

Abstract
Aim:
We examined whether variation in blood-based epigenome-wide association studies could be more completely explained by augmenting existing reference DNA methylation libraries.
Materials & methods:
We compared existing and enhanced libraries in predicting variability in three publicly available 450K methylation datasets that collected whole-blood samples. Models were fit separately to each CpG site and used to estimate the additional variability when adjustments for cell composition were made with each library.
Results:
Calculation of the mean difference in the CpG-specific residual sums of squares error between models for an arthritis, aging and metabolic syndrome dataset, indicated that an enhanced library explained significantly more variation across all three datasets (p < 10-3).
Conclusion:
Pathologically important immune cell subtypes can explain important variability in epigenome-wide association studies done in blood.
Keywords: : 450K methylation library, aging, arthritis, cell mixture deconvolution, cellular heterogeneity, confounding, differentially methylated regions, DNA methylation, epigenome-wide association study, inflammation, lymphocytes
Since 2010, there has been a rapidly increasing number of epigenome-wide association studies (EWAS), and many of these studies use various microarray methods including the evolving Illumina platforms [1]. With the recent release of a new generation of the IlluminaBeadchip platform for assessing DNA methylation, it is likely that these studies will continue to increase in number. Various health conditions and environmental exposures have been investigated using the array platforms, including aging, obesity, cancer and inflammatory-related diseases [1–6]. These studies also commonly use peripheral blood as a source of DNA to investigate associations between an exposure of interest and disease [1–6].
Cellular lineage and somatic differentiation are regulated by epigenetic mechanisms, including DNA methylation; thus, the pattern of methylation at phenotypically important CpG regions varies substantially across individual tissues and cell-types and specifically across the distinct leukocyte subtypes [7–12]. Moreover, since there are differentially methylated regions (DMRs) that demarcate the different types of leukocyte lineages and activation states, EWAS that used blood as a biospecimen for methylation profiling are likely to result in CpGs that merely reflect the differences in the cell composition and/or cell states, altered by the presence of disease or by immunomodulating exposures [1,4,8,10]. Specifically, any interpretation of differences in DNA methylation at CpG sites resulting from whole blood comparisons of DNA methylation between two or more disease states must include the possibility that such changes arise from variation in the leukocyte composition between study samples (occurring, e.g., as a result of inflammatory or other biologically related processes) [4,10]. Thus, methylation patterns associated with varying cell proportions or by the state of activation of any type of leukocyte in the blood can potentially be a confounder in the outcome of an EWAS [1,4,10,11].
We previously established a unique reference library of the DNA methylation profile for different leukocytes in peripheral blood and demonstrated that this library can inform a novel algorithm to estimate the relative abundance of the distinct leukocyte cell-types [7,11]. In addition, this approach has been widely adopted to adjust EWAS data, allowing one to discriminate DNA methylation differences reflecting changes in cell subpopulation from other possibly environmentally induced or disease-associated methylation events [7,11]. The operational approach to deconvoluting the cellular composition of whole relies upon a DNA methylation profile library of the individual cell-types comprising whole blood. Interestingly, a recent study has examined subtypes of B and T cells, specifically identifying methylation markers of naive and memory B cells; naive, memory and regulatory CD4+ T cells; and naive and memory CD8+ T cells [13]. This study has shown to estimate the proportions of these B- and T-cell subtypes using the methylation signatures and thus implicated that these signatures can be used for specific cell-type proportion estimates and adjustment for potential confounding in whole blood derived methylation studies [13].
Here, we hypothesized that there may be significant contributions made by normally, but pathologically important leukocyte subtypes in whole-blood DNA methylation signatures. Thus, in an effort to strictly determine if enlarging the size and scope of the input methylation library will explain additional variability in a discovery-driven EWAS analysis, we combined the library of Reinius et al. with additional novel input cell-type DNA methylation profiles that can supply preliminary (unvalidated) DMRs for additional specific immune cell subtypes [8,14–16]. This combined library included activated natural killer (NK) cells, dendritic cells, immature B cells, memory B cells, naive B cells, plasma B cells and naive Tregs (in addition to the cells profiled by Reinius et al.) [8,14–16]. Using novel statistical methods with this combined library, we explored whether these additional DMRs explained additional variability in the DNA methylation-disease associations from three selected studies: blood methylation profile in rheumatoid arthritis, normally aging individuals and in a family study of people at risk for metabolic syndrome [2,3,6].
Materials & methods
All of the following statistical analyses were performed in R software (R version 3.2.3).
Establishing an expanded 450K reference library & study selection
The following 450K methylation datasets were used to create a novel reference library of methylation markers for different white blood cell-types: two publicly available datasets from the Gene Expression Omnibus – GSE35069 and GSE45461, a set of analyzed 450K methylation from our prior and current research studies and a requested Blueprint methylation dataset from the European Genome-phenome Archive – EGAD00010000716 [8,14–16]. An established 450K reference library by Reinius et al. was used as the starting framework to create the new, updated library with more immune cell-types [8]. The DMRs of B cells from the Reinius et al. 450K library were removed and replaced with specific immature B-cell DMRs from the Lee et al. library and DMRs of memory, naive and plasma B cells from the EGA dataset [14,15]. DMRs of activated NK cells, dendritic cells and naive Tregs were added from our previous and ongoing studies. Thus, the final composition of immune cell-types in the new, updated 450K reference library included the following: T cells, immature B cells, memory B cells, naive B cells, plasma B cells, NK cells, monocytes, granulocytes, activated NK cells, dendritic cells and naive Treg cells. Three epigenetic datasets (GSE42861, GSE60132 and GSE40279) that used whole blood for methylation-disease association analyses were selected to apply the new 450K library and determine if potential cellular heterogeneity was better explained [2,3,6]. Studies used included Liu et al. (investigating methylation association with rheumatoid arthritis), Hannum et al. (normal aging) and Ali et al. (metabolic syndrome), respectively [2,3,6]. Sex chromosomes, sites with SNPs in probes, cross-reactive probes, polymorphic CpGs and CpGs with detection of p-values greater than 0.05 were excluded from the analysis [17].
Detecting specific DMRs for each immune cell-type in the library using dispersion separability criterion statistical approach
In order to create an optimal library with specific subsets of DMRs for each immune cell-type in the combined 450K reference library, the top CpGs that uniquely discriminated each cell-type from all other cell-types were selected. Specifically, for each cell-type the top 50 CpGs with largest absolute t-statistics were selected, which represented the top hypo- and hypermethylated loci between that cell-type and the remaining cell-types. The top CpGs of each cell-type were then pooled into a single list of N unique loci, which was established as the candidate DMR search space. Using the candidate DMR search space, an algorithm that has been developed by our research group called DMRSubsetFinder, was applied to randomly select subsets of varying sizes of DMRs from the candidate space [11]. For each of 1000 iterations, K < N CpGs were selected at random from the candidate DMR space, where K ranged from 50 to N – 50. For a given randomly selected DMR subset, the dispersion separability criterion (DSC) score was computed to assess the subset's power for discriminating the leukocyte subtypes. The DSC is a metric that was initially designed to quantify the amount of batch effects in microarray datasets, however, here we substitute ‘batch’ with the identity of a given cell-type. Specifically, the DSC is defined as Db/Dw, where Db is a measure of dispersion between cell-types, and Dw is a measure of dispersion within cell-types. Thus, the DSC was used here to quantify the dispersion ‘between’ and ‘within’ specific leukocyte subtypes for a given randomly selected DMR subset. Across the 1000 iterations of the DMRSubsetFinder algorithm, the DMR subset with the largest DSC score was selected so that the selected loci exhibited maximal heterogeneity between cell subtypes and homogeneity within cell subtypes with regard to their methylomic profile.
Deriving sum of squared errors to explain additional variability in the selected studies
Using the optimal DMR library selected via DMRSubsetFinder, the relative fraction of each cell-type was estimated for each sample in the arthritis, aging and metabolic syndrome datasets. The algorithm used to predict cell proportions has been previously determined by Houseman et al. [11]. Generally, this methodology identifies the CpGs that distinguish the leukocyte subtypes based on rank ordering CpGs from ANOVA models fit to a reference leukocyte DNA methylation dataset. The selected discriminative CpGs are then used as the basis for estimating cell-type distributions of samples comprising whole-blood derived DNA methylation data [11]. Using our optimal DMR library, the projected proportions were then used to adjust for cell-type composition in CpG-specific regression models fit to the arthritis, normal aging and metabolic syndrome datasets. A ‘null’ model adjusted only for the cell-types in the 450K reference library established by Reinius et al., and a ‘full’ model adjusted for the Reinius library and the additional leukocyte subtypes: immature B cells, memory B cells, naive B cells, plasma B cells, NK cells, activated NK cells, dendritic cells and naive Treg cells were considered in our analyses [8,14–16].
The null and full models were fitted for each CpG locus in the arthritis, normal aging and metabolic syndrome datasets and the residual sum of squared error (SSE) was computed for each locus. Specifically;
‘Null’ model:
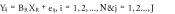 |
‘Full’ model:
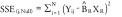 |
X R: Vector containing the cell-type estimates using the Reinius only reference library
X R+: Vector containing the cell-type estimates using the Reinius plus (+) new cell-types expanded library
where Yij represents the methylation beta-value for loci j in subject i. The sum of the SSEs over all loci for each model were calculated, and the sum of the SSEs for the null model was compared with that for the adjusted model; a smaller sum of SSE indicated a better fit, in other words, more variation in DNA methylation explained. As such, the difference in SSEs (defined as SSE_null – SSE_full) was calculated, and the significance of this difference was determined by performing a permutation test to determine the sampling distribution under the null hypothesis. For 1000 iterations, the methylation values from the arthritis, normal aging and metabolic syndrome datasets were permuted, and null and full models were fitted to the permuted data. The difference in the sum of the SSEs under the null and under the full model was calculated and the significance of the test statistic was determined based on this distribution. The application of this statistical derivation is based on previously optimized method to identify specific methylation libraries for cell mixture deconvolution [12].
Classification of rheumatoid arthritis cases & controls using expanded library
The null and full models were also used to predict and compare the classification of controls and cases in the arthritis dataset [3]. With the predicted probabilities obtained for the two models, receiver operating characteristic curves were constructed and the corresponding areas under the curve (AUC) were computed using the function colAUC in the R package caTools. The following notations depict the statistical computations for the AUC comparisons:
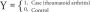 |
XR: Vector containing the cell-type estimates using the Reinius only reference library
XR+: Vector containing the cell-type estimates using the Reinius plus (+) new cell-types expanded library
‘Null’ model:
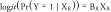 |
‘Full’ model:
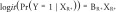 |
with X R and X R+ as previously defined.
Results
Using the DSC scores, the top loci for each specific cell-type were determined in order to create optimal subsets of the cell-types for the final 450K reference library (Figure 1). Overall, 74 loci were selected as an optimal set of DMRs (Supplementary Table 1). Three publicly available datasets (arthritis, normal aging and metabolic syndrome studies) were then interrogated to assess cell-type projections and variability (Table 1) [2,3,6]. After removing probes with missing values and probes with high detection p-values, the arthritis dataset contained 689 subjects and 390, 324 loci; the aging dataset contained 656 subjects and 413, 994 loci; and the metabolic syndrome dataset contained 192 subjects and 389, 426 loci.
Figure 1. . Heatmap of the top 74 loci in the optimal subset for each leukocyte subtype.

Table 1. . Whole blood 450K methylation datasets that were used to create the reference library and analyzed for variations.
| Study (year) | Title | Sample size | Study accession ID | Ref. |
|---|---|---|---|---|
| Kulis et al. (2015) |
Human B-cell differentiation methylation |
35 |
EGAD00010000716 |
[14] |
| Lee et al. (2013) |
A global DNA methylation and gene expression analysis of early human B-cell development |
22 |
GSE45461 |
[15] |
| Reinius et al. (2012) |
Differential DNA methylation in purified human blood cells |
60 |
GSE35069 |
[8] |
| Wiencke et al. (2015) |
Activated NKs, dendritics, Tregs |
|
|
[16] |
|
Whole blood methylation | ||||
| Ali et al. (2015) |
An epigenetic map of age-associated autosomal loci in northern European families at high risk for the metabolic syndrome |
192 |
GSE40279 |
[6] |
| Liu et al. (2013) |
Differential DNA methylation in rheumatoid arthritis |
689 |
GSE42861 |
[3] |
| Hannum et al. (2012) | Genome-wide methylation profiles reveal quantitative views of human aging rates | 656 | GSE40279 | [2] |
NK: Natural killer cell.
In order to determine whether the inclusion of additional immune cell-types improved the explanation of DNA methylation variability to the arthritis and aging datasets, the difference in sum of SSE between the null and full models and the null distribution of this statistic was computed over 1000 iterations. For the arthritis study, the difference in sum of SSE between the null models and adjusted models was 13.18, (95% value: 1.37 and 99% value: 2.10) (Table 2). In the aging study, the difference was 7.13, (95% value: 1.31 and 99% value: 2.13) (Table 2). Last in the metabolic syndrome study the difference was 8.40, (95% value: 2.97 and 99% value: 4.62) (Table 2). Based on the null sampling distribution, the sum of SSE from the full model was determined to be significantly lower than the sum of SSE from the null value for each dataset (p < 10-3).
Table 2. . Variation (sum of squared error) analyses on the arthritis, aging and metabolic syndrome datasets using the null and adjusted models.
| Study | Diff.SSE† | 95% distribution | 99% distribution | p-value |
|---|---|---|---|---|
| Arthritis |
13.18 |
1.37 |
2.1 |
<10-3 |
| Normal aging |
7.13 |
1.31 |
2.13 |
<10-3 |
| Metabolic syndrome | 8.4 | 2.97 | 4.62 | <10-3 |
†Diff.SSE: Difference in residual sum of squared errors.
We also determined if the fully adjusted model based on the Reinius library plus the additional cell-types would better classify cases and controls than the null model in the arthritis dataset [3]. Logistic regression models were fit to the arthritis dataset, in which one model controlled for the cell-type predictions generated from just the Reinius library and another model controlled for cell-type predictions generated from the Reinius plus additional cell-types library [8,14–16]. The constructed receiver operating characteristic curves and the corresponding AUC determined from each model (Figure 2) showed that the expanded library performed slightly better (AUC = 0.84) than the Reinius library alone (AUC = 0.82). Thus, this analysis shows that the expanded library slightly improved the prediction of cases and controls in the arthritis dataset [3].
Figure 2. . Area under the curve comparisons of classification sensitivity using expanding library and Reinius library alone.

AUC: Area under the curve; ROC: Receiver operating characteristic.
Discussion
Differences in DNA methylation seen when comparing cases versus controls in large association studies that apply platforms for gathering relatively large amounts of methylation data (such as the Illumina 450K platform) may reflect pathological changes in immune cell number, but it is important to note that these may also reflect immune cell activation. Statistical deconvolution methods have helped to interpret DNA methylation associated changes in datasets that have collected whole blood samples with mixed normal immune cell-types [1,7,8,11,12]. Hence, variations in DNA methylation changes detected in blood can potentially reflect variations in proportions of leukocytes [4,10]. However, little work has been done to discover if some of the additional remaining differences in the DNA methylation profile reflect disease associated changes in the immune system, such as activation of some specific cell-types.
From the SSE analyses, the differences between the null and adjusted models in both datasets were positive if the null model SSE values were greater than the adjusted model SSE values. The model with the smaller sum values of SSE indicated the better model, and a larger SSE sum value indicated more error. Thus, from our analyses, the adjusted model proved to be a significantly better fit when applied in the arthritis, normal aging and metabolic syndrome datasets (p < 10-3) [2,3,6]. Additional classification comparisons using AUC plots also indicated that the expanded library led to an improved ability to discriminate rheumatoid arthritis cases from controls compared with the existing Reinius library.
There were important limitations that must be kept in mind in creating and applying the combined, optimal 450Kmethylation library. The addition of more immune cell-types could result in an overfitting of the model and lead to more overlapping, nonspecific DMRs for all the immune cell-types in the model. Thus, the DSC approach was applied to extract efficiently the specific markers for each leukocyte subtype, even if it resulted in a lesser number of top loci for each cell. Whenever more specific leukocyte subtypes are added, one needs to account for potential overlap and nonspecificity and consequently compute the DSC scores to determine specific DMRs. Another limitation on the application of this library is that it can be used to explain significant variability in only studies with large sample size. Epigenetic studies with sample size (n) greater than 200 resulted in significant variability; while, studies with sample size of 100 or less did not show significant additional variability (Supplementary Table 2).
Separating cell populations from whole blood prior to epigenomic analyses may certainly validate or support results when accounting for different cell subtypes by quantitative methylation analyses. Usually, separating cell populations from whole blood involves purification of cells with antibody markers for each cell-type and analyses with flow cytometry; however, there are overlapping cell markers and often unspecific antibody markers for cell subtypes that may not result in ‘pure’ isolation of individual cell subtypes. Thus, there is an advantage in using quantitative methylation analyses since they are less dependent on getting efficient purification of cell subtypes but instead rely on specific cell lineage methylation signatures for each cell subtype to account for cell composition effects in whole blood methylation analyses.
Finally, we believe that it is crucially important to stress that the expanded library that we employed in this analysis has not been shown to predict, with accuracy, the cell-type proportions in whole blood methylation studies. That is, the library we have constructed, while able to explain additional variation in the context of an EWAS, has not been externally validated and thus, it should not be employed to directly assess immune cell-types in any epidemiologic context. While it is clearly conceivable that this could be done, it will require further validation, experimentally testing the accuracy and power of the expanded library to predict leukocyte subtype proportions, as has been previously done for prior libraries [7,8,11].
Conclusion
Thus, the more adjusted or ‘complete’ 450K library created with specific methylation markers for the additional immune cell-types explained, with robust statistical significance, additional variability in the arthritis, normal aging and metabolic syndrome epigenetic studies. Adding more cell-types to the library to account for cellular heterogeneity in these studies helped improve detection of specific methylation markers. Specifically, inclusion of markers for additional cell-types facilitated observation of the driving immune cell-types in these studies. Our findings are consistent with what has been previously reported, strongly arguing for the development, validation and use of enhanced libraries when investigating inflammatory diseases using blood-derived DNA. Future work should be focused on finding and adding unique DMRs for newly discovered immune cell subtypes to create an optimal DNA methylation library in order to facilitate applying it to epigenetic studies of environmental or otherwise acquired immune-related diseases.
Future perspective
The updated 450K reference library includes pathologically important leukocyte subtypes (such as dendritic cells, activated NK cells and Tregs, among others) that are known to have important implications in inflammatory-related diseases. In this study, the addition of the leukocyte subtypes explained significant additional variability in whole-blood DNA methylation signatures of inflammatory related diseases such as rheumatoid arthritis and metabolic syndrome. The next phase would be the validation of the expanded 450K library with prediction of different leukocyte subtype proportions, which would enhance the impact of this current study. We would need to validate the expanded library by extracting certain leukocyte subtypes and confirming methylation signatures. We did not aim for the expanded library to be used by others for prediction of cell subtype proportions but rather to show that an expanded library can explain additional significant variability when accounting for cell subtypes in whole blood methylation analyses. In the future, we aim for a validated, expanded 450K library that can be used to predict these additional leukocyte subtypes in whole-blood derived epigenetic studies. Studies that investigate specific pathological diseases can use the expanded 450K library that incorporates pathologically important leukocyte subtypes in order to account for adjustments for these cell composition effects in their studies.
Executive summary.
Epigenome-wide association studies (EWAS) frequently involve DNA methylation profiling of leukocyte-derived DNA (extracted from human peripheral blood) as a means of understanding the relationship between DNA methylation and human diseases and exposures.
Such investigations are highly susceptible to confounding by cell-type composition, both because of the cell-specificity of DNA methylation and since many conditions and exposures elicit an immune response, altering the immune landscape.
We hypothesized that usually uncommon, but pathologically important immune cell subtypes could account for non-negligible variation in the whole-blood DNA methylation profile of immune-associated conditions.
To test this hypothesis, we examined the extent to which variation in whole-blood-derived DNA methylation could be more completely explained by augmenting existing reference DNA methylation libraries comprised of normal leukocyte subtypes with the reference methylomes of immature B cells, memory B cells, plasma B cells, activated natural killer cells, dendritic cells and naive Treg cells.
Using our expanded reference DNA methylation library, we compared existing and our enhanced libraries in predicting additional epigenetic variability in whole-blood samples collected as part of three large, publicly available DNA methylation datasets: a study of DNA methylation in blood from rheumatoid arthritis patients; a study of normal individuals of varying age; and a study of metabolic syndrome.
Within each dataset, models were fitted separately to each CpG site and were used to estimate the additional variability in whole-blood DNA methylation signatures explained when adjustments for cell composition were carried out using our expanded library compared with an existing library. The within-dataset mean difference in the CpG-specific residual sums of squares error between the two models was computed.
In addition, the expanded library demonstrated better performance in terms of correctly classifying rheumatoid arthritis cases from disease-free controls compared with the existing normal library.
We conclude that uncommon, but pathologically important immune cell subtypes can explain important variability in EWAS done in blood and suggest that there is a need to define and validate methylation markers of specific immune cell methylomes in order to properly adjust for cell composition effects in EWAS.
Supplementary Material
Acknowledgements
The authors would like to thank the authors whose publicly available datasets have helped us to construct our optimal 450K reference library. The authors acknowledge and thank the helpful suggestions and discussions of R Butler.
Footnotes
Financial & competing interests disclosure
J Wiencke was supported by the Robert Magnin Newman endowment for Neurooncology. This work was also supported by the NIH grants: (1KL2TR000119 to DC Koestler) and the Kansas IDeA Network of Biomedical Research Excellence (K-INBRE) Bioinformatics Core supported in part by the National Institute of General Medical Science award P20GM103418. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Ethical conduct of research
The authors state that they have obtained appropriate institutional review board approval or have followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
References
- 1.Michels KB, Binder AM, Dedeurwaerder S, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat. Methods. 2013;10(10):949–955. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 2.Hannum G, Guinney J, Zhao L, et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y, Aryee MJ, Padyukov L, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013;31(2):142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15(2):R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Almén MS, Nilsson EK, Jacobsson JA, et al. Genome-wide analysis reveals DNA methylation markers that vary with both age and obesity. Gene. 2014;548(1):61–67. doi: 10.1016/j.gene.2014.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Ali O, Cerjak D, Kent JW, et al. An epigenetic map of age-associated autosomal loci in northern European families at high risk for the metabolic syndrome. Clin. Epigenet. 2015;7(1):12. doi: 10.1186/s13148-015-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Accomando WP, Wiencke JK, Houseman EA, Nelson HH, Kelsey KT. Quantitative reconstruction of leukocyte subsets using DNA methylation. Genome Biol. 2014;15(3):R50. doi: 10.1186/gb-2014-15-3-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reinius LE, Acevedo N, Joerink M, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE. 2012;7(7):e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khavari DA, Sen GL, Rinn JL. DNA methylation and epigenetic control of cellular differentiation. Cell Cycle Georget. Tex. 2010;9(19):3880–3883. doi: 10.4161/cc.9.19.13385. [DOI] [PubMed] [Google Scholar]
- 10.Houseman EA, Kim S, Kelsey KT, Wiencke JK. DNA methylation in whole blood: uses and challenges. Curr. Environ. Health Rep. 2015;2(2):145–154. doi: 10.1007/s40572-015-0050-3. [DOI] [PubMed] [Google Scholar]
- 11.Houseman EA, Accomando WP, Koestler DC, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koestler DC, Jones MJ, Usset J, et al. Improving cell mixture deconvolution by identifying optimal DNA methylation libraries (IDOL) BMC Bioinformatics. 2016;17:120. doi: 10.1186/s12859-016-0943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waite LL, Weaver B, Day K, et al. Estimation of cell-type composition including T and B cell subtypes for whole blood methylation microarray data. Front. Genet. 2016;7:23. doi: 10.3389/fgene.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulis M, Merkel A, Heath S, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat. Genet. 2015;47(7):746–756. doi: 10.1038/ng.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee S-T, Xiao Y, Muench MO, et al. A global DNA methylation and gene expression analysis of early human B-cell development reveals a demethylation signature and transcription factor network. Nucleic Acids Res. 2012;40(22):11339–11351. doi: 10.1093/nar/gks957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiencke JK, Butler R, Hsuang G, et al. The DNA methylation profile of activated human natural killer cells. Epigenetics. 2016;11(5):363–368. doi: 10.1080/15592294.2016.1163454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Lemire M, Choufani S, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8(2):203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.