

Abstract
Vibrio cholerae is an intestinal pathogen that causes the diarrheal disease cholera. Colonization of the intestine depends upon the expression of genes that allow V. cholerae to overcome host barriers, including low pH, bile acids, and the innate immune system. ToxR is a major contributor to this process. ToxR is a membrane-spanning transcription factor that coordinates gene expression in response to environmental cues. In previous work we showed that ToxR upregulated leuO expression in response to bile salts. LeuO is a LysR family transcription factor that contributes to acid tolerance, bile resistance, and biofilm formation in V. cholerae. Here, we investigated the function of ToxR and LeuO in cationic antimicrobial peptide (CAMP) resistance. We report that ToxR and LeuO contribute to CAMP resistance by regulating carRS transcription. CarRS is a two-component regulatory system that positively regulates almEFG expression. AlmEFG confers CAMP resistance by glycinylation of lipid A. We found that the expression of carRS and almEFG and the polymyxin B MIC increased in mutants lacking toxRS or leuO. Conversely, leuO overexpression decreased the polymyxin B MIC. Furthermore, we found that LeuO directly bound to the carRS promoter and that ToxR-dependent activation of leuO transcription regulated carRS transcription in response to bile salts. Our results suggest that LeuO functions downstream of ToxR to modulate carRS expression in response to environmental cues. This study extends the functional role of ToxR and LeuO in environmental adaptation to include cell surface remodeling and CAMP resistance.
INTRODUCTION
Vibrio cholerae is a Gram-negative facultative human pathogen and the causative agent of the diarrheal disease cholera. V. cholerae is native to aquatic ecosystems, where it often associates with chitinous aquatic organisms (reviewed in reference 1). People acquire V. cholerae by ingestion of contaminated food or water. Following ingestion, V. cholerae colonizes the small intestine, where it produces virulence factors that result in a severe secretory diarrhea that is the hallmark of the disease cholera. The secretory diarrhea then contributes to V. cholerae transmission to new hosts and dissemination into the aquatic ecosystem.
The ability of V. cholerae to rapidly transition between the aquatic ecosystem and the host is essential for its success as a pathogen. Colonization of the human gastrointestinal tract is mediated by transcriptional responses that facilitate adaptation to dynamic environments in the gastrointestinal tract. Following ingestion, V. cholerae activates the expression of virulence factors that are essential for both colonization and disease development. This includes the production of the enterotoxin cholera toxin (CT) and an adhesin called the toxin coregulated pilus (TCP) (2, 3). The expression of many V. cholerae virulence genes is coordinately regulated by the ToxR regulon (4). The ToxR regulon is divided into two branches: the ToxT-dependent branch and the ToxT-independent branch. In the ToxT-dependent branch, ToxR and TcpP, two membrane-associated regulators, function as coactivators of toxT expression in response to environmental cues (5, 6). ToxT then directly activates the expression of the gene encoding a number of virulence factors, including those for CT and TCP production (7). In the toxT-independent branch, ToxR regulates the expression of the ompU and ompT porins (8, 9). Recent studies in our laboratory have shown that ToxR also regulates the expression of the LysR family transcription factor leuO independent of toxT (10, 11).
ToxR is a membrane-associated regulator that contains a periplasmic domain that is connected to a cytoplasmic DNA binding domain by a single transmembrane-spanning domain (6, 12, 13). The periplasmic domain is thought to function as a sensor which transduces extracellular cues to effect the activity of the cytoplasmic DNA binding domain. ToxR has been shown to directly regulate ompU, ompT, and leuO expression in response to bile salts (10, 14). In the presence of bile salts, ToxR activates ompU expression while repressing ompT expression. OmpU production appears to contribute to bile resistance due to the properties of its pore, which restricts the diffusion of negatively charged molecules (e.g., bile salts) across the outer membrane (15, 16). ToxR-dependent expression of ompU also contributes to cationic antimicrobial peptide (CAMP) resistance. OmpU serves as a sensor for the sigma E membrane stress response, and cells that lack ompU (i.e., ompU or toxR mutants) fail to activate the sigma E membrane stress response upon membrane perturbation, a phenotype that has been linked to increased CAMP susceptibility (17, 18). ToxR also activates leuO transcription in response to bile salts and cyclic dipeptides (10, 11). LeuO has been linked to multiple phenotypes in V. cholerae, including biofilm production, virulence repression, acid tolerance, and bile resistance (10, 11, 19, 20). Taken together, these results suggest that in addition to regulating virulence factor production, ToxR also contributes to the regulation of numerous genes involved in environmental adaptation.
V. cholerae is exposed to a variety of antimicrobial compounds within the human gastrointestinal tract, including bile salts and products of the innate immune system, like CAMPs. V. cholerae growth in the presence of bile salts is dependent upon the expression of antimicrobial efflux systems combined with reduced cell permeability that is achieved by modulating porin production (21, 22). CAMP resistance is also mediated by multiple factors. CAMPs are short (∼12 to 50 amino acids) amphipathic and hydrophobic peptides that typically exhibit a net positive charge (23). In Gram-negative bacteria, the initial interaction between CAMPs and the cell surface is thought to be driven by the electrostatic attraction between positively charged CAMPs and the negatively charged lipopolysaccharide (LPS). The binding of CAMPs to LPS results in outer membrane disruption and further CAMP uptake. Once across the outer membrane, CAMPs can disrupt the cytoplasmic membrane and/or inhibit critical cytoplasmic processes, resulting in cell death. V. cholerae resistance to CAMPs is mediated by active efflux via the VexAB-TolC RND-efflux system (22), expression of the extracytoplasmic stress response (17), and covalent modification of LPS (24–27). LPS remodeling has been shown to be critical for high-level CAMP resistance. In V. cholerae the production of hexa-acylated lipid A via the MsbB acyltransferase confers resistance to polymyxin B (PXB), a CAMP antibiotic (28). Hexacylated lipid A can be further modified with glycine and diglycine residues by AlmEFG to effect high-level PXB resistance (25–27). Glycinylation of lipid A results in a net increase in the positive charge of lipid A which reduces the electrostatic interactions between CAMPs and lipid A, leading to CAMP resistance.
V. cholerae LPS glycinylation is regulated by CarRS (also known as VprAB), a two-component regulatory system (24, 25). CarRS was first identified as a calcium-responsive negative regulator of biofilm production (29). Two subsequent studies showed that CarRS also regulated CAMP resistance by positively regulating almEFG expression (24, 25). Mutations in carR or almEFG resulted in an ∼100-fold decrease in V. cholerae PXB resistance and attenuated intestinal colonization (24, 25), highlighting the importance of lipid A modification in V. cholerae. The molecular mechanisms controlling carRS expression are unknown. Previous studies showed that carRS expression was influenced by environmental cues, with carRS expression being activated by PXB and repressed by bile salts and calcium (25, 29).
In this study, we investigated the function of V. cholerae toxR and leuO in PXB resistance. We found that deletion of toxR or leuO resulted in increased PXB resistance. This phenotype was linked to carRS expression. Transcriptional reporter assays and electrophoretic mobility shift assays (EMSAs) revealed that LeuO functioned downstream of ToxR to directly repress carRS expression. This finding suggested that LeuO was a carRS repressor, a conclusion that was further supported by the results showing that leuO overexpression increased V. cholerae PXB susceptibility. We further found that both ToxR and LeuO were required for bile salt-dependent repression of carRS but did not influence carRS expression in response to PXB or calcium. Our collective results demonstrate that ToxR and LeuO effect cell surface remodeling and PXB resistance by repression of carRS. Our findings expand the function of ToxR and LeuO in environmental adaptation to include regulation of CAMP resistance via the CarRS two-component system.
MATERIALS AND METHODS
Strains, media, and growth conditions.
The bacterial strains used in this study are listed in Table 1. Escherichia coli strain SM10λpir was used for plasmid mobilization. E. coli strain EC100D+pir was used for the two-plasmid β-galactosidase reporter assays and for cloning experiments. E. coli strain ER2566 was used for purification of proteins for the EMSAs. The V. cholerae strains used in this study were derivatives of O1 El Tor strain N16961 (34). The V. cholerae N16961 ΔlacZ Smr strain was used as the wild-type (WT) control for all experiments. Bacterial strains were grown at 37°C in lysogeny broth (LB) or on LB agar. Bacterial stocks were maintained at −80°C in LB broth containing 25% glycerol. Culture medium was supplemented with carbenicillin (Cb) and streptomycin (Sm) at 100 μg/ml, kanamycin (Km) at 50 μg/ml, or chloramphenicol (Cm) at 25 μg/ml as required. Arabinose was added to growth medium at the concentrations indicated in the figure legends to induce expression from the arabinose-regulated promoter in pBAD18Km and pBAD33.
TABLE 1.
Strains, plasmids, and oligonucleotides used in this study
| Strain, plasmid, or oligonucleotide | Relevant characteristic(s) or sequence (5′ to 3′) | Source or reference |
|---|---|---|
| Strains | ||
| Escherichia coli | ||
| EC100D+pir | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 (λpirR6K) | Epicenter |
| SM10λpir | thi-1 thr leu tonA lacY supE recA::RP4-2-Tc::Mu kmR (λpirR6K) | Laboratory collection |
| ER2566 | F− glnV44(AS) galK2(Oc) rpsL704(strR) xylA5 mtl-1 argE3(Oc) thiE1 tfr-3 λ DE3 λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T gene1) i21 Δnin5 | New England BioLabs |
| Vibrio cholerae | ||
| JB3 | V. cholerae O1 El Tor strain N16961, Smr | Laboratory collection |
| JB58 | V. cholerae O1 El Tor strain N16961, Smr, ΔlacZ | Laboratory collection |
| XBV222 | JB58 ΔleuO | 11 |
| DT733 | JB58 ΔtoxRS | 11 |
| JB461 | JB3 ΔtoxRS | 30 |
| XBV302 | JB58 ΔalmE | This study |
| XBV532 | V. cholerae O1 El Tor strain N16961, Smr, ΔlacZ ΔompU | 17 |
| XBV534 | XBV532 ΔleuO | This study |
| XBV537 | XBV532 ΔtoxRS | This study |
| Plasmids | ||
| pTL61T | lacZ transcriptional reporter plasmid, Cbr | 31 |
| pXB266 | pTL61T containing the leuO promoter region | 11 |
| pVA289 | pTL61T containing the carRS promoter region | This study |
| pXB340 | pTL61T containing the carRS promoter region | This study |
| pXB342 | pTL61T containing the almEFG promoter region | This study |
| pBAD18Km | Arabinose-regulated expression plasmid, Kmr | 32 |
| pXB269 | pBAD18Km expressing VC2486 and leuO | 11 |
| pBAD33 | Arabinose-regulated expression plasmid, Cmr | 32 |
| pVA126 | pBAD33 expressing leuO | 19 |
| pXB302 | pBAD33 expressing toxRS | 10 |
| pMAL-c2 | IPTG-inducible expression vector for fusion of proteins to MBP and cytoplasmic expression, Cbr | New England BioLabs |
| pVA175 | pMAL-c2 expressing leuO | 19 |
| pWM91 | Suicide plasmid vector used for allelic exchange, Cbr | 33 |
| pWM91::ΔalmE | pWM91::ΔVC1579 | This study |
| Oligonucleotides | ||
| PcarRS-pVA289-F | AAACTCGAGAACACGCGGCGAGGAATTGAGTCAG | |
| PcarRS-pVA289-R | CGGGGATCCGATAATGTAGAGACTGGGTTGG | |
| P-vc1320-F-XhoI | ACCCTCGAGTAATCACTGAGAGTGTAGCC | |
| P-vc1320-R-XbaI | TTTTCTAGATGGGGACCTCGTATTTACGG | |
| P-vc1579-F-XhoI | AAACTCGAGATGTTGCGTCTATTGGCGCGCG | |
| P-vc1579-R-XbaI | AATTCTAGATCATGTCTTGATAGGTGT | |
| almE-F1 | CCCCCGGGCCACCAAGATACAAACTA | |
| almE-F2 | TACAATTCTGCGGCGAGTCAGACATA | |
| almE-R1 | ATGAGCTCGCTGCATCATGTCGGCTA | |
| almE-R2 | TGTCTGACTCGCCGCAGAATTGTATG | |
| carRS-F-EMSA | GCGGGAGTCGGCAGCGGGCGAGGAATTGAGTCAGAAGCC | |
| carRS-R-EMSA | GCGGGAGTCGGCAGCGGAGACTGGGTTGGTTAGACATGGGG | |
| 5′BIO | 5′-biotin-GCGGGAGTCGGCAGCG |
Plasmid and mutant construction.
Plasmids and oligonucleotides used in this study are listed in Table 1. The carRS-lacZ reporter, pVA289, was generated as follows. The PcarRS-pVA289-F and PcarRS-pVA289-R PCR primers were used to amplify the carRS promoter from the V. cholerae N16961 genome. The resulting PCR amplicon was digested with XhoI and BamHI restriction endonucleases before being ligated into similarly digested pTL61T to create pVA289. The carRS-lacZ reporter pXB340 was generated in an identical manner using the P-vc1320-F-XhoI and P-vc1320-R-XbaI PCR primers. The almEFG-lacZ reporter pXB342 was generated using P-vc1579-F-XhoI and P-vc1579-R-XbaI PCR primers. The almE (VC1579) deletion plasmid pWM91::ΔalmE was made by crossover PCR as previously described (35, 36). Briefly, the almE-F1/almE-R2 and almE-F2/almE-R1 primer pairs were used in separate PCRs with N16961 chromosomal DNA. The resulting ∼1-kb amplicons were purified and used as templates in a second PCR amplification with the flanking almE-F1/almE-R1 PCR primers. The resulting ∼2-kb amplicon was then digested with SacI and SmaI restriction endonucleases before being ligated into similarly digested pWM91 to generate pWM91::ΔalmE. The V. cholerae almE deletion mutant was constructed by conjugating pWM91::ΔalmE into JB58 with selection for resistance to Sm and Cb. Several Sm- and Cb-resistant cointegrants were then plated onto LB no-salt agar containing 5% sucrose. Sucrose-resistant and Cb-sensitive colonies were then screened by PCR to confirm deletion of almE using the almE-F1/almE-R1 primers to identify strain XBV302. The almE deletion in XBV302 was subsequently verified by DNA sequencing.
β-Galactosidase assays.
V. cholerae strains carrying the indicated lacZ reporter plasmids were cultured overnight in LB broth at 37°C with shaking. The cultures were then diluted 100-fold into fresh LB broth or LB containing 0.05% deoxycholate, 5 mM CaCl2, or polymyxin B at the indicated concentrations and grown at 37°C with shaking. Culture aliquots were then collected in triplicate at the indicated time points to quantify β-galactosidase production as previously described (37). E. coli containing an expression plasmid (pBAD33, pVA126, or pXB302) plus the carRS-lacZ reporter plasmid pVA289 was cultured overnight in LB broth with shaking at 37°C. The overnight cultures were then diluted 100-fold into fresh LB broth containing antibiotics and 0.005%, 0.01%, or 0.05% arabinose, and the cultures were incubated at 37°C with shaking. Culture aliquots were collected in triplicate at mid-exponential phase (optical density at 600 nm [OD600] of ∼0.5) to quantify β-galactosidase activity. All of the reporter experiments were performed independently at least three times. Gene expression from the β-galactosidase reporters were calculated and displayed as Miller units (MU).
Purification of the maltose binding protein (MBP) and LeuO-MBP.
Protein purification for the electrophoretic mobility shift assays (EMSAs) was done as previously described (19). Briefly, overnight cultures of E. coli ER2566 carrying pMAL-c2 or pMAL-c2::leuO (pVA175) were diluted 100-fold into fresh LB broth and incubated at 37°C with shaking until they reached an OD600 of ∼0.5 when isopropyl β-d-1-thiogalactopyranoside (IPTG) was added at 0.3 mM. The cultures were then incubated for an additional 2 h before the cells were harvested by centrifugation. The cell pellets were then resuspended in column buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA) plus 1 mM phenylmethylsulfonyl fluoride before being lysed with an M-11P Microfluidizer according to the manufacturer's instructions (Microfluidics). The resulting cell lysates were cleared of particulate matter by centrifugation at 9,900 rpm for 20 min at 4°C. The clarified supernatant was then diluted 1:6 in column buffer before being passed through a 0.8- by 7.0-cm chromatography column containing 1 ml of amylose resin (New England BioLabs). Following washing with column buffer, bound proteins were eluted from the resin using elution buffer (20 mM Tris-HCl, 200 mM NaCl, 1 mM EDTA, 10 mM maltose). Protein concentrations were determined using the Coomassie plus (Bradford) assay kit according to the manufacturer's instructions (Thermo Scientific). Protein purification was assessed by SDS-PAGE with Coomassie brilliant blue R-250 staining.
EMSAs.
EMSAs were performed as previously described (19). The DNA fragment containing the carRS promoter (the nucleotide sequence between −400 and +20 relative to the carR translational start site) was PCR amplified from the N16961 genome using carRS-F-EMSA and carRS-R-EMSA oligonucleotide primers. The resulting PCR fragment was then gel purified, and 100 ng was used as a template for a second PCR using the biotinylated 5′BIO oligonucleotide primer purchased from IDT to produce the end-labeled DNA probe. The vexR DNA fragment was previously described and consisted of the nucleotide sequence between −129 and −46 relative to the vexR translational start site (38). The biotin-labeled probes (1.5 nM carRS, 3 nM vexR) were incubated with purified LeuO-MBP or MBP in amounts ranging from 0 to 25 μM in binding buffer containing 10 mM Tris (pH 7.4), 150 mM KCl, 0.1 mM dithiothreitol (DTT), 0.1 mM EDTA (pH 8.0), and 200 μg/ml sheared salmon sperm. The 10-μl binding reaction mixtures were incubated at room temperature for 20 min before being subjected to electrophoresis on a nondenaturing 5% TBE-PAGE gel in 0.25× Tris-borate-EDTA (TBE) buffer at 200 V for 45 min. The gel was prerun at 100 V for 1 h in 0.25× TBE prior to sample addition. The DNA in the gel was then transferred to a nylon membrane in 0.5× TBE buffer at 380 mA for 1 h. The nylon membrane was then UV cross-linked at 120,000 μJ using a Stratalinker 1800 (Stratagene) before the biotin-labeled DNA fragments were detected using a chemiluminescent nucleic acid detection module (Thermo Scientific) and visualized using FluorChem E (Protein Simple).
Antimicrobial susceptibility assays.
Antimicrobial susceptibility was assayed by the Etest strip method according to the manufacturer's instructions (bioMérieux) or the gradient agar plate methods as previously described (22). Strains tested by the Etest strip method were grown in LB broth at 37°C overnight. These cultures were diluted in LB to an OD600 of 0.5 before being examined using Etest strips according to the manufacturer's directions. Mid-log-phase cultures were generated by inoculating overnight cultures at 1:100 into fresh LB broth, or LB supplemented with 0.05% deoxycholate, and growing the cultures at 37°C to an OD600 of 0.5 before use. The MIC for each respective strain was recorded at the PXB concentration displaying a clear zone of inhibition following overnight incubation at 37°C. The gradient agar plate method was used to test susceptibility for strains bearing pBAD18Km or pBAD18Km-leuO (39, 40). Overnight cultures of the respective strains were inoculated at 1:100 into fresh LB supplemented with 0.1% arabinose and grown at 37°C to an OD600 of 0.5 before being inoculated onto the PXB gradient agar plates supplemented with 0.1% arabinose. After overnight incubation, growth of each strain across the gradient was measured. The MIC for each strain was then calculated as the percentage of growth across the plate multiplied by the antimicrobial concentration used in the plate. The presented results are the means and standard deviations from three independent biological replicates. Statistical significance for the tested strains was determined using a one-sample Student's t test.
Polymyxin B killing assay.
Overnight cultures of V. cholerae WT strain JB58, ΔleuO strain XBV222, and ΔtoxRS strain JB461 were diluted 100-fold into fresh LB broth with or without 0.05% deoxycholate. The inoculated cultures were then incubated with aeration at 37°C until they reached an OD600 of ∼0.5. Culture aliquots were then collected by centrifugation and the cell pellets resuspended in phosphate-buffered saline (PBS) to an OD600 of 0.1. Serial dilutions of each strain were then plated onto LB agar plates to enumerate the cell titer at time zero (CFUinput). The remaining cells were then collected by centrifugation and resuspended in LB broth containing 500 μg/ml polymyxin B and incubated at 37°C with aeration for 60 min, when an aliquot was collected and washed in PBS before serial dilutions were plated onto LB agar to quantify the viable cells (CFUoutput). The percent survival of each strain was then calculated by dividing the number of cells recovered following 60 min of exposure to polymyxin B by the number of input cells (i.e., time zero). The reported data represent the averages ± standard deviations from four independent experiments.
RESULTS
ToxR and LeuO negatively regulate carRS expression.
During the course of our studies on V. cholerae LeuO, we observed that leuO deletion resulted in increased PXB resistance. This suggested that leuO regulates V. cholerae CAMP resistance. Since the carRS two-component regulatory system was recently shown to be critical for PXB resistance (24, 25), we examined whether leuO or its upstream positive regulator, toxR, affected carRS transcription. We therefore introduced a carRS-lacZ reporter (pXB340) into WT strain JB58 (ΔlacZ) and its isogenic ΔleuO and ΔtoxRS mutant strains. The resulting strains were then cultured to mid-logarithmic phase in LB broth when carRS-lacZ expression was quantified. The results showed an ∼2-fold increase in carRS expression in both the ΔleuO and ΔtoxRS mutants relative to the WT (Fig. 1A), suggesting that both ToxR and LeuO were negative regulators of carRS. The observation that carRS expression increased similarly in the ΔtoxRS mutant and the ΔleuO mutant suggested that ToxR indirectly regulates carRS via leuO.
FIG 1.
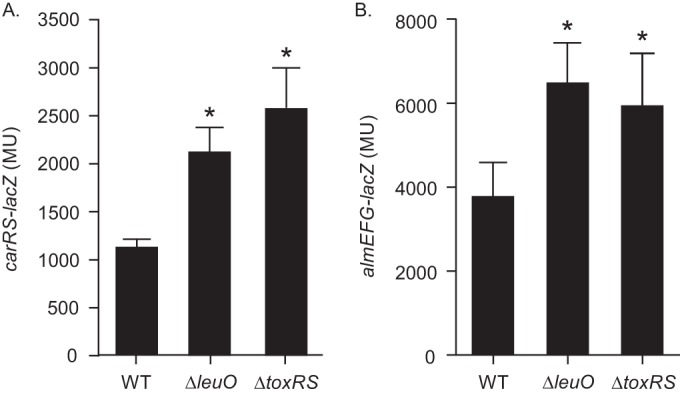
ToxR and LeuO regulate carRS and almEFG expression. WT strain JB58, ΔleuO strain XBV222, and ΔtoxRS strain DT733 carrying the carRS-lacZ reporter pXB340 (A) or the almEFG-lacZ reporter pXB342 (B) were grown in LB broth to mid-log phase, at which time the expression of the reporter genes was quantified using a β-galactosidase assay. The presented data are the means ± standard deviations from three independent experiments. Statistical significance was determined using one-way analysis of variance (ANOVA) with Dunnett's test, comparing each mean value to that of the WT. *, P < 0.01.
V. cholerae CarRS mediates PXB susceptibility by positively regulating the expression of the LPS remodeling genes almEFG (24). We hypothesized that if ToxR and LeuO were regulating PXB resistance via carRS repression, then deletion of toxRS or leuO should result in a corresponding increase in almEFG expression. To test this, we introduced an almEFG-lacZ reporter plasmid (pXB342) into the WT and the ΔleuO and ΔtoxRS mutant strains and quantified almEFG-lacZ expression as described above. As expected, almEFG expression increased by ∼2-fold in both the ΔleuO and ΔtoxRS mutants relative to the WT (Fig. 1B), supporting our hypothesis. Taken together, these results suggested that ToxR and LeuO modulate PXB resistance via repression of carRS.
LeuO directly represses carRS transcription.
The above-described reporter data indicated that both ToxR and LeuO negatively regulated carRS expression but did not address whether ToxR or LeuO acted directly at the carRS promoter. To examine this, we tested the effect of toxRS and leuO overexpression on carRS-lacZ expression in E. coli. We hypothesized that if either regulatory protein acted directly at the carRS promoter, then their overexpression in a heterologous host would repress expression of a carRS-lacZ reporter. We therefore cultured E. coli bearing pVA289 (carRS-lacZ) and either pBAD33 or pBAD33-leuO in the presence of various concentrations of arabinose and quantified carRS-lacZ expression. The results showed that the addition of arabinose did not affect carRS-lacZ expression in the negative-control cultures bearing pBAD33 (Fig. 2A). The addition of arabinose to the cultures bearing pBAD33-toxRS also did not affect carRS-lacZ expression, suggesting that ToxR represses carRS expression indirectly. In contrast, there was an arabinose dose-dependent reduction in carRS-lacZ expression in the cultures containing pBAD33-leuO, suggesting that LeuO directly represses carRS transcription.
FIG 2.
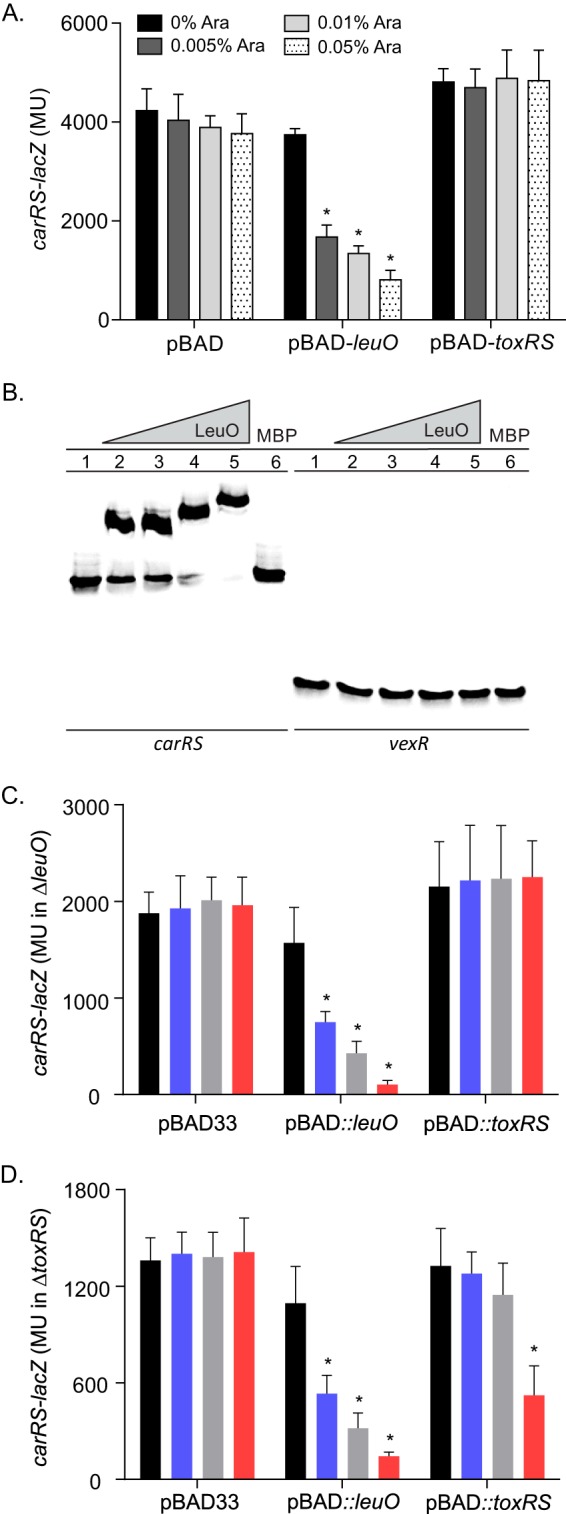
LeuO acts directly on the carRS promoter. (A) E. coli carrying either pBAD33, the pBAD33-leuO plasmid pVA126, or the pBAD33-toxRS plasmid pXB302 plus the carRS-lacZ reporter plasmid pVA289 was grown in LB broth containing the indicated amount of arabinose (Ara) to mid-logarithmic phase and assayed for β-galactosidase activity as described in Materials and Methods. The presented data are averages ± standard deviations from three independent experiments. Statistical analysis was conducted using two-way ANOVA with Tukey's posttest. *, P < 0.0001. (B) Electrophoretic mobility shift assay showing the binding of LeuO to the carRS promoter. Biotin-labeled DNA fragments containing carRS (1.5 nM) or vexR (3 nM) promoter were incubated with LeuO-MBP at 0 μM (lane 1), 2.5 μM (lane 2), 5 μM (lane 3), 10 μM (lane 4), or 25 μM (lane 5) or with MBP at 25 μM (lane 6) prior to electrophoresis. (C) V. cholerae ΔleuO strain carrying either pBAD33, pBAD33-leuO, or pBAD33-toxRS plus the carRS-lacZ reporter plasmid pVA289 was grown in LB broth containing 0% (black), 0.005% (purple), 0.05% (grey), or 0.01% (red) arabinose to mid-logarithmic phase and assayed for β-galactosidase activity as described in Materials and Methods. (D) Same as panel C, except the V. cholerae ΔtoxRS strain was used. Statistical analysis was conducted using two-way ANOVA with Tukey's posttest. *, P < 0.005.
The above-described results suggested that LeuO acts directly at the carRS promoter; however, they did not exclude the possibility that LeuO affected carRS expression via an intermediate protein that was conserved in E. coli and V. cholerae. To determine if LeuO was capable of acting directly on the carRS promoter, we performed an EMSA with MBP-LeuO and the carRS promoter as previously described (19). The results showed that incubation of MBP-LeuO with a DNA probe spanning the carRS promoter resulted in a mobility shift in the carRS probe starting at 2.5 μM MBP-LeuO (Fig. 2B, lane 2) and was further shifted with increasing concentrations of MBP-LeuO (lanes 4 and 5). It is unclear whether the additional shifting of the probe resulted from MBP-LeuO multimerization, as has been reported with LeuO in Salmonella enterica serovar Typhi (41). Incubation of the carRS probe with MBP alone did not result in a mobility shift (lane 6), indicating that LeuO-MBP binding to the carRS promoter probe was due to LeuO and not due to nonspecific binding by the MBP. To further test the specificity of LeuO binding at the carRS promoter, we repeated these experiments using a DNA probe derived from the V. cholerae vexR promoter. The results showed that neither MBP-LeuO (lanes 2 to 5) nor MBP (lane 6) affected the mobility of the vexR DNA probe. This result is consistent with the conclusion that MBP-LeuO binding to the carRS promoter was not due to nonspecific interactions between MBP-LeuO and DNA. Based on these results, we concluded that LeuO likely functioned downstream of ToxR to directly repress carRS transcription.
Our reporter data suggested that ToxR indirectly regulated carRS via LeuO (Fig. 2A). ToxR also directly regulates toxT expression in V. cholerae, but this phenotype was not replicated in E. coli due to the lack of a toxT coregulator (i.e., TcpPH) (42). Based on this fact, we could not rule out the possibility that ToxR directly regulated carRS in V. cholerae. To address this, we examined whether ToxR could activate carRS expression in V. cholerae mutants lacking leuO or toxRS. We introduced pXB340 (carRS-lacZ) and either pBAD33, pBAD33-leuO, or pBAD33-toxRS into XBV222 (ΔleuO mutant) and JB461 (ΔtoxRS mutant). The resulting strains were then cultured to mid-log phase in the presence of increasing concentrations of arabinose when carRS-lacZ expression was quantified. The results showed that the addition of arabinose did not have any effect on carRS expression in the empty vector control in either mutant background (Fig. 2C and D). Overexpression of leuO resulted in an arabinose concentration-dependent decrease in carRS expression in both mutants (Fig. 2C and D), providing additional support for the conclusion that LeuO directly represses carRS expression. Significantly, expression of leuO from pBAD33-leuO still repressed carRS in the toxRS-negative background, indicating that LeuO functions independently of ToxR. In contrast, overexpression of toxRS repressed carRS expression in the ΔtoxRS (leuO+) background but not in the ΔleuO mutant background. The observation that ToxR only represses carRS transcription in the presence of leuO is consistent with the conclusion that ToxR functions indirectly at the carRS promoter. This conclusion is further supported by the fact that the carRS promoter lacks a consensus ToxR binding site that is found in the promoter region of many ToxR-regulated genes, including leuO (10, 43).
LeuO regulates polymyxin B resistance in V. cholerae.
It has been reported that CarRS contributes to PXB resistance by positively regulating the expression of the almEFG lipid A glycinylation system (24, 25). Since our data described above indicated that LeuO was a carRS repressor (Fig. 1 and 2), we hypothesized that a leuO mutant should exhibit increased resistance to PXB as a result of carRS derepression. To test this, we determined the PXB MIC for mid-log-phase cultures of the WT and an isogenic ΔleuO mutant using Etests according the manufacturer's instructions (bioMérieux). The findings revealed that leuO deletion resulted in a 3-fold increase in the PXB MIC relative to that of the WT (Table 2), confirming that leuO negatively regulated PXB resistance. Since ToxR positively regulates leuO expression, we also examined a ΔtoxRS mutant. The ΔtoxRS mutant displayed a MIC that was identical to that of the ΔleuO mutant. Given that ToxR positively regulates leuO, this finding is consistent with the conclusion that ToxR indirectly represses carRS via leuO.
TABLE 2.
Polymyxin B susceptibility of V. cholerae strains
| Strain | Polymyxin B MIC (μg/ml)a |
|
|---|---|---|
| Log phase | Stationary phase | |
| WT | 128 | 32 |
| ΔleuO | 384 | 64 |
| ΔtoxRS | 384 | 2 |
| ΔompU | 128 | 3 |
| ΔompU ΔtoxRS | 384 | 3 |
| ΔompU ΔleuO | 384 | 3 |
The MICs for the indicated strains were determined using the Etest with LB broth cultures.
Production of the OmpU porin has been reported to confer a survival advantage to V. cholerae upon PXB exposure in killing assays due to its function in the activation of the sigma E extracytoplasmic stress response (17, 18). To determine whether toxRS and leuO affected the PXB MIC via the sigma E membrane stress response, we examined the effect of leuO and toxRS mutations on the PXB MIC in an ompU-negative strain which is deficient for sigma E activation. The PXB MIC results showed that mid-log-phase cultures of the respective ompU mutants phenocopied their respective ompU-positive parental strains (Table 2). The fact that ompU deletion did not affect PXB sensitivity when combined with deletions of leuO or toxRS indicates that leuO functioned independently of the sigma E membrane stress response to regulate V. cholerae PXB resistance. The fact that the PXB MIC increased in the ΔompU ΔtoxRS mutant, which cannot activate sigma E, suggests that ToxR has pleiotropic effects on PXB resistance, being required for both leuO expression and the induction of the sigma E membrane stress response via OmpU (11, 17).
We performed complementation experiments to confirm that leuO repressed PXB resistance in V. cholerae. In these experiments, we ectopically expressed leuO from the arabinose-regulated promoter in pBAD18Km in WT and ΔleuO strains before determining the PXB MIC of the resulting cultures on PXB gradient agar plates. The results showed that leuO expression decreased the PXB MIC in both the WT and the ΔleuO mutant (Table 3). Overexpression of leuO resulted in an ∼3-fold decrease in the PXB MIC in the WT and an ∼5-fold decrease in the PXB MIC in the ΔleuO mutant. The fact that leuO overexpression decreased the PXB MIC in both the WT and the ΔleuO mutant was consistent with the hypothesis that leuO regulates PXB resistance via carRS repression.
TABLE 3.
Polymyxin B susceptibility of V. cholerae strains overexpressing leuO at mid-log phase
| Strain | MICa (μg/ml) (SD) |
|---|---|
| WT (pBAD18Km) | 118 (13.4) |
| WT (pBAD18Km-leuO) | 40b (15.9) |
| ΔleuO (pBAD18Km) | 294 (45.6) |
| ΔleuO (pBAD18Km-leuO) | 56c (5.6) |
The MIC was determined using PXB gradient agar plates with mid-log-phase LB broth cultures.
P value of ≤0.01 relative to WT pBAD18Km.
P value of ≤0.01 relative to ΔleuO pBAD18Km.
Growth-phase-dependent regulation of PXB susceptibility.
During the course of our studies, we observed that V. cholerae susceptibility to PXB varied according to the growth phase. To confirm this observation, we repeated the PXB MIC studies using V. cholerae stationary-phase cultures. The results revealed a significant decrease in the PXB MIC in stationary-phase cells (Table 2). This was evidenced by 4-fold and 6-fold decreases in the PXB MIC for the WT and the ΔleuO mutant, respectively. The MIC for the ΔleuO mutant was still elevated relative to that of the WT, suggesting that leuO still contributed to PXB resistance at stationary phase (Table 2). Deletion of toxRS resulted in a 16-fold decrease in the PXB MIC relative to that of WT stationary-phase cells. This contrasts with mid-log-phase cells, where toxRS deletion increased the PXB MIC. To determine if the toxRS phenotype was related to its role in the induction of the sigma E stress response via ompU (17), we assessed an isogenic ΔompU mutant at stationary phase. The ompU mutant exhibited a PXB MIC that was similar to that of the ΔtoxRS mutant. Further deletion of toxRS or leuO in the ΔompU background did not affect the PXB MIC in stationary-phase cells. The observation that the ΔompU and ΔtoxRS mutants exhibited similar decreases in their PXB MICs in stationary-phase cells suggests that the OmpU-dependent activation of the sigma E extracytoplasmic stress response is important for PXB resistance at stationary phase. The fact that deletion of leuO in the ΔompU background did not affect the PXB MIC suggests that the contribution of OmpU, and likely the sigma E membrane stress response, is epistatic to leuO for PXB resistance at stationary phase. These results provide additional support for the conclusion that ToxR has pleiotropic effects on PXB resistance due to its requirement for the expression of both leuO and ompU.
LeuO represses carRS expression in response to bile salts.
Previous reports indicated that leuO and carRS were inversely regulated in response to bile salts, with leuO being upregulated and carRS being repressed (10, 25). This suggested the possibility that LeuO and, thus, ToxR are responsible for carRS repression by bile salts. To test this, we grew the WT, ΔleuO, and ΔtoxRS strains containing the carRS-lacZ reporter (pXB340) in LB broth with or without 0.05% deoxycholate. We then quantified carRS-lacZ expression at mid-log phase (4 h), late log phase (8 h), and stationary phase (20 h). We observed increased carRS expression in the leuO and toxRS mutants relative to the WT during growth in LB broth at all three time points (Fig. 3). The addition of deoxycholate repressed carRS expression in the WT at all three time points, which is consistent with previous reports (25). The deoxycholate-dependent repression of carRS was abrogated in both the toxRS and leuO mutants, a finding that is consistent with the notion that ToxR indirectly represses carRS expression via LeuO (10). Surprisingly, in the absence of leuO or toxRS, deoxycholate stimulated carRS expression in mid- and late-log-phase cultures by >2-fold relative to cells grown in LB broth alone (Fig. 3). This bile salt-dependent enhancement was lost in the stationary-phase cells. This suggests that deoxycholate can activate carRS expression by an unknown mechanism in the absence of leuO and toxRS. Although the mechanism by which this occurs is unknown, this regulatory signal could be important under environmental conditions where ToxR is diminished (44).
FIG 3.
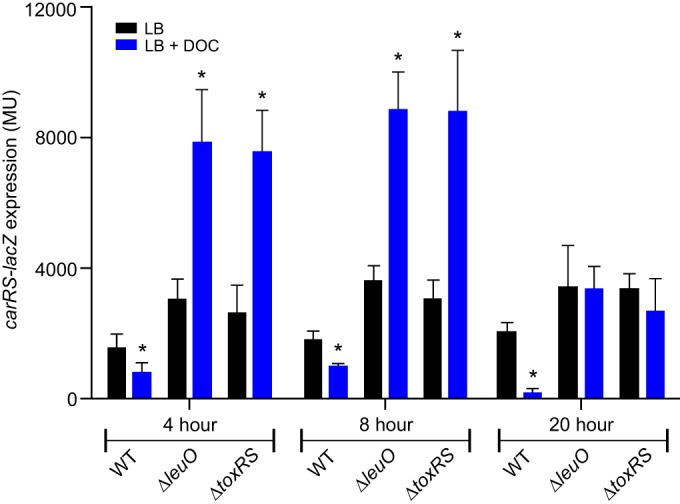
Expression of carRS is repressed by deoxycholate in a LeuO-dependent manner. V. cholerae WT strain JB58, ΔleuO mutant strain XBV222, and ΔtoxRS mutant strain DT733 carrying pXB340 (carRS-lacZ) were cultured in LB broth in the presence or absence of 0.05% deoxycholate (DOC) to mid-log phase (4 h), late-log phase (8 h), and stationary phase (20 h), and then β-galactosidase activity was quantified as described in Materials and Methods. The presented data are means ± standard deviations from three independent experiments. Statistical analysis was conducted using a paired t test. *, P < 0.05 relative to the control cultured in LB broth.
Our data suggested a model where bile salts activated the ToxR-dependent expression of leuO. LeuO then repressed carRS, which resulted in reduced almEFG expression, decreased LPS glycinylation, and increased PXB susceptibility. To test this model, we determined the PXB MIC for WT, ΔleuO, ΔtoxRS, ΔompU, ΔompU ΔleuO, and ΔompU ΔtoxRS strains following growth to mid-log phase in LB broth containing 0.05% deoxycholate (Table 4). The results showed that growth of the WT in deoxycholate decreased its PXB MIC by 4-fold relative to growth in LB broth (Tables 2 and 4). In contrast, deoxycholate did not affect the PXB MIC for the ΔleuO and ΔtoxRS mutants relative to their growth in LB broth alone. This indicated that the decreased PXB MIC observed in WT cells grown in deoxycholate was dependent on toxR-dependent expression of leuO in response to deoxycholate and not due to nonspecific effects of growth in deoxycholate. The PXB MIC for the ΔompU mutant was slightly lower than that for the WT, which was consistent with ompU contributing to PXB resistance. The MIC for the ΔompU ΔleuO mutant was unchanged relative to that of cells grown in the absence of deoxycholate. However, growth in deoxycholate reduced the PXB MIC for the ΔompU ΔtoxRS mutant (1.5-fold) relative to the same strain grown in the absence of deoxycholate. Since bile salts enhance ToxR activity at target promoters (10), the latter result suggests that ToxR contributes to PXB resistance by both leuO-dependent and leuO-independent mechanisms.
TABLE 4.
Sensitivity of V. cholerae strains to polymyxin B following exposure to deoxycholate
| Strain | PXB MICa (μg/ml) |
|---|---|
| WT | 16 |
| ΔleuO | 384 |
| ΔtoxRS | 384 |
| ΔompU | 12 |
| ΔompU ΔtoxRS | 256 |
| ΔompU ΔleuO | 384 |
The MICs for the indicated strains were determined using the Etest following growth to mid-log phase in LB broth containing 0.05% deoxycholate.
The above-described results suggested the possibility that the inverse regulation of leuO and carRS by deoxycholate affects V. cholerae survival upon challenge with CAMPs. To investigate this, we performed PXB survival assays with the WT and ΔleuO and ΔtoxRS mutants. An isogenic almE mutant was included as a negative control. The strains were independently cultured to mid-log phase in LB broth or LB broth plus 0.05% deoxycholate. The cells were then challenged by exposure to a high concentration of PXB (500 μg/ml) for 60 min, and then survival was assessed by plating for viable cells. The results showed a decrease in survival of the WT following growth in deoxycholate relative to cells grown in the absence of deoxycholate (Fig. 4). There was an ∼2-log drop in survival of the ΔleuO and ΔtoxRS mutants following growth in LB broth. Conversely, growth of the ΔleuO and ΔtoxRS mutants in deoxycholate imparted a >2-log increase in survival relative to cells grown in LB alone. These results appear to contradict the MIC results where growth of the ΔleuO and ΔtoxRS mutants in deoxycholate did not affect the PXB MIC. This discrepancy likely reflects the different experimental approaches used in each study. Killing assays assess the immediate physiological state of the cell and are not dependent on cell replication during PXB challenge. In contrast, MIC determination requires cell replication to visualize the results, which allows the expression of adaptive responses that dilute the physiological effects that were imparted to the cells during growth in the presence of deoxycholate. Finally, the almE mutant was not recovered under the test conditions, confirming previous reports that mutations into this locus resulted in a PXB hypersensitive phenotype (27).
FIG 4.
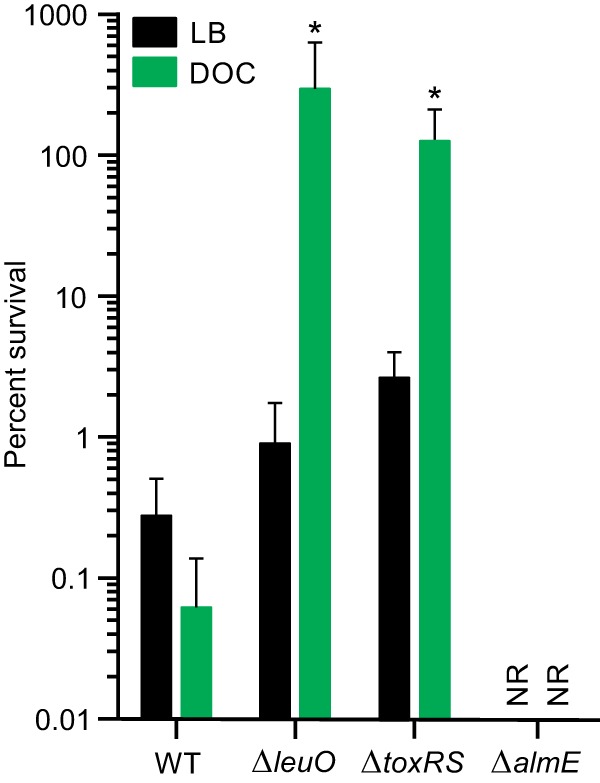
V. cholerae survival following PXB challenge. V. cholerae WT strain JB58, isogenic ΔleuO strain XBV222, ΔalmE strain XBV302, and ΔtoxRS strain JB461 were grown in LB broth or LB broth plus 0.05% deoxycholate (DOC) to mid-logarithmic phase before being incubated in LB broth containing a lethal concentration of polymyxin B. Aliquots were collected at 60 min after exposure to polymyxin B and plated onto LB agar to quantify the viable cells as described in Materials and Methods. The percent survival for each strain is reported relative to the input cell count. The data presented are averages ± standard deviations from four independent experiments. Statistical analysis was conducted using two-way ANOVA. *, P value of <0.05 relative to the input. NR, none recovered.
The results of the killing assays further supported the notion that leuO expression adversely affects PXB resistance. The fact that the ΔleuO and ΔtoxRS mutants showed a >2-log survival increase following growth in deoxycholate indicates the presence of an uncharacterized bile-dependent regulatory circuit that positively regulates CAMP resistance in the absence of leuO. This suggests that LeuO functions to antagonize this regulatory circuit in the presence of bile. This also suggests that the increased survival of the toxRS and leuO mutants following growth in deoxycholate is due to the deoxycholate-dependent upregulation of carRS.
LeuO does not regulate carRS expression in response to PXB or Ca2+.
In addition to being regulated by bile salts, carRS expression was also reported to be regulated in response to antimicrobial peptides and calcium (Ca2+) where antimicrobial peptides activated carRS expression (25) and extracellular Ca2+ repressed carRS expression (29). Since leuO regulated carRS in response to bile salts, we investigated whether LeuO also regulated carRS expression in response to these stimuli. We examined whether Ca2+ or PXB affected leuO expression. To do this, we cultured WT containing a leuO-lacZ reporter (pXB266) in the presence and absence of 5 mM CaCl2 or subinhibitory amounts of PXB (1/32 and 1/16 of its MIC) and quantified leuO expression. The results showed that PXB did not affect leuO expression at either concentration (Fig. 5), suggesting that leuO is not involved in regulating carRS in response to PXB. In contrast, growth of V. cholerae in the presence of 5 mM CaCl2 resulted in an ∼25% decrease in leuO expression. This indicates that leuO expression is regulated in response to Ca2+ in V. cholerae. However, as Ca2+ represses both leuO and carRS transcription, this result indicated that Ca2+-dependent repression of carRS occurs by a mechanism that is independent of leuO. Taken together, these results support the conclusion that leuO expression is modulated by calcium and suggest that LeuO is not involved in the PXB- or Ca2+-dependent regulation of carRS expression in V. cholerae.
FIG 5.
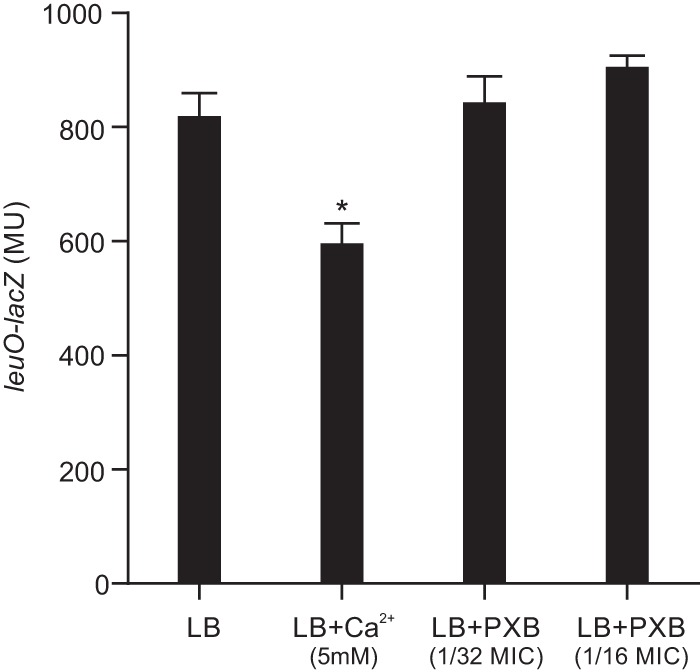
Expression of leuO is regulated by calcium but not polymyxin B. V. cholerae WT strain JB58 carrying the leuO-lacZ reporter plasmid pXB266 was cultured in LB broth in the presence or absence of 5 mM CaCl2 or the indicated concentrations of PXB to mid-logarithmic-growth phase before being assayed for β-galactosidase activity. The presented data are means ± SD from three independent experiments. Statistical analysis was conducted using a paired t test. *, P < 0.05.
DISCUSSION
V. cholerae is a natural inhabitant of aquatic ecosystems. Its entry into the human host represents a dramatic environmental shift where the cells are confronted with a number of barriers to colonization, including exposure to bile salts and products of the innate immune system. ToxR plays a critical role in the transition to the human host and overcoming these barriers (10, 11, 19, 45–47). In addition to regulating the production of essential virulence factors, ToxR also functions to modulate the composition of the outer membrane to fine-tune its barrier properties. In response to bile salts ToxR activates ompU expression while repressing ompT expression; the preferential expression of ompU enhances V. cholerae resistance to the toxic effects of bile salts (21). ToxR-dependent expression of ompU has also been linked to CAMP resistance due to the requirement for OmpU in the induction of the sigma E membrane stress response (17). In this study, we have shown that the function of ToxR extends beyond porin regulation to include the CarRS-AlmEFG lipid A glycinylation system (Fig. 6). We showed that ToxR indirectly regulated carRS and almEFG expression, and PXB resistance, via leuO. This was evident by the fact that deletion of toxRS or leuO increased V. cholerae resistance to PXB, while leuO overexpression decreased carRS and almEFG expression and increased PXB susceptibility. The bile salt deoxycholate stimulated the expression of this negative regulatory pathway in WT cells, but surprisingly it activated carRS expression and increased PXB resistance in cells lacking toxRS or leuO, which suggests that LeuO functions as a carRS antagonist in response to bile salts.
FIG 6.
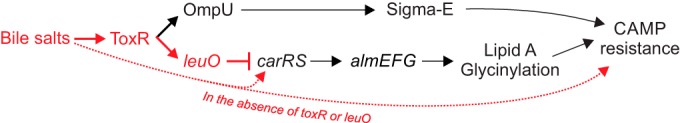
Model for the function of ToxR and LeuO in antimicrobial peptide resistance. ToxR-dependent transcription of leuO is activated in response to bile salts. LeuO then directly represses carRS by binding to its promoter. This results in decreased expression of almEFG, which reduces lipid A glycinylation and results in increased susceptibility to CAMPs. ToxR also activates expression of the major outer membrane porin ompU in response to bile salts. OmpU serves as a membrane stress sensor for activation of the sigma E membrane stress response, which contributes to the mitigation of the deleterious effects of CAMPs. Lastly, in the absence of ToxR and/or LeuO, bile salts activate carRS transcription and increase CAMP resistance by an unknown mechanism. Pathways shown in red were described in this work.
The linkage between ToxR, deoxycholate, and carRS expression suggests the intriguing possibility that ToxR regulates outer membrane remodeling in response to spatial cues in the host. Following ingestion, V. cholerae is initially localized to the intestinal lumen but then migrates to and colonizes the intestinal epithelium crypts. The intestinal crypts are separated from the lumen by a mucous layer which is thought to function as a diffusion barrier that retains CAMPs produced by enterocytes while limiting exposure of the intestinal epithelium to luminal bile salts (48). This would likely result in a bile salt and CAMP concentration gradient across the mucosa with CAMPs being concentrated near the epithelial surface and bile salts being most concentrated toward the lumen. In this scenario, it is conceivable that V. cholerae localized to the bile-rich, but CAMP-barren, lumen would downregulate carRS expression via ToxR-dependent activation of leuO. However, as V. cholerae traverses the mucus layer to colonize the crypts, it is likely to be exposed to decreasing levels of bile salts and increasing levels of CAMPs. Reduced bile salts exposure in the mucous layer would result in reduced leuO expression, derepression of carRS, and increased lipid A glycinylation by AlmEFG, which would mitigate the effects of the increased CAMP exposure near the epithelial surface. While this model is speculative, evidence does suggest that leuO, carRS, and almEFG are all expressed in vivo (11, 24, 25); however, additional work will be required to determine if ToxR differentially regulates its target genes between the lumen and epithelial surface.
ToxR indirectly regulated carRS transcription via LeuO in response to deoxycholate. Although ToxR is a major virulence regulator, toxR is also part of the ancestral chromosome, and its regulon extends beyond horizontally acquired virulence genes. This conclusion is supported by microarray studies which implicated ToxR in the regulation of >150 genes in V. cholerae, including genes involved in cellular transport, energy metabolism, motility, iron uptake, and outer membrane proteins (30). This conclusion is further supported by a recently published ToxR chromatin immunoprecipitation sequencing (ChIP-seq) study showing that ToxR can function as an H-NS antagonist (49). We previously showed that ToxR directly activated leuO expression in response to environmental cues by a process that was dependent upon the presence of the periplasmic domain of ToxR (10, 11). This suggested that the periplasmic domain of ToxR functioned as an environmental sensor to activate leuO expression in response to extracellular stimuli (e.g., bile salts). LeuO itself has been linked to diverse phenotypes in V. cholerae, including biofilm formation, virulence, bile resistance, and acid tolerance (10, 11, 19, 20). Since many of these phenotypes have also been associated with ToxR, these results suggest that ToxR regulates many of its gene targets indirectly. This conclusion is supported by the aforementioned ChIP-seq studies indicating that ToxR directly regulated just 39 genes (49). The gene encoding LeuO was among the 39 genes identified in the ChIP-seq data set, confirming our previous reports that ToxR directly regulated leuO transcription (10, 11). Collectively these findings suggest that the ToxT-independent branch of the ToxR regulon is a hierarchical regulatory system that likely plays a more extensive role in V. cholerae biology than previously appreciated.
The PXB MIC declined as V. cholerae entered stationary phase. The reason for this decline was unclear, but a similar phenomenon was also observed in V. cholerae O1 El Tor strain A1552 with both granulysin-derived peptides and PXB (50), which indicated that this was not a strain-specific phenotype. We noted that deletion of leuO or toxRS increased the PXB MIC in mid-log-phase cells but not in stationary-phase cells. This suggests that the effects of carRS derepression on PXB resistance in these mutants was abrogated at stationary phase. We also found that ompU mutation decreased the PXB MIC in stationary-phase cells and that additional deletion of leuO or toxRS in the ompU-negative strain did not further affect the PXB MIC. This contrasts with mid-log-phase cultures where ompU deletion did not affect the MIC and where ΔompU ΔleuO and ΔompU ΔtoxRS double mutants exhibited increased PXB MICs. These results indicate that unknown factors affect PXB resistance in stationary-phase cells. These results also suggest that OmpU production is critical for PXB resistance at stationary phase and that the ompU-dependent resistance trait was dominant in carRS derepression. Although we do not know the reasons for the ompU-dependent defect, we speculate that decreased PXB resistance in stationary-phase cells reflects the inability of ompU mutants to express the sigma E membrane stress response (17). The sigma E stress response has been shown to be activated predominantly at stationary phase when the cells experience deleterious effects caused by nutrient limitation, pH alterations, oxidative stress, and the accumulation of toxic metabolites (51). It is therefore possible that the deleterious environmental conditions that are present in stationary-phase cultures, and absent from mid-log-phase cultures, enhance the bactericidal activity of PXB on stationary-phase cells, resulting in a decrease in the PXB MIC.
ACKNOWLEDGMENTS
This research was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (NIH) under award numbers R01AI091845 and R21AI125799. M.F.H. was supported by NIH training grant T32AI049820.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
We acknowledge Ky Hoang for generating the almE mutant.
REFERENCES
- 1.Kaper JB, Morris JG Jr, Levine MM. 1995. Cholera. Clin Microbiol Rev 8:48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor RK, Miller VL, Furlong DB, Mekalanos JJ. 1987. Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc Natl Acad Sci U S A 84:2833–2837. doi: 10.1073/pnas.84.9.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmgren J, Svennerholm AM. 1977. Mechanisms of disease and immunity in cholera: a review. J Infect Dis 136(Suppl):S105–S112. doi: 10.1093/infdis/136.Supplement.S105. [DOI] [PubMed] [Google Scholar]
- 4.Childers BM, Klose KE. 2007. Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2:335–344. doi: 10.2217/17460913.2.3.335. [DOI] [PubMed] [Google Scholar]
- 5.Hase CC, Mekalanos JJ. 1998. TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc Natl Acad Sci U S A 95:730–734. doi: 10.1073/pnas.95.2.730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller VL, Taylor RK, Mekalanos JJ. 1987. Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell 48:271–279. doi: 10.1016/0092-8674(87)90430-2. [DOI] [PubMed] [Google Scholar]
- 7.DiRita VJ, Parsot C, Jander G, Mekalanos JJ. 1991. Regulatory cascade controls virulence in Vibrio cholerae. Proc Natl Acad Sci U S A 88:5403–5407. doi: 10.1073/pnas.88.12.5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford JA, Kaper JB, DiRita VJ. 1998. Analysis of ToxR-dependent transcription activation of ompU, the gene encoding a major envelope protein in Vibrio cholerae. Mol Microbiol 29:235–246. doi: 10.1046/j.1365-2958.1998.00925.x. [DOI] [PubMed] [Google Scholar]
- 9.Li CC, Crawford JA, DiRita VJ, Kaper JB. 2000. Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol Microbiol 35:189–203. doi: 10.1046/j.1365-2958.2000.01699.x. [DOI] [PubMed] [Google Scholar]
- 10.Ante VM, Bina XR, Howard MF, Sayeed S, Taylor DL, Bina JE. 2015. Vibrio cholerae leuO transcription is positively regulated by ToxR and contributes to bile resistance. J Bacteriol 197:3499–3510. doi: 10.1128/JB.00419-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bina XR, Taylor DL, Vikram A, Ante VM, Bina JE. 2013. Vibrio cholerae ToxR downregulates virulence factor production in response to cyclo(Phe-Pro). mBio 4:e00366-13. doi: 10.1128/mBio.00366-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller VL, Mekalanos JJ. 1985. Genetic analysis of the cholera toxin-positive regulatory gene toxR. J Bacteriol 163:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krukonis ES, Yu RR, Dirita VJ. 2000. The Vibrio cholerae ToxR/TcpP/ToxT virulence cascade: distinct roles for two membrane-localized transcriptional activators on a single promoter. Mol Microbiol 38:67–84. doi: 10.1046/j.1365-2958.2000.02111.x. [DOI] [PubMed] [Google Scholar]
- 14.Provenzano D, Schuhmacher DA, Barker JL, Klose KE. 2000. The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect Immun 68:1491–1497. doi: 10.1128/IAI.68.3.1491-1497.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duret G, Delcour AH. 2006. Deoxycholic acid blocks Vibrio cholerae OmpT but not OmpU porin. J Biol Chem 281:19899–19905. doi: 10.1074/jbc.M602426200. [DOI] [PubMed] [Google Scholar]
- 16.Duret G, Delcour AH. 2010. Size and dynamics of the Vibrio cholerae porins OmpU and OmpT probed by polymer exclusion. Biophys J 98:1820–1829. doi: 10.1016/j.bpj.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathur J, Davis BM, Waldor MK. 2007. Antimicrobial peptides activate the Vibrio cholerae sigmaE regulon through an OmpU-dependent signalling pathway. Mol Microbiol 63:848–858. doi: 10.1111/j.1365-2958.2006.05544.x. [DOI] [PubMed] [Google Scholar]
- 18.Mathur J, Waldor MK. 2004. The Vibrio cholerae ToxR-regulated porin OmpU confers resistance to antimicrobial peptides. Infect Immun 72:3577–3583. doi: 10.1128/IAI.72.6.3577-3583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ante VM, Bina XR, Bina JE. 2015. The LysR-type regulator LeuO regulates the acid tolerance response in Vibrio cholerae. Microbiology 161:2434–2443. doi: 10.1099/mic.0.000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moorthy S, Watnick PI. 2005. Identification of novel stage-specific genetic requirements through whole genome transcription profiling of Vibrio cholerae biofilm development. Mol Microbiol 57:1623–1635. doi: 10.1111/j.1365-2958.2005.04797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Provenzano D, Klose KE. 2000. Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc Natl Acad Sci U S A 97:10220–10224. doi: 10.1073/pnas.170219997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bina XR, Provenzano D, Nguyen N, Bina JE. 2008. Vibrio cholerae RND family efflux systems are required for antimicrobial resistance, optimal virulence factor production, and colonization of the infant mouse small intestine. Infect Immun 76:3595–3605. doi: 10.1128/IAI.01620-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 24.Bilecen K, Fong JC, Cheng A, Jones CJ, Zamorano-Sanchez D, Yildiz FH. 2015. Polymyxin B resistance and biofilm formation in Vibrio cholerae are controlled by the response regulator CarR. Infect Immun 83:1199–1209. doi: 10.1128/IAI.02700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrera CM, Crofts AA, Henderson JC, Pingali SC, Davies BW, Trent MS. 2014. The Vibrio cholerae VprA-VprB two-component system controls virulence through endotoxin modification. mBio 5:e02283-14. doi: 10.1128/mBio.02283-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henderson JC, Fage CD, Cannon JR, Brodbelt JS, Keatinge-Clay AT, Trent MS. 2014. Antimicrobial peptide resistance of Vibrio cholerae results from an LPS modification pathway related to nonribosomal peptide synthetases. ACS Chem Biol 9:2382–2392. doi: 10.1021/cb500438x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hankins JV, Madsen JA, Giles DK, Brodbelt JS, Trent MS. 2012. Amino acid addition to Vibrio cholerae LPS establishes a link between surface remodeling in gram-positive and gram-negative bacteria. Proc Natl Acad Sci U S A 109:8722–8727. doi: 10.1073/pnas.1201313109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matson JS, Yoo HJ, Hakansson K, Dirita VJ. 2010. Polymyxin B resistance in El Tor Vibrio cholerae requires lipid acylation catalyzed by MsbB. J Bacteriol 192:2044–2052. doi: 10.1128/JB.00023-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bilecen K, Yildiz FH. 2009. Identification of a calcium-controlled negative regulatory system affecting Vibrio cholerae biofilm formation. Environ Microbiol 11:2015–2029. doi: 10.1111/j.1462-2920.2009.01923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bina J, Zhu J, Dziejman M, Faruque S, Calderwood S, Mekalanos J. 2003. ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc Natl Acad Sci U S A 100:2801–2806. doi: 10.1073/pnas.2628026100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linn T, St Pierre R. 1990. Improved vector system for constructing transcriptional fusions that ensures independent translation of lacZ. J Bacteriol 172:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL. 1996. Conditionally replicative and conjugative plasmids carrying lacZ alpha for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1–13. doi: 10.1006/plas.1996.0001. [DOI] [PubMed] [Google Scholar]
- 34.Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O, Salzberg SL, Smith HO, Colwell RR, Mekalanos JJ, Venter JC, Fraser CM. 2000. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imai Y, Matsushima Y, Sugimura T, Terada M. 1991. A simple and rapid method for generating a deletion by PCR. Nucleic Acids Res 19:2785. doi: 10.1093/nar/19.10.2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bina JE, Mekalanos JJ. 2001. Vibrio cholerae tolC is required for bile resistance and colonization. Infect Immun 69:4681–4685. doi: 10.1128/IAI.69.7.4681-4685.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 38.Taylor DL, Ante VM, Bina XR, Howard MF, Bina JE. 2015. Substrate-dependent activation of the Vibrio cholerae vexAB RND efflux system requires vexR. PLoS One 10:e0117890. doi: 10.1371/journal.pone.0117890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryson V, Szybalski W. 1952. Microbial selection. Science 116:45–51. doi: 10.1126/science.116.3003.45. [DOI] [PubMed] [Google Scholar]
- 40.Taylor DL, Bina XR, Bina JE. 2012. Vibrio cholerae VexH encodes a multiple drug efflux pump that contributes to the production of cholera toxin and the toxin co-regulated pilus. PLoS One 7:e38208. doi: 10.1371/journal.pone.0038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guadarrama C, Medrano-Lopez A, Oropeza R, Hernandez-Lucas I, Calva E. 2014. The Salmonella enterica serovar Typhi LeuO global regulator forms tetramers: residues involved in oligomerization, DNA binding, and transcriptional regulation. J Bacteriol 196:2143–2154. doi: 10.1128/JB.01484-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higgins DE, DiRita VJ. 1994. Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol Microbiol 14:17–29. doi: 10.1111/j.1365-2958.1994.tb01263.x. [DOI] [PubMed] [Google Scholar]
- 43.Goss TJ, Morgan SJ, French EL, Krukonis ES. 2013. ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infect Immun 81:884–895. doi: 10.1128/IAI.00889-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Almagro-Moreno S, Kim TK, Skorupski K, Taylor RK. 2015. Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLoS Genet 11:e1005145. doi: 10.1371/journal.pgen.1005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mey AR, Craig SA, Payne SM. 2012. Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun 80:518–528. doi: 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miller VL, Mekalanos JJ. 1988. A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol 170:2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parsot C, Mekalanos JJ. 1990. Expression of ToxR, the transcriptional activator of the virulence factors in Vibrio cholerae, is modulated by the heat shock response. Proc Natl Acad Sci U S A 87:9898–9902. doi: 10.1073/pnas.87.24.9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dupont A, Heinbockel L, Brandenburg K, Hornef MW. 2014. Antimicrobial peptides and the enteric mucus layer act in concert to protect the intestinal mucosa. Gut Microbes 5:761–765. doi: 10.4161/19490976.2014.972238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kazi MI, Conrado AR, Mey AR, Payne SM, Davies BW. 2016. ToxR antagonizes H-NS regulation of horizontally acquired genes to drive host colonization. PLoS Pathog 12:e1005570. doi: 10.1371/journal.ppat.1005570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.da Silva AP, Unks D, Lyu SC, Ma J, Zbozien-Pacamaj R, Chen X, Krensky AM, Clayberger C. 2008. In vitro and in vivo antimicrobial activity of granulysin-derived peptides against Vibrio cholerae. J Antimicrob Chemother 61:1103–1109. doi: 10.1093/jac/dkn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Davis BM, Waldor MK. 2009. High-throughput sequencing reveals suppressors of Vibrio cholerae rpoE mutations: one fewer porin is enough. Nucleic Acids Res 37:5757–5767. doi: 10.1093/nar/gkp568. [DOI] [PMC free article] [PubMed] [Google Scholar]