of 0.14 ± 0.12 μM (Fig. 10c and d). On the basis of these values, we obtained a selectivity index (SI; CC50/IC50) of 96.2 ± 2.6, corresponding to the IC50(Tryp). The total number of cells, the number of infected cells, and the number of amastigotes per infected cell were counted. The treatment of infected cells with 0.5 µM MB17 diminished the infection index by 60%, indicating an IC50 of <0.5 μM by this criterion (Fig. 10e and f). These results indicate that treatment with submicromolar concentrations of MB17 interferes with proliferation and/or differentiation of intracellular stages (Fig. 10).
FIG 10.

Cytotoxicity effect of MB17 on amastigote replication and intracellular cycle of T. cruzi. The viability of CHO-K1 cells treated with different concentrations of MB17 (range, 1 μM to 100 μM) was assessed by MTT assay (a), and the corresponding dose-response curve is shown (b). The effect on the infectivity of trypomastigotes treated with MB17 (range, 0.1 to 5.0 μM) only during the period of infection was evaluated by counting the released parasites in a Neubauer chamber on the fifth day postinfection (c), and the corresponding dose-response curve was plotted (d). The effect on amastigote replication was measured using the infection index (percentage of infected cells × number of parasites per cell) of treated parasites compared to control levels (e). The percentages of infection index values compared to control values were also calculated (f). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Tukey posttest).
DISCUSSION
Initially, IC50 values for all three compounds were evaluated using their effect on the inhibition of epimastigote growth in the mid-exponential phase as the criterion for measuring. This measurement was complemented with a second evaluation performed by counting motile parasites (see Fig. S3 in the supplemental material) as previously reported (16). Based on these two evaluations, MB17 was the most potent inhibitor of T. cruzi epimastigote replication and mobility. To investigate the death mechanism(s) triggered by these drugs, we analyzed several parameters. When the integrity of the cytoplasmic membrane and phosphatidylserine exposure were evaluated, it was found that MB38 caused a loss of cytoplasmic membrane integrity, whereas MB17 and MB19 did not. In fact, none of the treatments caused exposure of phosphatidylserine to the external side of the plasma membrane. This excludes apoptosis-like cell death as a mechanism of action of these drugs. In addition, these data suggest different mechanisms of action for these compounds, indicating that MB38, unlike MB17 and MB19, induces necrosis in T. cruzi epimastigotes. MB17 and MB19 arrest parasite replication in the exponential phase of growth but do not induce damage to the plasma membrane. These observations led us to hypothesize that these compounds interfere with cell replication by impairing the cell cycle. As MB17 was a more efficient anti-T. cruzi agent than MB19 and MB38, we investigated its mechanism of action in more detail. Remarkably, in a previous study, MB17, which was the compound with the lowest IC50, also showed promising antibacterial activity against Gram-positive and Gram-negative bacterial strains compared to the activities of other compounds, including MB19 and MB38 (29). In addition to their antiparasitic effects, these compounds, due to their fluorescence properties, could also be considered promising probes for trypanosome-specific staining (30). As MB17 showed neither membrane permeabilization nor phosphatidylserine exposure but did inhibit epimastigote growth, we investigated the compound's effect on the parasite cell cycle. The binding activity of MB17 on gDNA and kDNA was consistent with findings of previous studies (10, 11, 31, 32). Furthermore, the presence of MB17-treated cells with two nuclei, two flagella, and one kinetoplast or with one nucleus and no kinetoplast supports our hypothesis that MB17 interferes with the epimastigote cell cycle. In fact, cytometry analysis confirmed this hypothesis, showing an accumulation of cells at the G1 phase, concomitant with a decrease in the percentage of cells in other phases of the cell cycle (S and G2/M) (Fig. 5). This result can be explained by the activity of this diamidine as a DNA-intercalating agent and probably as a DNA synthesis inhibitor, which is consistent with the fact that epimastigotes exhibited a cell cycle delay in G1 phase, as already shown in trypanosomatids (33). All of these results are summarized in Table 1.
TABLE 1.
Activity of MB17, MB19, and MB38
| Compound | IC50 (μM) in epimastigotes | IC50 (μM) in trypomastigotes | CC50 (μM) | Selectivity index | Causes loss of cytoplasmic membrane integrity | Alters ATP levels | Irreversibly inhibits growth | Affects amastigote replication | Affects host-cell infection | Causes DNA damage | Affects parasite cell cycle |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MB17 | 0.5 ± 0.13 | 0.14 ± 0.12 | Nob | Yesb | Yes | Yes | Yes | Yes | Yes | ||
| MB19 | 3.7 ± 0.29 | NDa | ND | ND | Noc | Noc | ND | ND | ND | ND | ND |
| MB38 | 8.3 ± 1.49 | ND | ND | ND | Yesc | Yesc | Yes | ND | ND | ND | ND |
ND, not determined.
After 24 h and 5 days.
After 5 days.
Notably, the increase in the percentage of cells with two flagella (1k2N) shown in Fig. 7 (see also Fig. S1 in the supplemental material) does not explain the decrease in the percentage of cells in the G2/M phase that is shown in Fig. 5: cells showing two flagella would be expected to have a DNA content equivalent to that of cells in the G2/M phase. The explanation for this result could be that the diamidine MB17 might inhibit kDNA synthesis but not prevent growth of the second flagellum or of cytokinesis. Thus, aberrant cell types (0k1N) resulting from cytokinesis of cells with two flagella (1k2N) are generated 5 days after treatment, as shown in Fig. 7. Similar results have already been reported in trypanosomatids after treatment with aphidicolin, rhizoxin (33), or acriflavine (34, 35). Furthermore, we observed for the parasite that the kDNA replication/segregation machinery is more sensitive to MB17 than the nuclear DNA replication/segregation machinery. MB17 likely interferes with kDNA due to its unique DNA network structure consisting of thousands of interconnected DNA circles and also due to its complex system of replication (12). The peculiarities of kDNA and its replication machinery may also explain the compound selectivity, as shown for other diamidine compounds (10, 11). However, it should be stressed that, until now, the mechanism disrupting kDNA replication remained elusive. MB17 likely binds specifically to kinetoplast DNA, which results in the inhibition of DNA-dependent enzymes (10, 11, 31). Alternatively, MB17 may directly inhibit transcription and replication enzymes (11, 31, 36–38). We propose that MB17 acts through its binding activity on the parasite DNA and not as a trypanocidal agent. We determined that this effect on parasite proliferation is irreversible and could be related to a global effect of the drug on mitochondrial functions, as suggested by the diminished amounts of total ATP content in treated cells. Indeed, it has been proposed that the trypanosoma mitochondrion is involved in the uptake of diamidines and in drug resistance mechanisms (39–41).
Remarkably, MB17 also caused a 50% reduction in the number of trypomastigotes released from infected cells. This effect was observed at low concentrations of MB17 and with a selectivity index of 96.2 ± 2.6. In addition, when infected cells were treated with 0.5 μM MB17, there was an approximately 60% reduction in the infection index. These results are particularly promising because both aspects, the establishment of the mammalian host infection and the release of trypomastigote forms, are required for the maintenance of the chronic phase of the disease. Nonetheless, in vivo studies will be necessary to validate the antiparasitic effect of MB17 (42, 43). This possibility should be carefully evaluated because similar compounds were effective (alone or in combination with other drugs) while other amidine derivatives showed high toxicity in animal models (43).
To conclude, the newly synthesized diamidines MB17, MB19, and MB38 showed in vitro antiparasitic effects. Taken together, these results indicate that MB17 is a remarkable anti-T. cruzi compound and, therefore, should be considered for further evaluation in vivo. As the kinetoplast is the preferred target, we propose that this diamidine has potential therapeutic activity for other diseases caused by kinetoplastid protozoans, such as Leishmania spp. and Trypanosoma brucei.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Ministry of Science, Education and Sports of the Republic of Croatia (grant number 053-0982914-2965 to M.B.), Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP grants 13/18970-6 to A.M.S. and 2013/07467-1 to M.C.E.), Instituto Nacional de Biologia Estrutural e Química Medicinal em Doenças Infecciosas (INBEQMeDI), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq grant 475741/2013-7 to A.M.S. and 306376/2012-1 to M.C.E.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01595-15.
REFERENCES
- 1.Bonney KM. 2014. Chagas disease in the 21st century: a public health success or an emerging threat? Parasite 21:11. doi: 10.1051/parasite/2014012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rassi A Jr, Rassi A, Marin-Neto JA. 2010. Chagas disease. Lancet 375:1388–1402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 3.Barrett MP, Burchmore RJ, Stich A, Lazzari JO, Frasch AC, Cazzulo JJ, Krishna S. 2003. The trypanosomiases. Lancet 362:1469–1480. doi: 10.1016/S0140-6736(03)14694-6. [DOI] [PubMed] [Google Scholar]
- 4.Brener Z. 1973. Biology of Trypanosoma cruzi. Annu Rev Microbiol 27:347–382. doi: 10.1146/annurev.mi.27.100173.002023. [DOI] [PubMed] [Google Scholar]
- 5.Alves MJ, Colli W. 2007. Trypanosoma cruzi: adhesion to the host cell and intracellular survival. IUBMB Life 59:274–279. doi: 10.1080/15216540701200084. [DOI] [PubMed] [Google Scholar]
- 6.Tyler KM, Engman DM. 2001. The life cycle of Trypanosoma cruzi revisited. Int J Parasitol 31:472–481. doi: 10.1016/S0020-7519(01)00153-9. [DOI] [PubMed] [Google Scholar]
- 7.Boscardin SB, Torrecilhas AC, Manarin R, Revelli S, Rey EG, Tonelli RR, Silber AM. 2010. Chagas' disease: an update on immune mechanisms and therapeutic strategies. J Cell Mol Med 14:1373–1384. doi: 10.1111/j.1582-4934.2010.01007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nunes MC, Dones W, Morillo CA, Encina JJ, Ribeiro AL. 2013. Chagas disease: an overview of clinical and epidemiological aspects. J Am Coll Cardiol 62:767–776. doi: 10.1016/j.jacc.2013.05.046. [DOI] [PubMed] [Google Scholar]
- 9.Baraldi PG, Bovero A, Fruttarolo F, Preti D, Tabrizi MA, Pavani MG, Romagnoli R. 2004. DNA minor groove binders as potential antitumor and antimicrobial agents. Med Res Rev 24:475–528. doi: 10.1002/med.20000. [DOI] [PubMed] [Google Scholar]
- 10.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. 2008. Antiparasitic compounds that target DNA. Biochimie 90:999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soeiro MN, Werbovetz K, Boykin DW, Wilson WD, Wang MZ, Hemphill A. 2013. Novel amidines and analogues as promising agents against intracellular parasites: a systematic review. Parasitology 140:929–951. doi: 10.1017/S0031182013000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen RE, Englund PT. 2012. Network news: the replication of kinetoplast DNA. Annu Rev Microbiol 66:473–491. doi: 10.1146/annurev-micro-092611-150057. [DOI] [PubMed] [Google Scholar]
- 13.Lukes J, Hashimi H, Zikova A. 2005. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet 48:277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- 14.Povelones ML. 2014. Beyond replication: division and segregation of mitochondrial DNA in kinetoplastids. Mol Biochem Parasitol 196:53–60. doi: 10.1016/j.molbiopara.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Lisvane Silva P, Mantilla BS, Barison MJ, Wrenger C, Silber AM. 2011. The uniqueness of the Trypanosoma cruzi mitochondrion: opportunities to identify new drug target for the treatment of Chagas disease. Curr Pharm Des 17:2074–2099. doi: 10.2174/138161211796904786. [DOI] [PubMed] [Google Scholar]
- 16.da Silva CF, Batista MM, Batista DDGJ, de Souza EM, da Silva PB, de Oliveira GM, Meuser AS, Shareef AR, Boykin DW, Soeiro MDN. 2008. In vitro and in vivo studies of the trypanocidal activity of a diarylthiophene diamidine against Trypanosoma cruzi. Antimicrob Agents Chemother 52:3307–3314. doi: 10.1128/AAC.00038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soeiro MDN, de Castro SL. 2011. Screening of potential anti-Trypanosoma cruzi candidates: in vitro and in vivo studies. Open Med Chem J 5:21–30. doi: 10.2174/1874104501105010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolic I, Miskovic K, Magdaleno A, Silber AM, Piantanida I, Bajic M, Glavas-Obrovac L. 2009. Effect of 3,4-ethylenedioxy-extension of thiophene core on the DNA/RNA binding properties and biological activity of bisbenzimidazole amidines. Bioorg Med Chem 17:2544–2554. doi: 10.1016/j.bmc.2009.01.071. [DOI] [PubMed] [Google Scholar]
- 19.Stolic I, Miskovic K, Piantanida I, Loncar MB, Glavas-Obrovac L, Bajic M. 2011. Synthesis, DNA/RNA affinity and antitumour activity of new aromatic diamidines linked by 3,4-ethylenedioxythiophene. Eur J Med Chem 46:743–755. doi: 10.1016/j.ejmech.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 20.Brener Z, Chiari E. 1965. Aspects of early growth of different Trypanosoma cruzi strains in culture medium. J Parasitol 51:922–926. doi: 10.2307/3275869. [DOI] [PubMed] [Google Scholar]
- 21.Tonelli RR, Silber AM, Almeida-de-Faria M, Hirata IY, Colli W, Alves MJ. 2004. l-Proline is essential for the intracellular differentiation of Trypanosoma cruzi. Cell Microbiol 6:733–741. doi: 10.1111/j.1462-5822.2004.00397.x. [DOI] [PubMed] [Google Scholar]
- 22.Magdaleno A, Ahn IY, Paes LS, Silber AM. 2009. Actions of a proline analogue, l-thiazolidine-4-carboxylic acid (T4C), on Trypanosoma cruzi. PLoS One 4:e4534. doi: 10.1371/journal.pone.0004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Damasceno FS, Barison MJ, Pral EM, Paes LS, Silber AM. 2014. Memantine, an antagonist of the NMDA glutamate receptor, affects cell proliferation, differentiation and the intracellular cycle and induces apoptosis in Trypanosoma cruzi. PLoS Negl Trop Dis 8:e2717. doi: 10.1371/journal.pntd.0002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mosmann T. 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 25.Duszenko M, Figarella K, Macleod ET, Welburn SC. 2006. Death of a trypanosome: a selfish altruism. Trends Parasitol 22:536–542. doi: 10.1016/j.pt.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Smirlis D, Soteriadou K. 2011. Trypanosomatid apoptosis: “apoptosis” without the canonical regulators. Virulence 2:253–256. doi: 10.4161/viru.2.3.16278. [DOI] [PubMed] [Google Scholar]
- 27.Smirlis D, Duszenko M, Ruiz AJ, Scoulica E, Bastien P, Fasel N, Soteriadou K. 2010. Targeting essential pathways in trypanosomatids gives insights into protozoan mechanisms of cell death. Parasit Vectors 3:107. doi: 10.1186/1756-3305-3-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ward CP, Wong PE, Burchmore RJ, de Koning HP, Barrett MP. 2011. Trypanocidal furamidine analogues: influence of pyridine nitrogens on trypanocidal activity, transport kinetics, and resistance patterns. Antimicrob Agents Chemother 55:2352–2361. doi: 10.1128/AAC.01551-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stolic I, Cipcic Paljetak H, Peric M, Matijasic M, Stepanic V, Verbanac D, Bajic M. 2015. Synthesis and structure-activity relationship of amidine derivatives of 3,4-ethylenedioxythiophene as novel antibacterial agents. Eur J Med Chem 90:68–81. doi: 10.1016/j.ejmech.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Giordani F, Munde M, Wilson WD, Ismail MA, Kumar A, Boykin DW, Barrett MP. 2014. Green fluorescent diamidines as diagnostic probes for trypanosomes. Antimicrob Agents Chemother 58:1793–1796. doi: 10.1128/AAC.02024-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson WD, Nguyen B, Tanious FA, Mathis A, Hall JE, Stephens CE, Boykin DW. 2005. Dications that target the DNA minor groove: compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr Med Chem Anticancer Agents 5:389–408. doi: 10.2174/1568011054222319. [DOI] [PubMed] [Google Scholar]
- 32.Daliry A, Da Silva PB, Da Silva CF, Batista MM, De Castro SL, Tidwell RR, Soeiro MDN. 2009. In vitro analyses of the effect of aromatic diamidines upon Trypanosoma cruzi. J Antimicrob Chemother 64:747–750. doi: 10.1093/jac/dkp290. [DOI] [PubMed] [Google Scholar]
- 33.Ploubidou A, Robinson DR, Docherty RC, Ogbadoyi EO, Gull K. 1999. Evidence for novel cell cycle checkpoints in trypanosomes: kinetoplast segregation and cytokinesis in the absence of mitosis. J Cell Sci 112:4641–4650. [DOI] [PubMed] [Google Scholar]
- 34.Manchester T, Cavalcanti DP, Zogovich M, W DES, Motta MC. 2013. Acriflavine treatment promotes dyskinetoplasty in Trypanosoma cruzi as revealed by ultrastructural analysis. Parasitology 140:1422–1431. doi: 10.1017/S0031182013001029. [DOI] [PubMed] [Google Scholar]
- 35.Kohl L, Robinson D, Bastin P. 2003. Novel roles for the flagellum in cell morphogenesis and cytokinesis of trypanosomes. EMBO J 22:5336–5346. doi: 10.1093/emboj/cdg518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peixoto P, Liu Y, Depauw S, Hildebrand MP, Boykin DW, Bailly C, Wilson WD, David-Cordonnier MH. 2008. Direct inhibition of the DNA-binding activity of POU transcription factors Pit-1 and Brn-3 by selective binding of a phenyl-furan-benzimidazole dication. Nucleic Acids Res 36:3341–3353. doi: 10.1093/nar/gkn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balana-Fouce R, Alvarez-Velilla R, Fernandez-Prada C, Garcia-Estrada C, Reguera RM. 2014. Trypanosomatids topoisomerase re-visited. New structural findings and role in drug discovery. Int J Parasitol Drugs Drug Resist 4:326–337. doi: 10.1016/j.ijpddr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zuma AA, Mendes IC, Reignault LC, Elias MC, de Souza W, Machado CR, Motta MC. 2014. How Trypanosoma cruzi handles cell cycle arrest promoted by camptothecin, a topoisomerase I inhibitor. Mol Biochem Parasitol 193:93–100. doi: 10.1016/j.molbiopara.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Basselin M, Denise H, Coombs GH, Barrett MP. 2002. Resistance to pentamidine in Leishmania mexicana involves exclusion of the drug from the mitochondrion. Antimicrob Agents Chemother 46:3731–3738. doi: 10.1128/AAC.46.12.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Macedo JP, Schumann Burkard G, Niemann M, Barrett MP, Vial H, Maser P, Roditi I, Schneider A, Butikofer P. 2015. An atypical mitochondrial carrier that mediates drug action in Trypanosoma brucei. PLoS Pathog 11:e1004875. doi: 10.1371/journal.ppat.1004875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mukherjee A, Padmanabhan PK, Sahani MH, Barrett MP, Madhubala R. 2006. Roles for mitochondria in pentamidine susceptibility and resistance in Leishmania donovani. Mol Biochem Parasitol 145:1–10. doi: 10.1016/j.molbiopara.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 42.da Gama Jaén Batista D, Batista MM, de Oliveira GM, do Amaral PB, Lannes-Vieira J, Britto CC, Junqueira A, Lima MM, Romanha AJ, Sales Junior PA, Stephens CE, Boykin DW, Soeiro Mde N. 2010. Arylimidamide DB766, a potential chemotherapeutic candidate for Chagas' disease treatment. Antimicrob Agents Chemother 54:2940–2952. doi: 10.1128/AAC.01617-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guedes-da-Silva FH, Batista DG, Meuser MB, Demarque KC, Fulco TO, Araujo JS, Da Silva PB, Da Silva CF, Patrick DA, Bakunova SM, Bakunov SA, Tidwell RR, Oliveira GM, Britto C, Moreira OC, Soeiro MN. 2016. In vitro and in vivo trypanosomicidal action of novel arylimidamides against Trypanosoma cruzi. Antimicrob Agents Chemother 60:2425–2434. doi: 10.1128/AAC.01667-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.