


Abstract
Repair of most modified and mispaired bases in the genome is initiated by DNA glycosylases, which bind to their respective targets and cleave the N-glycosyl bond to initiate base excision repair (BER). The mammalian homolog of the Escherichia coli MutY DNA glycosylase (MYH) cleaves adenine residues paired with either oxidized or non-modified guanines. MYH is crucial for the avoidance of mutations resulting from oxidative DNA damage. Multiple N-terminal splice variants of MYH exist in mammalian cells and it is likely that different variants result in the production of enzymes with altered properties. To investigate whether modifications in the N-terminus are consequential to MYH function, we overexpressed intact and N-terminal-deletion rat MYH proteins and examined their activities. We found that deletion of 75 amino acids, which perturbs the catalytic core that is conserved with E.coli MutY, abolished excision activity. In contrast, deletions limited to the extended mammalian N-terminal domain, differentially influenced steady-state excision rates. Notably, deletion of 50 amino acids resulted in an enzyme with a significantly lower Km favoring formation of excision products with 3′-OH termini. Our findings suggest that MYH isoforms divergent in the N-terminus influence excision rates and processing of abasic sites.
INTRODUCTION
Reactive oxygen species, the by-products of oxidative respiration, challenge cellular homeostasis and damage macromolecules, including DNA (1). To maintain genomic integrity, cells are equipped with DNA repair mechanisms with most oxidative DNA lesions repaired via the base excision repair (BER) process, which is initiated by DNA glycosylases (2,3). The key mammalian glycosylases for protection from the mutagenic consequences of oxidative lesions are orthologs of the bacterial MutM and MutY DNA glycosylases, OGG1 and MYH, respectively (4–6). MYH initiates base excision repair (BER) by catalyzing removal of adenine residues mispaired during erroneous replication with either oxidized or non-modified guanines (7). MYH is significantly larger than the bacterial protein and comprises the entire MutY sequence flanked by extended N- and C-terminal domains (8–11). These 50–60 amino acid terminal domains are involved in subcellular targeting of MYH and interactions with other proteins (9,12–14);e.g. replication protein A (RPA) and PCNA binding motifs map to the N- and C-terminus, respectively, suggesting the coupling of MYH to the DNA replication machinery (15). In addition, potential acetylation and phosphorylation sites in mammalian MYH sequences are clustered in both termini; defective phosphorylation has been implicated in MYH dysfunction in colorectal cancer cell lines (16), and in vitro, MYH glycosylase activity is modified by phosphorylation (17).
Interestingly, both the rat and mouse transcripts cloned to date (10,11,18) lack sequences encoding the first 14 N-terminal amino acids, which are present in human type α(1–3) MYH transcripts (9). In the human sequence, these 14 amino acids are a part of a putative mitochondria-targeting signal (12,13) as well as the proposed RPA binding motif (15). In this respect, the rodent cDNAs, are similar to type β human transcripts, which start with the second methionine in hMYHα(1–3). The cloning of multiple human transcripts divergent in the 5′ region suggested that corresponding protein isoforms might also exist (9), localize to distinct cellular compartments (12–14) and differentially interact with other proteins (15).
To date, the N-terminus of MYH has been studied mainly with a focus on subcellular localization and not with respect to catalytic properties, such as substrate preferences or differential processing of abasic sites. In mammalian cells, the primary enzyme for processing of abasic sites is the apurinic/ apyrimidinic (AP) endonuclease (APE) (2). Physical interactions between MYH and APE have been demonstrated (15) and in reconstituted systems, APE enhanced excision activity of DNA glycosylases (19,20) including MYH (10). Interestingly, moderate enhancement of MYH catalytic activity by APE can be mediated also via a mechanism that is independent of its endonuclease activity (10).
To investigate whether N-terminal modifications are consequential to MYH activity, the intact rat MYH (FL) and three consecutive N-terminal deletions (Δ25, Δ50, Δ75) were constructed (Figure 1) and overexpressed in cultured cells. The deletion of 25 N-terminal amino acids coincides with the predicted cleavage site for mitochondrial import (PSORT II server, http://psort.nibb.ac.jp) and also removes the putative RPA binding motif while Δ50 eliminates, in addition, several predicted targets for post-translational modifications. The consecutive Δ75 disrupts a stretch of amino acids homologous to the N-terminus of the Escherichia coli MutY protein, which is a component of the enzyme catalytic core (21,22). We found that the deletion of 75 amino acids abolished excision activity. In contrast, deletions of either 25 or 50 amino acids, retained glycosylase activity, but differentially influenced steady-state excision rates and subsequent processing of abasic sites. It should be noted that in our study, the tested proteins are likely to be post-translationally modified and their properties may differ from those of bacterially overexpressed proteins that are typically used in assays reconstituted with pre-selected components. Our data obtained in a mammalian system, in the presence of partner proteins, should provide complementary information, which is not readily attainable in typical reconstitution assays. The data raise the possibility that in the cell, MYH isoforms, divergent in the N-terminal region, may influence processing of abasic sites, which they generate.
Figure 1.
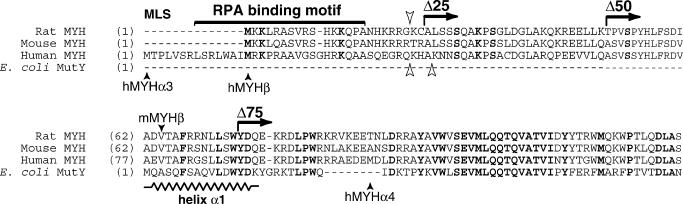
Sequence alignment of N-terminal amino acids of mammalian MYH and E.coli MutY proteins. Horizontal arrows indicate the N-terminal deletions in the rat sequence (Δ25, Δ50, Δ75). Closed vertical arrowheads indicate the human and mouse MYH variants. Open arrowheads indicate predicted cleavage sites for mitochondrial import in the rat (downwards), human (upwards left) and mouse (upwards right) proteins. Proposed mitochondrial localization signal (MLS) and RPA binding motif are marked. Potential phosphorylation and acetylation sites conserved in all mammalian proteins are highlighted in boldface. A stretch of amino acids corresponding to the α1 helix in MutY (22) is indicated.
MATERIALS AND METHODS
MYH expression constructs
Rat MYH cDNA AF478683 (11) was inserted into the pFLAG-CMV-4 vector (Sigma, St Louis, MO). The construct was named full-length (FL) MYH. Sequential 25 N-terminal deletions were named Δ25, starting with amino acid 25, Δ50 and Δ75. Constructs were confirmed by sequencing on both strands. Transfections of 293 human embryonic kidney cells were carried out with Fugene 6 (Roche, IN) according to the manufacturer's instructions.
Protein expression analysis: cell fractionation, western blotting and immunoprecipitation
Briefly, cell pellets were suspended in cold hypotonic buffer (10 mM HEPES, pH 7.6, 10 mM KCl, 0.1 mM EDTA, 0.1 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid (EGTA), 1 mM DTT, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2 μg/ml Pepstatin supplemented with the Complete Protease Inhibitor Cocktail Tablet (Roche Diagnostics) and incubated on ice for 30 min with occasional mixing. Homogenates were spun at 800 g for 3 min to pellet nuclei, which were then resuspended and washed. Supernatants were combined for preparation of mitochondrial and cytoplasmic fractions. Nuclear pellets were resuspended in high salt buffer (20 mM HEPES, pH 7.6, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 2 μg/ml Pepstatin) and incubated at 4°C for 30 min with occasional mixing. Nuclear extracts were cleared by centrifugation (18 000 g, 10 min) and aliquoted. Supernatants from the 800 g centrifugation steps were pooled and spun at 12 500 g for 8 min; the supernatant is the cytoplasmic fraction, the pellet, extracted in high salt buffer (20 mM HEPES, pH 7.6, 300 mM KCl, 1 mM EDTA, 1 mM DTT, 5% glycerol, 0.5% Triton X-100) is the mitochondrial fraction. Extracts (20 μg per well) were resolved by 10% SDS–PAGE; BenchMark Prestained Protein Ladder (Gibco BRL, Rockville, MD) was used for molecular mass reference. αFlag antibody was used at 1:20 000, αVDAC (Santa Cruz, Santa Cruz, CA) at 1:2000 and the enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia, Piscataway, NJ) was used according to the manufacturer's instructions. Immunoprecipitation of Flag fusion proteins was with monoclonal αFlag conjugated to agarose beads (Anti-Flag M2 Affinity Gel, Sigma, St Louis, MO). Elution of immunoprecipitated proteins was with excess Flag-Peptide (Sigma). Both procedures were done according to the manufacturer's instructions with minor modifications (23). Eluted fractions were brought to 30% glycerol and stored in aliquots at −80°C.
Electrophoretic mobility shift assay
End-labeled 33mer GO/A substrate (identical to the glycosylase assay) was used. Reactions were assembled in binding buffer (10 mM Tris–HCl, pH 7.4, 0.5 mM DTT, 5 mM EDTA, 20 mM NaCl, 75 μg/ml BSA, 1 μg/ml poly(dI–dC), 10% glycerol) with 5 μg nuclear extract and 10 fmol substrate, incubated 10 min at 23°C and resolved in 6% native polyacrylamide gel in Tris–Glycine buffer, pH 8.3. Substrate excision under these incubation conditions was <1% (data not shown). Specific and non-specific cold substrates were added at indicated molar ratios. Electrophoretic mobility shift assay (EMSA) with pull-down Flag-MYH fusion proteins: binding reactions were assembled with equivalent eluate volumes predetermined by western blotting analyses with αFlag antibody. Reactions were assembled as above, except that poly(dI–dC) was omitted and EDTA was reduced to 2.25 mM.
MYH glycosylase activity assay
DNA glycosylase activity was assayed either on a 33mer duplex substrate carrying 8-oxodG (O) or G (5′-CTG CT CAC ACT CGA CAC ACO/G AAT TGT GTA GTA GTG C-3′) paired with adenine at position 17 [3′-GA GTG TGA GCT GTG TGA TTA ACA CAT CAT CAC G-5′] (Midland Inc., Midland, TX). Adenine-containing oligo was 5′ end-labeled with T4 polynucleotide kinase in the presence of [γ-32P]ATP, twice purified through a G25 quick spin column and annealed with the GO- or G-containing matching oligomer. Glycosylase assays were done as described previously (24) with some modifications. Briefly, reactions were assembled in 20 μl volume with 0.25–1 nM end-labeled substrate, with or without addition of cold substrate, reaction buffer (10 mM Tris–HCl, pH 7.6, 0.5 mM DTT, 1.5% glycerol, 2.25 mM EDTA, 0.1 mg/ml BSA) with 10 μg nuclear extract and NaCl adjusted to 40 mM. When indicated, either EDTA was omitted or reactions were carried out in buffer containing MgCl2 (66 mM Tris–HCl pH 7.6, 1 mM DTT, 1 mM MgCl2, 0.2 mM EDTA, 0.1 mg/ml BSA). Incubation was at 37°C for the indicated time, reactions were terminated with 5× alkaline loading buffer (0.5 N NaOH, 97% formamide, 10 mM EDTA, pH 8, 0.025% bromophenol blue, 0.025% xylene cyanol) and heated for 3 min at 95°C. Reaction mixtures were resolved in 15% polyacrylamide–7 M urea denaturing gels. Electrophoresis was in Tris–Borate buffer, pH 8.3, at 14 mA for 2 h, products were visualized by autoradiography and quantified on Phosphorimager (Molecular Dynamics).
Analysis of gel migration of GO/A* excision products differing in 3′ termini
Products by MutY alone or by the combined action of MutY and either APE or Nfo endonucleases were generated in the presence or absence of MgCl2 (see glycosylase assay). Recombinant E.coli MutY, Endonuclease IV (Nfo) and recombinant human APE were used at 0.025, 0.5 and 0.25 U, respectively (Trevigen, Gaithersburg, MD). Reactions were terminated either with or without NaOH and resolved in 15% denaturing gels (Figure 2). MutY alone generated an abasic site, which was then hydrolyzed by a β-elimination reaction in the presence of NaOH (lane 2, arrow). Addition of APE (20-fold molar excess) in the absence of MgCl2 increased MutY product amount (lane 3), indicating that enhancement of MutY glycosylase activity can be independent of APE catalytic activity. On the other hand, addition of APE in the presence of MgCl2 and termination of reaction in the absence of alkali (97% formamide, 10 mM EDTA, pH 8, 0.025% bromophenol blue, 0.025% xylene cyanol), generated a fast migrating product, consistent with cleavage of the phosphodiester bond 5′ to the abasic site and generation of 3′-OH ends (lane 12, arrow). No 3′-OH ends were generated by MutY supplemented with APE, in the presence of EDTA (lane 9). In contrast, addition of Nfo, which does not require MgCl2, generated the fast migrating product consistent with 3′-OH ends, under all tested conditions (lanes 4, 7, 10 and 13). No product was obtained with Nfo alone (lane 1) or APE alone (lane 14) indicating that neither enzyme targets the intact GO/A substrate.
Figure 2.
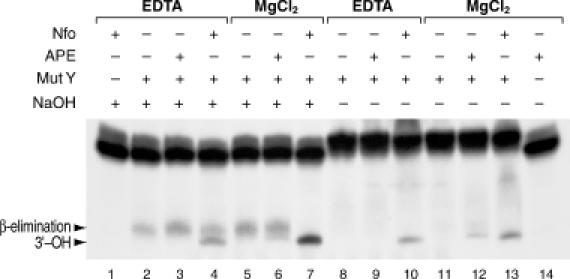
Gel migration analysis of 3′ termini generated by MutY, APE and Nfo. Autoradigram shows migration of GO/A excision products generated by MutY alone or in combination with either APE or Nfo and terminated either with or without NaOH. Migration of the product generated by MutY alone represents an abasic site hydrolyzed by NaOH-mediated β-elimination (lane 2, arrow). Addition of APE (20-fold molar excess) in the absence of MgCl2, increased MutY product amount (lane 3), whereas addition of APE in the presence of MgCl2 and termination without NaOH, generated a fast migrating product, consistent with APE cleavage of the phosphodiester bond 5′ to the abasic site and generation of 3′-OH ends (lane 12, arrow). In contrast, addition of Nfo, which is independent of MgCl2, generated the fast migrating product consistent with 3′-OH ends, under all tested conditions (lane 4, 7, 10, 13, arrow). Action of MutY alone generates an abasic site, which is not cleaved in absence of alkali and, accordingly, no product is observed (lane 8 and 11).
RESULTS
Excision products generated by full-length and N-terminal deletion MYH proteins differ on GO/A and G/A substrates
Western blotting analysis of cells transfected with the full-length (FL), Δ25, Δ50 and Δ75 rat MYH constructs showed that overexpressed MYH proteins preferentially localize to the nucleus with the exception of Δ75, which exhibits random distribution, suggesting a weakening of nuclear localization (Figure 3A). Lack of significant cross contamination among the different fractions is confirmed by western blotting with the mitochondrial channel protein VDAC (Figure 3A).
Figure 3.
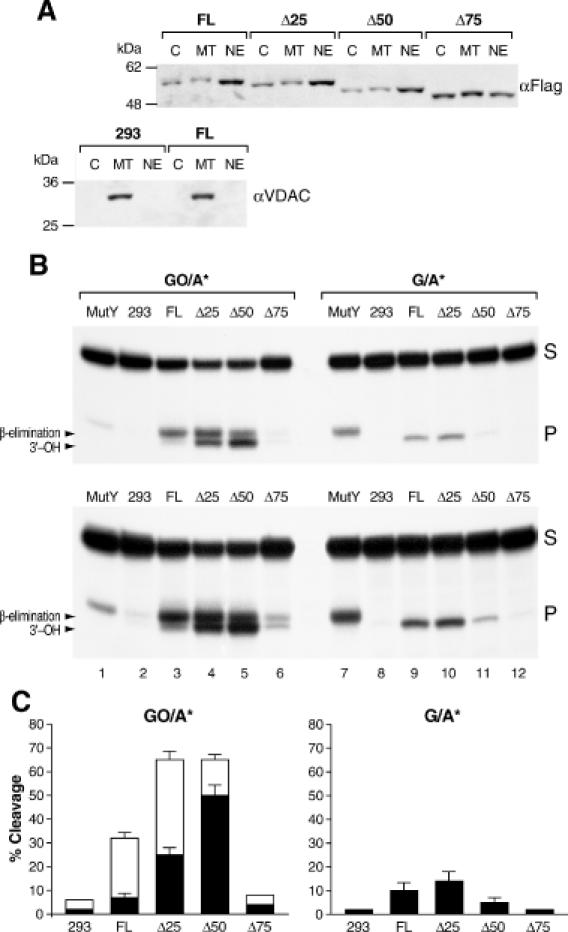
Excision products of FL and N-ter deletion-MYH proteins on GO/A and G/A substrates. (A) Expression and subcellular partition of FL and Δ25, Δ50 and Δ75 proteins: western blotting analysis of nuclear (NE), mitochondrial (MT) and cytoplasmic (C) fractions of FL, Δ25, Δ50 and Δ75 with the αFlag antibody and of non-transfected and FL-transfected 293 cells with αVDAC, as indicated. (B) Autoradiogram showing differences in migration and levels of excision products generated on GO/A and G/A substrates incubated with indicated nuclear extracts. Product generated by the E.coli MutY protein is included as size reference (lanes 1 and 7). Low level of excision is seen in naive 293 cells with both substrates (lanes 2 and 8, lower panel, long exposure). Mammalian MYH proteins generate higher levels of products on GO/A than on G/A, while the opposite is observed with E.coli MutY (lanes 1 and 7). On GO/A, the FL protein generates primarily a slowly migrating product (lane 3), Δ25 generates similar amounts of slow and fast products (lane 4), whereas Δ50 generates primarily the fast product (lane 5). Product levels for Δ75 are similar to those observed with naive 293 cells (lane 6). In all cases, only the fast migrating products are observed with the G/A substrate. (C) Bars represent fast (black) and slow (blank) cleavage products generated in three independent assays and expressed as mean ± SEM of radioactivity quantified on the Phosphorimager.
Excision activities of the FL and N-terminal-deletion proteins (Δ25, Δ50 and Δ75) were examined in parallel on GO/A and G/A substrates (Figure 3B). Nuclear extracts with similar levels of the different MYH proteins were identified by western blotting and used in excision assays. In agreement with previous observations with an overexpressed mammalian MYH protein (10), activity was higher on the GO/A than on the G/A substrate. Interestingly, on the G/A substrate, a significantly lower level of product was generated by Δ50 (lane 11) compared with FL and Δ25 (lanes 9–10), while on the GO/A substrate total product amounts for each of the three constructs were closer (lanes 3–5). Remarkably, all products obtained on G/A (lanes 8–11) migrated faster than the MutY product (lane 7), indicating that in nuclear extracts, MYH-generated abasic sites on the G/A substrate are readily processed to 3′-OH termini (see Figures 2 and 3). In comparison, robust amounts of products were generated on the GO/A substrate; each deletion, however, generated a different profile of excision products during a 2.5 h incubation without supplemental MgCl2. In the case of FL the predominant product migrated slowly (lane 3), similar to the product generated by the action of MutY alone (lane 1), i.e. consistent with abasic sites, hydrolyzed by alkali. In contrast, excision by Δ25 produced equal amounts of fast and slow migrating products (lane 4), whereas for Δ50 the fast migrating product was predominant (lane 5). The trace product levels generated by naive 293 cells (lane 2) and Δ75 (lane 6) indicated that catalytic activity is abolished when the 75 N-terminal amino acids of the rat MYH are deleted. Although a long exposure was required (lower panel) to visualize the trace product amounts generated with the naive 293 cells, it shows that endogenous MYH proteins generate both fast and slow migrating products. In combination the data show that both the N-terminus of MYH as well as the substrate, influence subsequent processing of MYH-generated abasic sites and, that given enough time, APE or APE-like activity modifies MYH-generated abasic sites also in the absence of added MgCl2.
Deletion of 75 N-terminal amino acids disrupts MYH binding to GO/A
The ability of overexpressed MYH proteins to bind the GO/A substrate was assessed by the formation of specific complexes in EMSAs (Figure 4). Binding reactions were carried out for 10 min at 23°C in presence of 5 mM EDTA, i.e. under conditions that do not favor cleavage of GO/A (data not shown). Nuclear extracts from the naive 293 cells generated several complexes; only one complex was competed with a 150-fold excess of the GO/A duplex (lanes 25–27). Nuclear extracts from 293 cells overexpressing MYH proteins generated robust specific complexes. The observed uniform intensities of specific complexes (Figure 4A) confirm that equivalent levels of overexpressed proteins are present in the corresponding nuclear extracts. Differential migration of complexes reflected the stepwise reduction in the molecular mass of deletion proteins. Specificity of complexes generated with the FL protein was confirmed by selective competition with an excess of the GO/A duplex and partial competition with excess of GO/C and G/A (lanes 2–7) but not with excess of G/C (lane 8). Interestingly, 150-fold molar excess of cold GO/A substrate completely abolished binding of Δ50 (lanes 12–13 and 19–20) but not of Δ25 (lanes 9–10) and FL proteins (lanes 2–3). Nuclear extract from cells overexpressing the Δ75 protein generated a faint complex (lane 22) similar to that observed with naive 293 cells (lane 25), suggesting that Δ75 is unable to form complexes with GO/A. To definitely test the ability of modified MYH proteins to bind the GO/A substrate, overexpressed proteins were immunoprecipitated with αFlag antibody and eluted with excess of the Flag-Peptide. Levels of eluted proteins were estimated by western blotting and equivalent amounts of the respective eluates were used for EMSAs. While substantial complexes were formed with the FL, Δ25 and Δ50 protein eluates, no specific complex was detectable with the Δ75 eluate (Figure 4B).
Figure 4.
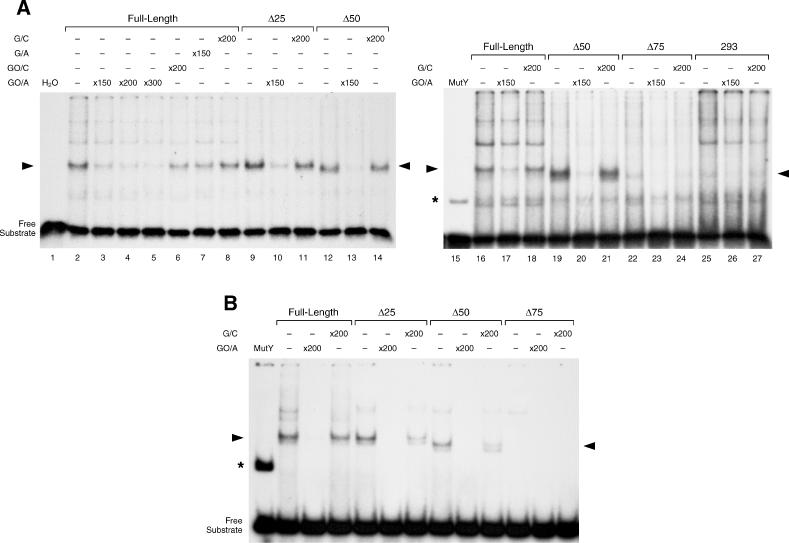
Binding of FL- and N-ter-deletion MYH proteins to GO/A substrate. (A) Autoradiograms show EMSAs with nuclear extracts from naive 293 cells and cells transfected with FL and N-terminal deletion MYH proteins. Complexes formed with MutY are shown for reference (asterisk). Arrowheads indicate specific complexes. Naive 293 cells generated several complexes; only one complex was competed with a 150-fold excess of the GO/A substrate (lanes 25–27). The complex generated with FL was challenged by competition with GO/A, GO/C G/A or G/C (lanes 2–8). A 150-fold molar excess of cold GO/A eliminated the complex formed with Δ50 (lanes 12–13, 19–20) and to a lesser degree complexes formed with Δ25 and FL MYH (lanes 9, 10 and 2, 3, respectively). Cells overexpressing Δ75 show a faint complex (lane 22) similar to that observed with naive 293 cells (lane 25). (B) Binding of immunoprecipitated FL- and N-ter-deletion MYH proteins. Autoradiogram shows complexes formed with eluates of immunoprecipitated FL, Δ25 and Δ50 proteins. Identity of complexes is confirmed by competition with excess specific and non-specific substrates, as indicated (arrowheads). No specific complexes are detectable with Δ75 protein.
Steady-state excision rates by FL and N-terminal deletion MYH proteins in presence or absence of MgCl2
Steady-state excision rates by overexpressed FL, Δ25 and Δ50 were determined over a 2000-fold range of GO/A* substrate concentrations (0.25–512 nM). Time courses for each construct were linear for at least 10 min (data not shown; accordingly, this time point was selected for measurement of steady-state excision rates.
To determine to what extent product release by MYH is rate limiting in nuclear extracts, excision by MYH was measured under conditions, which either favor (+MgCl2) or disfavor (−MgCl2) APE activity (equal levels of the APE protein and cleavage of abasic sites were verified in all extracts; data not shown). APE is thought to stimulate MYH via enhancement of product release (10). In the presence of MgCl2, excision rates by all MYH proteins exhibited Michaelis–Menten kinetics and Δ50 had the lowest apparent Km (∼15 nM) (Figure 5A and C, Table 1). In the absence of MgCl2, excision rates were significantly reduced (Figure 5B and C), and when reactions were done in the absence of MgCl2 and added EDTA, which may further inhibit APE, excision rates were indeed reduced further (Figure 5C, Table 1).
Figure 5.
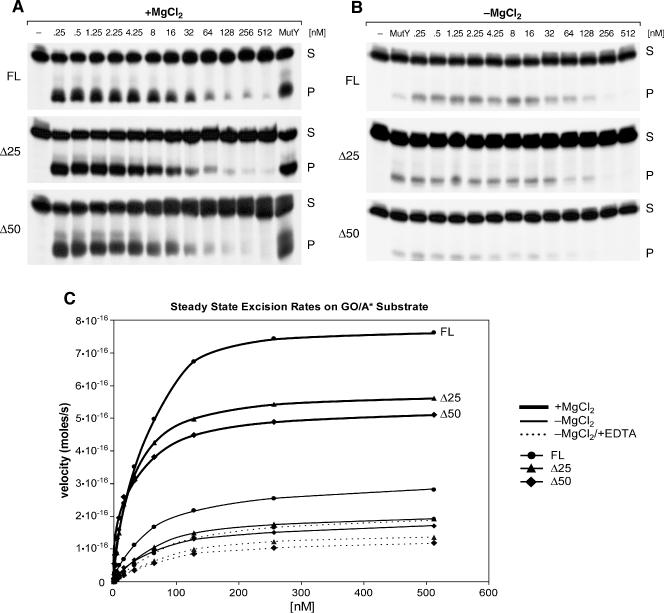
Steady-state kinetics of GO/A cleavage by MYH proteins. Representative autoradiograms show excision products generated on GO/A by nuclear extracts from 293 cells transfected with FL, Δ25 or Δ50 MYH. (A) Excision in the presence of MgCl2. Reactions included 0.25 nM end-labeled GO/A* supplemented with increasing concentrations of cold GO/A (0.25–512 nM) as indicated. Product generated by 0.1 U of E.coli MutY served as size reference. Reactions were carried out for 10 min at 37°C. (B) Excision in the absence of MgCl2. Reactions were carried out over the same range of substrate concentrations. E.coli MutY was used at 0.025 U. (C) Steady-state rates of GO/A Cleavage by FL- and N-ter-deletion MYH proteins. Heavy lines show cleavage in the presence of 1 mM MgCl2, regular lines in the absence of MgCl2 and dotted lines in absence of MgCl2 and presence of 2.25 nM EDTA. The values for FL protein are shown as circles, for Δ25 –triangles, and for Δ50 diamonds. Kinetic values represent means of four independent experiments. Standard errors for the data points were in the range of 3–10% (data not shown).
Table 1. Kinetic parameters of full-length, Δ25 and Δ50 MYH proteins.
| Condition | Construct | Apparent Vmax × 10−16 (mol/s) | Apparent Km (nM) | V/K |
|---|---|---|---|---|
| +MgCl2 | Full length | 8.5 ± 0.2 | 41 ± 3 | 0.21 ± 0.02 |
| Δ25 | 5.9 ± 0.04 | 25 ± 0.6 | 0.24 ± 0.01 | |
| Δ50 | 5.0 ± 0.1 | 15 ± 2 | 0.33 ± 0.04 | |
| −MgCl2 | Full length | 3.1 | 53 | 0.06 |
| Δ25 | 2.3 | 76 | 0.03 | |
| Δ50 | 1.9 | 57 | 0.03 | |
| −MgCl2 | Full length | 2.3 | 106 | 0.022 |
| +EDTA | Δ25 | 1.6 | 93 | 0.017 |
| Δ50 | 1.4 | 90 | 0.015 |
Excision assays were carried out with indicated nuclear extracts at 37°C for 10 min. Values were calculated from data points plotted in Figure 5C. The errors represent the standard errors for kinetic parameters for the Michaelis–Menten equation.
DISCUSSION
The existence of MYH isoforms divergent in the N-terminus is suggested by the cloning of human (9) and mouse (18) MYH splice-variants in this region and by purification of a low molecular mass MYH enzyme from calf liver mitochondria (24). Based on the reported human N-terminal splice variant MYHα4 (Figure 1), we initially generated a deletion with a translation start site corresponding to M107 in the human MYHα3 (9), which, however, did not exhibit excision activity (data not shown). Since the MYH transcripts reported so far are unlikely to represent the complete spectrum of MYH variants in mammalian tissues, to examine modifications in the N-terminus of MYH with respect to substrate preferences and processing of excision products, we generated three types of N-terminal deletions: (i) Δ75, which perturbs homology with the bacterial MutY protein; (ii) Δ50, which retains homology with the bacterial protein but eliminates most of the extended mammalian N-terminal domain and closely overlaps with the recently reported mouse splice variant, mMYHβ (18); (iii) Δ25, which coincides with predicted cleavage sites for mitochondrial import (PSORT II server, http://psort.nibb.ac.jp) for the rat, mouse and human MYH sequences (Figure 1).
Since homology with the bacterial MutY starts at position 62 within the rat sequence (Figure 1), Δ75 corresponds to a loss of 13 N-terminal amino acids in E.coli MutY. Earlier domain structure studies revealed that the 1–226 amino acid catalytic domain of MutY is sufficient for binding to the G/A mismatch (25); we find that the capacity of Δ75 to bind the GO/A substrate is abolished. Crystal structure of the catalytic core of the MutY protein (1–226 amino acids) revealed that 3–17 N-terminal amino acids form an alpha helix, a component of the [4F–4S] cluster domain and, therefore, are likely to be essential for MYH glycosylase activity (22). A recently solved, co-crystal structure of the MutY-A:8-oxoG-DNA lesion recognition complex confirms that these N-terminal amino acids constitute a component of the [4Fe–4S] cluster domain; we find that catalytic activity is abolished when the corresponding sequence in the rat MYH is disrupted. It is noteworthy that human MYH variants within the MutY homology region, which exhibit a reduced ability to recognize and excise GO/A adducts, and fail in MutY complementation assays, are associated with colorectal cancer (26). Notwithstanding, it is plausible that in the mammalian MYH, the extended N-terminus augments binding to target DNA, either directly or via cooperation with other proteins interacting at the N-terminus of MYH. We found, however, that the 13 N-terminal amino-acid–MutY homology region is essential for substrate binding by MYH in the context of nuclear extracts as well as with flag pull-down purified proteins. If MYH isoforms with N-terminal modifications that perturb homology to MutY, in fact, exist in mammalian cells, their biological functions remain to be elucidated.
Notably, we found that the catalytic properties of overexpressed FL, Δ25 and Δ50 MYH proteins, differ significantly as reflected in excision rates and product termini generated on substrates containing an adenine mispaired with an oxidized guanine (GO/A) as well as an adenine mispaired with a guanine (G/A). Higher product yields observed on GO/A compared to G/A may reflect differential substrate-binding affinities in agreement with specific recognition of the oxidized guanine mispaired with an adenine, by an array of contacts with residues in the C-terminal domain of MutY (21). Interestingly, on the G/A substrate, excision by all constructs resulted in the formation of the fast migrating product, suggesting that repair complexes on the GO/A substrate differ from those assembled on G/A, and that abasic sites generated on the G/A substrate are more readily processed than to those generated on GO/A.
It is generally thought that MutY and MYH lack an efficient lyase activity (27,28); in nuclear extracts, therefore, MYH-generated abasic sites are most likely to be processed by APE. Here, we observe two differentially migrating excision products, indicative of differential processing of MYH-generated abasic sites. In the case of FL the predominant product migrates slowly, consistent with unprocessed abasic sites, which are then hydrolyzed in alkaline termination buffer. In contrast, excision by Δ25 produces equivalent amounts of fast and slow migrating products, representing both unprocessed and processed abasic sites, whereas for Δ50 the fast migrating product, consistent with processed abasic sites, is predominant. Interestingly, two faint products at a ratio similar to that generated by Δ25 are observed in the naive 293 cells (Figure 3B). This suggests that endogenous isoforms similar to our deletion proteins may in fact exist in the 293 cells.
Similar to other DNA glycosylases (19), we observe that the rat MYH catalytic turnover is reduced when APE activity is diminished (i.e. no MgCl2 w/o EDTA). Thus, APE activity modifies the product of the glycosylase reaction, thereby accelerating the dissociation of MYH and alleviating product inhibition. Following omission of EDTA, a modest stimulation of MYH excision rates occurs. Enhancement of MYH activity and MYH dissociation from the product by addition of APE has been observed in reconstituted glycosylase assays (10). Importantly, the APE-binding motif within the MYH protein has been mapped to the central region of MYH (15), which is not perturbed by our N-terminal deletions. Here, we observe that Δ50 strongly favors the formation of the 3′-OH termini, i.e. the abasic site may be more readily available for the action of APE. We also demonstrate by EMSAs that binding of the Δ50 MYH to the GO/A substrate is more readily competed with excess cold substrate than that of the FL or Δ25 MYH.
It is plausible that the N-terminal deletions weaken association of MYH with its product and thereby facilitate dissociation of the enzyme, making the abasic site more readily available for the next step in the repair process, as suggested by the `hands off' mode of action proposed for the BER pathway (29,30). It is intriguing, whether the proposed accelerated substrate release by a truncated MYH may compromise fidelity of repair. In general, the low turnover of MYH is thought to be consistent with its biological function. It may be beneficial that MYH remains bound to its product since the abasic site in not instructive and prone to misinsertions. Bound MYH may also prevent action by other enzymes and shield the site until the next BER enzyme is recruited.
It is of note that our studies are carried out in nuclear extracts, i.e. in the presence of a full complement of proteins required for execution of BER. This process may be difficult to accurately reproduce in reconstituted systems assembled with pre-selected arrays of proteins. Moreover, in reconstituted assays, bacterially overexpressed proteins may lack post-translational modifications. It is conceivable that in cells, MYH isoforms may differentially integrate protein–protein and protein–DNA interaction, and thereby influence substrate preferences and product-binding affinities and consequently affect the BER process. Although speculative, if our deletion constructs mirror, to a certain extent, properties of naturally occurring MYH isoforms, it is plausible that in vivo, distinct isoforms might be recruited to initiate the BER process under different physiologic conditions, including excessive oxidative stress.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs William Beard for help with kinetic analyses, Tapas Hazra for vital advice, Kishor Bhakat for help with immunoprecipitation and Dr S. Mitra's group for insightful discussions. This work was supported by the National Institutes of Health R01-NS39449 and Shriners Hospitals for Children Grant SHC8670 to E.W.E.
REFERENCES
- 1.Beckman K.B. and Ames,B.N. (1997) Oxidative decay of DNA. J. Biol. Chem., 272, 19633–19636. [DOI] [PubMed] [Google Scholar]
- 2.Mitra S., Hazra,T.K., Roy,R., Ikeda,S., Biswas,T., Lock,J., Boldogh,I. and Izumi,T. (1997) Complexities of DNA base excision repair in mammalian cells. Mol. Cell, 7, 305–312. [PubMed] [Google Scholar]
- 3.Dianov G., Bischoff,C., Piotrowski,J. and Bohr,V.A. (1998) Repair pathways for processing of 8-oxoguanine in DNA by mammalian cell extracts. J. Biol. Chem., 273, 33811–33816. [DOI] [PubMed] [Google Scholar]
- 4.Michaels M.L., Cruz,C., Grollman,A.P. and Miller,J.H. (1992) Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc. Natl Acad. Sci. USA, 89, 7022–7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaels M.L., Tchou,J., Grollman,A.P. and Miller,J.H. (1992) A repair system for 8-oxo-7,8-dihydrodeoxyguanine. Biochemistry, 31, 10964–10968. [DOI] [PubMed] [Google Scholar]
- 6.Hazra T.K., Hill,J.W., Izumi,T. and Mitra,S. (2001) Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol., 68, 193–205. [DOI] [PubMed] [Google Scholar]
- 7.McGoldrick J.P., Yeh,Y.C., Solomon,M., Essigmann,J.M. and Lu,A.L. (1995) Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell. Biol., 15, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slupska M.M., Baikalov,C., Luther,W.M., Chiang,J.H., Wei,Y.F. and Miller,J.H. (1996) Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol., 178, 3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohtsubo T., Nishioka,K., Imaiso,Y., Iwai,S., Shimokawa,H., Oda,H., Fujiwara,T. and Nakabeppu,Y. (2000) Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res., 28, 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H., Clendenin,W.M., Wong,D., Demple,B., Slupska,M.M., Chiang,J.H. and Miller,J.H. (2001) Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res., 29, 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee H.M., Wang,C., Hu,Z., Greeley,G.H., Makalowski,W., Hellmich,H.L. and Englander,E.W. (2002) Hypoxia induces mitochondrial DNA damage and stimulates expression of a DNA repair enzyme, the Escherichia coli MutY DNA glycosylase homolog (MYH), in vivo, in the rat brain. J. Neurochem., 80, 928–937. [DOI] [PubMed] [Google Scholar]
- 12.Takao M., Aburatani,H., Kobayashi,K. and Yasui,A. (1998) Mitochondrial targeting of human DNA glycosylases for repair of oxidative DNA damage. Nucleic Acids Res., 26, 2917–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takao M., Zhang,Q.M., Yonei,S. and Yasui,A. (1999) Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res., 27, 3638–3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai-Wu J.J., Su,H.T., Wu,Y.L., Hsu,S.M. and Wu,C.H. (2000) Nuclear localization of the human mutY homologue hMYH. J. Cell Biochem., 77, 666–677. [PubMed] [Google Scholar]
- 15.Parker A., Gu,Y., Mahoney,W., Lee,S.H., Singh,K.K. and Lu,A.L. (2001) Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem., 276, 5547–5555. [DOI] [PubMed] [Google Scholar]
- 16.Parker A.R., O′Meally,R.N., Sahin,F., Su,G.H., Racke,F.K., Nelson,W.G., DeWeese,T.L. and Eshleman,J.R. (2003) Defective human MutY phosphorylation exists in colorectal cancer cell lines with wild-type MutY alleles. J. Biol. Chem., 278, 47937–47945. [DOI] [PubMed] [Google Scholar]
- 17.Gu Y. and Lu,A.L. (2001) Differential DNA recognition and glycosylase activity of the native human MutY homolog (hMYH) and recombinant hMYH expressed in bacteria. Nucleic Acids Res., 29, 2666–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichinoe A., Behmanesh,M., Tominaga,Y., Ushijima,Y., Hirano,S., Sakai,Y., Tsuchimoto,D., Sakumi,K., Wake,N. and Nakabeppu,Y. (2004) Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res., 32, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill J.W., Hazra,T.K., Izumi,T. and Mitra,S. (2001) Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res., 29, 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pope M.A., Porello,S.L. and David,S.S. (2002) Escherichia coli apurinic-apyrimidinic endonucleases enhance the turnover of the adenine glycosylase MutY with G:A substrates. J. Biol. Chem., 277, 22605–22615. [DOI] [PubMed] [Google Scholar]
- 21.Fromme J.C., Banerjee,A., Huang,S.J. and Verdine,G.L. (2004) Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature, 427, 652–656. [DOI] [PubMed] [Google Scholar]
- 22.Guan Y., Manuel,R.C., Arvai,A.S., Parikh,S.S., Mol,C.D., Miller,J.H., Lloyd,S. and Tainer,J.A. (1998) MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nature Struct. Biol., 5, 1058–1064. [DOI] [PubMed] [Google Scholar]
- 23.Bhakat K.K., Izumi,T., Yang,S.H., Hazra,T.K. and Mitra,S. (2003) Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J., 22, 6299–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parker A., Gu,Y. and Lu,A.L. (2000) Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic Acids Res., 28, 3206–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manuel R.C. and Lloyd,R.S. (1997) Cloning, overexpression, and biochemical characterization of the catalytic domain of MutY. Biochemistry, 36, 11140–11152. [DOI] [PubMed] [Google Scholar]
- 26.Chmiel N.H., Livingston,A.L. and David,S.S. (2003) Insight into the functional consequences of inherited variants of the hMYH adenine glycosylase associated with colorectal cancer: complementation assays with hMYH variants and pre-steady-state kinetics of the corresponding mutated E.coli enzymes. J. Mol. Biol., 327, 431–443. [DOI] [PubMed] [Google Scholar]
- 27.Yeh Y.C., Chang,D.Y., Masin,J. and Lu,A.L. (1991) Two nicking enzyme systems specific for mismatch-containing DNA in nuclear extracts from human cells. J. Biol. Chem., 266, 6480–6484. [PubMed] [Google Scholar]
- 28.Slupska M.M., Luther,W.M., Chiang,J.H., Yang,H. and Miller,J.H. (1999) Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J. Bacteriol., 181, 6210–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mol C.D., Izumi,T., Mitra,S. and Tainer,J.A. (2000) DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature, 403, 451–456. [DOI] [PubMed] [Google Scholar]
- 30.Wilson S.H. and Kunkel,T.A. (2000) Passing the baton in base excision repair. Nature Struct. Biol., 7, 176–178. [DOI] [PubMed] [Google Scholar]