

Abstract
Individuals with posttraumatic stress disorder (PTSD) typically exhibit altered hypothalamic-pituitary-adrenal (HPA) function and sympathetic nervous system (SNS) activity. The goals of this study were to determine whether HPA and SNS alterations in the immediate aftermath of trauma predict subsequent PTSD symptom development and whether inconsistencies observed between studies can be explained by key demographic and methodological factors. This work informs secondary prevention of PTSD by identifying subgroups of trauma survivors at risk for PTSD. This meta-analysis (26 studies, N = 5,186 individuals) revealed that higher heart rate measured soon after trauma exposure was associated with higher PTSD symptoms subsequently (r = .13). Neither cortisol (r = −.07) nor blood pressure (diastolic: r = −.01; systolic: r = .02) were associated with PTSD symptoms which may be influenced by methodological limitations. Associations between risk markers (heart rate, cortisol, systolic blood pressure) and PTSD symptoms were in the positive direction for younger samples and negative direction for older samples. These findings extend developmental traumatology models of PTSD by revealing an age-related shift in the presentation of early risk markers. More work will be needed to identify risk markers and pathways to PTSD while addressing methodological limitations in order to shape and target preventive interventions.
Keywords: traumatic stress, posttraumatic stress disorder, cortisol, heart-rate, blood pressure
1.1. Introduction
Traumatic life events involve exposure to actual or threatened serious injury, sexual violence or death (American Psychiatric Association, 2013), and for a sizeable minority of survivors, they are followed by intrusion, avoidance, cognitive, mood, and arousal symptoms that together comprise posttraumatic stress disorder (PTSD). For the majority of individuals coping with recent traumatic stress, these post-traumatic symptoms are short-lived and remit without need for treatment (Galatzer-Levy et al., 2013; McNally, Bryant, & Ehlers, 2003; Santiago et al., 2013). For a sizable minority of these individuals, however, these early symptoms will progress into a full-blown posttraumatic stress disorder and confer increased risk for debilitating physical and mental health outcomes (Hidalgo & Davidson, 2000). A variety of pretraumatic, peritraumatic and posttraumatic factors (genetic, neurobiological, cognitive, sociocultural, environmental) have been linked to the development of PTSD (for reviews, see Bomyea, Risbrough, & Lang, 2012; Brewin, Andrews, & Valentine, 2000; Elwood, Hahn, Olatunji, & Williams, 2009; McNally, 2003; Ozer, Best, Lipsey, & Weiss, 2003; Schmidt et al., 2015; Scott et al., 2015; Zoladz & Diamond, 2013). However, the neurobiological mechanisms of risk for PTSD operating during the early posttraumatic recovery phase are still poorly understood. Identifying the post-exposure predictors and mechanisms of risk could allow us to develop and efficiently target secondary prevention programs (Delahanty & Nugent, 2006; Morris & Rao, 2013), with the potential to prevent PTSD development following traumatic exposure.
This meta-analysis builds on prior reviews of PTSD neurobiology (de Kloet et al., 2006; Delahanty & Nugent, 2006; Klaassens et al., 2012; Meewisse et al., 2007; Miller et al., 2007; Morris et al., 2012; Pitman et al., 2012; Pole, 2007; Shalev & Segman, 2008; Yehuda, 2002; Zoladz & Diamond, 2013) by quantitatively focusing on longitudinal studies of peritraumatic risk markers within two major stress response systems: the sympathetic nervous system (SNS) and hypothalamic-pituitary-adrenal (HPA) axis. The SNS responds quickly to stressor exposure, with rapid release (within seconds) of norepinephrine and epinephrine from the adrenal medulla, triggering a number of peripherally measurable physiological changes (assessed in many PTSD studies by heart rate and/or blood pressure). The HPA axis also responds to threat or challenge, but reaches its peak 20 to 40 minutes following stress exposure via a hormonal cascade that culminates in the release of cortisol from the adrenal cortex (assessed in PTSD work by cortisol levels in saliva, plasma/serum and/or urine). The SNS plays a role in rapid, “fight-flight” responses to danger (Rodrigues, LeDoux, & Sapolsky, 2009). The HPA axis shapes longer-term patterns of stress response (Rodrigues et al., 2009). Both have been strongly implicated in the pathophysiology of PTSD and abnormalities in either or both may play a role in the development of PTSD following trauma exposure (Yehuda, 2002; Zoladz & Diamond, 2013). This review will evaluate the relevance of these stress response indicators for secondary prevention efforts by examining their associations with subsequent PTSD symptoms. Potential moderators of these associations will be tested to determine whether well-documented inconsistencies between studies could be explained by key methodological or demographic moderators.
1.2. Secondary prevention of PTSD: Relevance of HPA and SNS activity
PTSD offers more promise for preventive intervention than any other psychiatric disorder because it can only develop following an identifiable traumatic exposure, and a well-established and reliable percentage of people so exposed will develop the disorder if allowed to follow the natural history (McNally et al., 2003). If individuals at risk and their mechanistic pathways to illness can be identified, preventive interventions can be designed. Efforts to date have focused on SNS and HPA axis reactivity following trauma, and pharmacological prevention efforts have utilized glucocorticoids to alter HPA function and beta receptor blockers to alter SNS activity. Cortisol and noradrenergic neurotransmission play important, interacting roles in memory processes and may contribute to PTSD development through these effects (Rodrigues et al., 2009; Wolf, 2008).
Noradrenergic neurotransmission is essential for consolidation and long-term storage of memories, and a consolidation model of PTSD has suggested that sustained hyper-adrenergic states in the aftermath of trauma may create risk by a “searing in” or over-consolidation of the traumatic memories (de Quervain, Aerni, Schelling, & Roozendaal, 2009; Pitman, 1989; Pitman & Delahanty, 2005; Pitman, Orr, & Shalev, 1993; Yehuda & Harvey, 1997; Yehuda, McFarlane, & Shalev, 1998). There was early evidence that blocking this process by administering the beta receptor antagonist propranolol immediately after trauma exposure could reduce PTSD risk, but subsequent work has not supported its efficacy (see meta-analytic review by Sijbrandij, Kleiboer, Bisson, Barbui, & Cuipers, 2015). Timing may be critical, however, since there is a narrow consolidation window and these pharmacological intervention studies may not have administered propranolol soon enough to alter SNS-mediated memory consolidation processes (Hruska, Cullen, & Delahanty, 2014). The window of opportunity to alter (re)consolidation of memories appears to close within hours (McGaugh, 2000; Nader, Schafe, & LeDoux, 2000).
Preventive intervention using glucocorticoids has been more successful. A meta-analysis has found a large effect for acute, post-traumatic glucocorticoid administration in preventing subsequent onset of PTSD (Sijbrandij et al., 2015). This benefit could result from the impact of glucocorticoids on memory processes. Glucocorticoids can enhance memory consolidation; however, they impair memory retrieval and working memory (de Quervain et al., 2009). If administered within the consolidation window, as they have been in pharmacological prevention studies (Sijbrandij et al., 2015), glucocorticoids could in fact enhance traumatic memory consolidation and increase PTSD symptoms. Their efficacy suggests that any such impact might be overshadowed by inhibition of SNS-driven memory consolidation (Joëls, Fernandez, & Roozendaal, 2009; Ursin & Olff, 1993; Yehuda, 2002), but that pathway seems unlikely given the lack of efficacy of direct blockade of those SNS effects. This suggests that their benefits might occur through some other mechanism.
Newer models of PTSD have shifted emphasis from memory consolidation processes to deficits in extinction recall (Milad et al., 2009) and contextual modulation of memory (Garfinkel et al., 2014). According to context-processing models of PTSD, the persistence of fear, hypervigilance, and exaggerated physiological responses in ostensibly safe environments (Liberzon & Sripada, 2007) suggest impaired utilization of contextual cues to modulate fear expression (Garfinkel et al., 2014). The HPA axis likely plays a critical role in context-processing given the high density of mineralocorticoid and glucocorticoid receptors in the hippocampus and ventromedial prefrontal cortex (de Kloet, 2003), and there is evidence from animal studies that glucocorticoids, at very high levels, can interact with stress to produce a PTSD-like contextual-processing deficit (Kaouane et al., 2012). At more moderate levels, glucocorticoids can enhance hippocampal-dependent cognitive processes (Diamond, Bennett, Fleshner, & Rose, 1992). Those at risk for PTSD may have abnormally low cortisol levels (Yehuda, 2002), and glucocorticoid administration may bring them to the moderate levels needed for optimizing hippocampal-dependent learning processes. The preventive effects of cortisol administration on subsequent PTSD (Sribrandij et al., 2015) could thus perhaps be explained by its impact on hippocampal-prefrontal circuits and context-processing capacities, in which case the window for beneficial impact, and for the predictive relevance of endogenous cortisol levels, may be wider than that suggested by a consolidation model. Further exploration of parameters that could modify associations between HPA or SNS activity and PTSD symptoms is needed. This work should include a broader range of types and severity of traumatic exposures, since pharmacological prevention studies, to date, have generally included only severely ill inpatients (Sribandij et al., 2015). The present meta-analysis will inform the secondary prevention of PTSD by evaluating longitudinal studies conducted in the immediate aftermath of trauma (‘post-post’ designs; Schmidt et al., 2015) to determine whether acute HPA and SNS alterations are associated with subsequent PTSD symptoms.
1.3. Acute neurobiological responses to trauma
1.3.1. Cortisol levels
Individuals with PTSD often exhibit dysregulated HPA function in the form of enhanced glucocorticoid negative feedback (Morris, Compas, & Garber, 2012; Yehuda, 2002) and lower circulating cortisol levels compared to non-traumatized controls (Meewisse, Reitsma, De Vries, Gersons, & Olff, 2007; Miller, Chen, & Zhou, 2007; Morris et al., 2012; see also Klaassens, Giltay, Cuijpers, van Veen, & Zitman, 2012). However, it remains unclear whether HPA dysregulation is present in the immediate aftermath of trauma and contributes to elevated risk for developing PTSD (Delahanty & Nugent, 2006; Morris & Rao, 2013). Trauma-exposed individuals without PTSD also exhibit enhanced HPA negative feedback, like those with PTSD, but do not differ from non-traumatized controls in their daily cortisol output (Morris et al., 2012). Prospective studies indicate that individuals vulnerable to developing PTSD are characterized by dysregulations at different levels of the glucocorticoid signaling cascade (van Zuiden, Kavelaars, Geuze, Olff, & Heijnen, 2013), including lower circulating cortisol levels prior to (Steudte-Schmiedgen et al., 2015) – and in the early aftermath of – trauma exposure (Luo et al., 2012; van Zuiden et al., 2012). However, the strength and direction of findings from studies examining acute posttraumatic cortisol levels as markers of PTSD risk appears to be influenced by a variety of moderators including the timing of PTSD assessment, timing of cortisol measurement, gender, and developmental stage (Delahanty & Nugent, 2006; van Zuiden et al., 2013). To our knowledge, the association between acute posttraumatic cortisol levels and subsequent PTSD has not yet been investigated by a quantitative review. We hypothesized that lower cortisol levels in the immediate aftermath of the trauma would be associated with higher PTSD symptoms subsequently.
1.3.2. Heart rate
Individuals with PTSD often exhibit increased cardiac activity in the form of elevated resting heart rate and enhanced heart rate reactivity to startling sounds, standardized trauma cues and idiographic trauma cues (Pole, 2007). Meta-analytic evidence suggests that the association between heart rate reactivity to standardized trauma cues and PTSD (Pole, 2007) is comparable to the largest effect sizes reported for psychosocial variables including social support, peritraumatic distress, current stress levels, and perceived life threat (Brewin et al., 2000; Ozer et al., 2003). Heart rate reactivity to trauma cues is most strongly associated with the PTSD reexperiencing and hyperarousal symptoms (Costanzo et al., 2014) and appears to normalize in response to successful treatment (Keane & Kaloupek, 1982; Pitman et al., 1996; Pole & Bloomberg-Fretter, 2006). However, prospective studies indicate that pretraumatic cardiac activity is not associated with increased risk for PTSD (Pole, Neylan, Otte, Henn-Hasse, Metzler, & Marmar, 2009). Contradictory findings linking risk for PTSD to both higher (Shalev et al., 1998) and lower (Blanchard, Hickling, Galovski, & Veazey, 2002) heart rate measured in the emergency room has led some researchers to propose that elevated heart rate in the immediate aftermath of trauma is not a good biological marker for PTSD (e.g., Blanchard et al., 2002; Zoladz & Diamond, 2013). However, no quantitative reviews have examined the association between acute posttraumatic heart rate and subsequent PTSD symptoms and tested key moderators (e.g., sample characteristics, methodological factors) that may affect the strength and direction of this relation and account for the contradictory findings. We hypothesized that higher heart rate in the immediate aftermath of the trauma would be associated with higher subsequent PTSD symptoms when relevant moderators are appropriately controlled.
1.3.3. Blood pressure
Whereas systolic blood pressure reflects the force of blood in the circulatory system when the heart contracts, diastolic blood pressure reflects this force when the heart is at rest. Meta-analysis has shown that PTSD is associated with higher resting systolic and diastolic blood pressure and greater diastolic blood pressure responses to idiographic trauma cues. Based on larger unweighted effect sizes for diastolic blood pressure, the author concluded that “PTSD was more strongly related to blood pressure when the heart is at rest than when the heart contracts” (Pole, 2007, p. 741). Although individuals with PTSD have higher rates of hypertension and cardiovascular disease (O’Toole & Catts, 2008; Schnurr, Spiro, & Paris, 2000), no quantitative reviews have yet examined whether acute blood pressure responses to trauma are associated with subsequent development of PTSD. We hypothesized that higher peritraumatic systolic and diastolic blood pressure would be associated with higher subsequent PTSD symptoms.
1.4 Potential moderators
1.4.1. Age
Neurodevelopmental traumatology models posit that childhood trauma exerts a more devastating impact on physical and mental health outcomes than trauma exposure in adulthood due to the adverse consequences of dysregulated stress response systems on brain development during critical vulnerability periods (De Bellis et al., 1999, 2011). A longitudinal study of sexually-abused and non-abused females found that abuse was associated with higher morning cortisol levels in childhood and lower morning cortisol levels by early adulthood even after accounting for the time elapsed since trauma disclosure (Trickett, Noll, Susman, Shenk, & Putnam, 2010). Traumatology models highlight several factors that may contribute to a shift from cortisol hypersecretion in traumatized children to hyposecretion in traumatized adults, including developmental alterations and compensatory adaptations of the HPA axis (De Bellis et al., 2011). Developmental differences were found in a study of maltreated children with PTSD: duration of abuse was positively associated with pituitary volume in pre-pubertal youth and negatively associated with pituitary volume in pubertal and post-pubertal youth (Thomas & De Bellis, 2004). In addition, earlier onset of sexual abuse has been linked to smaller intracranial volume which, in turn, is associated with higher PTSD symptom severity (De Bellis et al., 1999). Vulnerability to traumatic stress throughout the first three decades of life may be affected by continuing brain development evident in synaptic elimination (Rabinowicz, 1986), myelination of the corpus callosum (Paus et al., 2001), and development of the prefrontal cortex (Fuster, 1989); moreover, elevations in catecholamine and cortisol levels may account, in part, for adverse brain development (De Bellis et al., 2011). Taken together, these findings suggest that the relation of trauma-exposure to PTSD symptoms may depend on ongoing brain developmental processes.
Developmental changes impact stress response systems and result in gradual increases in daily cortisol secretion during middle childhood and more rapid increases across adolescence (Walker, Walder, & Reynolds, 2001). Seeman and Robbins (1994, p. 233) observed that “age-related changes appear primarily in the re-setting of the HPA axis following a challenge.” A recent meta-analysis indicated that effect sizes for daily cortisol output were positively associated with age in individuals with comorbid PTSD and major depressive disorder (Morris et al., 2012). Psychosocial challenge studies have generally found elevated SNS reactivity in both trauma-exposed children (Kirsch et al., 2011; Saltzman, Holden, & Holahan, 2005) and adults (Liberzon, Anelson, Flagel, Raz, & Young, 1999) compared to non-exposed controls; however, findings regarding HPA reactivity in children have been equivocal (Gunnar, Frenn, Wewerka, & Van Ryzin, 2009; Ivanov et al., 2011). Age-related differences in the relation between peritraumatic cortisol levels and subsequent PTSD symptoms have been summarized in qualitative reviews; specifically, risk for PTSD has generally been linked to higher cortisol secretion in youth and lower cortisol secretion in adults (Delahanty & Nugent, 2006; Hruska et al., 2014; Morris & Rao, 2013). Unfortunately, it is impossible to parse the relative influence of developmental factors versus prior trauma exposure on neurobiological features of PTSD in child and adult survivors. Nevertheless, we sought to determine whether mean age of study participants impacted the strength or direction of effect sizes. We hypothesized that effect sizes for relations between cortisol levels in the immediate aftermath of trauma and PTSD symptoms would be more positive for studies including youth and more negative for studies of older adults. Based on evidence of elevated SNS reactivity in both trauma-exposed children and adults, we did not anticipate that age would moderate relations between heart rate or blood pressure and subsequent PTSD symptoms.
1.4.2. Sex
Women are twice as likely to develop PTSD during the lifetime (Kessler, Sonnega, Bromet, Hughes, & Nelson, 1995) and in response to trauma exposure (Giaconia, Reinherz, Silverman, Pakiz, Frost, & Cohen, 1995). Higher PTSD risk in women is likely explained by both psychosocial factors (e.g., greater exposure to interpersonal violence) and sex differences in neuroendocrine stress responses involving HPA dysregulation and/or SNS hyperactivity (Olff, Langeland, Draijer, & Gersons, 2007). A recent meta-analysis demonstrated higher evening cortisol levels in trauma-exposed women without PTSD than in their male counterparts and higher overall daily cortisol output in women compared to men with PTSD (Morris et al., 2012); however, it is unclear whether sex differences in HPA activity are observed shortly after trauma exposure. Elevated epinephrine following a motor vehicle accident was associated with higher PTSD symptoms at one- and six-month follow-up assessments in men but not women (Hawk, Dougall, Ursano, & Baum, 2000). In an exploratory fashion, we examined sex composition as a moderator of effect sizes.
1.4.3. Timing of PTSD assessment
Resilience in the face of trauma is the norm; however, posttraumatic symptoms are commonly reported by individuals during the first several months of recovery. In one study of rape survivors, threshold criteria for PTSD were already present in 94% of women after two weeks, but only 64% after five weeks, and 47% after 11 weeks (Foa & Riggs, 1995). If aberrant stress responses following trauma distinguish vulnerable from resilient individuals, clearer associations between risk markers and PTSD symptoms (i.e., a stronger signal-to-noise ratio) may emerge over time as resilient individuals recover. We examined whether the timing of PTSD assessments influenced the strength or direction of relations between acute stress responses to trauma and subsequent PTSD symptoms. We hypothesized that effect sizes would be larger (i.e., more negative for cortisol, more positive for heart rate and blood pressure) for PTSD assessments that occurred after longer lags from the focal traumatic event.
1.4.4. Timing and method of risk marker assessment
A recent meta-analysis did not find a preventative effect for propranolol in the development of PTSD symptoms (Sijbrandij et al., 2015), even though some studies administered it within the consolidation window (Hoge et al., 2012). The timing of administration may nevertheless still be important and other negative studies that missed the window for targeting SNS-mediated memory consolidation processes (Hruska et al., 2014) may have compromised the meta-analytic results. Timing may be important in the contribution of SNS activity and its disruption to PTSD-risk and preventive interventions. Acute heart rate in the aftermath of trauma has been assessed in a variety of contexts, with variable timing relative to the traumatic exposure. These include at the scene of an accident, in the ambulance, during emergency room (ER) admission, resting heart rate during the course of the ER visit, and at discharge. These measurement contexts all include trauma reminders and are likely perceived as stressful by most individuals; this ‘noise’ may interfere with signal detection (Shalev & Segman, 2008). However, given that the ‘golden hours’ for pharmacotherapeutic intervention appear to last up to six hours following trauma exposure (Vermetten, Zhohar, & Krugers, 2014), temporal dynamics may be important. Timing of heart rate assessment may influence the size and direction of effect sizes (Kirsch, Wilhelm, & Goldbeck, 2011). We hypothesized that studies including earlier heart rate assessments would show stronger and more positive correlations between heart rate and subsequent PTSD symptoms compared to those that measured heart rate at later times.
Longitudinal studies examining HPA risk markers for PTSD symptoms have measured cortisol levels in saliva, plasma/serum and/or urine. Cortisol secretion follows a diurnal rhythm and evidence from a prior meta-analysis suggests that time of measurement may impact effect sizes for cortisol levels obtained either in the morning or afternoon/evening (Morris et al., 2012). Timing of cortisol assessment is important but we could not examine this issue because all studies collected plasma/serum, saliva and/or urine samples during the ER visit, without describing the time of assessment. In an exploratory fashion, we investigated whether the type of cortisol measurement (saliva, plasma/serum, urine) was associated with effect sizes.
2. Method
2.1. Eligibility criteria and search strategy
Articles for this meta-analysis were identified through searches of Web of Knowledge, PsycINFO, PILOTS, and PubMed databases and included all studies published through April 2015. Unpublished literature (i.e., dissertation and masters theses, conference presentations) were excluded to ensure consistency with previous meta-analyses in this area (Brewin et al., 2000; Ozer et al., 2003; Pole, 2007). Initial searches crossed-referenced the keywords posttraumatic stress disorder or PTSD with keywords reflecting acute risk factors (acute, early, emergency room, ER, peritraumatic, pre-traumatic, resilience, resistance, risk factor, risk marker, vulnerability), indicators of HPA and SNS function (adrenocortical, blood pressure, cortisol, DBP, diastolic, glucocorticoid, heart rate, HR, HPA, SBP, systolic) and longitudinal designs (follow-up, longitudinal, pre-post, predicted, prediction, predictor, prospective, repeated measures). A flow diagram of this literature search is presented in Figure 1. In addition, we searched reference sections of qualifying articles as well as reviews on risk markers for PTSD for any studies not detected in our initial search (Delahanty & Nugent, 2006; Gillespie, Phifer, Bradley, & Ressler, 2009; Michopoulos, Norrholm, & Jovanovic, 2015; Morris & Rao, 2013; Ozer et al., 2003; Schmidt, Kaltwasser, & Wotjak, 2013; Schmidt et al., 2015; Walsh et al., 2013).
Figure 1.
Flowchart of study selection.
To be included in this meta-analysis, a study had to (a) enroll participants exposed to a traumatic life event, (b) include a standardized measure of PTSD symptoms or diagnosis, (c) assess risk markers (i.e., cortisol, heart rate, blood pressure) within 72 hours of the index trauma, (d) include a longitudinal design with at least one follow-up assessment of PTSD symptoms or diagnosis, (e) include sufficient data to compute effect sizes (or authors were contacted and provided requested data), and (f) published in an English-language scientific journal. Pharmacological prevention studies were excluded. When the results of a single study were reported in multiple articles, only effect sizes from the publication with the largest sample size were included. All studies meeting inclusion criteria were coded for participant and methodological features. Independent coding of a randomly selected 30% of the studies was done by a second coder. Reliabilities were calculated using intraclass correlations (r1) for continuous variables and kappa for categorical variables (Orwin, 1994). Disparities were resolved by consensus.
The following information was extracted from each study: (a) mean number of months from the focal traumatic event to the PTSD assessment (r1 = 1.00), (b) timing of heart rate or blood pressure assessment (i.e., at the scene or in the ambulance, at ER admission, resting heart rate during ER visit or on the day of discharge) (κ= 1.00), (c) method of cortisol assessment (saliva, plasma or serum, urine) (κ= 0.82), (d) percentage of females in the sample (r1 = 0.97), and (e) mean age of participants (r1 = 0.98). When studies reported multiple heart rate or blood pressure assessments but did not report separate correlations with PTSD symptoms (e.g., Bryant et al., 2008), the earliest assessment was coded for analyses.
2.2. Data Analytic Plan
Statistical analyses were conducted based on the recommendations of Lipsey and Wilson (2001) using SPSS Statistics 22 (SPSS Inc. Headquarters, Chicago, Illinois, USA). Forest plots and figures depicting meta-regression analyses were created using Comprehensive Meta-Analysis, Version 2 (Borenstein, Hedges, Higgins, & Rothstein, 2005). The Pearson product-moment correlation (r) was used to determine the strength and direction of the relation between acute posttraumatic risk markers and PTSD symptoms or disorder. For studies reporting data for pairings of continuous and dichotomous variables, including presence or absence of PTSD diagnosis at follow-up (Bryant et al., 2008; Coronas et al., 2011; De Young et al., 2007; Shalev & Freedman, 2005; Videlock et al., 2008; Yehuda et al., 1998), PTSD symptom cutoff scores (Alarcon et al., 2011; Gould et al., 2011), or cut-off scores for risk markers (Bryant et al., 2008; Zatzick et al., 2005), formulas were used to impute point-biserial correlations (Lipsey & Wilson, 2001). All r values were transformed using the Fisher’s Zr transform (Hedges & Olkin, 1985) and associated standard errors and inverse variance weights were computed for each effect size. Cohen’s (1988) guidelines were used to interpret small (r = .10), medium (r = .30), and large (r = .50) effect sizes.
To handle statistically-dependent effect sizes stemming from either repeated measures within longitudinal studies or multiple methods of cortisol assessment within studies (i.e., effect sizes nested within studies), an SPSS macro for robust variance estimation procedures in meta-regression was used (Hedges, Tipton, & Johnson, 2010). Two-level mixed effects models were specified to allow simultaneous estimation of within-study (level 1) and between-study (level 2) parameters. The intra-class correlation used to calculate variance components in the random effects model (ρ) was set at 0.7 after a sensitivity analysis indicated that re-estimating meta-regression models using a range of values for ρ between 0 and 1 had a negligible impact on findings. For dependent effects, meta-regression models including multiple effect sizes for each study, group mean centering was used to facilitate interpretation of coefficients by separating within- and between-study effects. Meta-regression models were used to test effect size moderators.
3. Results
3.1. Descriptive findings
A total of 26 studies with 5,186 participants were deemed eligible for inclusion (Table 1). Of these studies, 8 examined cortisol, 20 examined heart rate, 7 examined diastolic blood pressure, and 8 examined systolic blood pressure; 10 studies assessed more than one biomarker. Mean age of participants was 33.9 (SD = 11.8) and 46.3% were female. Studies included a wide range of timing of the PTSD assessments, ranging from within one month (n = 11), to 1 to 6 months (n = 20), 6 to 12 months (n = 4), and 12 to 24 months (n = 3) after the focal traumatic event. Study characteristics including sample size, trauma type, percentage female, mean age, PTSD measure, timing of PTSD assessment, acute posttraumatic HPA/SNS risk markers assessed, and timing of SNS assessment are presented in an online data supplement.
Table 1.
Characteristics of studies included in meta-analysis.
| PTSD Assessment
|
HPA/SNS Assessment
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Study | N | Trauma type | Female (%) | Mean Age | Measure | Timing post trauma* | Type of HPA/SNS measurement | Timing of SNS assessment |
| Alarcon et al. (2011) | 1386 | Injury, assault | 35 | 43 | PCL-C | Mean follow-up 24 days | HR, SBP | ER admission |
| Blanchard et al. (2002) | 68 | MVA | 82 | 39 | CAPS | 1 and 13 months | HR | ER resting |
| Bryant et al. (2003) | 87 | MVA | 41 | 31 | CIDI-PTSD | ~7 days, 6 and 24 months | HR, DBP, SBP | ER discharge |
| Bryant et al. (2008) | 1105 | Injury, assault | 26 | 38 | CAPS | 3 months | HR, DBP, SBP | At the scene or ER admission |
| Buckley et al. (2004)a | 65 | MVA | 38 | 36 | IES | 1 month | HR, DBP, SBP | Ambulance, ER admission, ER resting, ER discharge |
| Coronas et al. (2011) | 119 | MVA | 51 | 38 | SCID-PTSD | 1 month | HR, DBP, SBP | Ambulance, ER admission |
| Cremeans-Smith et al. (2012) | 110 | TKR surgery | 68 | 69 | IES | 3 months | HR, SBP, DBP | ER resting (<1 hour post-surgery) |
| Delahanty et al. (2005)c | 58 | Injury | 32 | 13 | CAPS-CA | 6 weeks | Urinary cortisol | N/A |
| Delahanty et al. (2000)a | 48 | MVA | 38 | 36 | IES | 1 month | Urinary cortisol | N/A |
| De Young et al. (2007) | 101 | Injury | 35 | 11 | SCID-PTSD | 6 months | HR | ER admission, ER resting |
| Ehring et al. (2008) | 53 | MVA | 26 | 34 | PDS | 2 weeks, 6 months | Salivary cortisol, HR, DBP, SBP | ER admission, ER resting |
| Gould et al. (2011) | 145 | Injury (burn) | 28 | 40 | DTS | 1, 6, 12, 24 months | HR | Ambulance, ER admission |
| Kassam-Adams et al. (2005) | 190 | MVA | 25 | 11 | CAPS-CA | Mean of 6.4 months | HR | ER admission |
| Kraemer et al. (2008) | 255 | Injury (accident) | 33 | 41 | CAPS | 6 months | HR | ER admission, ER discharge |
| Kuhn et al. (2006) | 50 | MVA | 60 | 39 | CAPS | 0.5, 1, 3, and 6 months | HR | ER admission |
| Mouthann et al. (2014) | 291 | Injury, assault | 36 | 43 | CAPS | 6 weeks, 6 months | Plasma cortisol | N/A |
| Nishi et al. (2007) | 106 | MVA | 32 | 38 | CAPS | 6 months | HR | ER admission |
| Nugent et al. (2006)c | 66 | Injury | 32 | 13 | CAPS-CA | 6 weeks, 6 months | HR | Ambulance, ER admission, ER resting |
| O’Donnell et al. (2007) | 176 | Injury | 22 | 36 | CAPS | 12 months | HR | At the scene |
| Price et al. (2014) | 55 | Injury, assault | 65 | 31 | PSS-I | 1 and 3 months | HR, salivary cortisol | ER admission |
| Shalev & Freedman, (2005)d | 224 | MVA | 49 | 30 | CAPS | 4 months | HR | ER admission |
| Shalev & Freedman, (2005)d | 23 | Terrorist attacks | 51 | 30 | CAPS | 4 months | HR | ER admission |
| Shalev et al. (2008)b | 137 | MVA, terrorist attacks, other | 41 | 31 | CAPS | 5 months | Salivary, plasma, and urinary cortisol | N/A |
| Videlock et al. (2008)b | 119 | MVA, terrorist attacks, other | 41 | 31 | CAPS | 5 months | HR, DBP, SBP | ER resting |
| Walsh et al. (2013) | 193 | Sexual assault | 100 | 26 | PSS-PR | 6 weeks, 3 and 6 months | Serum cortisol | N/A |
| Yehuda et al. (1998) | 20 | Sexual assault | 100 | 30 | Foa PTSD scale | 27–157 days post rape | Plasma cortisol | N/A |
| Zatzick et al. (2005) | 161 | Injury | 34 | 36 | PCL | Post-surgery, 1, 5, and 12 months | HR | ER admission |
Note: letters indicate which studies were based on similar samples;
only time points for which we could compute effect sizes are included; PTSD = posttraumatic stress disorder; MVA= motor vehicle accident; CAPS= Clinician-Administered PTSD Scale; CAPS-CA= Clinician-Administered PTSD Scale-Children and Adolescents; CIDI-PTSD= Composite International Diagnostic Interview-PTSD; SCID-PTSD= Structured Clinical Interview for DSM-IV-PTSD scale; IES= Impact of Event Scale; ASD= acute distress disorder; PDS= Posttraumatic Diagnostic Scale; DTS= Davidson Trauma Scale; PCL-C= PTSD Checklist-Civilian; FPA= Foa PTSD Scale; PSS-PR= PTSD Symptom Scale-Self Report; PSS-I = Posttraumatic Symptom Scale-Interview Version; K-SADS= Kiddie-Schedule for Affective Disorders and Schizophrenia; HPA= hypothalamic-pituitary-adrenal axis; SNS= sympathetic nervous system; HR= heart rate; DBP= diastolic blood pressure; SBP= systolic blood pressure; ER= emergency room; TKR = total knee replacement.
3.2. Cortisol levels
The weighted average correlation from 8 studies (combined n = 837) assessing the relationship between cortisol and subsequent PTSD symptoms was not significantly different from zero (Table 2). Individual effect sizes ranged from −.50 to .32. Meta-regression models showed that the strength of the relation between cortisol and PTSD symptoms varied significantly according to the mean age of the sample (Figure 2); cortisol levels were positively associated with PTSD symptoms for youth and became increasingly negatively associated with PTSD symptoms for samples with a mean age of approximately 30 years or older. Exploratory analyses examined the weighted average correlation separately for samples with mean ages above and below 30 years. The weighted average correlation between cortisol and PTSD symptoms was .15 (p = .203) in the younger samples and −.19 (p = .053) in the older samples. The relationship between cortisol and PTSD symptoms did not differ by the method of cortisol measurement (i.e., saliva, plasma or serum, urine), nor the time elapsed between focal trauma and PTSD assessment, nor the percentage of females in the sample. Because all studies collected samples in the ER, at varying durations following the traumatic event, the impact of timing of measurement could not be assessed for cortisol.
Table 2.
Summary of Robust Variance Estimation Meta-Regression Findings across Studies Assessing Correlations between Cortisol and PTSD Symptoms.
| r | k | ES | SE | 95% CI | p | ρ | |
|---|---|---|---|---|---|---|---|
| Overall Correlation | −.067 | 8 | 15 | .084 | −.266, +.132 | .449 | 0.7 |
| Moderators | |||||||
| Method | .064 | 8 | 15 | .149 | −.299, +.428 | .680 | 0.7 |
| Months since trauma | .000 | 8 | 13 | .002 | −.006, +.006 | .872 | 0.7 |
| Percent female | .003 | 8 | 13 | .002 | −.002, +.009 | .208 | 0.7 |
| Mean age | −.019 | 8 | 13 | .007 | −.037, −.001 | .044 | 0.7 |
Notes. PTSD = Posttraumatic Stress Disorder; method (1 = saliva; 2 = plasma/serum; 3 = urine); r = effect size; k = number of studies; ES = number of effect sizes; SE = standard error; CI = confidence interval; ρ = intraclass correlation.
Figure 2.
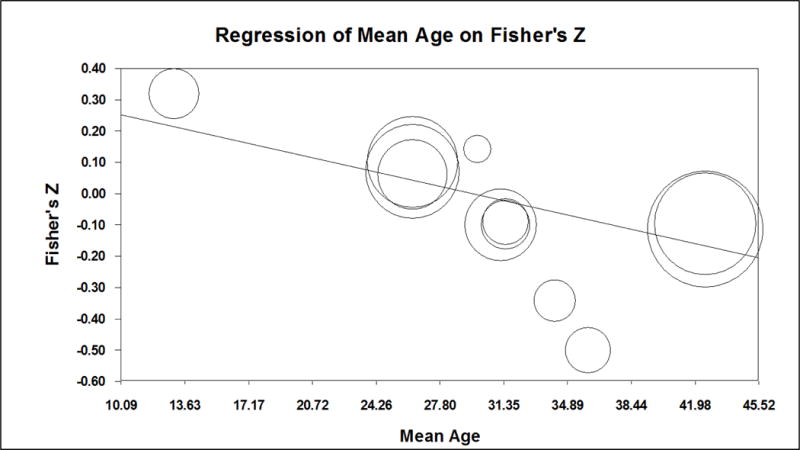
Meta-regression of effect sizes for the association between cortisol and PTSD symptoms on mean age of participants.
3.3. Heart rate
The weighted average correlation from 20 studies (combined n = 4,656) assessing the relationship between heart rate and subsequent PTSD symptoms was .13, a statistically significant but small effect size (Table 3, Figure 3). This positive correlation indicates that higher heart rates were associated with higher PTSD symptoms, on average, at follow-up. Individual effect sizes ranged from −.22 to .70. Meta-regression models showed that the strength of the relation between heart rate and PTSD symptoms varied significantly according to the mean age of the sample (Figure 4); heart rate was positively associated with PTSD symptoms for youth (individual effect sizes ranged from .17 to .45) and became increasingly negatively associated with PTSD symptoms for older samples (= −.004). There was a non-significant trend for the relation between heart rate and subsequent PTSD symptoms to be stronger when more time had elapsed between trauma exposure and symptom assessment (r = .004, p = .090). The relationship between heart rate and PTSD symptoms did not differ by the timing of heart rate measurement (i.e., scene of accident or in ambulance, ER admission, mean HR during ER visit or ER discharge), nor the percentage of females in the sample.
Table 3.
Summary of Robust Variance Estimation Meta-Regression Findings across Studies Assessing Correlations between Heart Rate and PTSD Symptoms.
| r | k | ES | SE | 95% CI | p | ρ | |
|---|---|---|---|---|---|---|---|
| Overall Correlation | .125 | 20 | 52 | .039 | +.043, +.207 | .005 | 0.7 |
| Moderators | |||||||
| Timing | −.021 | 20 | 52 | .036 | −.096, +.054 | .565 | 0.7 |
| Months since | .004 | 20 | 37 | .002 | −.001, +.008 | .090 | 0.7 |
| Percent female | −.001 | 20 | 37 | .003 | −.007, +.005 | .700 | 0.7 |
| Mean age | −.004 | 20 | 37 | .001 | −.007, −.001 | .009 | 0.7 |
Notes. PTSD = Posttraumatic Stress Disorder; timing (1 = scene of accident or in ambulance; 2 = ER admission; 3 = mean HR during visit or ER discharge); r = effect size; k = number of studies; ES = number of effect sizes; SE = standard error; CI = confidence interval; ρ = intraclass correlation.
Figure 3.
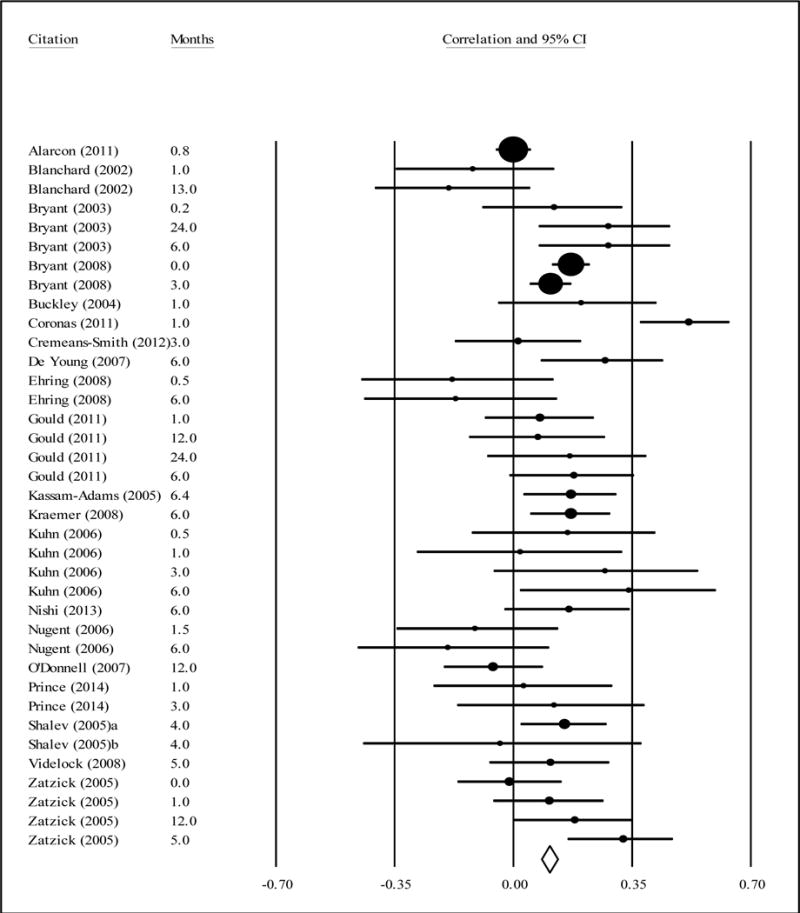
Correlations between heart rate (earliest measurement timing) and subsequent PTSD symptoms (with 95% CI).
Figure 4.
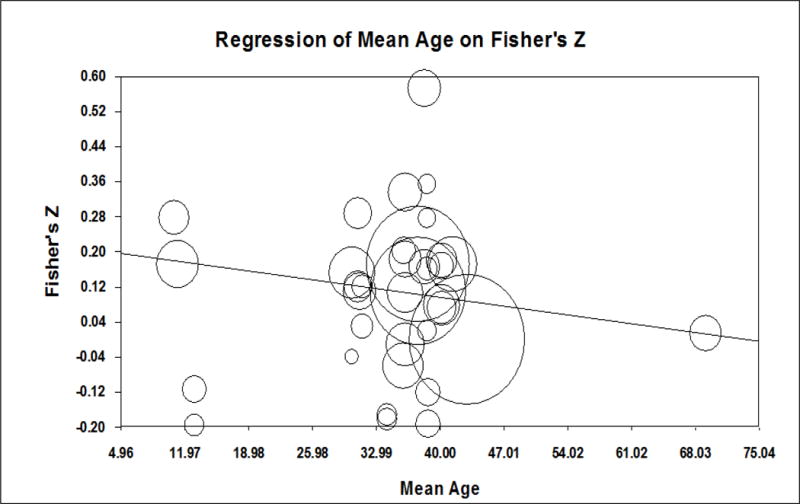
Meta-regression of effect sizes for the association between heart rate and PTSD symptoms on mean age of participants.
3.4. Blood pressure
The weighted average correlations from 7 studies (combined n = 1,530) assessing the relationship between diastolic blood pressure and subsequent PTSD symptoms (Table 4) and 8 studies (combined n = 2,916) assessing the relationship between systolic blood pressure and PTSD symptoms (Table 5) were not significantly different from zero. Individual effect sizes ranged from −.45 to .39 for diastolic blood pressure and from −.26 to .31 for systolic blood pressure. There was a non-significant trend for weaker relations between systolic blood pressure and subsequent PTSD symptoms in older samples (r = −.003, p = .081). Meta-regression models showed that the relationships between diastolic or systolic blood pressure and PTSD symptoms did not differ by the timing of blood pressure measurement (i.e., scene of accident or in ambulance, ER admission, mean HR during ER visit or ER discharge), nor the time elapsed since the focal trauma, nor the percentage of females in the sample.
Table 4.
Summary of Robust Variance Estimation Meta-Regression Findings across Studies Assessing Correlations between Diastolic Blood Pressure and PTSD Symptoms.
| r | k | ES | SE | 95% CI | p | ρ | |
|---|---|---|---|---|---|---|---|
| Overall Correlation | −.006 | 7 | 14 | .053 | −.135, +.122 | .907 | 0.7 |
| Moderators | |||||||
| Timing | −.070 | 7 | 14 | .070 | −.249, +.109 | .360 | 0.7 |
| Months since trauma | −.003 | 7 | 10 | .003 | −.012, +.006 | .366 | 0.7 |
| Percent female | .005 | 7 | 10 | .008 | −.015, +.024 | .547 | 0.7 |
| Mean age | −.002 | 7 | 10 | .003 | −.009, +.004 | .383 | 0.7 |
Notes. PTSD = Posttraumatic Stress Disorder; timing (1 = scene of accident or in ambulance; 2 = ER admission; 3 = during ER assessment or mean HR during visit; 4 = ER discharge); r = effect size; k = number of studies; ES = number of effect sizes; SE = standard error; CI = confidence interval; ρ = intraclass correlation.
Table 5.
Summary of Robust Variance Estimation Meta-Regression Findings across Studies Assessing Correlations between Systolic Blood Pressure and PTSD Symptoms.
| r | k | ES | SE | 95% CI | p | ρ | |
|---|---|---|---|---|---|---|---|
| Overall Correlation | .018 | 8 | 15 | .036 | −.066, +.102 | .630 | 0.7 |
| Moderators | |||||||
| Timing | −.030 | 8 | 15 | .034 | −.113, +.053 | .411 | 0.7 |
| Months since trauma | .018 | 8 | 11 | .012 | −.013, +.050 | .197 | 0.7 |
| Percent female | .003 | 8 | 11 | .005 | −.009, +.014 | .586 | 0.7 |
| Mean age | −.003 | 8 | 11 | .001 | −.007, +.001 | .081 | 0.7 |
Notes. PTSD = Posttraumatic Stress Disorder; timing (1 = scene of accident or in ambulance; 2 = ER admission; 3 = during ER assessment or mean HR during visit; 4 = ER discharge); r = effect size; k = number of studies; ES = number of effect sizes; SE = standard error; CI = confidence interval; ρ = intraclass correlation.
4. Discussion
Researchers have argued that biological markers of PTSD “lack predictive power” and “drown” in the “sea of other biological, environmental, and behavioral occurrences that underlay the occurrence of the disorder” (Shalev & Segman, 2008, p. 188). The present meta-analysis takes an important step toward embracing the importance of context and “embedding the noise” (Shalev & Segman, 2008, p. 194) by capitalizing on heterogeneity in effect sizes across studies to examine key demographic and methodological moderators of the relations between biological markers and PTSD symptoms. Despite the low conditional risk for PTSD observed in longitudinal studies anchored by a focal traumatic event (e.g., Breslau, Kessler, Chilcoat, Schultz, Davis, & Andreski, 1998) and considerable between-study variability in trauma type and measurement approaches, the present findings highlight several small but non-trivial effects of biological risk markers and moderators.
4.1 Age
One of the most robust findings of this meta-analysis was that the mean age of study participants moderated relations between risk markers and subsequent PTSD symptoms in a fairly consistent manner. Prior reviews have noted that increased risk for developing PTSD is linked to higher circulating cortisol levels in children and lower circulating cortisol levels in adults (Delahanty & Nugent, 2006; Hruska et al., 2014; Morris & Rao, 2013; Pervanidou, 2008). The present findings extend these observations by identifying a developmental shift: higher cortisol levels were positively associated with PTSD symptoms in samples with a mean age less than 30 years and negatively associated with PTSD symptoms in samples with a mean age greater than 30 years. This pattern is remarkable given the importance of brain maturation in developmental traumatology models (De Bellis et al., 1999) and evidence that synaptic elimination (Rabinowicz, 1986), corpus callosum myelination (Paus et al., 2001), and prefrontal cortex development (Fuster, 1989) continue throughout the first three decades of life. Trauma exposure may be particularly devastating during adolescence, when its impact on the developing frontal cortex can produce long-lasting changes in HPA responses (Lupien, McEwen, Gunnar, & Heim, 2009).
Another perspective is that it is not age per se, but rather trauma history, that determines whether PTSD vulnerability is linked to enhanced or diminished cortisol secretion: recently-traumatized adults are more likely to have prior trauma-exposure and to have progressed further from HPA hyper- to hypo-activity than recently-traumatized children (Miller et al., 2007; Pervanidou, 2008). Supporting this hypothesis, a recent study found that hair cortisol concentrations were negatively correlated with number of prior traumatic life events in trauma-exposed individuals with or without PTSD (Steudte et al., 2013). However, abuse has been associated with higher morning cortisol levels in childhood and lower morning cortisol levels by early adulthood even after accounting for the time elapsed since trauma disclosure (Trickett et al., 2010). We propose an integrative framework for understanding PTSD risk that draws from developmental traumatology (De Bellis et al., 1999) and trauma history (Miller et al., 2007; Pervanidou, 2008) models. In younger individuals for whom the focal traumatic event is more likely to be their first exposure, HPA hyperactivity signals greater potential for disrupted brain maturation and, consequently, increased risk for PTSD onset. In older individuals outside this critical brain maturation window, HPA hypoactivity signals greater cumulative lifetime trauma exposure, adaptation of the HPA axis, and risk for PTSD onset.
To our knowledge, this study is the first to show a similar developmental progression for heart rate and systolic blood pressure (non-significant trend) during the acute posttraumatic phase: both heart rate and systolic blood pressure were positively associated with PTSD symptoms for youth and became negatively associated with PTSD symptoms for older adults. This shift is likely influenced by changes in brain maturation (De Bellis et al., 2011) and cumulative trauma exposure (Miller et al., 2007; Pervanidou, 2008). Elevated noradrenergic activity is associated with increased risk for developing PTSD in youth recently exposed to traumatic stress (Pervanidou et al., 2007). One caveat in interpreting the findings for heart rate is meta-analytic evidence suggesting that older adults exhibit lower heart rate reactivity to laboratory stressors than younger adults (Uchino, Birmingham, & Berg, 2010).
4.2 Cortisol levels
Contrary to our hypothesis, cortisol levels in the acute post-traumatic phase were not associated with higher subsequent PTSD symptoms for all studies. Instead, as described above, age moderated the relationship between cortisol and risk for PTSD symptoms. These findings suggest that secondary prevention interventions for PTSD may require different criteria for defining high-risk groups based on HPA activity in youth and adults.
A second important factor is the timing of cortisol assessment. Consolidation models posit that lower circulating cortisol levels enhance SNS-driven consolidation of the traumatic memory, suggesting that measurement of cortisol, or administration of exogenous glucocorticoids, must be done within 6 hours of the event to have a bearing on symptomatic outcomes (Joëls et al., 2009; Ursin & Olff, 1993; Yehuda, 2002). If, however, an interaction between glucocorticoid levels and receptor sensitivity might shape outcomes (Yehuda et al., 2015) via impact on context-processing capacities (Kaouane et al., 2012), the window within which cortisol levels (endogenously measured or exogenously manipulated) could impact outcomes might be much wider.
It is important to note that HPA function is influenced by a multitude of factors that we were unable to examine, including timing and context of sample collection, menstrual cycle phase and medication use, which may have amplified the noise and diminished the signal. Variability in the time of day when cortisol was measured is particularly critical given the diurnal rhythm of the HPA axis. Also, because cortisol levels are highly variable, multiple samples are essential to minimize variability. The lack of an overall relationship between simple and poorly controlled measures of HPA activity and subsequent PTSD symptoms does not mean that HPA axis function before, during or in the aftermath of trauma is not relevant to symptomatic outcomes and to secondary prevention efforts. Additionally, we cannot rule out the possibility that null findings for cortisol were due to a smaller sample size compared to analyses examining heart rate for which the sample size was 5.5 times larger.
4.3 Heart rate
Consistent with our hypothesis, acute posttraumatic measurement of heart rate was significantly and positively associated with subsequent PTSD symptoms. The weighted average effect size was small (r = .13) but comparable to effect sizes reported in prior meta-analyses by Buckley and Kaloupek (2001; weighted effect size r = .18) and Pole (2007; weighted effect size r = .18), examining cross-sectional associations between heart rate and chronic PTSD. These findings suggest that ambulatory heart rate assessed at the scene of an accident or during an ER visit – whether indicative of resting activity or reactivity to trauma cues – could represent a useful early risk marker for PTSD and for refining secondary prevention efforts. However, elevated heart rate has been found to predict a broad range of trauma-related psychiatric disorders, including panic disorder, agoraphobia and social phobia, suggesting limited predictive specificity for predicting PTSD (Bryant, 2006; Bryant, Creamer, O’Donnell, Silove, & McFarlane, 2011). Though researchers have posited that elevated SNS activity in the immediate aftermath of trauma increases risk for PTSD by facilitating ‘over-consolidation’ of the traumatic memory (Pitman, 1989; Pitman & Delahanty, 2005; Pitman et al., 1993; Yehuda & Harvey, 1997; Yehuda et al., 1998), no studies directly examined this hypothesis. The processes by which higher heart rate in the acute posttraumatic phase influence subsequent PTSD symptoms remain unclear. Preliminary evidence suggests that prolonged exposure therapy delivered within hours of trauma exposure is associated with lower PTSD severity at 4 and 12 weeks compared to an assessment-only condition; importantly, this early intervention included stress management components (i.e., breathing retraining) that could have influenced peritraumatic heart rate (Rothbaum et al., 2012).
4.4 Blood pressure
Cross-sectional studies reveal high rates of hypertension and cardiovascular disease among individuals with PTSD (O’Toole & Catts, 2008; Schnurr et al., 2000) and prospective studies indicate that individuals with PTSD are at elevated risk for developing cardiovascular illnesses (Boscarino, 2008). We found no evidence that systolic or diastolic blood pressure in the acute posttraumatic phase is associated with subsequent PTSD symptoms. Meta-analyses examining associations between blood pressure and chronic PTSD (Buckley & Kaloupek, 2001; Pole, 2007) also reported small and non-significant weighted average effect sizes for systolic (r’s < .05) and diastolic (r’s < .10) blood pressure. Despite evidence of small correlations between blood pressure indices and heart rate assessed during hospitalization (e.g., Bryant et al., 2003), these findings suggest that – unlike heart rate – measures of systolic and diastolic blood pressure obtained shortly after a traumatic event are not useful risk markers for PTSD. One caveat is that differences in sample sizes (e.g., the sample size for analysis on heart rate was three times that of diastolic blood pressure) could have influenced these findings. A second caveat concerns measurement error: whereas heart rate measurement is continuous, non-invasive blood pressure measurements are frequently obtained at discrete intervals and have raised concerns regarding their accuracy (Skirton, Chamberlain, Lawson, Ryan, & Young, 2011).
The polyvagal theory proposed by Porges (2007) offers a more comprehensive model for understanding cortisol, heart rate and blood pressure changes associated with the vulnerability for PTSD. According to this theory, the myelinated component of the vagus nerve or the ventral vagal complex which is phylogenetically more evolved in mammals serves as a brake on the sinoatrial node (the primary cardiac pacemaker), resulting in a resting heart rate lower than the intrinsic rate of the pacemaker alone. Under threatening conditions, however, its inhibitory influence on the sinoatrial node diminishes. Simultaneously, less-evolved components of the autonomic nervous system become overactive to support survival functions. For example, sympathetic-adrenal system activation elicits increases in heart rate and blood pressure that support defensive/fight or escape/flight behaviors. Alternatively, the unmyelinated component of the vagus nerve or the dorsal vagal complex elicits decreases in heart rate and blood pressure that support immobility/freeze behaviors. Heart rate variability (HRV) provides an index of autonomic nervous system function that reflects the relative influence of sympathetic versus parasympathetic cardiac modulation. Lower HRV characterizes individuals with PTSD (e.g., Agorastos et al., 2012; Cohen, Kotler, Matar, Kaplan, Miodownik, & Cassuto, 1997; Mellman, Knorr, Pigeon, Leiter, & Akay, 2004) and predicts the onset of PTSD among motor vehicle accident survivors (al Arab et al., 2012). Importantly, the ventral vagal complex also exerts an inhibitory influence on the HPA axis (Porges, 2007). Hence, the polyvagal theory provides an integrative framework for linking acute heart rate, blood pressure and HPA features to unique and potentially adaptive responses to traumatic life events (i.e., fight, flight, freeze).
4.5 Sex
Theoretical models have proposed that sex differences in neuroendocrine stress responses may account, in part, for elevated PTSD risk in women (Olff et al., 2007). However, we did not find that sex had a moderating effect on the relations between cortisol, heart rate or blood pressure responses to trauma and subsequent PTSD. Two factors could have contributed to this null finding. First, these analyses focused on the percentage of females in the sample and could not examine within-individual relations between risk markers and PTSD symptoms. Second, many of the longitudinal studies included in this meta-analysis focused on traumatic events that did not involve interpersonal violence (e.g., a majority of the studies included motor vehicle accidents). Previous studies suggest that sex differences in risk for PTSD are primarily due to the higher incidence of interpersonal violence in women (Breslau, 2009), which is associated with higher conditional risk for PTSD in this population (Kessler et al., 1995).
4.6 Timing of PTSD assessment
Empirically-supported treatment approaches based on information and emotional processing theories (Foa, Steketee, & Rothbaum, 1989; Lang, 1977) or social-cognitive models (Resick, Monson, & Chard, 2007) conceptualize PTSD as a disorder of ‘non-recovery’ in which the majority of individuals exposed to a traumatic stressor will experience transient symptoms that resolve naturally within several months and that PTSD is the result of non-recovery. Our hypothesis that clearer relations between acute neurobiological responses to trauma and subsequent PTSD symptoms would emerge over time as resilient individuals recover was not supported for cortisol, heart rate or blood pressure, though there was a non-significant trend in the expected direction for stronger relations between baseline heart rate and PTSD symptoms assessed at longer follow-up intervals.
4.7 Timing and method of risk marker assessment
Contrary to our hypothesis, heart rate measures obtained closer to the time of the traumatic event did not correlate more strongly with subsequent PTSD symptoms. This is inconsistent with the emphasis of consolidation models of PTSD on the first 6 hours of recovery as potentially critical for disrupting SNS-mediated trauma memory consolidation processes (Hruska et al., 2014; Vermetten et al., 2014). Unfortunately, studies in the present meta-analysis did not routinely assess potentially critical moderating factors – including peritraumatic dissociation and the presence of social support (Ozer et al., 2003).
4.8 Limitations and future directions
Limitations of the current meta-analysis provide directions for future research. First, we cannot rule out the possibility that trauma history, and not age per se, influenced relations between risk markers and subsequent PTSD symptoms (Delahanty & Nugent, 2006; Hruska et al., 2014). We were unable to disentangle mean age and prior trauma exposure due to differences in the manner in which trauma history was assessed. Studies variously reported mean childhood trauma exposure score (e.g., Price, Kearns, Houry, & Rothbaum, 2014), mean number of prior traumatic events (e.g., Delahanty, Raimonde, Spoonster, & Cullado, 2003), and percentage of participants reporting prior trauma exposure (e.g., Shalev, Videlock, Peleg, Segman, Pitman, & Yehuda, 2008). To address this critical question, we recommend that future studies report the mean number of prior traumatic life events, percentage of participants with prior trauma history, age of prior trauma exposure, and a measure of childhood traumatic events that distinguishes trauma subtypes.
Second, most studies did not report when risk marker measurements were obtained during the ER visit. In order to more rigorously assess neurobiological models of PTSD and to integrate findings with the secondary prevention literature (e.g., Sijbrandij et al., 2015), we recommend that future studies report the mean duration from trauma to risk marker assessment, and whenever possible to assess SNS risk markers within 6 hours of trauma-exposure. HPA measures may need to be assessed serially, to track response and recovery, to see more meaningful relationships. Third, the non-significant trend for stronger associations between heart rate and PTSD symptoms assessed at longer follow-up intervals suggest that future studies should consider including PTSD assessments outside of the 3-month ‘normal recovery’ window and should include a narrower range of time points for PTSD assessment than the studies reported to date.
Fourth, the weighted effect sizes reported are considered small (Cohen, 1988), though they are comparable to those reported in other meta-analyses of psychophysiological correlates (Buckley & Kaloupek, 2001; Pole, 2007) and psychosocial risk markers of PTSD (Ozer et al., 2003). Given our narrow focus on early biomarkers of PTSD risk and the likely importance of additional factors (i.e., features of the traumatic event, individual characteristics, peri- and post-traumatic moderators), the small magnitude of these effects is not surprising. A significant challenge for examining neurobiological mechanisms of risk for PTSD is that not all at-risk individuals exhibit dysregulated HPA or SNS function as assessed by currently used biomarkers (e.g., Hopper, Spinazzola, Simpson, & van der Kolk, 2006). It remains unclear whether the field has yet identified ideal ways to mark salient function within these systems.
Fifth, we did not examine prospective studies assessing individuals before and after trauma-exposure (pre-post designs). Instead, we focused on longitudinal studies following individuals after trauma-exposure (post-post designs; Schmidt et al., 2015). Therefore, we cannot determine whether early risk markers such as elevated heart rate were present prior to the traumatic event and if they represent relatively stable stress response features that could be used to inform primary prevention programs (Bomyea et al., 2012; DiGangi et al., 2013). Sixth, the present findings were based on studies that included participants exposed to a wide variety of traumatic life events. Although these events share many common features, they may be associated with unique appraisals, physiological responses and conditional risk for developing PTSD. Seventh, we acknowledge that the number of studies included in analyses of cortisol and blood pressure indices were relatively small. Nevertheless, we believe this reflects the challenge of conducting research on recently-traumatized individuals and that the potential benefits of identifying early risk markers for PTSD justify this meta-analysis.
4.9 Summary and conclusions
Although no biomarkers have yet demonstrated clinical applicability for PTSD, researchers have suggested that “an aggregate or pattern of multiple factors will ultimately constitute a biomarker for PTSD” (Yehuda, Neylan, Flory, & McFarlane, 2013, p. 1913). This meta-analysis represents a step toward identifying this pattern. A significant association was found between elevated heart rate in the early aftermath of trauma and subsequent PTSD symptoms. In addition, age emerged as a robust moderator of relations between risk markers and subsequent PTSD symptoms. Clarifying the necessary conditions and mechanisms by which early HPA and SNS activity influence risk or resilience for PTSD is critical given contrasting findings for the role of each system reported by longitudinal ‘post-post’ studies and by pharmacological secondary prevention and psychotherapeutic augmentation studies (Sijbrandij et al., 2015; Yehuda et al., 2015).
Highlights.
Chronic PTSD is linked to alterations in major stress response systems.
Neurobiological mechanisms of risk for developing PTSD are not well understood.
A meta-analysis of 26 studies (N = 5,186 individuals) was conducted.
Higher heart rate predicted higher PTSD symptoms with a small effect size.
Age of participants moderated associations between predictors and PTSD symptoms.
Acknowledgments
This research was funded in part by grants from the National Institute of Health (G12 RR003032/MD007586, K01 MH101403), and by the Betsey R. Bush Endowed Professorship in Behavioral Health at the University of Tennessee (Uma Rao). These funding agencies had no further role in the study design, data collection, analysis or interpretation of data, writing of the report, or the decision to submit the manuscript for publication. We gratefully acknowledge the authors who generously provided their data for this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no conflicts of interest.
References
References marked with an asterisk indicate studies included in the meta-analysis.
- Agorastos A, Boel JA, Heppner PS, Hager T, Moeller-Bertam T, Haji U, Motazedi A, Yanagi MA, Baker DG, Stiedl O. Dimished vagal activity and blunted diurnal variation of heart rate dynamics in posttraumatic stress disorder. Stress. 2012;3:300–310. doi: 10.3109/10253890.2012.751369. [DOI] [PubMed] [Google Scholar]
- al Arab AS, Guedon-Moreau L, Ducrocq F, Molenda S, Duhem S, Salleron J, Chaudieu I, Bert D, Libersa C, Vaiva G. Temporal analysis of heart rate variability as a predictor of posttraumatic stress disorder in road traffic accidents survivors. Journal of Psychiatric Research. 2012;46:790–796. doi: 10.1016/j.jpsychires.2012.02.006. [DOI] [PubMed] [Google Scholar]
- *.Alarcon HL, Germain A, Clontz AS, Roach E, Nicholas DH, Zenati MS, Peitzman AB, Sperry JL. Predictors of acute posttraumatic stress disorder symptoms following civilian trauma: Highest incidence and severity of symptoms after assault. Journal of Trauma. 2011;72:629–637. doi: 10.1097/TA.0b013e31824416aa. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. Washington, DC: Author; 2013. [Google Scholar]
- *.Blanchard EB, Hickling EJ, Veazey CH, Buckley TC, Freidenberg BM, Walsh JD, Keefer L. Treatment-related changes in cardiovascular reactivity to trauma cues in motor vehicle accident-related PTSD. Behavior Therapy. 2002;33:417–426. [Google Scholar]
- Bomyea J, Risbrough V, Lang AJ. A consideration of select pre-trauma factors as key vulnerabilities in PTSD. Clinical Psychology Review. 2012;32:630–641. doi: 10.1016/j.cpr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borenstein M, Hedges L, Higgins J, Rothstein H. Comprehensive Meta Analysis, Version 2. Englewood, NH: Biostat; 2005. [Google Scholar]
- Boscarino JA. A prospective study of PTSD and early-age heart disease mortality among Vietnam veterans: Implications for surveillance and prevention. Psychosomatic Medicine. 2008;70:668–676. doi: 10.1097/PSY.0b013e31817bccaf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslau N. Trauma and mental health in US inner-city populations. General Hospital Psychiatry. 2009;31:501–502. doi: 10.1016/j.genhosppsych.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Breslau N, Kessler RC, Chilcoat HD, Schultz LR, Davis GC, Andreski P. Trauma and posttraumatic stress disorder in the community – The 1996 Detroit Area Survey of Trauma. Archives of General Psychiatry. 1998;55:626–632. doi: 10.1001/archpsyc.55.7.626. [DOI] [PubMed] [Google Scholar]
- Brewin CR, Andrews B, Valentine JD. Meta-analysis of risk factors for posttraumatic stress disorder in trauma-exposed adults. Journal of Consulting and Clinical Psychology. 2000;68:748–766. doi: 10.1037//0022-006x.68.5.748. [DOI] [PubMed] [Google Scholar]
- Bryant RA. Longitudinal psychophysiological studies of heart rate: Mediating effects and implications for treatment. Annals of the New York Academy of Sciences. 2006;1071:19–26. doi: 10.1196/annals.1364.002. [DOI] [PubMed] [Google Scholar]
- *.Bryant RA, Creamer M, O’Donnell M, Silove D, McFarlane AC. A multisite study of initial respiration rate and heart rate as predictors of posttraumatic stress disorder. Journal of Clinical Psychiatry. 2008;69:1694–1701. doi: 10.4088/jcp.v69n1104. [DOI] [PubMed] [Google Scholar]
- Bryant RA, Creamer M, O’Donnell M, Silove D, McFarlane AC. Heart rate after trauma and the specificity of fear circuitry disorders. Psychological Medicine. 2011;41:2573–2580. doi: 10.1017/S0033291711000948. [DOI] [PubMed] [Google Scholar]
- *.Bryant RA, Harvey AG, Guthrie RM, Moulds ML. Acute psychophysiological arousal and posttraumatic stress disorder: A two-year prospective study. Journal of Traumatic Stress. 2003;16:439–443. doi: 10.1023/A:1025750209553. [DOI] [PubMed] [Google Scholar]
- Buckley TC, Kaloupek DG. A meta-analytic examination of basal cardiovascular activity in posttraumatic stress disorder. Psychosomatic Medicine. 2001;63:585–594. doi: 10.1097/00006842-200107000-00011. [DOI] [PubMed] [Google Scholar]
- *.Buckley B, Nugent N, Sledjeski E, Raimonde AJ, Spoonster E, Bogart LM, Delahanty DL. Evaluation of initial posttruama cardiovascular levels in association with acute PTSD symptoms following a serious motor vehicle accident. Journal of Traumatic Stress. 2004;17:317–324. doi: 10.1023/B:JOTS.0000038480.87290.4a. [DOI] [PubMed] [Google Scholar]
- Cohen H, Kotler M, Matar MA, Kaplan Z, Miodownik H, Cassuto Y. Power spectral analysis of heart rate variability in posttraumatic stress disorder patients. Biological Psychiatry. 1997;41:627–629. doi: 10.1016/s0006-3223(96)00525-2. [DOI] [PubMed] [Google Scholar]
- Cohen J. Statistical power analyses for the behavioral sciences. 2nd. Hillsdale, NJ: Lawrence Erlbaum Associates, Inc; 1988. [Google Scholar]
- *.Coronas R, Gallardo O, Moreno MJ, Suarez D, Garcia-Pares G, Menchon JM. Heart rate measured in the acute aftermath of trauma can predict post-traumatic stress disorder: A prospective study in motor vehicle accident survivors. European Psychiatry. 2011;26:508–512. doi: 10.1016/j.eurpsy.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Costanzo ME, Leaman S, Jovanovic T, Norrholm SD, Rizzo AA, Taylor P, Roy MJ. Psychophysiological response to virtual reality and subthreshold posttraumatic stress disorder symptoms in recently deployed military. Psychosomatic Medicine. 2014;76:670–677. doi: 10.1097/PSY.0000000000000109. [DOI] [PubMed] [Google Scholar]
- *.Cremeans-Smith JK, Krupko TA, Greene K, Delahanty DL. Predicting symptoms of post-traumatic stress among patients undergoing orthopedic surgery on the basis of routinely collected cardiovascular data. Journal of Health Psychology. 2012;18:55–64. doi: 10.1177/1359105312438110. [DOI] [PubMed] [Google Scholar]
- De Bellis MD. The neurobiology of posttraumatic stress disorder across the life cycle. In: Soares JC, Gershon S, editors. The Handbook of Medical Psychiatry. NY: Marcel Dekker, Inc; 2003. pp. 449–466. [Google Scholar]
- De Bellis MD, Baum AS, Birmaher B, Keshavan MS, Eccard CH, Boring AM, Jenkins FJ, Ryan ND. Developmental traumatology part I: Biological stress systems. Biological Psychiatry. 1999;45:1259–1270. doi: 10.1016/s0006-3223(99)00044-x. [DOI] [PubMed] [Google Scholar]
- De Bellis MD, Spratt EG, Hooper SR. Neurodevelopmental biology associated with childhood sexual abuse. Journal of Child Sexual Abuse. 2011;20:548–587. doi: 10.1080/10538712.2011.607753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet CS, Vermetten E, Geuze E, Kavelaars A, Heijnen CJ, Westenberg HGM. Assessment of HPA-axis function in posttraumatic stress disorder: Pharmacological and non-pharmacological challenge tests, a review. Journal of Psychiatric Research. 2006;40:550–567. doi: 10.1016/j.jpsychires.2005.08.002. [DOI] [PubMed] [Google Scholar]
- de Kloet ER. Hormones, brain and stress. Endocrine Regulations. 2003;37:51–68. [PubMed] [Google Scholar]
- Delahanty DL, Nugent NR. Predicting PTSD prospectively based on prior trauma history and immediate biological responses. Annals of the New York Academy of Sciences. 2006;1071:27–40. doi: 10.1196/annals.1364.003. [DOI] [PubMed] [Google Scholar]
- *.Delahanty DL, Raimonde J, Spoonster E. Initial posttraumatic urinary cortisol levels predict subsequent PTSD symptoms in motor vehicle accident victims. Biological Psychiatry. 2000;48:940–947. doi: 10.1016/s0006-3223(00)00896-9. [DOI] [PubMed] [Google Scholar]
- Delahanty DL, Raimonde AJ, Spoonster E, Cullado M. Injury severity, prior trauma history, urinary cortisol levels, and acute PTSD in motor vehicle accident vicitims. Journal of Anxiety Disorders. 2003;17:149–164. doi: 10.1016/s0887-6185(02)00185-8. [DOI] [PubMed] [Google Scholar]
- de Quervain DJF, Aerni A, Schelling G, Roozendaal B. Glucocorticoids and the regulation of memory in health and disease. Frontiers in Neuroendocrinology. 2009;30:358–370. doi: 10.1016/j.yfrne.2009.03.002. [DOI] [PubMed] [Google Scholar]
- *.De Young MC, Kenardy JA, Spence SH. Elevated heart rate as a predictor of PTSD six months following accidental pediatric injury. Journal of Traumatic Stress. 2007;20:751–756. doi: 10.1002/jts.20235. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Bennett MC, Fleshner M, Rose GM. Inverted-U relationships between the level of peripheral corticosterone and the magnitude of hippocampal primed burst potentiation. Hippocampus. 1992;2:421–430. doi: 10.1002/hipo.450020409. [DOI] [PubMed] [Google Scholar]
- DiGangi JA, Gomez D, Mendoza L, Jason LA, Keys CB, Koenen KC. Pretrauma risk factors for posttraumatic stress disorder: A systematic review of the literature. Clinical Psychology Review. 2013;33:728–744. doi: 10.1016/j.cpr.2013.05.002. [DOI] [PubMed] [Google Scholar]
- *.Ehring T, Ehlers A, Cleare AJ, Glucksman E. Do acute psychological and psychobiological responses to trauma predict subsequent symptom severities of PTSD and depression? Psychiatry Research. 2008;161:67–75. doi: 10.1016/j.psychres.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwood LS, Hahn KS, Olatunji BO, Williams NL. Cognitive vulnerabilities to the development of PTSD: A review of four vulnerabilities and the proposal of an integrative vulnerability model. Clinical Psychology Review. 2009;29:87–100. doi: 10.1016/j.cpr.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Foa EB, Riggs DS. Posttraumatic-stress-disorder following assault – Theoretical considerations and empirical-findings. Current Directions in Psychological Science. 1995;4:61–65. [Google Scholar]
- Foa EB, Steketee G, Rothbaum BO. Behavioral/cognitive conceptualization of post-traumatic stress disorder. Behavior Therapy. 1989;20:155–176. [Google Scholar]
- Fuster JM. The prefrontal cortex: Anatomy, physiology and neuropsychology of the frontal lobe. New York: Raven Press; 1989. [Google Scholar]
- Galatzer-Levy IR, Ankri Y, Freedman S, Israeli-Shalev Y, Roitman P, Gilad M, Shalev AY. Early PTSD symptom trajectories: Persistence, recovery, and response to treatment: Results from the Jerusalem Trauma Outreach and Prevention Study (J-TOPS) PLoS ONE. 2013;8:e70084. doi: 10.1371/journal.pone.0070084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel SN, Abelson JL, King AP, Sripada RK, Wang X, Gaines LM, Liberzon I. Impaired contextual modulation of memories in PTSD: An fMRI and psychophysiological study of extinction retention and fear renewal. The Journal of Neuroscience. 2014;34:13435–13443. doi: 10.1523/JNEUROSCI.4287-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaconia RM, Reinherz HZ, Silverman AB, Pakiz B, Frost AK, Cohen E. Traumas and posttraumatic-stress-disorder in a community population of older adolescents. Journal of the American Academy of Child and Adolescent Psychiatry. 1995;34:1369–1380. doi: 10.1097/00004583-199510000-00023. [DOI] [PubMed] [Google Scholar]
- Gillespie CF, Phifer J, Bradley B, Ressler KJ. Risk and resilience: Genetic and environmental influences on development of the stress response. Depression and Anxiety. 2009;26:984–992. doi: 10.1002/da.20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Gould NF, McKibben JB, Hall R, Corry NH, Amoyal NA, Mason ST, McCann UD, Fauerbach JA. Peritraumatic heart rate and posttraumatic stress disorder in patients with severe heart burns. Journal of Clinical Psychiatry. 2011;72:539–547. doi: 10.4088/JCP.09m05405blu. [DOI] [PubMed] [Google Scholar]
- Gunnar MR, Frenn K, Wewerka SS, van Ryzin MJ. Moderate versus sever early life stress: Associations with stress reactivity and regulation in 10–12-year-old children. Psychoneuroendocrinology. 2009;34:62–75. doi: 10.1016/j.psyneuen.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk LW, Dougall AL, Ursano RJ, Baum A. Urinary catecholamines and cortisol in recent-onset posttraumatic stress disorder after motor vehicle accidents. Psychosomatic Medicine. 2000;62:423–434. doi: 10.1097/00006842-200005000-00016. [DOI] [PubMed] [Google Scholar]
- Hedges LV, Olkin I. Statistical methods for meta-analysis. Orlando, FL: Academic Press; 1985. [Google Scholar]
- Hedges LV, Tipton E, Johnson MC. Robust variance estimation in meta-regression with dependent effect size estimates. Research Synthesis Methods. 2010;1:39–65. doi: 10.1002/jrsm.5. [DOI] [PubMed] [Google Scholar]
- Hidalgo RB, Davidson JRT. Diagnostic psychopharmacologic aspects of posttraumatic stress disorder. Psychiatric Annals. 2004;34:834–844. [Google Scholar]
- Hoge EA, Worthington JJ, Nagurney JT, Chang Y, Kay EB, Feterowski CM, Katzman AR, Goetz JM, Rosasco ML, Lasko N, Zusman RM, Pollack MH, Orr SP, Pitman RK. Effect of acute posttrauma propranolol on PTSD outcome and physiological responses during script-driven imagery. CNS Neuroscience & Therapeutics. 2012;18:21–27. doi: 10.1111/j.1755-5949.2010.00227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper JW, Spinazzola J, Simpson WB, van der Kolk BA. Preliminary evidence of parasympathetic influence on basal heart rate in posttraumatic stress disorder. Journal of Psychosomatic Research. 2006;60:83–90. doi: 10.1016/j.jpsychores.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Hruska B, Cullen PK, Delahanty DL. Pharmacological modulation of acute trauma memories to prevent PTSD: Considerations from a developmental perspective. Neurobiology of Learning and Memory. 2014;112:122–129. doi: 10.1016/j.nlm.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov I, Yehuda R, Greenblatt E, Davidow J, Makotkine I, Alfi L, Newcorn JH. The effect of trauma on stress reactivity in aggressive youth. Psychiatry Research. 2011;189:396–402. doi: 10.1016/j.psychres.2011.05.046. [DOI] [PubMed] [Google Scholar]
- Joëls M, Fernadez G, Roozendaal B. Stress and emotional memory: a matter timing. Trends in Cognitive Sciences. 2009;15:280–288. doi: 10.1016/j.tics.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Kaouane N, Porte Y, Vallée M, Brayda-Bruno L, Mons N, Calandreau L, Marighetto A, Piazza PV, Desmedt A. Glucocorticoids can induce PTSD-like memory impairments in mice. Science. 2012;335:1510–1513. doi: 10.1126/science.1207615. [DOI] [PubMed] [Google Scholar]
- *.Kassam-Adams N, Garcia-Espana F, Fein JA, Winston FK. Heart rate and posttraumatic stress in injured children. Archives of General Psychiatry. 2005;62:335–340. doi: 10.1001/archpsyc.62.3.335. [DOI] [PubMed] [Google Scholar]
- Keane TM, Kaloupek DG. Imaginal flooding in the treatment of a post-traumatic stress disorder. Journal of Consulting and Clinical Psychology. 1982;50:138–140. doi: 10.1037//0022-006x.50.1.138. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Archives of General Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Kirsch V, Wilhelm FH, Goldbeck L. Psychophysiological characteristics of PTSD in children and adolescents: A review of the literature. Journal of Traumatic Stress. 2011;24:146–154. doi: 10.1002/jts.20620. [DOI] [PubMed] [Google Scholar]
- Klaassens ER, Giltay EJ, Cuijpers P, van Veen T, Zitman FG. Adulthood trauma and HPA-axis functioning in healthy subjects and PTSD patients: A meta-analysis. Psychoneuroendocrinology. 2012;37:317–331. doi: 10.1016/j.psyneuen.2011.07.003. [DOI] [PubMed] [Google Scholar]
- *.Kraemer B, Moergeli H, Roth H, Hepp U, Schnyder U. Contribution of initial heart rate to the prediction of posttraumatic stress symptom level in accident victims. Journal of Psychiatric Research. 2008;42:158–162. doi: 10.1016/j.jpsychires.2006.11.005. [DOI] [PubMed] [Google Scholar]
- *.Kuhn E, Blanchard EB, Fuse T, Hickling EJ, Broderick J. Heart rate of motor vehicle accident survivors in the emergency department, peritraumatic psychological reactions, ASD, and PTSD severity: A 6-month prospective study. Journal of Traumatic Stress. 2006;19:735–740. doi: 10.1002/jts.20150. [DOI] [PubMed] [Google Scholar]
- Lang PJ. Imagery in therapy: An information processing analysis of fear. Behavior Therapy. 1977;8:862–886. doi: 10.1016/j.beth.2016.08.011. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Abelson JL, Flagel SB, Raz J, Young EA. Neuroendocrine and psychophysiological responses in PTSD: A symptom provocation study. Neuropsychopharmacology. 1999;21:40–50. doi: 10.1016/S0893-133X(98)00128-6. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Sripada CS. The functional neuroanatomy of PTSD: A critical review. Progress in Brain Research. 2007;167:151–169. doi: 10.1016/S0079-6123(07)67011-3. [DOI] [PubMed] [Google Scholar]
- Lipsey M, Wilson D. Practical Meta-Analysis. SAGE Publications; Thousand Oaks: 2001. [Google Scholar]
- Luo H, Hu X, Liu X, Ma X, Guo W, Qiu C, Wang Y, Wang Q, Zhang X, Zhang W, Hannum G, Zhang K, Liu X, Li T. Hair cortisol level as a biomarker for altered hypothalamic-pituitary-adrenal activity in female adolescents with posttraumatic stress disorder after the 2008 Wenchuan earthquake. Biological Psychiatry. 2012;72:65–69. doi: 10.1016/j.biopsych.2011.12.020. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nature Reviews Neuroscience. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Stress hormones and amygdala regulate long-term memory storage. International Journal of Psychology. 2000;35:405–405. [Google Scholar]
- McNally RJ. Psychological mechanisms in acute response to trauma. Biological Psychiatry. 2003;53:779–788. doi: 10.1016/s0006-3223(02)01663-3. [DOI] [PubMed] [Google Scholar]
- McNally RJ, Bryant RA, Ehlers A. Does early psychological intervention promote recovery from posttraumatic stress? Psychological Science in the Public Interest. 2003;4:45–79. doi: 10.1111/1529-1006.01421. [DOI] [PubMed] [Google Scholar]
- Meewisse ML, Reitsma JB, De Vries GJ, Gersons BPR, Olff M. Cortisol and post-traumatic stress disorder in adults – Systematic review and meta-analysis. British Journal of Psychiatry. 2007;191:387–392. doi: 10.1192/bjp.bp.106.024877. [DOI] [PubMed] [Google Scholar]
- Mellman TA, Knorr BR, Pigeon WR, Leiter JC, Akay M. Heart rate variability during sleep and the early development of posttraumatic stress disorder. Biological Psychiatry. 2004;55:953–956. doi: 10.1016/j.biopsych.2003.12.018. [DOI] [PubMed] [Google Scholar]
- Michopoulos V, Norrholm SD, Jovanovic T. Diagnostic Biomarkers for posttraumatic stress disorder: Promising horizon form Translational Neuroscience Research. Biological Psychiatry. 2015;78:344–353. doi: 10.1016/j.biopsych.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MR, Pitman RK, Ellis CB, Gold AL, Shin LM, Lasko NB, Zeidan MA, Handwerger K, Orr SP, Rauch SL. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biological Psychiatry. 2009;66:1075–1082. doi: 10.1016/j.biopsych.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GE, Chen E, Zhou ES. If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychological Bulletin. 2007;133:25–45. doi: 10.1037/0033-2909.133.1.25. [DOI] [PubMed] [Google Scholar]
- Morris MC, Compas BE, Garber J. Relations among posttraumatic stress disorder, comorbid major depression, and HPA function: A systematic review and meta-analysis. Clinical Psychology Review. 2012;32:301–315. doi: 10.1016/j.cpr.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MC, Rao U. Psychobiology of PTSD in the acute aftermath of trauma: Integrating research on coping, HPA function and sympathetic nervous system activity. Asian Journal of Psychiatry. 2013;6:3–21. doi: 10.1016/j.ajp.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Mouthaan J, Sijbrandij M, Luitse JSK, Goslings JC, Gersons BPR, Olff M. The role of acute cortisol and DHEAS in predicting acute and chronic PTSD symptoms. Psychoneuroendocrinology. 2014;45:179–186. doi: 10.1016/j.psyneuen.2014.04.001. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, LeDoux JE. The liable nature of consolidation theory. Nature Reviews Neuroscience. 2000;1:216–219. doi: 10.1038/35044580. [DOI] [PubMed] [Google Scholar]
- *.Nishi D, Noguchi H, Yonemoto N, Nakajimi S, Kim Y, Matsuoka Y. Incidence and prediction of post-traumatic stress disorder at 6 months after motor vehicle accidents in Japan. Psychosomatics. 2013;54:263–271. doi: 10.1016/j.psym.2012.07.010. [DOI] [PubMed] [Google Scholar]
- *.Nugent NR, Christopher NC, Delahanty D. Emergency medical service and in-hospital vital signs as predictors of sequent PTSD symptom severity in pediatric injury patients. Journal of Child Psychology and Psychiatry. 2006;47:919–926. doi: 10.1111/j.1469-7610.2006.01648.x. [DOI] [PubMed] [Google Scholar]
- *.O’Donnell ML, Creamer M, Elliott P, Bryant R. Tonic and phasic heart rate as predictors of posttraumatic stress disorder. Psychosomatic Medicine. 2007;69:256–261. doi: 10.1097/PSY.0b013e3180417d04. [DOI] [PubMed] [Google Scholar]
- Olff M, Langeland W, Draiger N, Gersons BPR. Gender differences in posttraumatic stress disorder. Psychological Bulletin. 2007;133:183–204. doi: 10.1037/0033-2909.133.2.183. [DOI] [PubMed] [Google Scholar]
- Orwin RG. Evaluating coding decisions. In: Cooper H, Hedges LV, editors. The handbook of research synthesis. New York: Russell Sage Foundation; 1994. pp. 139–162. [Google Scholar]
- O’Toole BI, Catts SV. Trauma, PSTD, and physical health: An epidemiological study of Australian Vietnam veterans. Journal of Psychosomatic Research. 2008;64:33–40. doi: 10.1016/j.jpsychores.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Ozer EJ, Best SR, Lipsey TL, Weiss DS. Predictors of posttraumatic stress disorder and symptoms in adults: A meta-analysis. Psychological Bulletin. 2003;129:52–73. doi: 10.1037/0033-2909.129.1.52. [DOI] [PubMed] [Google Scholar]
- Paus T, Collins DL, Evans AC, Leonard G, Pike B, Zijdenbos A. Maturation of white matter in the human brain: A review of magnetic resonance studies. Brain Research Bulletin. 2001;54:255–266. doi: 10.1016/s0361-9230(00)00434-2. [DOI] [PubMed] [Google Scholar]
- Pervanidou P. Biology of post-traumatic stress disorder in childhood and adolescence. Journal of Neuroendocrinology. 2008;20:632–638. doi: 10.1111/j.1365-2826.2008.01701.x. [DOI] [PubMed] [Google Scholar]
- Pervanidou P, Kolaitis G, Charitaki S, Lazaropoulou C, Papassotiriou L, Hindmarsh P, Bakoula C, Tsiantis J, Chrousos GP. The natural history of neuroendocrine changes in pediatric posttraumatic stress disorder (PTSD) after motor vehicle accidents: Progressive divergence of noradrenaline and cortisol concentrations over time. Biological Psychiatry. 2007;62:1095–1102. doi: 10.1016/j.biopsych.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Pitman RK. Post-traumatic stress disorder, hormones and memory. Biological Psychiatry. 1989;26:221–223. doi: 10.1016/0006-3223(89)90033-4. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Delahanty DL. Conceptually driven pharmacologic approaches to acute trauma. CNS Spectrums. 2005;10:99–106. doi: 10.1017/s109285290001943x. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Orr SP, Altman B, Longpre RE, Poire RE, Macklin ML. Emotional processing during eye movement desensitization and reprocessing therapy of Vietnam veterans with chronic posttraumatic stress disorder. Comprehensive Psychiatry. 1996;37:419–429. doi: 10.1016/s0010-440x(96)90025-5. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Orr SP, Shalev AY. Once bitten, twice shy – Beyond the conditioning model of PTSD. Biological Psychiatry. 1993;33:145–146. doi: 10.1016/0006-3223(93)90132-w. [DOI] [PubMed] [Google Scholar]
- Pitman RK, Rasmussion AM, Koenen KC, Shin LM, Orr SP, Gilbertson MW, Milad MR, Liberzon I. Biological studies of post-traumatic stress disorder. Nature Reviews Neuroscience. 2012;13:769–787. doi: 10.1038/nrn3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pole N. The psychophysiology of posttraumatic stress disorder: A meta-analysis. Psychological Bulletin. 2007;133:725–746. doi: 10.1037/0033-2909.133.5.725. [DOI] [PubMed] [Google Scholar]
- Pole N, Bloomberg-Fretter P. Using control mastery therapy to treat major depression and posttraumatic stress disorder. Clinical Case studies. 2006;5:53–70. [Google Scholar]
- Pole N, Neylan TC, Otte C, Henn-Hasse C, Metzler TJ, Marmar CR. Prospective prediction of posttraumatic stress disorder symptoms using fear potentiated auditory startle responses. Biological Psychiatry. 2011;65:235–240. doi: 10.1016/j.biopsych.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porges SW. The polyvagal perspective. Biological Psychology. 2007;74:116–143. doi: 10.1016/j.biopsycho.2006.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Price M, Kearns M, Houry D, Rothbaum BO. Emergency department predictors of posttraumatic stress reduction for trauma-exposed individuals with and without early intervention. Journal of Consulting and Clinical Psychology. 2014;82:336–341. doi: 10.1037/a0035537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowicz T. The differentiated maturation of the cerebral cortex. In: Falkner F, Tanner JM, editors. Human Growth, Volume 2. New York: Plenum; 1986. pp. 385–410. [Google Scholar]
- Resick PA, Monson CM, Chard KM. Cognitive processing therapy: Veteran/military version. Department of Veterans’ Affairs; Washington, DC: 2007. [Google Scholar]
- Rodrigues SM, LeDoux JE, Sapolsky RM. The influence of stress hormones on fear circuitry. Annual Review of Neuroscience. 2009;32:289–313. doi: 10.1146/annurev.neuro.051508.135620. [DOI] [PubMed] [Google Scholar]
- Rothbaum BO, Kearns MC, Price M, Malcoun E, Davis M, Ressler KJ, Lang D, Houry D. Early intervention may prevent the development of posttraumatic stress disorder: A randomized pilot civilian study with modified prolonged exposure. Biological Psychiatry. 2012;72:957–963. doi: 10.1016/j.biopsych.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman KM, Holden GW, Holahan CJ. The psychobiology of children exposure to marital violence. Journal of Clinical Child and Adolescent Psychology. 2005;34:129–139. doi: 10.1207/s15374424jccp3401_12. [DOI] [PubMed] [Google Scholar]
- Santiago PN, Ursano RJ, Gray CL, Pynoos RS, Spiegel D, Lewis-Fernandez R, Feidman MJ, Fullerton CS. A systematic review of PTSD prevalence and trajectories in DMS-5 defined trauma exposed populations: Intentional and non-intentional traumatic events. PLoS ONE. 2013;8:e59236. doi: 10.1371/journal.pone.0059236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U, Kaltwasser SF, Wotjak CT. Biomarkers in posttraumatic stress disorder: Overview and implications for future research. Disease Markers. 2013;35:43–54. doi: 10.1155/2013/835876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U, Willmund GD, Holsboer F, Wotjak CT, Gallinat J, Kowalski JT, Zimmermann P. Searching for non-genetic molecular and imaging PTSD risk and resilience markers: Systematic review of literature and design of the German Armed Forces PTSD biomarker study. Psychoneuroendocrinology. 2015;51:444–458. doi: 10.1016/j.psyneuen.2014.08.020. [DOI] [PubMed] [Google Scholar]
- Schnurr PP, Ford JD, Friedman MJ, Green BL, Dain BJ, Sengupta A. Predictors and outcomes of posttraumatic stress disorder in World War II veterans exposed to mustard gas. Journal of Consulting and Clinical Psychology. 2000;68:258–268. [PubMed] [Google Scholar]
- Schnurr PP, Spiro A, Paris AH. Physician-diagnosed medical disorders in relation to PTSD symptoms in older male military veterans. Health Psychology. 2000;19:91–97. doi: 10.1037//0278-6133.19.1.91. [DOI] [PubMed] [Google Scholar]
- Scott JC, Matt GE, Wrocklage KM, Crnich C, Jordan J, Southwick SM, Krystal JH, Schweinsburg BC. A quantitative meta-analysis of neurocognitive functioning in posttraumatic stress disorder. Psychological Bulletin. 2015;141:105–140. doi: 10.1037/a0038039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman TE, Robbins RJ. Aging and hypothalamic-pituitary-adrenal response to challenge in humans. Endocrine Reviews. 1994;15:233–260. doi: 10.1210/edrv-15-2-233. [DOI] [PubMed] [Google Scholar]
- *.Shalev AY, Freedman S. PTSD following terrorist attacks: A prospective evaluation. American Journal of Psychiatry. 2005;162:1188–1191. doi: 10.1176/appi.ajp.162.6.1188. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Sahar T, Freedman S, Peri T, Glick N, Brandes D, Orr SP, Pitman RK. Archives of General Psychiatry. 1998;55:553–559. doi: 10.1001/archpsyc.55.6.553. [DOI] [PubMed] [Google Scholar]
- Shalev AY, Segman RH. Commentary: biological findings in PTSD – too much or too little? Progress in Brain Research. 2008;167:187–199. doi: 10.1016/S0079-6123(07)67013-7. [DOI] [PubMed] [Google Scholar]
- *.Shalev AY, Videlock EJ, Peleg T, Segman R, Pitman RK, Yehuda R. Stress hormones and post-traumatic stress disorder in civilian trauma victims: A longitudinal study. Part I: HPA axis responses. International Journal of Neuropsychopharmacology. 2008;11:365–372. doi: 10.1017/S1461145707008127. [DOI] [PubMed] [Google Scholar]
- Sijbrandij M, Kleiboer A, Bisson JI, Barbui C, Cuijpers P. Pharmacological prevention of post-traumatic stress disorder and acute stress disorder: A systematic review and meta-analysis. Lancet Psychiatry. 2015;2:413–421. doi: 10.1016/S2215-0366(14)00121-7. [DOI] [PubMed] [Google Scholar]
- Skirton H, Chamberlain W, Lawson C, Ryan H, Young E. A systematic review of variability and reliability of manual and automated blood pressure readings. Journal of Clinical Nursing. 2011;20:602–614. doi: 10.1111/j.1365-2702.2010.03528.x. [DOI] [PubMed] [Google Scholar]
- Steudte S, Kirschbaum C, Gao W, Alexander N, Schönfeld S, Hoyer J, Stalder T. Hair cortisol as a biomarker of traumatization in healthy individuals and posttraumatic stress disorder patients. Biological Psychiatry. 2013;74:639–646. doi: 10.1016/j.biopsych.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Steudte-Schmiedgen S, Stalder T, Kirschbaum C, Weber F, Hoyer J, Plessow F. Trauma exposure is associated with increased context-dependent adjustments of cognitive control in patients with posttraumatic stress disorder and healthy controls. Cognitive Affective & Behavioral Neuroscience. 2014;14:1310–1319. doi: 10.3758/s13415-014-0299-2. [DOI] [PubMed] [Google Scholar]
- Thomas LA, De Bellis MD. Pituitary volumes in pediatric maltreatment-related PTSD. Biological Psychiatry. 2004;55:752–758. doi: 10.1016/j.biopsych.2003.11.021. [DOI] [PubMed] [Google Scholar]
- Trickett PK, Noll JG, Susman EJ, Shenk CE, Putnam PW. Attenuation of cortisol across development for victims of sexual abuse. Development and Psychopathology. 2010;22:165–175. doi: 10.1017/S0954579409990332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino BN, Birmingham W, Berg CA. Are older adults less or more physiologically reactivity? A meta-analysis of age-related differences in cardiovascular reactivity to laboratory tasks. Journal of Gerontology: Psychological Sciences. 2010;65:154–162. doi: 10.1093/geronb/gbp127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursin H, Olff M. Aggression, defense, and coping factors in humans. Aggressive Behavior. 1993;19:35–36. [Google Scholar]
- van Zuiden M, Hijnen CJ, Mass M, Amarouchi K, Vermetten E, Geuze E, Kavelaars A. Glucocorticoid sensitivity of leukocytes predicts PTSD, depressive and fatigue symptoms after military deployment: A prospective study. Psychoneuroendocrinology. 2012;37:1822–1836. doi: 10.1016/j.psyneuen.2012.03.018. [DOI] [PubMed] [Google Scholar]
- van Zuiden M, Kavelaars A, Geuze E, Olff M, Heijnen CJ. Predicting PTSD: Pre-existing vulnerabilities in glucocorticoid-signaling and implications for preventive interventions. Brain Behavior and Immunity. 2013;30:12–21. doi: 10.1016/j.bbi.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Vermetten E, Zhohar J, Krugers HJ. Pharmacotherapy in the aftermath of trauma: Opportunities in the ‘golden hours’. Current Psychiatry Reports. 2014;16:455. doi: 10.1007/s11920-014-0455-y. [DOI] [PubMed] [Google Scholar]
- *.Videlock EJ, Peleg T, Segman R, Yehuda R, Pitman RK, Shalev AY. Stress hormones and post-traumatic stress disorder in civilian trauma victims: A longitudinal study. Part II: The adrenergic response. International Journal of Neuropsychopharmacology. 2008;11:373–380. doi: 10.1017/S1461145707008139. [DOI] [PubMed] [Google Scholar]
- Walker EF, Walder DJ, Reynolds F. Developmental changes in cortisol secretion in normal and at risk youth. Development and Psychopathology. 2001;13:721–732. doi: 10.1017/s0954579401003169. [DOI] [PubMed] [Google Scholar]
- *.Walsh K, Nugent NR, Kotte A, Amstadter AB, Wang S, Guille C, Acierno R, Kilpatrick DG, Resnick HS. Cortisol at the emergency room rape visit as a predictor of PTSD and depression symptoms over time. Psychoneuroendocrinology. 2013;38:2520–2528. doi: 10.1016/j.psyneuen.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf OT. The influence of stress hormones on emotional memory: Relevance for Psychopathology. Acta Psychologica. 2008;127:513–531. doi: 10.1016/j.actpsy.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Current status of cortisol findings in post-traumatic stress disorder. Psychiatric clinics of North America. 2002;25:2002. doi: 10.1016/s0193-953x(02)00002-3. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Bierer LM, Pratchett LC, Lehrner A, Koch EC, Van Manen JA, Flory JD, Makotkine I, Hildebrandt T. Cortisol augmentation of a psychological treatment for warfighters with posttraumatic stress disorder: Randomized trial showing improved treatment retention and outcome. Psychoneuroendocrinology. 2015;51:589–597. doi: 10.1016/j.psyneuen.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Harvey P. Relevance of neuroendocrine alterations in PTSD to memory-related impairments of trauma survivors. In: Read D, Lindsay S, editors. Recollections of trauma: Scientific research and clinical practice. New York: Plenum Press; 1997. pp. 221–252. [Google Scholar]
- *.Yehuda R, McFarlane AC, Shalev AY. Predicting the development of posttraumatic stress disorder from the acute response to a traumatic event. Biological Psychiatry. 1998;44:1305–1313. doi: 10.1016/s0006-3223(98)00276-5. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Neylan TC, Flory JD, McFarlane AC. The use of biomarkers in the military: From theory to practice. Psychoneuroendocrinology. 2013;38:1912–1922. doi: 10.1016/j.psyneuen.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Zatzick DF, Russo J, Pitman RK, Rivara F, Jurkovich G, Roy-Byrne P. Reevaluating the association between emergency department heart rate and the development of posttraumatic stress disorder: A public health approach. Biological Psychiatry. 2005;57:91–97. doi: 10.1016/j.biopsych.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Zoladz PR, Diamond DM. Current status on behavioral and biological markers of PTSD: A search for clarity in a conflicting literature. Neuroscience and Biobehavioral Reviews. 2013;37:860–895. doi: 10.1016/j.neubiorev.2013.03.024. [DOI] [PubMed] [Google Scholar]