

Abstract
Background
The Rhynchosporium species complex consists of hemibiotrophic fungal pathogens specialized to different sweet grass species including the cereal crops barley and rye. A sexual stage has not been described, but several lines of evidence suggest the occurrence of sexual reproduction. Therefore, a comparative genomics approach was carried out to disclose the evolutionary relationship of the species and to identify genes demonstrating the potential for a sexual cycle. Furthermore, due to the evolutionary very young age of the five species currently known, this genus appears to be well-suited to address the question at the molecular level of how pathogenic fungi adapt to their hosts.
Results
The genomes of the different Rhynchosporium species were sequenced, assembled and annotated using ab initio gene predictors trained on several fungal genomes as well as on Rhynchosporium expressed sequence tags. Structures of the rDNA regions and genome-wide single nucleotide polymorphisms provided a hypothesis for intra-genus evolution. Homology screening detected core meiotic genes along with most genes crucial for sexual recombination in ascomycete fungi. In addition, a large number of cell wall-degrading enzymes that is characteristic for hemibiotrophic and necrotrophic fungi infecting monocotyledonous hosts were found. Furthermore, the Rhynchosporium genomes carry a repertoire of genes coding for polyketide synthases and non-ribosomal peptide synthetases. Several of these genes are missing from the genome of the closest sequenced relative, the poplar pathogen Marssonina brunnea, and are possibly involved in adaptation to the grass hosts. Most importantly, six species-specific genes coding for protein effectors were identified in R. commune. Their deletion yielded mutants that grew more vigorously in planta than the wild type.
Conclusion
Both cryptic sexuality and secondary metabolites may have contributed to host adaptation. Most importantly, however, the growth-retarding activity of the species-specific effectors suggests that host adaptation of R. commune aims at extending the biotrophic stage at the expense of the necrotrophic stage of pathogenesis. Like other apoplastic fungi Rhynchosporium colonizes the intercellular matrix of host leaves relatively slowly without causing symptoms, reminiscent of the development of endophytic fungi. Rhynchosporium may therefore become an object for studying the mutualism-parasitism transition.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3299-5) contains supplementary material, which is available to authorized users.
Keywords: CAZymes, Effectors, Host specificity, Leotiomycetes, Non-ribosomal peptide synthetases, Phylogenetic evolution, Polyketide synthases, Rhynchosporium, Sex-related genes, Whole genome sequencing
Background
The genus Rhynchosporium (order: Leotiales) consists of haploid fungi that are pathogenic to grass species (Poaceae) including cereal crops. In an early review article [1], only two species were described, R. secalis and R. orthosporum, differing both in the shape of their two-celled conidia and in their host specificities. The name of the fungal genus originates from the beak (Greek: rhynchos) carried by one of the spore cells of R. secalis. In contrast, R. orthosporum produces cylindrical spores (Greek: ortho, straight). R. secalis was first isolated from rye (Secale cereale; [2]; cited in [1, 3]). Nevertheless, the major host worldwide is barley (Hordeum vulgare), on which the fungus causes the economically important disease leaf blotch or scald. R. orthosporum is a pathogen of orchard grass (cocksfoot, Dactylis glomerata).
It is usually assumed that plant pathogens originated on direct ancestors of their modern crop hosts and coevolved with them during domestication [4]. Therefore the center of origin of a pathogen is usually also the center of genetic diversity [5]. Gene diversity analysis of Rhynchosporium suggests, however, that the fungus did not originate from the Fertile Crescent [5], where its host plants barley and rye were domesticated about 10,000 years ago [6]. Instead, the genetic history of the fungus as it was reconstructed using the nucleotide sequence variation of the effector gene NIP1 [7, 8] along with sequences from neutral RFLP loci [9] and microsatellite markers [10] suggests that Rhynchosporium emerged in Northern Europe following a host switch from a wild grass onto cultivated barley and rye after arrival of these plants about 5000–3000 years ago. From there the fungus spread globally most likely due to anthropogenic dispersal of infected host material [9–11], reaching regions such as South Africa, California, Australia and New Zealand only during the last few hundred years [10].
The occurrence of host-specialized ‘races’ of R. secalis was reported early on [1]. Nevertheless, for a long time there were conflicting reports about the host range of Rhynchosporium isolates [12]. Recently, multigene-sequencing and phylogenetic analysis (genealogical concordance phylogenetic species recognition; [13, 14]) demonstrated that R. secalis was actually a complex of at least three closely related host-specialized species [15]. Given that the fungus was initially described on rye, the name R. secalis was retained for the species infecting rye and triticale (x Triticosecale Wittmack). The species colonizing cultivated barley and other Hordeum spp. as well as brome grass (Bromus diandrus) was named R. commune, whereas R. agropyri is pathogenic to couch grass (Elymus repens; syn. Agropyron repens). Finally, fungal isolates from perennial ryegrass (Lolium perenne) were found to represent yet another species, R. lolii [16]. The cylindrical shape of the spores as well as phylogenetic analysis based on nucleic acid sequences position the latter species closer to R. orthosporum (cylindrical conidia group, CCG) than to the other three species (beaked conidia group, BCG). Due to its economic importance the majority of studies has been carried out on the barley-infecting fungal species, R. commune (in the older literature called R. secalis).
Phylogenetic concordance analyses based on the sequences of four housekeeping genes (ITS1-5.8S-rRNA-ITS2, EF1-α, α-tubulin, β-tubulin) and neutral RFLP loci using several hundred fungal isolates suggested that the CCG and the BCG branches separated 14,000–35,000 years ago, hence long before the advent of agriculture, with R. lolii diverging from R. orthoporum 4300–7200 years ago [16]. In contrast, R. commune, R. secalis and R. agropyri originated from a common ancestor only 1200–3600 years ago [8, 9, 16]. Furthermore, reconstructions of demographic growth indicate that populations of the three BCG species evolved in parallel. After the host shift, which was soon followed by the split of the three fungal lineages, they experienced a bottleneck due to their reproductive isolation. Specific adaptation to their modern hosts [15] led to the recovery of the sizes of all 3 populations in particular during the last 250 years [10, 17]. Thus, worldwide dissemination and population increase of the pathogen coincide with intensification and global expansion of cereal production [17, 18].
In evolutionary terms, the Rhynchosporium BCG species are very young and closely related. Thus it can be expected that their genomes did not diverge substantially yet, except for those genes that are involved in adaptation to the different hosts and/or environments. Hence, they represent an excellent basis to address the question of host specialization by comparative genomics. Genes under diversifying selection may be uncovered through a fully unbiased approach by scanning the genomes of several isolates from the different species. Alternatively, crucial processes associated with host adaption of a pathogen and epitomized in the concept of (quantitative) virulence may be defined a priori. This would allow the identification of patterns of adaptive evolution such as species-specifically occurring genes or of gene family expansion.
What are the factors that contribute to fungal virulence? Rhynchosporium development in planta starts with germination of conidia on the surface of host leaves and the penetration of fungal hyphae through the cuticle, frequently without forming obvious appressoria [19, 20]. Then, a long latent period of a few days up to several months [21, 22] without any visible disease symptoms follows. During this stage fungal mycelia are established between the cuticle and the outer wall of epidermis cells. Finally, after formation of a dense subcuticular fungal stroma necrotic lesions occur. New spores are formed both during the latent period and in the lesion areas. Penetration as well as subcuticular proliferation in the host tissue require the secretion of hydrolytic enzymes to degrade the cuticle and cell wall structures and to release carbohydrate nutrients form the plant cell wall polysaccharides. In addition, the physiology of the host needs to be redirected towards tolerating and accommodating the pathogen. For this purpose (hemibiotrophic) fungal pathogens secrete small molecules (secondary metabolites; [23]) as well as effector proteins. Therefore, the genes coding for cell wall-degrading enzymes, key secondary biosynthetic enzymes or protein effectors are prime targets of diversification/diversifying selection during adaptive evolution.
Finally, at the base of any considerations regarding evolutionary processes is the mating system of an organism. For Rhynchosporium, no teleomorph has been identified to date despite the presence of mating-type (MAT) genes [24]. Nevertheless, ‘the signature of sex’ [25], i.e., mating type equilibrium in most populations at least of the BCG species and evidence for MAT gene expression [5, 26, 27], high genotype diversity, gametic equilibrium among neutral markers and low levels of clonality, was found in fungal populations worldwide [5, 9, 18, 28, 29]. These results suggest that sexual recombination occurs cryptically but regularly.
This paper presents a comparative analysis of the genomes of isolates from four of the five Rhynchosporium species described to date. The intra-genus evolution and the broader phylogenetic relationship to other fungal species are re-analyzed. Furthermore, the possibility of sexual recombination is assessed using a genomics approach by screening for the presence of sex-related genes known to be involved in meiosis and recombination in other organisms. Finally, genes that may be involved in controlling host specificity, including those coding for key enzymes of fungal secondary metabolism or effector proteins are identified.
Results and discussion
Genome statistics
Three isolates of R. commune and one isolate each of R. secalis, R. agropyri, R. lolii and R. orthosporum were sequenced using a whole-genome shotgun strategy [30] with the Roche 454 GS FLX and the Illumina GAIIx platforms. De novo assembly resulted in a total sequence length of 52–58 Mb. Overall sequencing and assembly statistics are presented in Table 1. The assembled genome of R. commune, isolate UK7, consisting of only 163 scaffolds was established as the reference genome. Gene models for R. commune, isolate UK7, R. secalis and R. agropyri were generated by three de novo prediction programs with different matrices. Completeness of genomic predictions was assessed by BLASTp comparison to a set of 246 highly conserved fungal single-copy protein-coding homologs (SCPCH, [31]) and to a set of 248 core genes from the core Eukaryotic genes mapping approach (CEGMA, [32]; Table 2). The predicted coding sequences comprise about 30–33% of the genomic sequences, the estimated gene numbers range from 10,254 to 13,674 with about 50% coding for proteins of unknown function. Repetitive DNA elements were identified and categorized. The total coverage of repetitive sequences is relatively high as compared to other fungi reaching 31% in R. commune, but lower than in the close relative Marssonina brunnea f.sp. ‘multigermtubi’ (see below; 42% [33]).
Table 1.
Genome sequencing and assembly
| Parameter | R. commune (UK7) | R.c. (AU2) | R.commune (13-13) | R. secalis | R. agropyri | R. ortho-sporum | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primary seq. data | Illumina PE (GAIIx) | Illumina PE (GAIIx) | Illumina MP (GAIIx) | 454 | Illumina PE (GAIIx) | Illumina PE (GAIIx) | 454a | Illumina PE (GAIIx) | 454 | Illumina PE (GAIIx) | Illumina PE (GAIIx) | Illumina PE (HiSeq) | Illumina MP (HiSeq) | Illumina PE (GAIIx) |
| No. of reads | 18 Mio. | 35 Mio. | 6.5 Mio | 2.37 Mio | 32.1 Mio. | 25.23 Mio. | 2.21 Mio | 34.7 Mio. | 1.86 Mio. | 34 Mio. | 104 Mio. | 26 Mio. | 24 Mio. | 38 Mio. |
| Read length | 2 × 36 bp | 2 × 151 bp, 270 bp dist. | 2 × 151 bp, 3 kb dist. | 377 bp | 2 × 101 bp | 2 × 76 bp | 366.5 bp | 2 × 101 bp | 410 bp | 2 × 101 bp, 300 bp dist. | 2 × 100 bp, 180 bp dist. | 2 × 151 bp, 270 bp dist. | 2 × 151 bp, 3 kb dist. | 2 × 101 bp |
| Total length | 1.29 Gb | 9.7 Gb | 1.4 Gb | 894 Mb | 6.48 Gb | 1.92 Gb | 810 Mb | 7.0 Gb | 746 Mb | 6.9 Gb | 20.8 Gb | 7.8 Gb | 7.3 Gb | 7.6 Gb |
| Nominal seq. depth | 26x | 194x | 29x | 18x | 130x | 25x | 15x | 140x | 15x | 137x | 420x | 156x | 146x | 150x |
| Assembly software | ALLPATHS-LG + SSPACE | Velvet | Velvet + Newbler + Minimus2 | Newbler | ALLPATHS-LG + SSPACE | Velvet | ||||||||
| Total seq. length (LFI) | 55.59 Mb | 54.32 Mb | 53.93 Mb | 56.80 Mb | 52.18 Mb | 58.16 Mb | ||||||||
a 0.5 run 20 kb PE library, 1.5 runs 0.8 kb library
Table 2.
Genome characteristics
| R. commune UK7 | R. secalis | R. agropyri | R. ortho-sporum | M. brunnea | |
|---|---|---|---|---|---|
| Assembly statistics | |||||
| Total scaffold length (Mb) | 55.59 | 56,80 | 52.18 | 58.16 | 51.95 |
| N50 contig (kb) | 49.6 | 47.5 | 61.6 | ||
| N50 scaffold (kb) | 800.5 | 113.7 | 303.4 | 104.3 | |
| Scaffolds/Chromosomes | 163 | 2129 | 508 | 29183 | 89 |
| GC-content (%) | 42.33 | 43.91 | 42.9 | 41.1 | 42.72 |
| - coding (%) | 49.86 | 49.85 | 49.75 | 54.12 | |
| - non-coding (%) | 39.09 | 40.87 | 39.52 | 38.12 | |
| Coding sequence | |||||
| Percent coding (%) | 30.06 | 33.84 | 33.08 | 28.78 | |
| Average gene size (bp) | 1367.7 | 1271.6 | 1269.4 | 1491 | |
| Average gene density (gene/kb) | 0.22 | 0.27 | 0.26 | 0.19 | |
| Protein-coding genes | 12212 | 13151 | 13674 | 10027 | |
| Exons | 38363 | 39806 | 41720 | 33423 | |
| Average exon size | 434.4 | 419.1 | 415.1 | 394.6 | |
| Exons/gene | 3.1 | 3 | 3.1 | 3.9 | |
| tRNA genes | 113 (29 mta) | 117 (30 mta) | 107 (29 mta) | nd | |
| Validation of prediction | |||||
| SCPCH (246) | 246 | 246 | 246 | 246 | |
| CEGMAS (248) | 248 | 248 | 247 | 247 | |
| Secretome | |||||
| Secreted proteins ≥300aa | 711 | 690 | 724 | 509 | |
| Small secreted proteins <300aa | 457 | 574 | 655 | 429 | |
| Non-classical secreted proteins ≥300aa | 77 | 90 | 89 | 61 | |
| Small non-classical secr. proteins <300aa | 63 | 167 | 196 | 63 | |
| non-secreted protein | 10904 | 11630 | 12010 | 8965 | |
| All proteins | 12212 | 13151 | 13674 | 10027 | |
| Non-coding sequence | |||||
| Introns | 26151 | 26655 | 28046 | 24747 | |
| Introns/gene | 2.6 | 2.5 | 2.5 | 2.9 | |
| Average intron length (base) | 80.5 | 82.4 | 83.1 | 118.5 | |
| Average intergenic distance (bp) | 3013 | 2319 | 2398 | 3398 | |
| Repeat sequences | |||||
| DNA Transposon | 8.04% | 4.27% | 6.89% | 7.64% | |
| LINE | 0.39% | 0.97% | 0.44% | 1.56% | |
| SINE | 0.07% | 0.11% | 0.07% | 0.15% | |
| LTR Retrotransposon | 17.06% | 7.84% | 10.56% | 27.41% | |
| Unclassified nonLTR-Retrotransposon | 0.02% | 0.03% | 0.00% | 0.07% | |
| Unclassified Retrotransposon | 2.56% | 3.35% | 2.69% | 2.54% | |
| Unclassfied | 0.22% | 0.60% | 0.42% | 1.36% | |
| Total TE class | 28.21% | 16.68% | 20.88% | 40.33% | |
| Simple sequence repeats | 0.90% | 1.03% | 0.94% | 2.57% | |
| Total excl. Tandem repeats | 29.16% | 17.79% | 21.89% | 43.07% | |
| Tandem repeats | 8.27% | 5.05% | 7.38% | 11.46% | |
| Total repeat coverage | 31.23% | 20.09% | 24.00% | 45.64% | |
a mitochondrial
Phylogenetic relationship
Phylogenetic analysis using multilocus DNA sequence data (concatenated α-tubulin, β-tubulin and ITS sequences) identified R. secalis as the closest sister species to R. commune, whereas the CCG species R. orthosporum is more distantly related [15, 17]. This relationship was supported in the present study by comparing the Rhynchosporium rDNA gene structures (Fig. 1). The genes of R. commune and R. secalis are highly similar lacking any introns, whereas an intron is present in the 28S gene of R. agropyri. The CCG species R. orthosporum and the recently described R. lolii [16] carry two introns of the same size in the 18S gene and single introns differing in length by one nucleotide in the 28S gene. The latter are homologous to the 28S intron of R. agropyri and located at the same site, but their sequences are 99 bp and 87 bp shorter at the 5′ and the 3′ side, respectively. In contrast to these results, phylogenetic analysis based on concatenated sequences of 13 core mitochondrial genes resulted in R. agropyri and R. secalis as being most closely related [30].
Fig. 1.
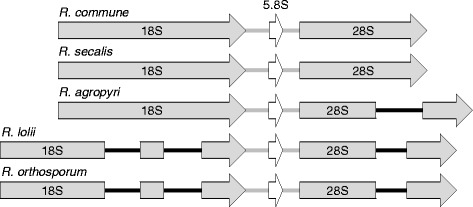
Rhynchosporium rDNA structures. The sequences of the rDNA regions of all Rhynchosporium species were obtained by Sanger sequencing. 18S, 5.8S and 28S genes are separated by ITS1 and ITS2, respectively (grey lines). The introns (black lines) in the 18S and 28S genes of R. lolii and R. orthosporum are identical except for 1 additional nucleotide in the R. lolii 28S intron. The 28S intron of R. agropyri is highly similar to those of the CCG species (87% identity) except for 5′- and 3′-terminal extensions of 99 and 87 bp, respectively
To better resolve the Rhynchosporium phylogeny a two-step analysis was carried out in the present study. The first step aimed at confirming the general position of the Rhynchosporium genus in the Leotiomycetes class of Ascomycota [34]. To this end, the sequences of three protein-coding genes (EF1-α, RNA polymerase II subunits RPB1 and RPB2) and of almost complete rDNA regions (18S rDNA, ITS1-5.8S rDNA-ITS2, 28S rDNA; [35]) from 12 members of the Leotiomycetes class were used in lack of complete genome sequences of these fungi other than Botryotinia fuckeliana [36]. The resulting phylogeny shows the Rhynchosporium BCG branch with R. commune and R. agropyri as the closest sister species alongside the CCG branch (Fig. 2a, Additional file 1: Figure S1). In a second step, to resolve the BCG evolution in more detail, single nucleotide polymorphisms (SNPs; Additional file 2: Table S1) were identified after aligning the genomes of BCG species and isolates (Fig. 2b). The phylogenetic tree inferred from the concatenated SNPs again confirms that R. agropyri branched off “shortly” before R. commune and R. secalis diverged. According to the published evolutionary time scale the former occurred 1200–3600 years ago [8, 9, 16], the latter about 500 years later. In contrast, the age of the 3 R. commune isolates is probably around 100 years or less.
Fig. 2.
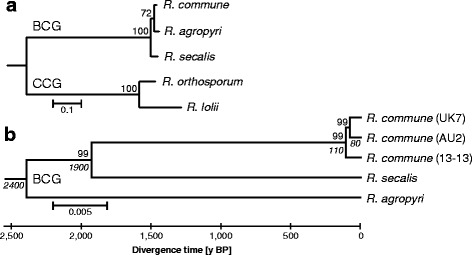
Rhynchosporium evolutionary relationships. a Rhynchosporium subtree of the Leotiomycetes phylogeny (cf. Additional file 1: Figure S1). The nucleotide sequences of 18S rDNA, 28S rDNA, ITS region, elongation factor EF1-a and RNA polymerase II subunits RPB1 and RPB2 were concatenated. b SNP-based phylogeny of the BCG species including the three R. commune isolates UK7, AU2 and 13-13. The evolutionary history was inferred using the Minimum Evolution method (optimal tree with sum of branch length = 1.022). The tree is drawn to scale. All nucleotide positions containing gaps and missing data were eliminated, leaving a final dataset of 5,904,161 positions. Scale: number of substitutions per site and SNPs per position, respectively. Bootstrap numbers are given above branching points, divergence times in italics below branching points. BCG, beaked conidia group; CCG, cylindrical conidia group
Using the amino acid sequences of the three proteins EF1-α, RPB1 and RPB2 the relationship of Rhynchosporium to more distantly related plant-interacting fungal species was established (Fig. 3). The closest relative of Rhynchosporium spp., the poplar pathogen M. brunnea f.sp. ‘multigermtubi’ [33], belongs to the Dermataceae, a family of one of the most diverse ascomycete orders, the Helotiales. This confirms a previous ITS-based phylogenetic analysis, which located R. commune and R. orthosporum between the Dermataceae species Tapesia yallundae (syn. Oculimacula yallundae) and Pyrenopeziza brassicae, pathogens of grasses and oil seed rape (Brassica napus), respectively [34].
Fig. 3.
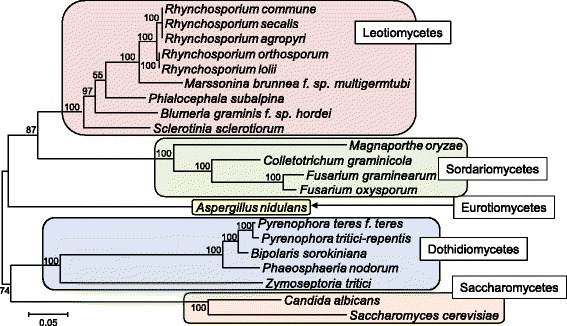
Integration of the genus Rhynchosporium into the fungal systematics. The concatenated amino acid sequences of elongation factor EF1-a and of the polymerase II subunits RPB1 and RPB2 from 21 taxa were used to construct the phylogenetic tree. Numerals on the nodes represent the percentages from 500 bootstraps. Scale: number of substitutions per nucleotide
All host species of Rhynchosporium belong to the Pooideae subfamily of Poaceae [37, 38]. However, while the fungal BCG species, R. commune, R. secalis and R. agropyri, infect closely related grass species from genera of the subtribe Hordeinae (supertribe Triticodae, tribe Hordeeae), the CCG members, R. lolii and R. orthosporum, are pathogenic to species from the more distantly related subtribes Loliinae and Dactylidinae (supertribe Poodae; Fig. 4). Nevertheless there are exceptions to this rule. Firstly, from Bromus diandrus, a grass species belonging to the Bromeae tribe of the Triticodae supertribe, a few isolates were collected that grouped into the R. commune lineage [17]. Interestingly, in an early report isolates from brome grass were unable to infect other grass species tested including barley [1]. Secondly, triticale became susceptible to Rhynchosporium about 30 years after its introduction as a crop [39]. This cereal species originated from hybridizing the non-host species wheat and the host species rye and was assigned to the Triticinae subtribe of the Hordeae tribe (supertribe Triticodae). Rhynchosporium isolates collected from triticale grouped into the R. secalis lineage [17]. Lastly, two strains of R. commune were isolated from Italian ryegrass (Lolium multiflorum) that were also pathogenic to barley [16] and, thus, able to colonize hosts from both Triticodae and Poodae supertribes. Despite these exceptions, however, the relationship found between the Rhynchosporium species corresponds largely to host systematics.
Fig. 4.
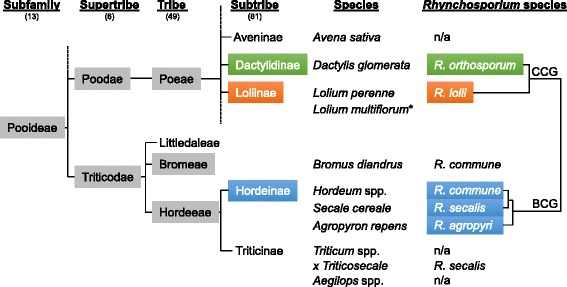
Rhynchosporium species and their hosts from the Poaceae family. The Rhynchosporium systematics is contrasted with the section of the grass systematics [37] containing all known host species. Numbers in brackets indicate the total number in this order. *Two R. commune isolates were described to be able to cross the Poodae-Triticodae border, being pathogenic to Lolium multiflorum and Hordeum vulgare [16]. BCG: beaked conidia group, CCG: cylindrical conidia group, n/a: not a host species
Genes related to sexual reproduction
Work since the 1930s has failed to demonstrate a sexual cycle in any Rhynchosporium species. Availability of the Rhynchosporium genomes allows now a genomic interrogation to assess whether the genetic machinery required for sexual reproduction is present or not. Such insights into the pathogen life cycle are important as this impacts on the possibility for recombination and evolution of the pathogen [40]. Despite the apparent absence of a teleomorph R. commune isolates have been shown to possess the mating type loci MAT1-1 or MAT1-2, which are typical for heterothallic ascomycetes [24]. MAT1-1 is characterized by genes coding for proteins with two different DNA-binding domains, an α-box motif in MAT1-1-1 and a high-mobility group (HMG) DNA-binding motif in MAT1-1-3. Both loci were also found in the other BCG species, R. secalis and R. agropyri, whereas the CCG species R. orthosporum and R. lolii lack MAT1-2 [27]. In the present study, only the R. agropyri isolate had the MAT1-2 genotype, whereas all others carried the MAT1-1 locus. Closer inspection of the MAT1-1 locus revealed the presence of an additional ORF coding for a protein with about 30% similarity to the MAT1-1-5 protein from B. cinerea and several Sclerotinia species (Fig. 5). An additional ORF with homology to a hypothetical gene from the poplar pathogen M. brunnea f.sp. ‘multigermtubi’ was detected at the MAT1-2 locus. However, this ORF codes for a protein with no homology to a functional protein from other organisms. Sequence comparison of the MAT loci demonstrated that the idiomorphic region comprises most of the MAT1-1-1 gene and the genes MAT1-1-5 and MAT1-1-3 in the MAT1-1 locus (c. 4150 bp). In MAT1-2 it spans from the truncated MAT1-1-1 gene to the 3′ end of the MAT1-2-1 gene (c. 3600 bp). Both MAT loci are flanked by the genes SLA2 and APN2 coding for a cytoskeletal protein and a DNA lyase, respectively. The positions of these genes are highly conserved across Pezizomycotina species [41–44].
Fig. 5.
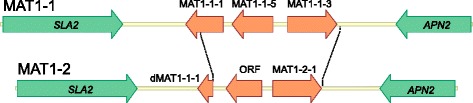
Rhynchosporium MAT gene loci. MAT1-1, R. commune UK7 (13,701 bp); MAT1-2, R. agropyri (12,801 bp). Dotted lines mark the idiomorphic regions. The genes flanking the MAT loci code for a cytoskeletal protein (SLA2) and a DNA lyase (APN2), respectively
Meiosis is “at the heart of sexual reproduction” of Eukaryotes [45]. Therefore, searching for genes involved in meiosis and sexual reproduction can identify the capacity for cryptic sexuality. To this end, the Rhynchosporium genomes were screened using a “meiosis detection tool kit” [46], which comprises a set of genes defining a “core meiotic recombination machinery” (Table 3, cf. Additional file 3: Table S2A) [47, 48]. The products of these genes are involved in generating meiotic double-strand breaks, crossing over and cohesion of sister chromatids and homologous chromosomes. Bidirectional BLASTp analysis detected 26 out of the 29 core meiotic genes in the Rhynchosporium (with the exception of R. lolii) genomes with sequence similarities mostly exceeding 50%. For two additional proteins, HOP2 and MND1, the similarity to Rhynchosporium proteins is low (c. 30%), although the functional domains were identified. Many genes required for meiosis also have a role in mitosis. However, nine meiosis-specific genes are regarded to reliably predict the capacity of a sexual cycle [48]. Three of these genes that code for elements of the synaptonemal complex, HOP1, HOP2 and MND1, are missing in Rhynchosporium (HOP1) or their products show only low similarity with Rhynchosporium proteins (HOP2, MND1). Nevertheless this does not argue against sexual reproduction, because these genes are also absent from the genomes of several fungi with known sexuality (Neurospora crassa, Gibberella zeae, Magnaporthe grisea, Ustilago maydis) and of other sexual organisms such as Drosophila melanogaster [48]. Hence, meiosis appears to be possible without these gene products. Transcripts for all but two (DMC1, MSH5) of the 26 core meiotic genes have been identified in germinated conidia and/or cDNA libraries from infected barley leaves (Table 3). This confirms that the core meiotic genes are not only present in the Rhynchosporium genomes but are actively transcribed.
Table 3.
Core meiotic genes
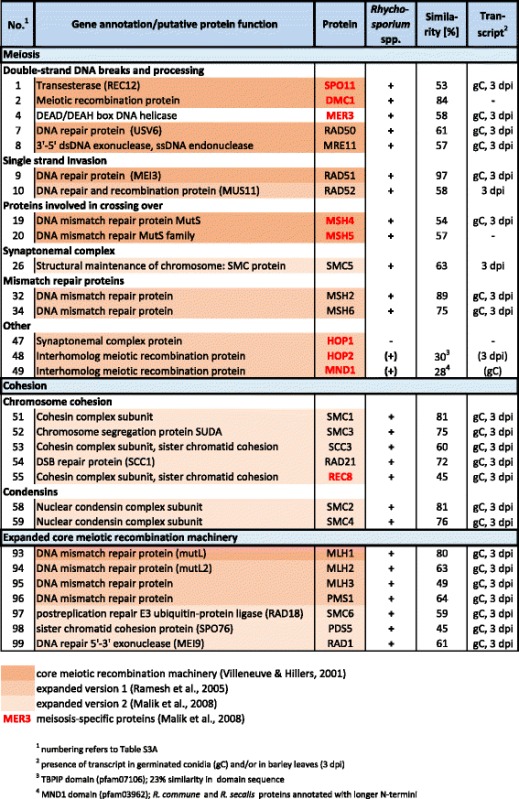
Of an additional set of 70 genes involved in different aspects of meiosis and recombination in N. crassa, Saccharomyces cerevisiae and Podospora anserina, 68 were found to be maintained in the genomes of R. commune, R. agropyri and R. orthosporum (Additional file 3: Table S2A). Two genes, KEX1 and RAM1, were missing only from the genome of R. secalis. Transcripts for 63 of these genes, including those coding for meiotic recombination protein REC4 and all pheromone proteins essential for mating, have been identified in germinated conidia and/or cDNA libraries from infected barley leaves. Finally, 74 genes known to be involved in different aspects of sexual reproduction in the aspergilli such as sensing of environmental signals that control sexual cycle induction, intracellular signal transduction, transcription control and ascospore production were examined (Additional file 3: Table S2B; [49]). Outside the MAT genes and the genes coding for pheromones and their receptors, only 5 genes were not found in the Rhynchosporium genomes, whereas for 12 gene products a relatively weak similarity with the A. nidulans proteins (<40%, 4 with <30%) was observed. Transcripts for 64 of the 65 genes, including MAT1 and esdC, have been identified in germinated conidia and/or cDNA libraries from infected barley leaves. Taken together these results provide strong additional evidence for the presence and expression of the genetic machinery required for sexual reproduction to occur in the Rhynchosporium life cycle.
Enzymes involved in cell wall degradation
Plant cell walls are built up of polysaccharides (cellulose, hemicellulose, pectin), (glyco-) proteins and aromatic polymers (lignin). Phytopathogenic fungi need to degrade this barrier to penetrate and colonize host tissues as well as to release nutrients for colony growth. To this end, they produce many carbohydrate-active enzymes (CAZymes) that cleave or modify oligo- and polysaccharides as well as other glycoconjugates. A recent genome comparison comprising 103 fungal species from different classes revealed a great diversity in the cell wall-degrading capacity [50]. In phytopathogens, CAZyme numbers correlate by and large with the fungal life style. In both hemibiotrophic and necrotrophic fungi large numbers of CAZymes are found, whereas obligately biotrophic fungi such as Blumeria graminis have smaller numbers. Furthermore, polygalacturonases of the GH28 family have been found to be less abundant in monocot- as compared to dicot-infecting fungi. When the CAZy database [51, 52] was used to annotate the Rhynchosporium BCG genomes, R. commune, R. secalis and R. agropyri had CAZyme numbers characteristic for monocot-infecting hemibiotrophic and necrotrophic fungi [50] (Additional file 4: Figure S2, Additional file 5: Table S3).
To reach their substrates, the cell wall-degrading enzymes (CWDEs) need to be secreted to the apoplast. In addition to the classical signal peptide-based secretion pathway through the ER/Golgi system, mechanisms independent of this system exist in Eukaryotes for unconventional secretion of proteins lacking a signal peptide [53, 54]. A sequence-based prediction method, SecretomeP, has been developed that is capable of identifying proteins secreted through the latter pathway [55]. Therefore, the Rhynchosporium CAZymes were tested for the presence of a classical N-terminal secretion signal and for the likelihood of unconventional secretion. Furthermore, enzyme numbers were corrected for modular enzymes, which are characterized by more than one annotation. In most cases (28 out of 39 proteins), an enzyme domain was combined with one or more carbohydrate binding domains (CBMs). As a result, about 330 CWDEs were identified for each Rhynchosporium species (Table 4, Additional file 5: Table S3, Fig. 6a). They group into glycoside hydrolases (GH, c. 54%), carbohydrate esterases (CE, c. 19%), polysaccharide lyases (PL, c. 3%), and auxiliary redox enzymes involved in lignocellulose breakdown (AA, c. 18%).
Table 4.
Cell wall degrading enzymes
| CAZy family | R. commune | R. secalis | R. agropyri | |||
| Number | [%] | Number | [%] | Number | [%] | |
| GHa | 182 | 53.8 | 176 | 53.7 | 183 | 54.3 |
| CEb | 66 | 19.5 | 62 | 18.9 | 64 | 19.0 |
| PLc | 9 | 2.7 | 9 | 2.7 | 9 | 2.7 |
| CBMd only | 20 | 5.9 | 20 | 6.1 | 20 | 5.9 |
| AAe | 61 | 18.0 | 61 | 18.6 | 61 | 18.1 |
| Ʃ | 338 | 99.9 | 328 | 100.0 | 337 | 100.0 |
| CWDE substrate | R. commune | R. secalis | R. agropyri | |||
| Number | [%] | Number | [%] | Number | [%] | |
| Cutin | 14 | 4.1 | 14 | 4.3 | 14 | 4.2 |
| Cellulose | 64 | 18.9 | 64 | 19.5 | 64 | 19.0 |
| Hemicellulose | 107 | 31.7 | 101 | 30.8 | 108 | 32.0 |
| Lignin | 18 | 5.3 | 18 | 5.5 | 18 | 5.3 |
| Pectin | 27 | 8.0 | 27 | 8.2 | 27 | 8.0 |
| FCWf | 70 | 20.7 | 68 | 20.7 | 70 | 20.8 |
| unknown | 38 | 11.2 | 36 | 11.0 | 36 | 10.7 |
| Ʃ | 338 | 99.9 | 328 | 100.0 | 337 | 100.0 |
aglycoside hydrolases
bcarbohydrate esterases
cpolysaccharide lyases
dcarbohydrate-binding modules
eauxiliary activities
ffungal cell wall
Fig. 6.
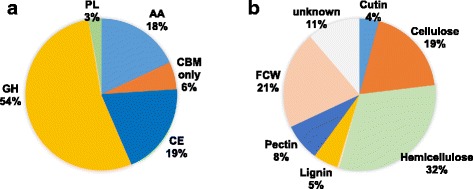
Cell wall-degrading enzymes of R. commune. a Genes coding for secreted CAZyme. AA, auxiliary activities, CBM only, proteins with carbohydrate-binding modules but lacking known enzyme activities, CE, carbohydrate esterases, GH, glycoside hydrolases, PL, polysaccharides lyases. b Genes coding for enzymes that target the different cell wall components
In addition to enzymes, proteins possessing one or more carbohydrate-binding modules but lacking domains for enzyme activity were found (CBM only, c. 6%). Among these the LysM motif (CBM50) has attracted most attention [56]. This module has general N-acetylglucosamine binding properties and is therefore able to bind to chitin or chitin-like carbohydrates [57]. In fungi, LysM domains are predominantly found in secreted LysM effector proteins and in subgroup C chitinases. In the Rhynchosporium genomes seven proteins are encoded that contain one or more LysM domains (LysM type A [57]; LysM1, LysM5 and LysM7 with one domain, LysM2 with two domains, LysM3 with three domains, LysM4 and LysM6 with four domains). In addition, two genes code for enzymes that contain LysM domain pairs (Additional file 6: Table S4A), a subgroup C chitinase (CAZy family GH18; LysM type B; see below), which also possesses a different chitin-binding motif (CBM18), and a putative peptidoglycan lytic transglycosidase (CAZy family GH23). Four of the LysM proteins are secreted through the classical pathway, whereas three of the proteins, LysM5, LysM6 and LysM7 and the GH23 enzyme may be secreted via the unconventional pathway. A central transmembrane domain predicted for LysM7 may cause this protein to reside in the plasma membrane with the LysM domain oriented outwards. Interestingly, in M. brunnea a family of 24 genes was identified that code for effector proteins of 142–151 aa with 1 or 2 LysM motifs [58]. This family is completely missing in Rhynchosporium. Two representative members of this gene family were shown to weaken the chitin-induced defense response when expressed in Arabidopsis thaliana. As proposed for the effector proteins ECP6 from Cladosporium fulvum [59], Mg3LysM from Mycosphaerella graminicola (Zymoseptoria tritici; [60]) and Slp1 from Magnaporthe oryzae [61], this effect is probably due to the sequestration of chitin oligosaccharides that occur as breakdown products of fungal cell walls during invasion and that are able to trigger host immunity. Other LysM effectors including Avr4 from C. fulvum [62], Mg1LysM and Spl1 prevented hyphal lysis by plant chitinases [60, 61].
When the Rhynchosporium CWDEs were assigned to their putative substrates (cellulose, hemicellulose, pectin, lignin, other polysaccharides), about 21% were found to act or are likely to act on the fungal cell wall, whereas c. 64% presumably target the host cell wall (Table 4, Additional file 5: Table S3, Fig. 6b). For about 11% of the enzymes their substrate is unknown or ambiguous. Fourteen enzymes (4%) are putative cutinases. These enzymes, albeit no proper CWDEs, are required at a very early stage of pathogenesis to allow or facilitate the fungal penetration of the plant surface.
The physiological role of the CAZymes targeting the fungal cell wall is thought to be in cell wall remodeling during fungal morphogenesis (septum formation, hyphal branching, spore germination). The cell walls of filamentous fungi are mainly built up of β-1,3-glucan (65–90%), glycoproteins (20–30%) and chitin (10–20%; [63]). Accordingly, glucan-degrading enzymes constitute the biggest group of hydrolytic enzymes encoded in the Rhynchosporium genomes with at least 22 members (CAZy families GH17, GH55, GH81, GH128, GH72, possibly supplemented by members of families GH3, GH16 and GH131 [64]). Eight enzymes presumably target glycoproteins (GH20, GH114, GH125; Additional file 6: Table S4). In addition, 17 chitinolytic enzymes were identified (GH18, GH75, GH89; CE4; Additional file 6: Table S4). Eight of the 14 chitinases (GH18) belong to subgroup A (40–50 kDa, no CBMs; [65]). The biggest of these chitinases is predicted for unconventional secretion. One member of this subgroup was found to lack a signal peptide and, hence, to be located intracellularly (cf. [66]). Three chitinases belong to subgroup B (30–90 kDa, CBMs), but two additional enzymes may fall into this group despite their high molecular mass (c. 120 kDa). Subgroup C chitinases were defined to have a high molecular mass (140–170 kDa) and to carry a chitin-binding domain (CBM18) and LysM motifs. These criteria are fulfilled by one Rhynchosporium enzyme of almost 150 kDa. This enzyme may not be fully released into the apoplast, but rather tethered to the outside of the plasma membrane as suggested by a predicted C-terminal transmembrane helix.
In Trichoderma atroviride the gene coding for subgroup C chitinase TAC6 is located next to the gene encoding the TAL6 protein, which contains 7 LysM motifs and was considered to inhibit spore germination [67]. TAC6 appears to be inactivated by a deletion in the catalytic domain and hence to be a chitin-binding protein. Interestingly, a tac6 deletion mutant was reported to grow faster than the wild type, suggesting a growth decelerating function of the protein and thus a function in the regulation of fungal development. Similar to T. atroviride, the subgroup C chitinase-encoding gene of R. commune is located next to a gene coding for a protein with 4 LysM motifs, LysM4. Furthermore, chitinase-LysM gene pairs are also found for both high molecular mass subgroup B chitinases. Their genes are located next to the genes coding for LysM1 and for a protein that lacks a LysM domain but has high similarity to a LysM protein from Zymoseptoria brevis [68], respectively. This chitinase-LysM gene arrangement may therefore point to the participation of LysM proteins in a more wide-spread mechanism to control fungal growth in planta.
Regrouping of the plant CWDEs according to the different types of cell wall substrates suggested that c. 32% are involved in hemicellulose degradation and about 19% target cellulose (Fig. 6b). The enzymes acting on pectin (8%) and lignin (5%) constitute smaller groups, thus reflecting the low content of these materials in the cell walls of grasses. Due to their large number and the functional redundancy of enzyme activities involved in cell wall degradation, the likelihood is generally presumed to be small that any one of these enzymes is responsible for host specificity. Recently, however, a combination of comparative genomics and transcriptomics revealed that members of CWDE families from Z. tritici are differentially transcribed during pathogenesis [69]. This suggested the specific occurrence of at least some CWDEs during relevant stages of the fungal life cycle rather than their general functional redundancy. In addition, selection analysis yielded evidence that a few cutinases, cellulases and hemicellulases may be involved in host adaptation or evasion of host recognition.
Based on the genome sequences, a very coarse intra-genus comparison was done for the Rhynchosporium CAZymes, which considered the presence or absence of genes in a family. In several of the CAZy families (e.g., arabinose family GH93, poorly characterized family GH89 [70]) single members are missing from one of the three BCG species. It remains to be shown however whether these differences reflect differences in the composition of the targeted cell walls and, thus, adaptations to the host.
Secondary metabolism
Generally, secondary metabolites can be regarded as tools that play a role in the interaction of the producing organism with its environment. Fungal secondary products are not only notorious toxins, but also mediators of chemical communication with other microbes and the environment or defense agents [71]. Hence, they also play a more or less prominent role in the interaction of phytopathogenic fungi with their host plants. Most fungal secondary metabolites belong to one of four structural classes, polyketides, non-ribosomal peptides, cyclic terpenes or tryptophan-derived indole alkaloids [72]. Among the fungal compounds with known effect in plant-pathogen interactions are several of the host-specific toxins of the Dothideomycetes (for review s. [73]). T-toxin from Cochliobolus heterostrophus and PM-toxin from Mycosphaerella zeae-maydis are linear polyketides. Victorin from Cochliobolus victoriae, AM-toxin from Alternaria alternata and HC-toxin from Cochliobolus carbonum are cyclic non-ribosomal peptides, whereas HS-toxin from Bipolaris sacchari is a sesquiterpene galactoside. Several fungi produce perylenequinone toxins (e.g. cercosporin from Cercospora spp.). These are cyclic polyketides with non-host-specific activity. Some secondary metabolites have a less pronounced effect in the interaction with host plants. Examples are the polyketide botcinic acid and the sesquiterpene botrydial, which play a redundant role in the virulence of Botrytis cinerea on bean leaves [74]. Finally, many secondary products have an impact on human and livestock health; these include the ergot indole alkaloids with their tryptophan-derived lysergic acid moiety [75] or the sesquiterpenoid trichothecenes [76].
The only secondary metabolite described to date from a Rhynchosporium species is orthosporin, a polyketide from R. orthosporum [77]. This isocoumarin was also isolated from Drechslera siccans, a pathogen of oats (Avena sativa), perennial ryegrass (L. perenne) and Italian ryegrass (L. multiflorum). It is phytotoxic causing necrosis on leaves of several plants, but not on perennial ryegrass [78], and it has an inhibitory effect on root growth of the host plant, orchard grass (D. glomerata) and lettuce (Lactuca sativa).
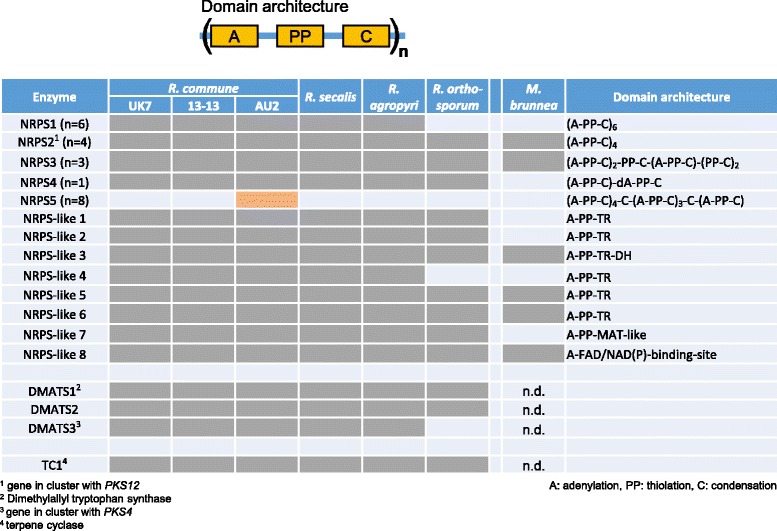
The four major fungal secondary metabolite classes are synthesized via four characteristic key enzymes [72, 79], polyketide synthases (PKS), non-ribosomal peptide synthetases (NRPS), terpene cyclases (TC) or dimethylallyl tryptophane synthases (DMATS). In addition, several hybrid compounds are known, which are either synthesized by PKS-NRPS hybrid enzymes or by two separate enzymes that are encoded in a single gene cluster. The Rhynchosporium genomes were screened for the presence of these key enzyme genes to find clues for their possible involvement in determining host specificity. In almost all isolates/species three DMATS genes and one TC gene were identified, whereas DMATS3 was missing only in R. orthosporum, suggesting that a host-specific role of these enzymes is unlikely.
PKS genes constitute the largest group of key enzyme genes of secondary metabolism. PKS catalyze the synthesis of a large and structurally diverse group of chemical substances from acyl-CoA precursors. Fungal PKS are usually multidomain enzymes that elongate and modify their products iteratively (type I PKS; [80]). Three domains are essential for polyketide synthesis, ketoacyl CoA synthase (KS), acyltransferase (AT) and a phosphopantetheine attachment site (PP). Several other enzymatic motifs such as the three β-keto processing domains ketoreductase (KR), dehydratase (DH) and enoyl reductase (ER) and a methyl transferase domain (MT) are not found in all PKS. Lack of all or some of the reducing domains yields non-reducing or partially reducing PKS. The polyketide product is usually released from the enzyme through thioreductase (TR), thioesterase (TE) or transferase activities. Twelve PKS genes (PKS1-12) including 2 PKS-NRPS genes (PKS1, PKS11) occur in all BCG species (Table 5). In contrast, PKS13 was not found outside R. commune isolates and appears to be species-specific, whereas an additional gene, PKS14, was present only in the genome of R. commune isolate AU2. Orthologs of eight of the PKS genes were also identified in R. orthosporum along with four unique genes including two PKS-NRPS hybrid genes (PKS15, PKS16). In the closely related M. brunnea, orthologs of only 6 Rhynchosporium genes were identified along with two unique PKS genes. As the conclusion, the specific occurrence of several of the genes may suggest their involvement in the biosynthesis of polyketides with a role in interaction specificity at the species and isolate level.
Table 5.
Polyketide synthases
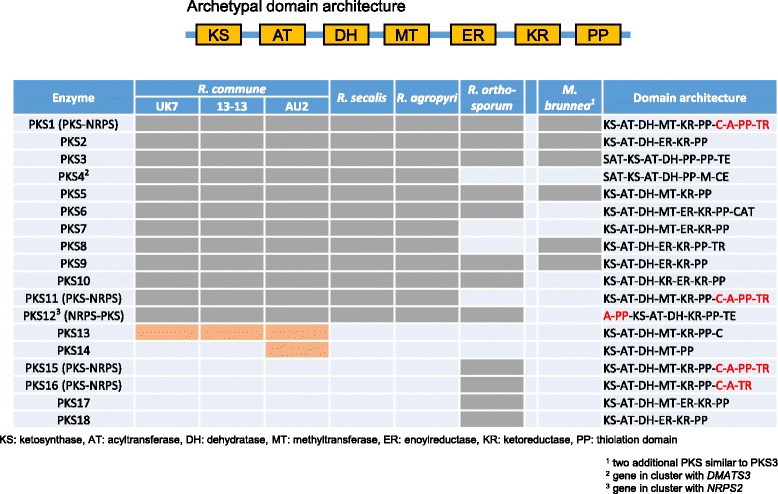
The phylogeny of type I PKS from ascomycetes and bacteria as inferred from KS domain relationship revealed three main clades comprising fungal reducing PKSs, fungal non-reducing (NR) PKSs and bacterial PKSs [81]. The fungal clades are further subdivided into four reducing subclades (I-IV), three NR subclades (NR I-III) and a subclade basal to NR subclades I and II (NR b I + II). In addition, the bacterial PKS clade contains two subclades of fungal PKS. This phylogeny was first reconstructed based on the KS domain sequences from 86 ascomycete PKS (including eight from M. brunnea [33]), ten bacterial PKS and two animal fatty acid synthetases. Subsequently, it was complemented with the KS domain sequences of the 18 Rhynchosporium PKS (Fig. 7), which were predicted by BLASTp analysis [82] and comparison to the PFAM 28.0 database (Table 5; [83]). The archetypal PKS domain architecture KS-AT-DH-MT-ER-KR-PP [81] was found in only three Rhynchosporium PKS, RcPKS6, RcPKS7 and RoPKS17 (Table 5), which are members of reducing subclades I and IV, respectively. Both subclades contain additional Rhynchosporium PKS lacking the MT domain (I: PKS8, IV: PKS10, PKS18). Most Rhynchosporium PKS were in reducing subclade II, which lacks the ER domain and contains all PKS-NRPS hybrid enzymes. Notably, R. commune-specific PKS13 appears to be closely related to LovB, one of the enzymes involved in the biosynthesis of lovastatin [84] in Aspergillus terreus (47.3% identity, 63.5% similarity, 6.7% gaps), suggesting that both enzymes catalyze similar reactions. Lovastatin is an inhibitor of 3-hydroxy-3-methylglutaryl-CoA reductase, which catalyzes a crucial step in the biosynthetic pathway leading to ergosterol (antifungal properties) or cholesterol (hypolipidemic agent; [85]). Two Rhynchosporium enzymes, PKS3 and PKS4, occur in the non-reducing clades II and III, respectively. Like all other Rhynchosporium PKS, however, the latter contain the DH reduction domain and therefore should be called partially reducing PKSs. Finally, PKS12 groups with C. heterostrophus PKS24 in the bacterial PKS clade, both being NRPS-PKS hybrids (cf. also NRPS below) and suspected of being horizontally transmitted from bacteria to fungi [86].
Fig. 7.
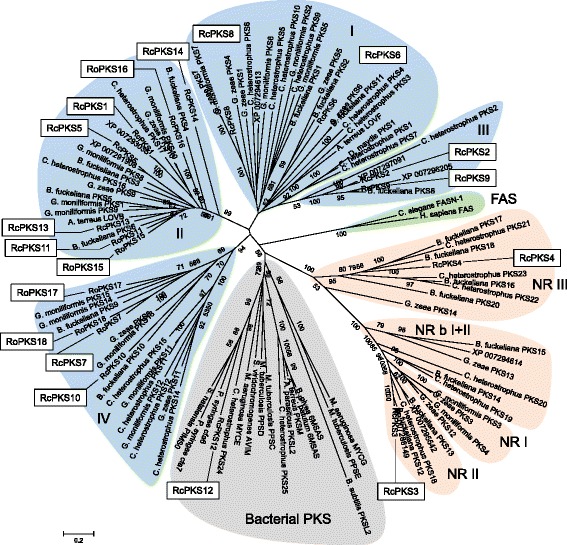
PKS phylogenetic tree. The amino acid sequence of the KS domains from 114 fungal and bacterial PKS were used to construct the phylogenetic tree. Numerals on the nodes represent the percentages from 500 bootstraps. Numerals <50 were omitted. Scale: number of substitutions per nucleotide. Colored backgrounds indicate enzyme groups: blue, reducing PKS clades I-IV; orange, non-reducing PKS clades NRI-III and NR bI + II [81]; grey, bacterial PKS; green, fatty acid synthases (FAS)
Among the key genes of secondary biosynthesis in Rhynchosporium those coding for NRPS and NRPS-like proteins represent the second largest group. These enzymes typically consist of one or more modules comprising an amino acid-adenylation domain (A), a thiolation domain (PP) and a condensation domain (C). Mono-modular NRPS4, tri-modular NRPS3 and tetra-modular NRPS2 were present in all species, whereas hexa-modular NRPS1 was missing in R. orthosporum (Table 6). Remarkably, R. commune isolate AU2 was again unique by carrying a specific gene coding for the most complex enzyme, the octa-modular NRPS5. Orthologs of the eight genes coding for NRPS-like (NPSL) proteins were found in all Rhynchosporium species with the exception of NPLS4, which was missing in R. orthosporum. In comparison to Rhynchosporium, closely related M. brunnea had orthologs of only six of the NRPS and NPSL genes. Only one NRPS gene shows isolate-specific occurrence indicating a possible role of its product in host specificity at the isolate level. In addition, several genes may be involved in BCG specificity.
Table 6.
Nonribosomal peptide synthetases
Neither structures nor functions of the NRPS products in Rhynchosporium spp. are known. Therefore, the domain structures and, in particular, the A domains of the Rhynchosporium NRPS and NRPS-like proteins were compared to those of other fungi and bacteria. In a previous phylogenomic analysis of fungal NRPS and NRPS-like (NPSL) proteins [86] two main enzyme groups had been identified, one group containing seven subfamilies of primarily mono- and bi-modular fungal enzymes along with the bacterial clade (Fig. 8), the other group containing two subfamilies of primarily multi-modular and exclusively fungal (Euascomycetes, EAS; Fig. 9) enzymes. Members of the latter are presumed to be of more recent origin, most of them having less conserved structures suggesting the biosynthesis of products with more specific functions in niche adaptation including virulence. In contrast, members of the former group are presumably of more ancient origin, exhibiting more conserved domain architectures suggestive of more basic functions.
Fig. 8.
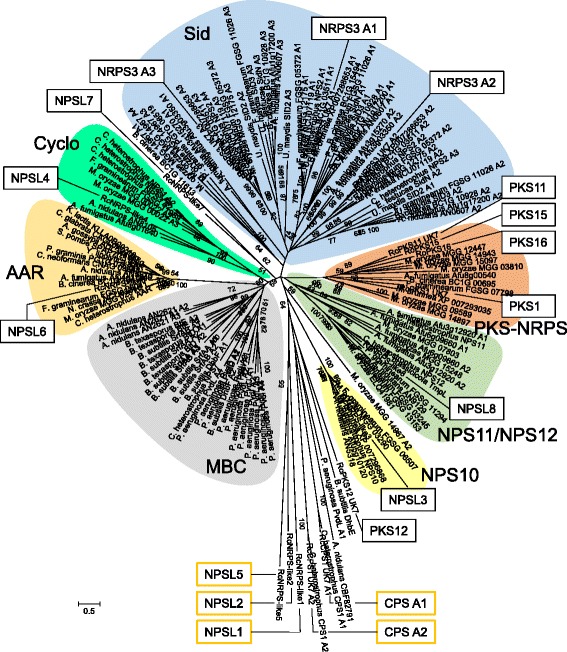
NRPS phylogenetic tree 1. The amino acid sequences of the A domain from 153 mostly mono-modular fungal and bacterial NRPS were used to construct the phylogenetic tree. Numerals at the nodes represent the percentages from 500 bootstraps. Numeral <50 were omitted. Scale: number of substitutions per nucleotide. Colored backgrounds indicate enzyme groups: Sid, siderophore synthetases; NPS11/NPS12, NPS11/12-like NRPS and ETP toxin synthetases; Cyclo, cyclosporine synthetases; MBC, major bacterial clade; AAR, α-amino-adipate reductases; NPS10, NRPS 10-like NRPS; PKS-NPS, hybrid enzymes. Orange-framed boxes mark members of outgroups
Fig. 9.
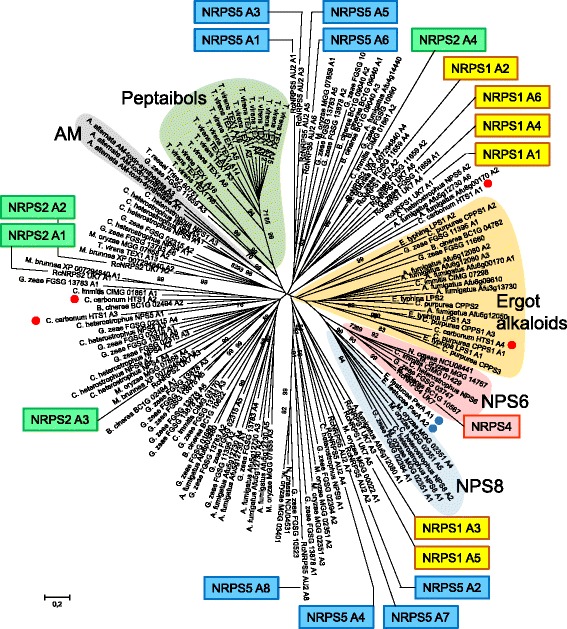
NRPS phylogenetic tree 2. The amino acid sequences of the A domain from 151 mostly oligo-modular specifically fungal NRPS were used to construct the phylogenetic tree. Numerals at the nodes represent the percentages from 500 bootstraps. Numerals <50 were omitted. Colored backgrounds indicate enzyme groups: blue, NPS8-like; pink, NPS6-like, orange, ergot alkaloid synthetases, green, peptaibol synthetase TEX1; grey, AM-toxin synthetase. Red and blue dots mark the modules of HC-toxin and peramine synthetase, respectively. Coloured boxes indicate modules of the 4 Rhynchosporium NRPS
Following the previous analysis [86], the A domains of NRPS from Rhynchosporium were compared to those of other fungi including the closely related species M. brunnea and bacteria. Most NRPS subfamilies contain at least one member from the Rhynchosporium genus. Two proteins, tri-modular RcNRPS3 and mono-modular RcNRPS4, are predicted to be involved in the biosynthesis of two types of Fe3+-chelating hydroxamate-type siderophores required for iron uptake and storage. RcNRPS3 and its M. brunnea ortholog (XP_007288653) group with type-IV siderophore synthetases [87] (Fig. 8). Each of its three A domains joins a sub-branch with one of the A domains of C. heterostrophus NPS2, a ferrichrome synthetase. Ferrichromes, which are involved in intracellular iron storage, are typically cyclic hexapeptides built up of three N 5-acetyl-N 5-hydroxy-L-ornithines (AHO) and three small amino acids, an obligatory Gly and one to two Ser, Gly or Ala [87]. RcNRPS3 and its M. brunnea ortholog share the domain architecture with C. heterostrophus NPS2 including the C-terminal C-PP-C-PP module, which is predicted to function in ring closure of the nascent peptide [88]. In addition, the amino acid residues suggested to determine the substrate specificity of the A domains are almost identical in both enzymes and very similar to those of the A domains of C. heterostrophus NPS2 (Table 7). The NRPS2 product, ferricrocin, is an intracellular siderophore involved in iron storage, oxidative stress resistance, germination and sexual development in A. nidulans and other fungi [89]. This molecule is made up of two glycines and one serine in addition to the three AHO [90]. According to the 10-aa code for amino acid binding, the A1 domain of the NRPS from the three fungi binds glycine and the A2 domain binds serine [87, 91]. In the A3 domains of RcNRPS3 and its M. brunnea ortholog an L239 → M exchange has occurred compared to the A4 domain of C. heterostrophus NPS2. Nevertheless, the domain appears to be responsible for AHO binding (10-aa code: 9/10, 13-aa code: 11/13, 17-aa key positions: 14/17). Therefore, despite the presence of the second presumably glycine-binding domain A3 in C. heterostrophus NPS2, the siderophore product of RcNRPS3 and its M. brunnea ortholog is predicted to be identical with the NPS2 product, ferricrocin.
Table 7.
Amino acid code of siderophore synthases
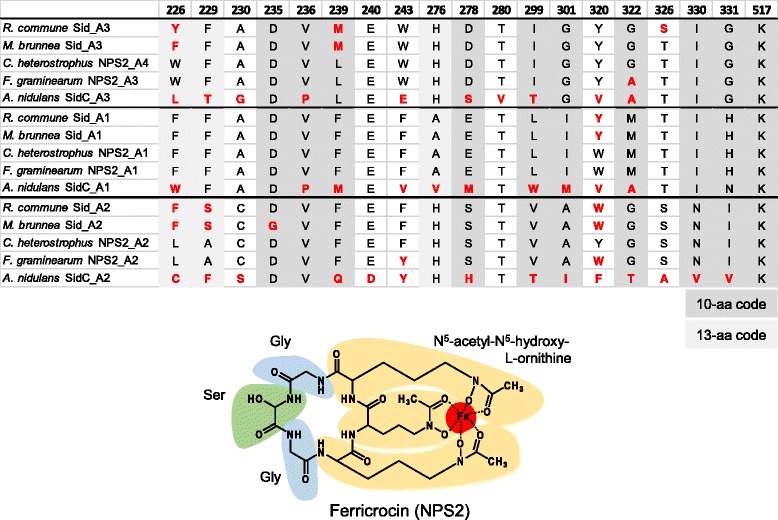
RcNRPS4 was in the most conserved clade of the EAS group exemplified by C. heterostrophus NPS6 [86] coding for a different type of siderophore synthetases (Fig. 9). No M. brunnea homolog was identified. RcNRPS4 has the typical domain structure of this enzyme group, a single A-PP-C module followed by a module with a degenerate A domain (dA-PP-C). NPS6 of C. heterostrophus was shown to be responsible for the synthesis of extracellular siderophores of the coprogen family [92]. Deletion of the NPS6 gene and its orthologs in different plant pathogenic fungi resulted in reduced virulence and hypersensitivity to H2O2.
Mono-modular RcNPSL3 and its M. brunnea ortholog belong to the NPS10 subfamily, which also has a conserved domain structure. The A-PP domain is followed by a thioester reductase (TR)-like domain and a dehydrogenase domain. RcNPSL4 has a single A-PP-TR module and groups together with mono-modular enzymes from A. nidulans (AN8105) and A. fumigatus (Afu8g01640) with the cyclosporin synthetases.
RcNPSL6 is in a clade with high bootstrap support that comprises α-aminoadipate reductases (AAR) such as Lys2 from S. cerevisiae and Lys1+ from S. pombe. AAR are evolutionary related to but not identical with NRPS and catalyze a step in the fungi-specific biosynthesis of lysine [93]. Neither RcNPSL7 nor B. cinerea protein BC1G_11613 fits into one of the clades described. Both proteins have the same domain architecture. The A and PP domains are followed by a domain that makes up the C-terminal half of the protein, is predicted to contain three pairs of transmembrane domains (TMDs) and is exclusively found in NRPS.
RcNPSL8 is in the same clade as C. heterostrophus NPS12 and Alternaria brassicicola TmpL (with high bootstrap support). These proteins lack thiolation (PP) and condensation (C) domains and, hence, are no true NRPS proteins. Instead, they are characterized by an A domain and a C-terminal FAD/NAD(P)-binding domain, which are separated by putative TMDs. The number of TMDs is predicted to be between 5 and 7 (depending on the exclusion or inclusion of two TMDs with low probability following TMD1). Therefore, membrane orientation of these proteins needs to be verified experimentally. TmpL was shown to be involved in virulence through regulation of intracellular ROS concentrations and tolerance to external ROS [94]. The biochemical function of the TmpL-like proteins is not known, but they are suggested to possess an enzymatic function involving FAD/NAD(P) binding like other flavoenzymes such as enzymes from the ferric reductase group.
The basic architecture of NPSL1-6 is very similar with A- and PP-domains followed by a TR domain. However, the A domains of RcNPSL3, RcNPSL4 and RcNPL6 are structurally similar to the A domains of RcNRPS1-5 (NCBI PSSM-ID CD05930), but deviate from those of RcNPSL1 (CD04433), RcNPSL2 (CD05907) and RcNPSL5 (CD05936). As a consequence, the latter along with RcPKS12 (NRPS-PKS hybrid, CD04433) form a phylogeny outgroup. Other outgroup A domains comprise the Rhynchosporium ortholog of CPS1, a protein consisting of two A domains (A1: CD05931, A2: CD05905), which was shown to be a virulence factor of C. heterostrophus and Gibberella zeae on maize and of C. victoriae on oats [95]. Furthermore, A domains from bacterial proteins, B. subtilis 2,3-dihydroxybenzoate-AMP ligase (DhbE, CD5920) and P. aeruginosa pyoverdine synthetase (PvdL A1, CD05931), as well as an A. nidulans NPSL protein, CBF82791 (CD05906), were used as outgroup.
In the mostly multi-modular EAS group (Fig. 9), little information can be deduced from the phylogenetic position of the A domains regarding the function of NRPS products. However, occurrence of the A domains of tri-modular AM-toxin synthetase from A. alternata or 14 of the 19 modules of peptaibol synthetase Tex1 from Trichoderma virens in a single subclade with high bootstrap support is suggestive of an evolutionary origin of the enzymes by duplication events. A similar mechanism may have played a role in the evolution of Rhynchosporium NRPS enzymes. Three of the A domains of hexa-modular RcNRPS1 occur in one subclade, two in another subclade and one in a third subclade. Similarly, the A domains of octa-modular RcNRPS5 are in three subclades with four, three and one members, respectively. In contrast, the A domains of tetra-modular RcNRPS2 and its M. brunnea ortholog are spread over four different subclades indicating a recombination-type mode of molecular evolution.
Finally, the four PKS-NRPS (PKS1, PKS11, PKS15, PKS16) from Rhynchosporium and the PKS1 ortholog from M. brunnea, which all belong to (partially) reducing PKS clade II (cf. PKS above), are found in the PKS-NRPS subfamily of the phylogenetic tree (Fig. 8). They all have the same domain architecture except for RcPKS16, which lacks the C-terminal PP domain (for review s. [96, 97]).
The basic structures formed by the key secondary biosynthesis enzymes are usually modified by decorating enzymes such as oxidoreductases, glycosyl or prenyl transferases. Their genes are frequently found in contiguous clusters, i.e., most or all genes of a biosynthetic pathway, possibly along with genes responsible for regulation and transport, are co-localized and co-regulated [98]. For instance, the cluster for the biosynthetic pathway of the polyketide asperfuranone in A. nidulans comprises genes coding for two fungal PKSs, five additional enzymes and a transcriptional activator [99]. Similarly, putative PKS gene clusters were detected in the Rhynchosporium BCG genomes (Additional file 7: Table S5). Two clusters contain genes coding for the key biosynthetic enzymes of two different secondary metabolite classes: the PKS4 cluster includes the DMATS3 gene (Fig. 10) and the NRPS2 cluster harbors the PKS12 gene. DMATS3 might be a transferase required for the prenylation of a PKS4-derived polyketide. Alternatively, the gene pairs might be involved in the biosynthesis of mixed compounds through separate assembly lines.
Fig. 10.
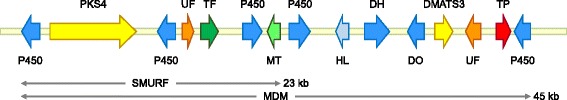
PKS4 gene cluster of R. commune. The cluster harbors genes coding for two secondary metabolism key enzymes, PKS4 and DMATS3, along with several decorating enzymes, a putative transporter and a regulatory gene. DH, dehydrogenase; DO, dioxygenase; HL, hydrolase; MT, methyl transferase; P450, cytochrome P-450 enzymes; TF, transcription factor; TP, transporter; UF, unknown function. SMURF and MDM, see text
Small secreted proteins: necrosis-inducing proteins
Secreted effector molecules have been recognized as crucial for host colonization and pathogenicity [100]. In both pathogens and mutualists they function by deregulating the host immune response. In addition, they have a role in self-defense and competition with other microbes [101, 102]. Furthermore, they influence the physiology of the host in favor of the pathogen’s needs, thus enhancing virulence [103]. Finally, they contribute to host adaptation and to establishing host specificity, thereby driving microbial speciation [104].
In R. commune, the genes NIP1, NIP2 and NIP3 code for effector proteins that contribute quantitatively to the virulence of R. commune depending on the host cultivar [105]. Therefore, the sequenced genomes of all Rhynchosporium species were screened for the presence of orthologous genes using the NIP gene sequences from R. commune isolate UK7 as probes. In this context, only those R. lolii genes that have homologs in R. orthosporum could be included. NIP1 and NIP3 occur as single genes in the genomes. NIP3 orthologs coding for highly similar proteins are present in the three BCG species, whereas NIP1 orthologs are found in R. commune, albeit not in all isolates (cf. [106]), and in the CCG species R. orthosporum (Fig. 11). NIP2 is also a single gene in the two CCG species. In contrast, NIP2 families of 7-10 members are present in the BCG species (NIP2.1-NIP2.11).
Fig. 11.
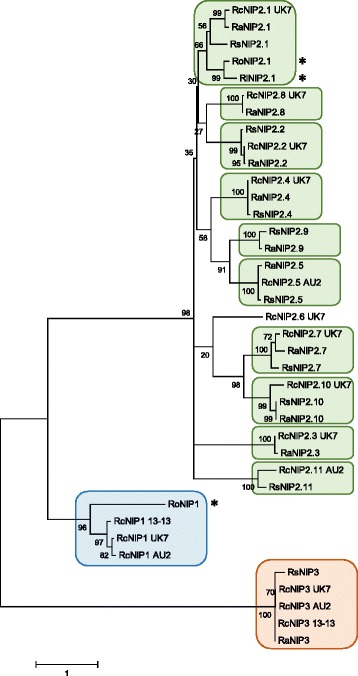
Phylogeny of NIPs. The amino acid sequences of 39 mature NIPs were aligned using the MUSCLE algorithm. For clarity reasons and due to their high similarity only one NIP2 protein from the 3 R. commune isolates was taken into consideration. The tree is drawn to scale. Numerals on the nodes represent the percentages from 500 bootstraps. Asterisks mark the CCG species. Scale: number of substitutions per nucleotide
Comparison of the deduced NIP2 amino acid sequences using Multiple Expectation Maximization for Motif Elicitation (MEME, version 4.9.1; [107]) revealed three conserved protein motifs, a 40-amino acid stretch spanning the N-terminal half of the protein, followed by a 15-amino acid domain around an absolutely conserved CRS motif, and near the C-terminus of about 50% of the proteins another 15-amino acid sequence. In addition, BLASTp analysis identified genes coding for proteins with similarity to NIP2 in the BCG genomes. These NIP2-like proteins (NLPs) differ from NIP2 by a smaller degree of domain conservation, but most strikingly by the absence of the CRS motif, which is absolutely conserved in NIP2 (Fig. 12). All NIP2 and NLP carry six cysteine residues at homologous positions. Interestingly, all proteins except NIP2.2 have a cysteine at the C-terminus. This structural feature was also found in NIP1. The phylogenetic tree of the mature NIP2 proteins demonstrates that the different isoforms are more similar between fungal species than they are to each other. This implies that duplication of the NIP2 gene occurred in a common ancestor before the three BCG species diverged, possibly to allow or facilitate host jump and colonization of new cereal crop species.
Fig. 12.
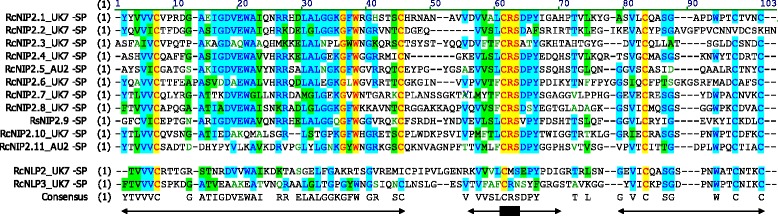
Sequence comparison of NIP2 and NIP2-like proteins (NLP). For clarity reasons the amino acid sequences of the mature proteins (-SP) from R. commune UK7 were aligned with the sequences only available in other isolates/species. Arrows indicate the domains identified by MEME. The CRS domain in position 61-63 (marked by the black box on the arrow) differentiates the two protein groups
To date no biochemical function could be assigned to NIP2. In contrast, NIP1 and NIP3 both stimulate the host plant plasma membrane H+-ATPase [108]. This enzyme generates a cellular membrane potential, which may affect nutrient accumulation and fungal membrane transport processes. In addition, acidification of the apoplast may create more convenient growth conditions for the fungus [109]. The originally reported induction of necrosis by the purified proteins [110] may therefore be a consequence of over-stimulation of the H+-ATPase due to severe overloading of the host leaf tissue [23]. An amino acid sequence comparison of NIP1 and NIP3 revealed that a stretch of six amino acids, -G45EGGNG50-, which is present in exposed loop III near the C-terminus of RcNIP1 [111], but absent from RoNIP1 (50.8% similarity, 39.7% identity with RcNIP1), is almost identically found near the C-terminus of NIP3 as -G82EGGEG87-. Remarkably, G45 is essential for NIP1 activity [7, 112], which suggests that this amino acid motif could be critical for the common function of NIP1 and NIP3.
NIP1 from R. commune is targeted by the barley resistance gene Rrs1 [7], thereby triggering defense reactions in the host [113]. To be able to infect Rrs1-barley the fungus therefore evolved two strategies to prevent recognition of NIP1, namely introduction of mutations into the NIP1 gene and its complete deletion [106, 112]. As the consequence, NIP1 was only found in about half of the R. commune isolates analyzed and inactive NIP1 isoforms were identified [7, 112]. Nevertheless, the importance of NIP1 as a virulence factor of R. commune was shown in a recent study on the quantitative virulence of 14 fungal isolates from each of nine global field populations worldwide. As the result, “on average isolates carrying a functional NIP1 protein had significantly higher virulence than isolates where the protein was missing or non-functional” [114]. In contrast to NIP1, NIP3 was found in almost all R. commune isolates studied [106] as well as in R. secalis and R. agropyri. Hence, at least one of the H+-ATPase stimulators, NIP1 or NIP3, appears to be present in a given fungal species/isolate.
To give a résumé, NIP1 and NIP3 appear to be specific for R. commune and the BCG species, respectively. The presence of an – albeit possibly “inactive” – NIP1 in R. orthosporum indicates that the gene may have been lost in the other species. In contrast, each of the five Rhynchosporium species harbors at least one NIP2 gene. However, evolution of the NIP2 gene family by gene duplication and sequence diversification [115] suggests an involvement of this protein in the adaptation of the BCG species to their hosts.
Small secreted proteins: specific occurrence of effector candidates in R. commune
The NIP effectors had been identified originally through their necrosis-inducing activity [110]. With the genome sequences available, additional candidate effector genes were obtained through an in silico approach. To this end, several general criteria based on existing knowledge about protein effectors from other fungal pathogens were established that ought to be fulfilled by candidate effector proteins (cf. [116, 117]). Effectors are secreted, i.e. most of them carry a signal peptide while lacking an additional transmembrane domain. Furthermore, they are usually of small size and cysteine-rich. In addition, they should be specific for the genus Rhynchosporium and expressed during pathogenesis, probably during the stage of fungal establishment in planta. Of particular interest are those effector genes that are present exclusively in the genome of a single species and, hence, may be involved in determining host specificity of this fungal species.
The genome of R. commune, isolate UK7, was screened for candidate genes coding for proteins that match the general criteria (Fig. 13). The subgroup containing ≥2% cysteine residues contained 139 proteins. The occurrence of these genes was probed (BLASTn) in the genomes of the other Rhynchosporium species (with the exception of R. lolii) to identify candidates, which may be crucial for host specificity. In R. commune, seven gene models (RcSP1, RcSP2, RcSP3, RcSP4, RcSP5, RcSP6, RcSP9) were found to be specifically present, among them the NIP2.6 paralog (RcSP6). When the OrthoMCL algorithm [118] was applied to the Rhynchosporium genomes, 29 R. commune proteins were found to lack orthologs in the other species. BLASTn analysis identified the seven RcSP genes and two additional genes in the <2%-cysteine fraction (RcSP7, RcSP8). No candidate proteins were identified in the 201–350 amino acid size fraction.
Fig. 13.
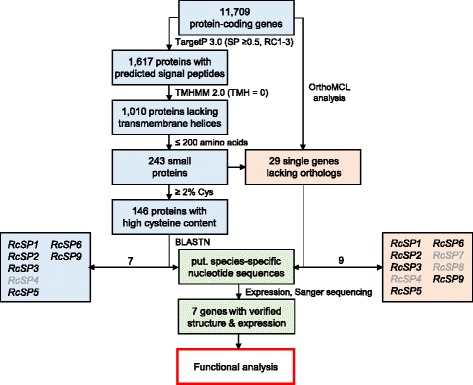
Flow chart for the identification of candidate effector genes in the R. commune genome (for details see text). RcSP7 and RcSP8 did not match the ≥ 2% Cys criterion. RcSP4 (mis-annotated), RcSP7 (very low expression) and RcSP8 (no expression) were not submitted to functional analysis
The gene models as predicted by the annotation algorithms were verified using EST libraries from R. commune isolate UK7 grown in liquid culture (A. Navarro-Quezada and W. Knogge, unpublished), from germinated conidia of British R. commune isolate 214 and from epidermal strips of barley leaves 3 dpi with British R. commune isolate 2LA (A. Avrova, unpublished). In addition, RT-PCR was carried out using gene-specific primers and RNA from ex planta grown R. commune isolate UK7 as a template. Sanger sequencing of the amplification products confirmed the structures of seven of the R. commune genes (RcSP1, RcSP2, RcSP3, RcSP5, RcSP6, RcSP7, RcSP9). In contrast, RcSP4 had been annotated incorrectly, while RcSP8 was not expressed. More detailed BLASTn and OrthoMCL analysis detected paralogs of RcSP1, RcSP2, RcSP3 and RcSP5 in R. commune and homologs in other Rhynchosporium species (Table 8, Additional file 8: Table S6). The original genes were renamed accordingly.
Table 8.
RcSP homologs
| R. commune protein | Identity [%] | Orthologs | ||
|---|---|---|---|---|
| R. secalis | R. agropyri | R.orthosporum | ||
| RcSP1.1 | 100 | |||
| RcSP1.2 | 50.0 | |||
| RcSP1.3 | 27.6 | RsSP1.3 (44.9) | ||
| RcSP2.1 | 100 | |||
| RcSP2.2 | 44.4 | RaSP2.2 (82.1) | ||
| RcSP3.1 | 100 | RaSP3.1 (63.4) | RoSP3.1 (47.5) | |
| RcSP3.2 | 46.5 | RsSP3.2 (45.5) | RaSP3.2 (46.6) | RoSP3.2a (31.3) RoSP3.2b (42.4) |
| RcSP3.3 | 57.4 | RaSP3.3 (98.1) | RoSP3.3 (47.0) | |
| RcSP5.1 | 100 | |||
| RcSP5.2 | 28.0 | RaSP5.2a (98.2) RaSP5.2b (51.0) RaSP5.2c (55.8) | ||
| RcSP5.3 | 25.3 | RaSP5.3 (52.5) | RoSP5.3 (37.2) | |
| RcSP5.4 | 26.7 | RsSP5.4 (78.9) | RaSP5.4 (75.0) | |
Numbers in brackets indicate the amino acid identity (%) with the respective R. commune protein
Expression of the seven R. commune candidate effector genes during pathogenesis was quantified using RNA extracted from inoculated leaves of susceptible barley cultivar ‘Ingrid’ at different time-points post inoculation with R. commune isolate UK7 [105]. Transcript amounts of the target genes were determined relative to the transcript amounts of a constitutively expressed fungal gene, GPD, by qRT-PCR [119]. As observed for the NIP effector genes [105], the RcSP genes were all transiently expressed early during fungal infection of susceptible host leaves. Between 10 and 14 dpi, when disease symptom occur, RcSP mRNAs virtually can no longer be detected (Fig. 14). RcSP2.1, RcSP5 and RcSP9 showed a maximum transcript abundance at 3 dpi, whereas RcSP1.1 and RcSP3.1 mRNA levels reached a maximum at 6 dpi. However, RcSP1.1 mRNA abundance accounted for only about 4% of that of RcSP3.1. RcSP6 and RcSP7 mRNA levels were even lower, peaking at 3–6 dpi. When the infection process of the hemibiotroph Colletotrichum higgensianum on Arabidopsis thaliana was studied successive waves of effector gene expression could be observed during the consecutive developmental stages from unpenetrated spores through penetrated biotrophic hyphae to the biotrophy to necrotrophy switch [120]. The different stages of Rhynchosporium development cannot be discerned as clearly as in C. higgensianum. Nevertheless, maximum transcript levels for some genes (RcSP2.1, RcSP5, RcSP9, NIP1) occur at 1-3 dpi, for others (RcSP1.1, RcSP3.1, NIP2, NIP3) at 6 dpi suggesting a similar sequence of gene expression waves.
Fig. 14.
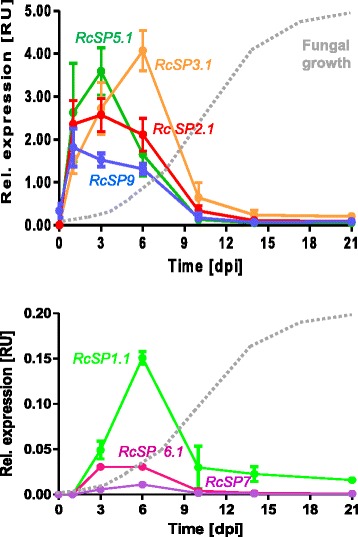
Expression of RcSP genes during pathogenesis. Relative RNA abundance was measured by qRT-PCR during growth of fungal isolate UK7 on barley cv. ‚Ingrid’. Dotted line indicates the development of fungal biomass
To assess the role of the R. commune-specific effector genes during pathogenesis, deletion mutants were generated. Using split-marker recombination [121] single deletion mutants were obtained for RcSP1.1 and RcSP6, two independent mutants for RcSP5.1 and three for RcSP2.1, RcSP3.1 and RcSP9. After inoculation of susceptible barley cultivar ‘Ingrid’ none of the deletion mutants caused a phenotype deviating substantially from that caused by wild type isolate UK7 at 14 dpi (not shown). However, when fungal biomass in planta was quantified by qPCR 10 of the 13 individual RcSP deletion mutants had grown to a significantly greater amount (1.7–4.5-fold) than the wild type UK7 (Fig. 15, Additional file 9: Figure S3), whereas deviations from wild type did not occur during fungal growth on agar plates (not shown). Their overlapping 95% confidence intervals allowed to combine the data of the independent deletion mutants of single RcSP genes (Fig. 15). The weakest growth response occurred with the ΔRcSP5 mutant, whereas the largest confidence interval was found with the ΔRcSP6 mutant. The latter finding may be due to the fact that RcSP6 is identical with NIP2.6, which is part of a gene family with seven additional members in R. commune isolate UK7. The observed growth increase of the deletion mutants is in contrast to the NIP gene deletion mutants, which did not differ from wild type (ΔNIP1, ΔNIP2) or grew substantially slower (ΔNIP3) than the wild type on highly susceptible cultivar ‘Ingrid’. (All three mutants showed strong growth reduction on moderately susceptible barley cultivar ‘Atlas’; [105]). When growth of the three RcSP9 deletion mutants and that of wild type were compared in more detail during pathogenesis, mutant biomasses deviated significantly from that of isolate UK7 from about 6 dpi on (Fig. 16, Additional file 10: Figure S4). In addition, close inspection of disease phenotype development revealed that the first symptoms became visible already at 8–10 dpi with the deletion mutants, whereas wild type symptoms did not occur before 10 dpi (Fig. 17; Additional file 11: Figure S5). At 14 dpi the mutant phenotype appeared more severe than the wild type phenotype; almost the entire upper blade of the mutant-inoculated leaves was necrotic, whereas green tissue remained between the lesions of wild type-infected leaves.
Fig. 15.
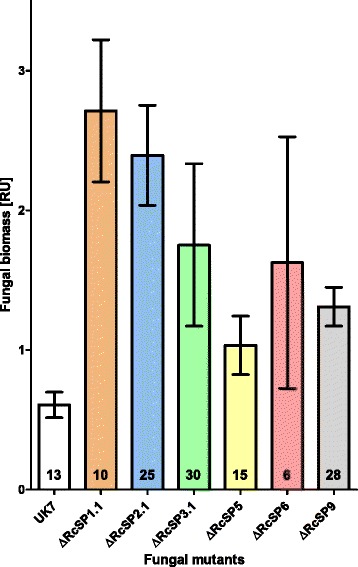
Growth acceleration of deletion mutants. Relative biomass of fungal deletion mutants and wild-type isolate UK7 was determined by qPCR at 14 dpi on barley cv. ‘Ingrid’. Results from independent mutants are combined (cf. Additional file 9: Figure S3). Bars represent the 95% confidence intervals. n-values are given at the base of the bars
Fig. 16.
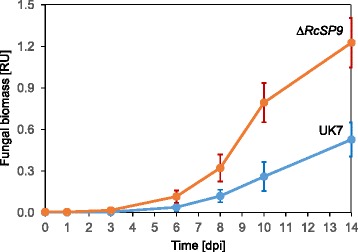
Growth of ΔRcSP9 mutants. Relative biomasses of three independent deletion mutants and wild-type isolate UK7 were determined by qPCR during pathogenesis on barley cv. ‘Ingrid’. Bars represent 95% confidence intervals. nΔRcSP9 = 9, nUK7 = 4
Fig. 17.

Disease phenotype of RcSP9 deletion mutants. Primary leaves of barley cv. ‘Ingrid’ were inoculated with spores of wild-type isolate UK7 or of the mutants (#1036, #1067, #1231) and photos were taken at indicated times post inoculation. C, mock inoculation
In summary, significant quantitative effects on fungal growth and symptom development became apparent upon deletion of the six R. commune-specific genes. Notably, the deletion-caused growth acceleration suggested that the fungus is not adapted for the most rapid development in planta. This is similar to mutants of the grass endophyte Epichloë festucae, which had switched from mutualistic to pathogenic growth including a dramatically increased fungal biomass on perennial ryegrass. Different types of non-effector proteins were inactivated in the mutants; a MAP kinase (SakA, [122]), an NADPH oxidase (NoxA, [123]), which is regulated by NoxR [124] through interaction with small GTP binding protein RacA [125], and a siderophore-synthesizing NRPS (SidN, [126]). Therefore, it was postulated that functional stress signaling, regulated ROS synthesis and siderophore-mediated iron homeostasis are crucial for restricting fungal growth as prerequisite for maintaining the mutualistic interaction with the host. It is tempting to speculate that the R. commune RcSPs fulfil an analogous role in affecting hyphal growth, probably by interacting with plant compounds, thereby establishing an endophyte-like growth in host leaves during the early symptomless period of pathogenesis.
It remains to be shown whether the effects caused by the RcSPs contribute to host specialization. However, the occurrence of specific effector genes in the genome of one Rhynchosporium species and their absence from others species represents the simplest model to explain host specificity. Alternatively, effector genes, albeit present in several Rhynchosporium genomes, may be specifically expressed only in the interaction of one of them with its host plant and not in the specific interactions of other species. Finally, host specificity may originate from structural differences of effector proteins rather than from presence or expression of specific genes. In addition, small molecules such as host-specific toxins originating from fungal secondary metabolism may play a role in colonizing a particular host species.
Conclusions
Phylogenetic analysis confirmed that the Rhynchosporium genus belongs to the Leotiomycetes clade of fungal systematics, most likely to the Dermataceae family, the BCG and CCG species clearly forming separate branches of the Rhynchosporium genus. Genome-wide SNP analysis along with the structure of the rDNA regions suggested that during BCG evolution R. agropyri branched off shortly before R. commune and R. secalis diverged. At least in the BCG species, complete mating type loci exist. In addition, core meiotic genes are present and expressed along with most genes required for meiosis and sexual recombination in other (ascomycete) fungi, providing molecular evidence for the existence of a cryptic sexual cycle.
Bioinformatic analysis revealed a large number and types of CWDEs, which are characteristic for hemibiotrophic and necrotrophic fungi infecting monocotyledonous hosts, but failed to provide evidence for their involvement in host specialization. In contrast, the occurrence of genus-, species- and isolate-specific PKS and NRPS genes suggests a possible role in host adaptation in particular of those genes, which are missing from the genome of the closest sequenced relative, the poplar pathogen M. brunnea. The most important finding was, however, that six R. commune-specific effector proteins contribute to decelerating fungal development in planta. Host adaptation of this hemibiotrophic fungus therefore appears to aim at stabilizing the biotrophic growth stage in favor of the necrotrophic destructive stage. Unlike haustoria-forming fungi, apoplastic fungi like R. commune colonize the intercellular matrix of their hosts relatively slowly, usually without triggering a fast hypersensitive cell death response. The long biotrophic stage of Rhynchosporium development resembles endophytic fungal growth, rendering this fungus a possible object for studying the mutualism-parasitism transition.
Methods
Fungal strains
The following fungal isolates were subjected to genome sequencing; for R. commune UK isolates UK7 and 13-13 and Australian isolate AU2 [21], for R. secalis Swiss isolate 02CH4-6a.1 [17], for R. agropyri Swiss isolate 04CH-RAC-A.6.1 (B. McDonald, ETH Zurich), for R. lolii UK isolate 15lp11 [16], and for R. orthosporum Swiss isolate 04CH-BAR-A.1.1.3 (B. McDonald, ETH Zurich).
Fungal cultures, DNA extraction, genome sequencing and assembly
Fungal culture conditions, DNA extraction and genome sequencing were described previously [30]. Briefly, the genomes of R. commune, isolates UK7 and 13-13 and R. secalis were sequenced using the Roche 454 Genome Sequencer FLX with GS FLX Titanium series reagents, fosmids and the lllumina Genome Analyzer IIx (paired-end and mate-pair libraries). Paired-end libraries of R. commune, isolate AU2, R. agropyri (along with a mate-pair library) and R. orthosporum were sequenced using Illumina GAIIx and HiSeq. To establish the genome of R. commune, isolate UK7, as the reference genome, contigs were assembled using the ALLPATHS-LG software package [127] and pre-assembled contigs were combined into scaffolds using the SSPACE algorithm [128]. The same strategy was followed to assemble the genome of R. agropyri, whereas genome assembly for R. commune, isolate AU2 and for R. orthosporum was carried out using the Velvet algorithm package [129], and for R. secalis using the Newbler software package. For R. commune isolate 13-13, 454 and Illumina GAIIx reads were assembled independently using the Newbler and Velvet algorithm packages, respectively, followed by combined assembly using Minimus2 [130]. The genome of isolate 15lp11 from the fifth species, R. lolii [16], was sequenced by Richard Harrison (Illumina MiSeq, East Malling Research, East Malling, UK) and a draft assembly using the VELVET algorithms [129] was obtained by David Hughes (University of Hertfordshire, Hatfield, UK). Sequence reads of this isolate were mapped to the R. orthosporum genomic contigs. Hence, differential sequence information could be identified only for R. lolii genes that are orthologous to R. orthosporum genes.
Structural annotation
Gene models for R. commune UK7, R. secalis and R. agopyri were generated by three de novo prediction programs: 1) Fgenesh [131] with different matrices (trained on Aspergillus nidulans, Neurospora crassa and a mixed matrix based on different species), 2) GeneMark-ES [132] and 3) Augustus [133] with R. commune ESTs as training sets. Annotation was aided by exonerate [134] hits of protein sequences from B. cinerea T4, B. cinerea B05.10, S. sclerotinia, R. commune UK7, R. secalis and R. agropyri, respectively, to uncover gene annotation gaps. EST transcripts were assembled and mapped on the genome using Blat, a final cleaning was done with Perl Scripts. The different gene structures and evidences (exonerate mapping and EST transcripts) were visualized in GBrowse [135], allowing manual validation of coding sequences. The best fitting model per locus was selected manually and gene structures were adjusted by splitting or fusion of gene models or redefining exon-intron boundaries if necessary. The final call sets comprises 12,212 (R. commune UK7), 13,151 (R. secalis) and 13,674 (R. agropyri) protein coding genes. In addition, 91 (+22 mitochondrial) tRNA-encoding genes are predicted for R. commune UK7, 95 (+22 mitochondrial) for R. secalis and 107 for R. agropyri using tRNAscan-SE [136]. The predicted protein sets were searched for highly conserved single (low) copy genes to assess the completeness of the genomic sequences and gene predictions. Orthologous genes to all 246 single copy genes were identified for all three proteomes by Blastp comparisons (eVal: 10−3) against the single-copy families from all 21 species available from the FUNYBASE [137]. In addition, all of the 248 core genes commonly present in higher eukaryotes (CEGs) could be identified by Blastp comparisons (eVal: 10−3) for two proteomes, whereas only one protein was missing in the R. agropyri proteome [138].
Identification of repetitive DNA elements
Determination of repeat sequences involved first the calculation of repeat families de novo followed by assessment of known repeat elements in a second step. Families of previously unknown interspersed repeat elements were identified by RepeatScout [139]. Repeat families were included when they comprised more than 10 repeats and when their consensus sequence length was longer than 50 bp. In addition, low complexity and simple sequence repeats were determined with the tools NSEG [140] and Tandem Repeats Finder [141], which are part of the RepeatScout procedure and remove them from the interspersed repeat library.
The RepBase database [142] was used to detect previously published families of transposable elements, pseudogenes and retroviruses. In order to determine the exact locations of the repetitive elements on the genome we used the RepBase library and the calculated library of interspersed repeat families as input for RepeatMasker [143]. RepeatMasker was also used to find and mask genomic regions of low complexity. We applied the automated classification tool TEclass [144] to categorize the predicted repeat sequences into the four main transposable element categories DNA transposon, long interspersed nuclear element (LINE), short interspersed nuclear element (SINE) and retrotransposon with long terminal repeats (LTRs).
Functional annotation of predicted open reading frames and data repositories
The protein coding genes were analyzed and functionally annotated using the PEDANT system [145]. Data sets are accessible at http://pedant.helmholtz-muenchen.de/genomes.jsp?category=fungal. The genome and annotation data were submitted to the European Nucleotide Archive (ENA, R. commune, isolate UK7: http://www.ebi.ac.uk/ena/data/view/FJUW01000001-FJUW01000164; R. secalis: http://www.ebi.ac.uk/ena/data/view/FJVC01000001-FJVC01002130; R. agropyri: http://www.ebi.ac.uk/ena/data/view/FJUX01000001-FJUX01000508).
Phylogenetic analyses
Sequence data for phylogenetic estimations of fungal relationships were generated using standard PCR protocols and primers that were (a) designed according to the 18S rDNA sequence from R. commune, isolate 788 (GenBank accession no. AY038583.1, [34]; Additional file 12: Table S7) or (b) described in a previous study [35]. After amplification and cloning into pJET (Thermo Scientific, Schwerte, Germany) Sanger sequencing (LGC Genomics, Berlin) was done on the nearly full-length rDNA region (18S rDNA-ITS1-5.8 S rDNA-ITS2-28S rDNA) using five (R. commune, R. secalis), six (R. agropyri) or seven (R. orthosporum) overlapping primer pair combinations starting on the 5′ side with primer 18SrDNA1f and ending on the 3′ side with primer LR7 (Additional file 12: Table S7). The sequences coding for elongation factor 1-α (EF1-α) and the RNA polymerase II subunit RPB1 and RPB2 were extracted from the genome database.
To integrate the Rhynchosporium genus into the estimated fungal phylogeny [35], sequences coding for 18S rRNA, 28S rRNA, the ITS region including the 5.8S rRNA, elongation factor 1-α (EF1-α), RPB1 and RPB2 were obtained from GenBank (genome projects, AFTOL data) for 12 taxa of the Leotiomycetes class (Additional file 13: Table S8). Sequences for each gene were aligned using the ClustalW algorithm implemented in BioEdit (v7.2.5) and ambiguous regions were excluded. Gap-bridging sequences in the alignments were removed. Accordingly, an alignment utilizing the implemented ClustalW algorithm of MEGA (v6.06; [146] was generated, where sequences differ in single nucleotide polymorphisms (SNP) and in additional sequences of up to three consecutive nucleotides per site. The conflict that the RPB1 sequences of the species Coccomyces dentatus, Potebniamyces pyri, Monilinia fructicola and Lachnum virgineum are not accessible was ignored. Data were combined in one matrix with concatenated trimmed sequences in the order 18S rDNA-28S rDNA-ITS-EF1a-RPB1-RPB2. For phylogenetic estimation the nucleotide sequences were imported into MEGA (v6.06) and fitted to a general-time-reversible model with a proportion of Invariant sites and Gamma distribution rates (four rate classes, GTR + I + Γ). The Maximum Likelihood method was applied using 1000 bootstrap replicates (MLBS). Gaps and missing data were treated as complete deletion. Nearest-Neighbor-Interchange (NNI) served as the Maximum Likelihood heuristic method. The MEGA-estimated phylogeny was verified with an independent MLBS estimation in PHYML [147], also under the GTR model with gamma distribution in four rate classes. 250 bootstraps were done and tree improvement used NNI and subtree pruning and regrafting (SPR).
In a second phylogenetic estimation, a tree was generated that includes more distantly related fungal species. To this end, the amino acid sequences of EF1-α, RBP1 and RBP2 from R. commune were submitted to BLASTp searches [148] to identify the homologous genes in 17 different taxa (Additional file 13: Table S8). An RBP1 sequence from B. fuckeliana was not found. The combined and concatenated sequence data were fitted into a single EF1-α/RPB1/RPB2 matrix. No trimming was performed due to the availability of complete coding sequence for all proteins investigated. Amino acid sequences were aligned using the ClustalW algorithm in MEGA (v6.06). Thus, phylogenetic estimation was carried out under the LG substitution model [149] utilizing gamma distributed rates with invariant sites (G + I, four discrete gamma categories). Maximum Likelihood analyses took 500 bootstrap replications of heuristic searches into account (MLBS). Partial deletion was estimated for gaps and missing data and tree improvement were accomplished using NNI. The MEGA-estimated phylogeny was verified by MLBS estimation in PHYML using the LG method with four substitution rate classes. 250 bootstraps were performed and tree improvement used NNI and SPR.
Genome-wide single nucleotide polymorphisms (SNPs) were used to infer the phylogeny of the closely related BCG species. To this end, the genomes of R. commune, isolates UK7, AU2 and 13-13, R. secalis, R. agropyri and R. orthosporum were aligned using the MAUVE algorithm (version 2.4.0; Darling lab, University of Technology, Sydney). The obtained collinear genomic sequence (24.85 Mb) represented 45–50% of the genomes. Homologous sites containing SNPs at least in one of the genomes were identified and concatenated. A multiple sequence alignment was generated and a phylogenetic tree was constructed under the Tamura-Nei substitution model ([150], G + I, four discrete categories). Maximum Likelihood analysis was done with NNI and 500 bootstrap replications. Gaps and missing data were eliminated. Divergence times were calculated for all branching points using the RelTime method [151]. Relative times were translated into absolute times based on previous Bayesian calculations [17].
To construct the phylogenetic tree of the NIPs the amino acid sequences of the mature proteins were used. For the phylogenetic trees of polyketide synthases and non-ribosomal peptide synthases, amino acid sequences from other mostly fungal taxa were obtained from the NCBI databases. Enzymatic domains were identified by BLASTp analysis using NCBI’s Conserved Domain Database [82] and aligned with MUSCLE [152]. Evolutionary analyses were done in MEGA (v6.06). The evolutionary history was inferred by using the Maximum Likelihood method based on the LG substitution model [149]. In each case the tree with the highest log likelihood is shown. Initial trees for the heuristic search were obtained by applying the Neighbor-Joining-Interchange (NNI) method to a matrix of pairwise distances estimated using a JTT model. To model evolutionary rate differences among sites Gamma distribution (four discrete categories) with Invariant sites (G + I) was used. All positions with <95% site coverage were eliminated, i.e., <5% alignment gaps, missing data and ambiguous amino acids were allowed at any position. 500 bootstraps were done to assess the robustness of the phylogeny.
Identification of CWDEs
Genes coding for enzymes with carbohydrate substrates were identified using the CAZy database [51, 52]. CWDEs belong to CAZy families GH, CE, PL and AA. Therefore, all members of these families were subjected to secretion analysis by using the SecretomeP algorithm [55]. A neural network score of ≥0.6 was taken to predict secreted CAZymes, i.e. CWDEs.
Identification of gene clusters
To predict putative secondary metabolism gene clusters in silico two different algorithms were used, the Secondary Metabolite Unique Regions Finder (SMURF, [153]) and the Motif Density Method (MDM, [154]). With SMURF predictions are based on PFAM and TIGRFAM domain content and chromosomal position of a gene. MDM uses the density of occurrences of cluster-specific regulatory motifs in the promoter regions as an additional criterion to distinguish cluster from non-cluster genes. Application of the two algorithms to the Rhynchosporium BCG genomes yielded putative PKS gene clusters with varying degrees of overlap.
Identification of effector candidates
Candidate effector genes were identified by screening the genome for gene models whose products fulfill the criteria (1) secretion, (2) small size, (3) high cysteine content [117]. The TargetP 1.1 [155] and WoLF PSort [156] protein localization algorithms (NN-based SP score >0.5, prediction confidence from RC = 1 to RC = 3) and transmembrane helix prediction (TMHMM 2.0, TMH = 0; [157] were used to predict secreted proteins. Sorting of the candidates obtained according to length and cysteine content was done to yield proteins with ≤ 200 amino acids and ≥ 2% cysteine residues. The nucleotide sequences of these genes were used as queries in a BLASTn analysis to identify candidates, for which homologous sequences do not exist in the genomes of the other species. In addition, OrthoMCL analysis [118] was carried out on the three BCG genomes for orthology detection. Those proteins that lack any orthologs and paralogs were compared to the ≤200-amino acid protein fraction. Finally, using the single-gene sequences coding for small, cysteine-rich proteins as a query, BLASTn search of the other genomes was done to identify genes that are unique for a given genome.
EST libraries
Three EST libraries were generated from R. commune mycelia, from germinated conidia and from epidermal strips of inoculated barley leaves. Mycelia of isolate UK7 were grown for 14 d in liquid culture, subsequently filtered, air-dried and ground to a fine powder using glass beads (acid washed, 150–212 μm, SIGMA) to efficiently disrupt fungal hyphae [30]. Total RNA was extracted from mycelia using the Trizol method [158], then treated with DNAse (Roche), quality-controlled on a 1.2% agarose gel and shipped for cDNA synthesis using the SMART technology [159]; Evrogen Technologies, Moscow, Russia). After normalization using the duplex-specific nuclease technology [160] the cDNA was 454-sequenced, assembled and trimmed (GATC Biotech, Konstanz, Germany).
Conidia of isolate 214 were incubated in sterile distilled water for 24 h and mRNA was extracted from germinated conidia using Dynabeads Oligo (dT)25 (Invitrogen Dynal AS, Oslo, Norway). RNA yield was measured using a NanoDrop Micro Photometer (NanoDrop Technologies, Inc., Rockland, USA). Prior to cDNA synthesis, RNA samples were treated with DNaseI using the Ambion DNA-free kit following the manufacturer’s protocol. First strand cDNA for sequencing was synthesized from 0.5 μg of mRNA and PCR-amplified for 11 cycles using the SMART PCR cDNA synthesis kit (Clontech Laboratories/Takara Bio Europe SAS, Saint-Germain-en-Laye, France) following the manufacturer’s protocol. The non-normalized cDNA library was 454-sequenced, trimmed and assembled at the University of Liverpool, UK.
Barley plants were inoculated with spores of UK isolate 2LA. At 3 dpi epidermal strips were prepared and total RNA was extracted using an RNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the protocol supplied by the manufacturer. After testing RNA integrity by agarose gel electrophoresis, total RNA was sent to the University of Exeter, UK, for mRNA isolation, lllumina sequencing, trimming and assembling. Finally, all ESTs were mapped on the R. commune genome.
Quantitative PCR
Relative fungal biomass and fungal gene expression during pathogenesis were quantified by qPCR with DNA as template and by qRT-PCR with cDNA as template using the efficiency calibrated model [119] as described previously [105, 161]. For qPCR the fungal target gene GPD was quantified using the primers GPDRT2s and GPDRT2as, the barley reference gene TSP (GenBank accession no. AK359241) using the primer combination TSP_RT_F/TSP_RT_R (Additional file 12: Table S7). For qRT-PCR, expression of GPD or sterol C4-methyl oxidase served as reference (primer pairs GPDRT2s/GPDRT2as), whereas RcSP expression was quantified using the primer combinations RT-RcSP1f/RT-RcSP1r (RcSP1), RT-RcSP2f/RT-RcSP2r (RcSP2), RT-RcSP3f/RcSP3r (RcSP3), RT-RcSP5f/RcSP5r (RcSP5), RT-NIP2.6-UK7f/RT-NIP26-UK7r (RcSP6), RT-RcSP7f/RT-RcSP7r (RcSP7) and RT-RcSP9f/RT-RcSP9r (RcSP9; Additional file 12: Table S7).
Generation of fungal RcSP deletion mutants
The RcSP genes were re-sequenced using the primer pairs RcSPn-seq-s/RcSPn-seq-as (LGC Genomics, Berlin; Additional file 12: Table S7). Constructs for the disruption of RcSP genes were generated by fusion PCR [161, 162] and deletion mutants were obtained through split-marker recombination by replacing the RcSP genes with a hph resistance cassette [121]. To this end, 1000 bp of RcsP 5′ and 3′ flanking sequence were amplified in a first step using the primer pairs fusion1_s/fusion2_as and fusion3_s/fusion4_as, respectively, (Additional file 12: Table S7) and genomic DNA as template. After amplifying the hph cassette with the primers hph_fusion_s and hph_fusion_as and the vector pAN7-1 [163] as template, in the second step the complete deletion construct was generated with the primers fusionA-s and fusionB-as using the 1000-bp flanking sequences and the hph cassette as a template. In the third step, the split-marker constructs overlapping in the hph gene by 50 bp were generated using the primer pairs fusionA_s/HPH-r and HPH50-f/fusion-as. Fungal transformation and mutant selection was described previously [105, 161, 164]. Successful deletion of the genes RcSP1, RcSP2, RcSP5 and RcSP6 was analyzed using the primer pairs RcSPn_dt_s/RcSPn_dt_as, whereas deletion of RcSP3 and RcSP9 was shown with the primer pairs RcSPn-seq-s/RcSPn-seq-as. Integration of the disruption construct was proven by amplifying both ends using the primer pairs fusion1_s/gpdA_KO-as and trpC_KO_s/fusion4-as with mutant DNA as template.
Acknowledgements
The authors would like to thank B. McDonald, ETH Zurich, for providing the Swiss fungal isolates and for critically reading the manuscript.
Funding
This work was funded by the Leibniz Association in the framework of the Competition Committee’s competitive procedure and by the Scottish Government Rural and Environment Science and Analytical Services (RESAS).
Availability of data and materials
Datasets used for phylogenetic analyses are available from the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.qm130). Sequence data sets are accessible at http://pedant.helmholtz-muenchen.de/genomes.jsp?category=fungal. The genome and annotation data were submitted to the European Nucleotide Archive (ENA) R. commune UK7: http://www.ebi.ac.uk/ena/data/view/FJUW01000001-FJUW01000164; R. secalis: http://www.ebi.ac.uk/ena/data/view/FJVC01000001-FJVC01002130; R. agropyri: http://www.ebi.ac.uk/ena/data/view/FJUX01000001-FJUX01000508.
Authors’ contribution
MF, ST and MP performed the genome sequencing and assembly except for isolate 13-13. KP, KA and AA sequenced and assembled the 13-13 genome and transcriptome and made these data available. MM and UG conducted the genome annotation and all large-scale genome analyses. DP and SK carried out general lab work, inoculation experiments and phylogeny analyses and provided the RcSP gene expression data. In addition, DP participated in gene annotation and collected the NIP2 gene data. CW and WK carried out the bioinformatics analyses of secondary metabolism enzymes. TW and ES carried out the SMURF- and MDM-based gene cluster analyses. JG, JH, NW and SK generated and characterized the RcSP deletion mutants. KMK, RH, DJH and BDLF made the R. lolii data available. DP was involved in drafting the manuscript, which was mainly written by WK with contributions from AA and MM. All authors provided intellectual input, all principal investigators read and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
Not applicable.
Abbreviations
- BCG
Beaked conidia group
- BLAST
Basic local alignment search tool
- CAZyme
Carbohydrate-active enzyme
- CBM
Carbohydrate-binding motif
- CCG
Cylindrical conidia group
- CEGMA
Core Eukaryotic genes mapping approach
- CWDE
Cell wall degrading enzymes
- DMATS
Dimethylallyl tryptophane synthase
- EAS
Euascomycetes
- EST
Expressed sequence tag
- ETF1-α
Eukaryotic translation termination factor 1-α
- GPD
Glyceraldehyde-3-phosphate dehydrogenase
- MAT
Mating type
- NIP
Necrosis inducing protein
- NLP
NIP2-like protein
- NPSL
NRPS-like protein
- NRPS
Non-ribosomal peptide synthetase
- PKS
Polyketide synthase
- RcSP
R. commune secreted protein
- ROS
Reactive oxygen species
- RPB1/2
RNA polymerase II subunits B1/2
- SCPCH
Single-copy protein-coding homolog
- TC
Terpene cyclase
Additional files
The Leotiomycetes class of Ascomycetes. The concatenated nucleotide sequences of 18S rDNA, 28S rDNA, ITS region, elongation factor EF1-a and RNA polymerase II subunits RPB1 and RPB2 from Rhynchosporium and twelve Leotiomycetes species were used to construct the phylogenetic tree. An enlargement of the Rhynchosporium subtree is shown in Fig. 1. Most of the species live a saprobic lifestyle (S). Three are plant pathogens (P) causing brown rot blossom blight disease on stone fruit and occasionally on pome fruit trees (M. laxa), grey mold disease on many plant species (B. fuckeliana) and the Phacidiopycnis post-harvest fruit rot of pear (P. pyri), respectively. Numerals on the nodes represent the percentages from 500 bootstraps. Scale: number of substitutions per nucleotide (PPTX 50 kb)
SNPs in organism pairs. (XLSX 11 kb)
(A) Core meiotic recombination machinery genes. (B) Genes involved in sexual reproduction in the aspergilli. (XLSX 43 kb)
CAZyme numbers in different fungal species. GH, glycosyl hydrolases; CE, carbohydrate esterases; PL, pectate lyases; GT, glycosyl transferases; CBM, carbohydrate binding motifs (modified after [50]). (PPTX 72 kb)
CAZymes. (XLSX 24 kb)
(A) Cell wall degrading enzymes. (B) CWDE numbers according to their substrates. (XLSX 72 kb)
Secondary metabolism gene clusters. (XLSX 43 kb)
RcSP homologs (gene IDs). (XLSX 12 kb)
Growth acceleration of single deletion mutants. Relative biomass of fungal deletion mutants and wild-type isolate UK7 was determined by qPCR at 14 dpi on barley cv. ‘Ingrid’. Wild-type references are associated to the mutants to the right. Bars represent the 95% confidence intervals. n-values are given above bars. (PDF 14 kb)
Growth of DRcSP9 mutants. Relative biomass of three independent deletion mutants and wild-type isolate UK7 were determined by qPCR during pathogenesis on barley cv. ‘Ingrid’. Bars represent 95% confidence intervals. nmutants = 3, nUK7 = 4. (PPTX 145 kb)
Disease phenotype of DRcSP9 mutants. Primary leaves of barley cv. ‘Ingrid’ were inoculated with spores of wild-type isolate UK7 or of the mutants and photos were taken at indicated times post inoculation. C, mock inoculation. (PPTX 382 kb)
Primers used in this study. (DOCX 17 kb)
Genbank and internal numbers (Rhynchosporium) for taxa used in this study. (XLSX 13 kb)
Contributor Information
Daniel Penselin, Email: daniel.penselin@ipb-halle.de.
Martin Münsterkötter, Email: m.muensterkoetter@web.de.
Susanne Kirsten, Email: skirsten@ipb-halle.de.
Marius Felder, Email: mfelder@fli-leibniz.de.
Stefan Taudien, Email: stau@fli-leibniz.de.
Matthias Platzer, Email: mplatzer@fli-leibniz.de.
Kevin Ashelford, Email: ashelfordke@cardiff.ac.uk.
Konrad H. Paskiewicz, Email: k.h.paszkiewicz@exeter.ac.uk
Richard J. Harrison, Email: richard.harrison@emr.ac.uk
David J. Hughes, Email: david.hughes@rothamsted.ac.uk
Thomas Wolf, Email: thomas.wolf@leibniz-hki.de.
Ekaterina Shelest, Email: ekaterina.shelest@leibniz-hki.de.
Jenny Graap, Email: jey0112@web.de.
Jan Hoffmann, Email: jan.hoffmann@teleos-web.de.
Claudia Wenzel, Email: claudia.wenzel@agroscope.admin.ch.
Nadine Wöltje, Email: nadine.woeltje@web.de.
Kevin M. King, Email: kevin.king@rothamsted.ac.uk
Bruce D. L. Fitt, Email: b.fitt@herts.ac.uk
Ulrich Güldener, Email: u.gueldener@tum.de.
Anna Avrova, Email: anna.avrova@hutton.ac.uk.
Wolfgang Knogge, Email: wknogge@ipb-halle.de.
References
- 1.Caldwell RM. Rhynchosporium scald of barley, rye, and other grasses. J Agric Res. 1937;55(3):175–198. [Google Scholar]
- 2.Oudemans CAJA. Observations mycologiques. Verslag Wis- en Natuurk Afd K Acad Wetensch Amsterdam. 1897;6:86–92. [Google Scholar]
- 3.Frank AB. Über die Zerstörung der Gerste durch einen neuen Getreidepilz. Wochenschr Brau. 1897;42:518–520. [Google Scholar]
- 4.Stukenbrock EH, McDonald BA. The origins of plant pathogens in agro-ecosystems. Annu Rev Phytopathol. 2008;46:75–100. doi: 10.1146/annurev.phyto.010708.154114. [DOI] [PubMed] [Google Scholar]
- 5.Zaffarano PL, McDonald BA, Zala M, Linde CC. Global hierarchical gene diversity analysis suggests the Fertile Crescent is not the center of origin of the barley scald pathogen Rhynchosporium secalis. Phytopathology. 2006;96(9):941–950. doi: 10.1094/PHYTO-96-0941. [DOI] [PubMed] [Google Scholar]
- 6.Salamini F, Ozkan H, Brandolini A, Schafer-Pregl R, Martin W. Genetics and geography of wild cereal domestication in the Near East. Nat Rev Genet. 2002;3(6):429–441. doi: 10.1038/nrg817. [DOI] [PubMed] [Google Scholar]
- 7.Rohe M, Gierlich A, Hermann H, Hahn M, Schmidt B, Rosahl S, Knogge W. The race-specific elicitor, NIP1, from the barley pathogen, Rhynchosporium secalis, determines avirulence on host plants of the Rrs1 resistance genotype. EMBO J. 1995;14(17):4168–4177. doi: 10.1002/j.1460-2075.1995.tb00090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunner PC, Schürch S, McDonald BA. The origin and colonization history of the barley scald pathogen Rhynchosporium secalis. J Evol Biol. 2007;20(4):1311–1321. doi: 10.1111/j.1420-9101.2007.01347.x. [DOI] [PubMed] [Google Scholar]
- 9.Zaffarano PL, McDonald BA, Linde CC. Phylogeographical analyses reveal global migration patterns of the barley scald pathogen Rhynchosporium secalis. Mol Ecol. 2009;18(2):279–293. doi: 10.1111/j.1365-294X.2008.04013.x. [DOI] [PubMed] [Google Scholar]
- 10.Linde CC, Zala M, McDonald BA. Molecular evidence for recent founder populations and human-mediated migration in the barley scald pathogen Rhynchosporium secalis. Mol Phylogenet Evol. 2009;51(3):454–464. doi: 10.1016/j.ympev.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Zaffarano PL, Linde CC, Brunner PC, McDonald BA. Rhynchosporium secalis is composed of host-specialized species that originated recently in northern Europe. Phytopathology. 2007;97(7):S128. [Google Scholar]
- 12.Shipton WA, Boyd WJR, Ali SM. Scald of barley. Rev Plant Pathol. 1974;53:839–861. [Google Scholar]
- 13.Taylor JW, Jacobson DJ, Kroken S, Kasuga T, Geiser DM, Hibbett DS, Fisher MC. Phylogenetic species recognition and species concepts in fungi. Fungal Genet Biol. 2000;31(1):21–32. doi: 10.1006/fgbi.2000.1228. [DOI] [PubMed] [Google Scholar]
- 14.Cai L, Giraud T, Zhang N, Begerow D, Cai GH, Shivas RG. The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers. 2011;50(1):121–133. doi: 10.1007/s13225-011-0127-8. [DOI] [Google Scholar]
- 15.Zaffarano PL, McDonald BA, Linde CC. Two new species of Rhynchosporium. Mycologia. 2011;103:195–202. doi: 10.3852/10-119. [DOI] [PubMed] [Google Scholar]
- 16.King KM, West JS, Brunner PC, Dyer PS, Fitt BDL. Evolutionary relationships between Rhynchosporium lolii sp. nov. and other Rhynchosporium species on grasses. PLoS ONE. 2013;8(10):e72536. doi: 10.1371/journal.pone.0072536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zaffarano PL, McDonald BA, Linde CC. Rapid speciation following recent host shifts in the plant pathogenic fungus Rhynchosporium. Evolution. 2008;62(6):1418–1436. doi: 10.1111/j.1558-5646.2008.00390.x. [DOI] [PubMed] [Google Scholar]
- 18.McDonald BA. How can research on pathogen population biology suggest disease management strategies? The example of barley scald (Rhynchosporium commune) Plant Pathol. 2015;64(5):1005–1013. doi: 10.1111/ppa.12415. [DOI] [Google Scholar]
- 19.Ayesu-Offei EN, Clare BG. Processes in the infection of barley leaves by Rhynchosporium secalis. Aust J Biol Sci. 1970;23:299–307. [Google Scholar]
- 20.Hosemans D, Branchard M. Étude in vitro de la Rhynchosporiose de l’orge: cycle de reproduction du parasite - histopathologie de l’hôte. Phytopath Z. 1985;112(2):127–142. doi: 10.1111/j.1439-0434.1985.tb04821.x. [DOI] [Google Scholar]
- 21.Lehnackers H, Knogge W. Cytological studies on the infection of barley cultivars with known resistance genotypes by Rhynchosporium secalis. Can J Bot. 1990;68:1953–1961. doi: 10.1139/b90-257. [DOI] [Google Scholar]
- 22.Zhan J, Fitt BDL, Pinnschmidt HO, Oxley SJP, Newton AC. Resistance, epidemiology and sustainable management of Rhynchosporium secalis populations on barley. Plant Pathol. 2008;57(1):1–14. [Google Scholar]
- 23.Möbius N, Hertweck C. Fungal phytotoxins as mediators of virulence. Curr Opin Plant Biol. 2009;12(4):390–398. doi: 10.1016/j.pbi.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Foster SJ, Fitt BDL. Isolation and characterisation of the mating-type (MAT) locus from Rhynchosporium secalis. Curr Genet. 2003;44(5):277–286. doi: 10.1007/s00294-003-0445-9. [DOI] [PubMed] [Google Scholar]
- 25.Zaffarano PL, Queloz V, Duo A, Gruenig CR. Sex in the PAC: A hidden affair in dark septate endophytes? BMC Evol Biol. 2011;11:282. doi: 10.1186/1471-2148-11-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linde CC, Zala M, Ceccarelli S, McDonald BA. Further evidence for sexual reproduction in Rhynchosporium secalis based on distribution and frequency of mating-type alleles. Fungal Genet Biol. 2003;40(2):115–125. doi: 10.1016/S1087-1845(03)00110-5. [DOI] [PubMed] [Google Scholar]
- 27.King KM, West JS, Fitt BDL, Dyer PS. Differences in MAT gene distribution and expression between Rhynchosporium species on grasses. Plant Pathol. 2015;64(2):344–354. doi: 10.1111/ppa.12265. [DOI] [Google Scholar]
- 28.McDonald BA, Zhan J, Burdon JJ. Genetic structure of Rhynchosporium secalis in Australia. Phytopathology. 1999;89(8):639–645. doi: 10.1094/PHYTO.1999.89.8.639. [DOI] [PubMed] [Google Scholar]
- 29.Salamati S, Zhan J, Burdon JJ, McDonald BA. The genetic structure of field populations of Rhynchosporium secalis from three continents suggests moderate gene flow and regular recombination. Phytopathology. 2000;90(8):901–908. doi: 10.1094/PHYTO.2000.90.8.901. [DOI] [PubMed] [Google Scholar]
- 30.Torriani SFF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, McDonald BA, Brunner PC. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion initiated by recent lateral intron transfer. Fungal Genet Biol. 2014;62:34–42. doi: 10.1016/j.fgb.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Feau N, Decourcelle T, Husson C, Desprez-Loustau ML, Dutech C. Finding single copy genes out of sequenced genomes for multilocus phylogenetics in non-model Fungi. PLoS One. 2011;6(4):e18803. doi: 10.1371/journal.pone.0018803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parra G, Bradnam K, Korf I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics. 2007;23(9):1061–1067. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 33.Zhu S, Cao Y-Z, Jiang C, Tan B-Y, Wang Z, Feng S, Zhang L, Su X-H, Brejova B, Vinar T, et al. Sequencing the genome of Marssonina brunnea reveals fungus-poplar co-evolution. BMC Genomics. 2012;13:382. doi: 10.1186/1471-2164-13-382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodwin SB. The barley scald pathogen Rhynchosporium secalis is closely related to the discomycetes Tapesia and Pyrenopeziza. Mycol Res. 2002;106(6):645–654. doi: 10.1017/S0953756202006007. [DOI] [Google Scholar]
- 35.James TY, Kauff F, Schoch CL, Matheny PB, Hofstetter V, Cox CJ, Celio G, Gueidan C, Fraker E, Miadlikowska J, et al. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature. 2006;443(7113):818–822. doi: 10.1038/nature05110. [DOI] [PubMed] [Google Scholar]
- 36.Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP, Dyer PS, Fillinger S, et al. Genomic analysis of the necrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genetics. 2011;7(8):e1002230. doi: 10.1371/journal.pgen.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soreng RJ, Davidse G, Peterson PM, Zuloaga FO, Judziewicz EJ, Filgueiras TS, Morrone O, Romaschenko K. A world-wide phylogenetic classification of Poaceae (Gramineae) 2012. [Google Scholar]
- 38.Aliscioni S, Bell HL, Besnard G, Christin PA, Columbus JT, Duvall MR, Edwards EJ, Giussani L, Hasenstab-Lehman K, Hilu KW, et al. New grass phylogeny resolves deep evolutionary relationships and discovers C4 origins. New Phytol. 2012;193(2):304–312. doi: 10.1111/j.1469-8137.2011.03972.x. [DOI] [PubMed] [Google Scholar]
- 39.Welty RE, Metzger RJ. First report of scald of triticale caused by Rhynchosporium secalis in North America. Plant Dis. 1996;80(11):1220–1223. doi: 10.1094/PD-80-1220. [DOI] [Google Scholar]
- 40.McDonald BA, Linde C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol. 2002;40:349–379. doi: 10.1146/annurev.phyto.40.120501.101443. [DOI] [PubMed] [Google Scholar]
- 41.Debuchy R, Turgeon BG. Mating-type structure, evolution, and function in Euascomycetes. In: Kües U, Fischer R, editors. The Mycota I: growth, differentiation and sexuality. Berlin: Springer; 2006. pp. 293–323. [Google Scholar]
- 42.Rydholm C, Dyer PS, Lutzoni F. DNA sequence characterization and molecular evolution of MAT1 and MAT2 mating-type loci of the self-compatible ascomycete mold Neosartorya fischeri. Eukaryot Cell. 2007;6(5):868–874. doi: 10.1128/EC.00319-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poeggeler S, O'Gorman CM, Hoff B, Kueck U. Molecular organization of the mating-type loci in the homothallic Ascomycete Eupenicillium crustaceum. Fungal Biol. 2011;115(7):615–624. doi: 10.1016/j.funbio.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 44.Hughes TJ, O'Donnell K, Sink S, Rooney AP, Mercedes Scandiani M, Luque A, Bhattacharyya MK, Huang X. Genetic architecture and evolution of the mating type locus in fusaria that cause soybean sudden death syndrome and bean root rot. Mycologia. 2014;106(4):686–697. doi: 10.3852/13-318. [DOI] [PubMed] [Google Scholar]
- 45.Villeneuve AM, Hillers KJ. Whence meiosis? Cell. 2001;106(6):647–650. doi: 10.1016/S0092-8674(01)00500-1. [DOI] [PubMed] [Google Scholar]
- 46.Schurko AM, Logsdon JM., Jr Using a meiosis detection toolkit to investigate ancient asexual “scandals” and the evolution of sex. Bioessays. 2008;30(6):579–589. doi: 10.1002/bies.20764. [DOI] [PubMed] [Google Scholar]
- 47.Ramesh MA, Malik SB, Logsdon JM. A phylogenomic inventory of meiotic genes: evidence for sex in Giardia and an early eukaryotic origin of meiosis. Curr Biol. 2005;15(2):185–191. doi: 10.1016/j.cub.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Malik S-B, Pightling AW, Stefaniak LM, Schurko AM, Logsdon JM., Jr An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis. PLoS ONE. 2008;3(8):e2879. doi: 10.1371/journal.pone.0002879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dyer PS, O'Gorman CM. Sexual development and cryptic sexuality in fungi: insights from Aspergillus species. FEMS Microbiol Rev. 2012;36(1):165–192. doi: 10.1111/j.1574-6976.2011.00308.x. [DOI] [PubMed] [Google Scholar]
- 50.Zhao Z, Liu H, Wang C, Xu J-R. Comparative analysis of fungal genomes reveals different plant cell wall degrading capacity in fungi. BMC Genomics. 2013;14:274. doi: 10.1186/1471-2164-14-274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(D1):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nickel W. Pathways of unconventional protein secretion. Curr Opin Biotechnol. 2010;21(5):621–626. doi: 10.1016/j.copbio.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 54.Nickel W, Seedorf M. Unconventional mechanisms of protein transport to the cell surface of Eukaryotic cells. Annu Rev Cell Dev Biol. 2008;24:287–308. doi: 10.1146/annurev.cellbio.24.110707.175320. [DOI] [PubMed] [Google Scholar]
- 55.Bendtsen JD, Jensen LJ, Blom N, von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17(4):349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 56.Akcapinar GB, Kappel L, Sezerman OU, Seidl-Seiboth V. Molecular diversity of LysM carbohydrate-binding motifs in fungi. Curr Genet. 2015;61(2):103–113. doi: 10.1007/s00294-014-0471-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Jonge R, Thomma BPHJ. Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol. 2009;17(4):151–157. doi: 10.1016/j.tim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 58.Jiang C, He B, Huang R, Huang M, Xu L. Identification and functional analysis of LysM effectors from Marssonina brunnea. Australas Plant Pathol. 2014;43(6):615–622. doi: 10.1007/s13313-014-0316-5. [DOI] [Google Scholar]
- 59.de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MHAJ, Thomma BPHJ. Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science. 2010;329(5994):953–955. doi: 10.1126/science.1190859. [DOI] [PubMed] [Google Scholar]
- 60.Marshall R, Kombrink A, Motteram J, Loza-Reyes E, Lucas J, Hammond-Kosack KE, Thomma BPHJ, Rudd JJ. Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 2011;156(2):756–769. doi: 10.1104/pp.111.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mentlak TA, Kombrink A, Shinya T, Ryder LS, Otomo I, Saitoh H, Terauchi R, Nishizawa Y, Shibuya N, Thomma BPHJ, et al. Effector-mediated suppression of chitin-triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell. 2012;24(1):322–335. doi: 10.1105/tpc.111.092957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van den Burg HA, Harrison SJ, Joosten MHAJ, Vervoort J, de Wit PJGM. Cladosporium fulvum Avr4 protects fungal cell walls against hydrolysis by plant chitinases accumulating during infection. Mol Plant-Microbe Interact. 2006;19(12):1420–1430. doi: 10.1094/MPMI-19-1420. [DOI] [PubMed] [Google Scholar]
- 63.Bowman SM, Free SJ. The structure and synthesis of the fungal cell wall. Bioessays. 2006;28(8):799–808. doi: 10.1002/bies.20441. [DOI] [PubMed] [Google Scholar]
- 64.Mouyna I, Hartl L, Latge J-P. β-1,3-glucan modifying enzymes in Aspergillus fumigatus. Front Microbiol. 2013;4:81. doi: 10.3389/fmicb.2013.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seidl V. Chitinases of filamentous fungi: a large group of diverse proteins with multiple physiological functions. Fungal Biol Rev. 2008;22:36–42. doi: 10.1016/j.fbr.2008.03.002. [DOI] [Google Scholar]
- 66.Kim DJ, Baek JM, Uribe P, Kenerley CM, Cook DR. Cloning and characterization of multiple glycosyl hydrolase genes from Trichoderma virens. Curr Genet. 2002;40(6):374–384. doi: 10.1007/s00294-001-0267-6. [DOI] [PubMed] [Google Scholar]
- 67.Seidl-Seiboth V, Zach S, Frischmann A, Spadiut O, Dietzsch C, Herwig C, Ruth C, Rodler A, Jungbauer A, Kubicek CP. Spore germination of Trichoderma atroviride is inhibited by its LysM protein TAL6. FEBS J. 2013;280(5):1226–1236. doi: 10.1111/febs.12113. [DOI] [PubMed] [Google Scholar]
- 68.Grandaubert J, Bhattacharyya A, Stukenbrock EH. RNA-seq-based gene annotation and comparative genomics of four fungal grass pathogens in the genus Zymoseptoria identify novel orphan genes and species-specific invasions of transposable elements. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brunner PC, Torriani SFF, Croll D, Stukenbrock EH, McDonald BA. Coevolution and life cycle specialization of plant cell wall degrading enzymes in a hemibiotrophic pathogen. Mol Biol Evol. 2013;30(6):1337–1347. doi: 10.1093/molbev/mst041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blackman LM, Cullerne DP, Hardham AR. Bioinformatic characterisation of genes encoding cell wall degrading enzymes in the Phytophthora parasitica genome. BMC Genomics. 2014;15:785. doi: 10.1186/1471-2164-15-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fox EM, Howlett BJ. Secondary metabolism: regulation and role in fungal biology. Curr Opin Microbiol. 2008;11(6):481–487. doi: 10.1016/j.mib.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 72.Keller NP, Turner G, Bennett JW. Fungal secondary metabolism - From biochemistry to genomics. Nat Rev Microbiol. 2005;3(12):937–947. doi: 10.1038/nrmicro1286. [DOI] [PubMed] [Google Scholar]
- 73.Stergiopoulos I, Collemare J, Mehrabi R, de Wit PJGM. Phytotoxic secondary metabolites and peptides produced by plant pathogenic Dothideomycete fungi. FEMS Microbiol Rev. 2013;37(1):67–93. doi: 10.1111/j.1574-6976.2012.00349.x. [DOI] [PubMed] [Google Scholar]
- 74.Dalmais B, Schumacher J, Moraga J, Le Pecheur P, Tudzynski B, Gonzalez Collado I, Viaud M. The Botrytis cinerea phytotoxin botcinic acid requires two polyketide synthases for production and has a redundant role in virulence with botrydial. Mol Plant Pathol. 2011;12(6):564–579. doi: 10.1111/j.1364-3703.2010.00692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eaton CJ, Cox MP, Scott B. What triggers grass endophytes to switch from mutualism to pathogenism? Plant Sci. 2011;180(2):190–195. doi: 10.1016/j.plantsci.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 76.McCormick SP, Stanley AM, Stover NA, Alexander NJ. Trichothecenes: from simple to complex mycotoxins. Toxins. 2011;3(7):802–814. doi: 10.3390/toxins3070802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ichihara A, Hashimoto M, Hirai T, Takeda I, Sasamura Y, Sakamura S, Sato R, Tajimi A. Structure, synthesis, and stereochemistry of (+)-orthosporin, a phytotoxic metabolite of Rhynchosporium orthosporum. Chem Lett. 1989;8:1495–1498. doi: 10.1246/cl.1989.1495. [DOI] [Google Scholar]
- 78.Hallock YF, Clardy J, Kenfield DS, Strobel GA. De-O-methyldiaporthin, a phytotoxin from Drechslera siccans. Phytochemistry. 1988;27(10):3123–3125. doi: 10.1016/0031-9422(88)80012-8. [DOI] [Google Scholar]
- 79.Hoffmeister D, Keller NP. Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep. 2007;24(2):393–416. doi: 10.1039/B603084J. [DOI] [PubMed] [Google Scholar]
- 80.Hertweck C. The biosynthetic logic of polyketide diversity. Angew Chem Int Ed. 2009;48(26):4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 81.Kroken S, Glass NL, Taylor JW, Yoder OC, Turgeon BG. Phylogenomic analysis of type I polyketide synthase genes in pathogenic and saprobic ascomycetes. Proc Natl Acad Sci U S A. 2003;100(26):15670–15675. doi: 10.1073/pnas.2532165100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, et al. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43(D1):D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(D1):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Campbell CD, Vederas JC. Biosynthesis of lovastatin and related metabolites formed by fungal iterative PKS enzymes. Biopolymers. 2010;93(9):755–763. doi: 10.1002/bip.21428. [DOI] [PubMed] [Google Scholar]
- 85.Keller NP. Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol. 2015;11(9):671–677. doi: 10.1038/nchembio.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bushley KE, Turgeon BG. Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol. 2010;10:26. doi: 10.1186/1471-2148-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bushley KE, Ripoll DR, Turgeon BG. Module evolution and substrate specificity of fungal nonribosomal peptide synthetases involved in siderophore biosynthesis. BMC Evol Biol. 2008;8:328. doi: 10.1186/1471-2148-8-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee BN, Kroken S, Chou DYT, Robbertse B, Yoder OC, Turgeon BG. Functional analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot Cell. 2005;4(3):545–555. doi: 10.1128/EC.4.3.545-555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eisendle M, Schrettl M, Kragl C, Muller D, Illmer P, Haas H. The intracellular siderophore ferricrocin is involved in iron storage, oxidative-stress resistance, germination, and sexual development in Aspergillus nidulans. Eukaryot Cell. 2006;5(10):1596–1603. doi: 10.1128/EC.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Oide S, Krasnoff SB, Gibson DM, Turgeon BG. Intracellular siderophores are essential for ascomycete sexual development in heterothallic Cochliobolus heterostrophus and homothallic Gibberella zeae. Eukaryot Cell. 2007;6(8):1339–1353. doi: 10.1128/EC.00111-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schwecke T, Gottling K, Durek P, Duenas I, Kaufer NF, Zock-Emmenthal S, Staub E, Neuhof T, Dieckmann R, von Dohren H. Nonribosomal peptide synthesis in Schizosaccharomyces pombe and the architectures of ferrichrome-type siderophore synthetases in fungi. Chembiochem. 2006;7(4):612–622. doi: 10.1002/cbic.200500301. [DOI] [PubMed] [Google Scholar]
- 92.Oide S, Moeder W, Krasnoff S, Gibson D, Haas H, Yoshioka K, Turgeon BG. NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell. 2006;18(10):2836–2853. doi: 10.1105/tpc.106.045633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zabriskie TM, Jackson MD. Lysine biosynthesis and metabolism in fungi. Nat Prod Rep. 2000;17(1):85–97. doi: 10.1039/a801345d. [DOI] [PubMed] [Google Scholar]
- 94.Kim K-H, Willger SD, Park S-W, Puttikamonkul S, Grahl N, Cho Y, Mukhopadhyay B, Cramer RA, Jr, Lawrence CB. TmpL, a transmembrane protein required for intracellular redox homeostasis and virulence in a plant and an animal fungal pathogen. PLoS Pathogens. 2009;5(11):e1000653. doi: 10.1371/journal.ppat.1000653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu SW, Kroken S, Lee BN, Robbertse B, Churchill ACL, Yoder OC, Turgeon BG. A novel class of gene controlling virulence in plant pathogenic ascomycete fungi. Proc Natl Acad Sci U S A. 2003;100(10):5980–5985. doi: 10.1073/pnas.0931375100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Boettger D, Hertweck C. Molecular diversity sculpted by fungal PKS-NRPS hybrids. ChemBioChem. 2013;14(1):28–42. doi: 10.1002/cbic.201200624. [DOI] [PubMed] [Google Scholar]
- 97.Fisch KM. Biosynthesis of natural products by microbial iterative hybrid PKS-NRPS. RSC Adv. 2013;3(40):18228–18247. doi: 10.1039/c3ra42661k. [DOI] [Google Scholar]
- 98.Keller NP, Hohn TM. Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol. 1997;21(1):17–29. doi: 10.1006/fgbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 99.Chiang Y-M, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CCC. A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, asperfuranone, in Aspergillus nidulans. J Am Chem Soc. 2009;131(8):2965–2970. doi: 10.1021/ja8088185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hogenhout SA, Van der Hoorn RAL, Terauchi R, Kamoun S. Emerging concepts in effector biology of plant-associated organisms. Mol Plant-Microbe Interact. 2009;22(2):115–122. doi: 10.1094/MPMI-22-2-0115. [DOI] [PubMed] [Google Scholar]
- 101.Rafiqi M, Jelonek L, Akum NF, Zhang F, Kogel K-H. Effector candidates in the secretome of Piriformospora indica, a ubiquitous plant-associated fungus. Front Plant Sci. 2013;4:228. doi: 10.3389/fpls.2013.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rovenich H, Boshoven JC, Thomma BPHJ. Filamentous pathogen effector functions: of pathogens, hosts and microbiomes. Curr Opin Plant Biol. 2014;20:96–103. doi: 10.1016/j.pbi.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 103.Stergiopoulos I, de Wit PJGM. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 104.Stukenbrock EH. Evolution, selection and isolation: a genomic view of speciation in fungal plant pathogens. New Phytol. 2013;199(4):895–907. doi: 10.1111/nph.12374. [DOI] [PubMed] [Google Scholar]
- 105.Kirsten S, Navarro-Quezada A, Penselin D, Wenzel C, Matern A, Leitner A, Baum T, Seiffert U, Knogge W. Necrosis-inducing proteins of Rhynchosporium commune, effectors in quantitative disease resistance. Mol Plant-Microbe Interact. 2012;25(10):1314–1325. doi: 10.1094/MPMI-03-12-0065-R. [DOI] [PubMed] [Google Scholar]
- 106.Schürch S, Linde CC, Knogge W, Jackson LF, McDonald BA. Molecular population genetic analysis differentiates two virulence mechanisms of the fungal avirulence gene NIP1. Mol Plant-Microbe Interact. 2004;17(10):1114–1125. doi: 10.1094/MPMI.2004.17.10.1114. [DOI] [PubMed] [Google Scholar]
- 107.Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Res. 2015;43(W1):W39–W49. doi: 10.1093/nar/gkv416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wevelsiep L, Rüpping E, Knogge W. Stimulation of barley plasmalemma H+-ATPase by phytotoxic peptides from the fungal pathogen Rhynchosporium secalis. Plant Physiol. 1993;101:297–301. doi: 10.1104/pp.101.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Elmore JM, Coaker G. The role of the plasma membrane H+-ATPase in plant–microbe interactions. Mol Plant. 2011;4(3):416–427. doi: 10.1093/mp/ssq083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wevelsiep L, Kogel K-H, Knogge W. Purification and characterization of peptides from Rhynchosporium secalis inducing necrosis in barley. Physiol Mol Plant Pathol. 1991;39:471–482. doi: 10.1016/0885-5765(91)90013-8. [DOI] [Google Scholar]
- 111.van’t Slot KAE, van den Burg HA, Kloks CPAM, Hilbers CW, Knogge W, Papavoine CHM. Solution structure of the plant disease resistance-triggering protein NIP1 from the fungus Rhynchosporium secalis shows a novel b-sheet fold. J Biol Chem. 2003;278(46):45730–45736. doi: 10.1074/jbc.M308304200. [DOI] [PubMed] [Google Scholar]
- 112.Fiegen M, Knogge W. Amino acid alterations in isoforms of the effector protein NIP1 from Rhynchosporium secalis have similar effects on its avirulence- and virulence-associated activities on barley. Physiol Mol Plant Pathol. 2002;61(5):299–302. doi: 10.1006/pmpp.2002.0442. [DOI] [Google Scholar]
- 113.Steiner-Lange S, Fischer A, Böttcher A, Rouhara I, Liedgens H, Schmelzer E, Knogge W. Differential defense reactions in leaf tissues of barley in response to infection by Rhynchosporium secalis and to treatment with a fungal avirulence gene product. Mol Plant-Microbe Interact. 2003;16(10):893–902. doi: 10.1094/MPMI.2003.16.10.893. [DOI] [PubMed] [Google Scholar]
- 114.Stefansson TS, Willi Y, Croll D, McDonald BA. An assay for quantitative virulence in Rhynchosporium commune reveals an association between effector genotype and virulence. Plant Pathol. 2013;63(2):405–414. doi: 10.1111/ppa.12111. [DOI] [Google Scholar]
- 115.Conant GC, Wolfe KH. Turning a hobby into a job: how duplicated genes find new functions. Nat Rev Genet. 2008;9(12):938–950. doi: 10.1038/nrg2482. [DOI] [PubMed] [Google Scholar]
- 116.Syme RA, Hane JK, Friesen TL, Oliver RP. Resequencing and comparative genomics of Stagonospora nodorum: Sectional gene absence and effector discovery. G3 (Bethesda) 2013;3(6):959–969. doi: 10.1534/g3.112.004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Condon BJ, Leng YQ, Wu DL, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, MohdZainudin N, et al. Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 2013;9(1):e1003233. doi: 10.1371/journal.pgen.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kleemann J, Rincon-Rivera LJ, Takahara H, Neumann U, van Themaat EVL, van der Does HC, Hacquard S, Stueber K, Will I, Schmalenbach W, et al. Sequential delivery of host-induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum. PLoS Pathogens. 2012;8(4):e1002643. doi: 10.1371/journal.ppat.1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Catlett NL, Lee BN, Yoder OC, Turgeon BG. Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newslett. 2003;50:9–11. [Google Scholar]
- 122.Eaton CJ, Cox MP, Ambrose B, Becker M, Hesse U, Schardl CL, Scott B. Disruption of signaling in a sungal-grass symbiosis leads to pathogenesis. Plant Physiol. 2010;153(4):1780–1794. doi: 10.1104/pp.110.158451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tanaka A, Christensen MJ, Takemoto D, Park P, Scott B. Reactive oxygen species play a role in regulating a fungus-perennial ryegrass mutualistic interaction. Plant Cell. 2006;18(4):1052–1066. doi: 10.1105/tpc.105.039263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Takemoto D, Tanaka A, Scott B. A p67(Phox)-like regulator is recruited to control hyphal branching in a fungal-grass mutualistic symbiosis. Plant Cell. 2006;18(10):2807–2821. doi: 10.1105/tpc.106.046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Tanaka A, Takemoto D, Hyon GS, Park P, Scott B. NoxA activation by the small GTPase RacA is required to maintain a mutualistic symbiotic association between Epichloe festucae and perennial ryegrass. Mol Microbiol. 2008;68(5):1165–1178. doi: 10.1111/j.1365-2958.2008.06217.x. [DOI] [PubMed] [Google Scholar]
- 126.Johnson LJ, Koulman A, Christensen M, Lane GA, Fraser K, Forester N, Johnson RD, Bryan GT, Rasmussen S. An extracellular siderophore is required to maintain the mutualistic interaction of Epichloe festucae with Lolium perenne. PLoS Pathogens. 2013;9(5):e1003332. doi: 10.1371/journal.ppat.1003332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 129.Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18(5):821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Salamov AA, Solovyev VV. Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000;10(4):516–522. doi: 10.1101/gr.10.4.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 2008;18(12):1979–1990. doi: 10.1101/gr.081612.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34:W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Donlin MJ. Using the Generic Genome Browser (GBrowse). Curr Protoc Bioinform. 2009;28:9.9:9.9.1-9.9.25. [DOI] [PubMed]
- 136.Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Aguileta G, Marthey S, Chiapello H, Lebrun MH, Rodolphe F, Fournier E, Gendrault-Jacquemard A, Giraud T. Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol. 2008;57(4):613–627. doi: 10.1080/10635150802306527. [DOI] [PubMed] [Google Scholar]
- 138.Parra G, Bradnam K, Ning Z, Keane T, Korf I. Assessing the gene space in draft genomes. Nucleic Acids Res. 2009;37(1):289–297. doi: 10.1093/nar/gkn916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:I351–I358. doi: 10.1093/bioinformatics/bti1018. [DOI] [PubMed] [Google Scholar]
- 140.Wootton JC, Federhen S. Statistics of local complexity in amino acid sequences and sequence databases. Comput Chem. 1993;17(2):149–163. doi: 10.1016/0097-8485(93)85006-X. [DOI] [Google Scholar]
- 141.Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. RepBase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 143.Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0. 2010. pp. 1996–2010. [Google Scholar]
- 144.Abrusán G, Grundmann N, DeMester L, Makalowski W. TEclass-a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics. 2009;25(10):1329–1330. doi: 10.1093/bioinformatics/btp084. [DOI] [PubMed] [Google Scholar]
- 145.Walter MC, Rattei T, Arnold R, Gueldener U, Muensterkoetter M, Nenova K, Kastenmueller G, Tischler P, Woelling A, Volz A, et al. PEDANT covers all complete RefSeq genomes. Nucleic Acids Res. 2009;37:D408–D411. doi: 10.1093/nar/gkn749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guindon S, Dufayard JF, Hordijk W, Lefort V, Gascuel O. PhyML: fast and accurate phylogeny reconstruction by Maximum Likelihood. Infect Genet Evol. 2009;9(3):384–385. [Google Scholar]
- 148.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Le SQ, Gascuel O. An improved general amino acid replacement matrix. Mol Biol Evol. 2008;25(7):1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 150.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 151.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S. Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci U S A. 2012;109(47):19333–19338. doi: 10.1073/pnas.1213199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:1–19. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47(9):736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wolf T, Shelest V, Shelest E. Motif-based method for the genome-wide prediction of eukaryotic gene clusters. In: Petrosino A, Maddalena L, Pala P, editors. New Trends in Image Analysis and Processing – ICIAP 2013. Berlin, Heidelberg: Springer; 2013. pp. 389–398. [Google Scholar]
- 155.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat Protoc. 2007;2(4):953–971. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 156.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 158.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162(1):156–159. doi: 10.1016/0003-2697(87)90021-2. [DOI] [PubMed] [Google Scholar]
- 159.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART (TM) approach for full-length cDNA library construction. Biotechniques. 2001;30(4):892–897. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- 160.Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucl Acid Res. 2004;32(3):e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Siersleben S, Penselin D, Wenzel C, Albert S, Knogge W. PFP1, a gene encoding an Epc-N domain-containing protein, is essential for pathogenicity of the barley pathogen Rhynchosporium commune. Eukaryot Cell. 2014;13(8):1026–1035. doi: 10.1128/EC.00043-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kuwayama H, Obara S, Morio T, Katoh M, Urushihara H, Tanaka Y. PCR-mediated generation of a gene disruption construct without the use of DNA ligase and plasmid vectors. Nucleic Acids Res. 2002;30(2):e2. doi: 10.1093/nar/30.2.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Punt PJ, Oliver RP, Dingemanse MA, Pouwels MA, van den Hondel CAMJJ. Transformation of Aspergillus based on the hygromycin B resistance marker from E. coli. Gene. 1987;56:117–124. doi: 10.1016/0378-1119(87)90164-8. [DOI] [PubMed] [Google Scholar]
- 164.Rohe M, Searle J, Newton AC, Knogge W. Transformation of the plant pathogenic fungus, Rhynchosporium secalis. Curr Genet. 1996;29:587–590. doi: 10.1007/BF02426964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Datasets used for phylogenetic analyses are available from the Dryad Digital Repository (http://dx.doi.org/10.5061/dryad.qm130). Sequence data sets are accessible at http://pedant.helmholtz-muenchen.de/genomes.jsp?category=fungal. The genome and annotation data were submitted to the European Nucleotide Archive (ENA) R. commune UK7: http://www.ebi.ac.uk/ena/data/view/FJUW01000001-FJUW01000164; R. secalis: http://www.ebi.ac.uk/ena/data/view/FJVC01000001-FJVC01002130; R. agropyri: http://www.ebi.ac.uk/ena/data/view/FJUX01000001-FJUX01000508.