

Abstract
We study the electrophoretic transport of single-stranded RNA molecules through 1.5-nm-wide pores of carbon nanotube membranes by molecular dynamics simulations. From ≈170 individual RNA translocation events analyzed at full atomic resolution of solvent, membrane, and RNA, we identify key factors in membrane transport of biopolymers. RNA entry into the nanotube pores is controlled by conformational dynamics, and exit by hydrophobic attachment of RNA bases to the pores. Without electric field, RNA remains hydrophobically trapped in the membrane despite large entropic and energetic penalties for confining charged polymers inside nonpolar pores. Differences in RNA conformational flexibility and hydrophobicity result in sequence-dependent rates of translocation, a prerequisite for nanoscale separation devices.
Biopolymer translocation across membranes is essential in many important biological processes, such as gene expression and protein targeting. Electrostatic membrane potentials play a critical role in biopolymer transport, as demonstrated for the import of unfolded proteins into the mitochondrial matrix (1). Electrostatically driven membrane translocation is also increasingly used to measure the properties of single polymers (2–6). The blockage of ionic currents during electric-field-driven translocation of individual nucleic acid molecules through membrane-inserted α-hemolysin channels (7) was shown to depend on length, base composition, and sequence (2, 3), suggesting possible applications in ultrafast and single-molecule sequencing of nucleic acids. However, the transport of polymers through membrane-bound protein channels (2–6) is complicated by specific molecular interactions with the highly structured pores. Nonbiological membranes and pores (8, 9), such as carbon nanotubes assembled into hexagonally packed two-dimensional arrays (10, 11), provide simple, controllable, and potentially more robust systems to study fundamental aspects of membrane translocation. Computer simulations suggest that carbon nanotubes accommodate rapid water (12–14) and proton (15) flow and take up nucleic acids (16), despite their highly restricted pore size and low polarity. Water filling of nanotubes (17, 18), as well as the flow of an aqueous electrolyte through carbon nanotube membranes (11) and the transport of DNA (19) through a single carbon nanotube were also observed experimentally.
Here, we report the results of all-atom molecular dynamics simulations of RNA translocation through carbon nanotube membranes in explicit solvent. These simulations allow us to study membrane translocation at atomic detail, extending earlier studies of coarse-grained polymer translocation models (20–23). By including detailed descriptions of water and ions, the simulations capture electrostatic and hydrophobic solvation effects on the translocation processes and permit a detailed study of sequence dependences. As we will show, hydrophobic interactions of the bases with the nanotube pores can transiently trap RNA at the pore walls. To analyze the resulting multiphasic kinetics, we use a trap-diffusion model that combines diffusive RNA motion along the pore with random trapping. We will show that the model motivated by our simulations can also account for detailed experimental measurements of DNA translocation through α-hemolysin pores (5). By varying the strength of the transmembrane potential, we will study translocation of RNA at different driving forces. The simulations also give us access to the RNA structure, which will allow us to study the role of conformational dynamics in RNA translocation. Finally, we will discuss the effects of RNA sequence on the overall translocation rate and implications on molecular separation devices.
Methods and Theory
Simulations. Molecular dynamics simulations at a constant pressure of 1 bar and a temperature of T = 300 K were performed with the namd program (24), the assisted model building with energy refinement 94 (AMBER 94) force field (25) for the RNA, and the transferable intermolecular potential 3 point (TIP3P) model of water (26). Potential parameters for single-wall carbon nanotubes are based on sp2 carbon parameters in the AMBER 94 force field (12, 25).
In our molecular dynamics simulations, single-stranded RNA molecules A6 and U6 with 6 bases of adenine and uracil, respectively, are driven across membranes of hexagonally packed single-wall carbon nanotubes by electric fields of up to 0.5 V/nm (Fig. 1). To form a nanotube membrane (13) under periodic boundary conditions, four (14, 14) “armchair”-type nanotubes (1.33-nm length and 1.87-nm carbon-to-carbon diameter with a pore size of ≈1.5 nm) were packed into a hexagonal lattice in the x–y plane, with their tube axes aligned with the z axis of the simulation cell. The pore diameter of the nanotubes is comparable to the constriction region of the protein channel α-hemolysin (7) and to the diameter reported for carbon nanotube bundles used as fluid-flow sensors (27).
Fig. 1.

Schematic of simulations. (A) Translocation of negatively charged single-stranded RNA through carbon nanotube membranes is driven by the electric field E along the z axis normal to the membranes. Water molecules and counterions are not shown for clarity. (B) Position z(t) of the geometric center of A6 (blue) and U6 (red) RNA as a function of time t from five simulation runs each at E0 = 0.4 V/nm. Periodic boundary conditions in all three spatial dimensions result in a system of fluid layers separated by regularly spaced nanotube membranes (shaded regions) through which RNA flows under the influence of the external electric field. Differences in membrane translocation times result in a gradual separation of the “ensembles” of A6 and U6 RNA, analogous to gel electrophoresis, as indicated by the superimposed single-molecule trajectories.
Five initial configurations each of A6 and U6 RNA were taken from two 5-ns simulations in free solution, starting from single strands in A-form RNA duplexes (28). The RNA molecules, together with five neutralizing K+ counterions (29), were then inserted into the water phase of equilibrated nanotube/water interfacial systems. The resulting RNA/nanotube/water systems with 2,267 water molecules were further equilibrated for 1 ns without electric field, and then 100 ps with field. The simulation boxes were ≈4.4, 3.8, and 5.0 nm long in x, y, and z directions, respectively. We performed 30 RNA/nanotube/water simulations of 20–28 ns duration at electric fields of E0 = 0.3, 0.4, and 0.5 V/nm (5 simulations each for A6 and U6 RNA) and 10 simulations without field covering between 11 and 26 ns.
Electric Fields. Particle-mesh Ewald summation (30) with conducting boundary conditions was used to calculate long-range electrostatic interactions. The electric field averaged over the simulation cell is then equal to the external electric field E0 (31), but the local electric field in the bulk water phase and inside the nanotube pores deviates significantly from E0. To estimate the local electric field across the water/nanotube interface, we calculated the electrostatic potential along the z direction from simulations of water/nanotube interfaces without RNA. The potential Φ(z) averaged parallel to the membrane is calculated from the charge density ρq(z) through Gauss' law, 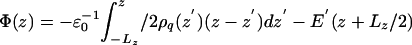 , with ε0 the vacuum permittivity and Lz the length of the simulation cell in the z direction. The integration constant E′ is chosen to enforce the boundary condition Φ(0) - Φ(Lz) = E0Lz for Ewald summation under conducting boundary conditions (31, 32). From the slopes of the electrostatic potentials, we estimate average electric fields of 0.15, 0.17, and 0.19 V/nm in the bulk phase, and 0.56, 0.83, and 1.09 V/nm inside the nanotube membrane at external fields of E0 = 0.3, 0.4, and 0.5 V/nm, respectively. The electric field in the bulk water region calculated from the slope of Φ(z) and the orientation of water dipoles are consistent with the previously determined relationship between the polarization and the electric field of TIP3P bulk water (32).
, with ε0 the vacuum permittivity and Lz the length of the simulation cell in the z direction. The integration constant E′ is chosen to enforce the boundary condition Φ(0) - Φ(Lz) = E0Lz for Ewald summation under conducting boundary conditions (31, 32). From the slopes of the electrostatic potentials, we estimate average electric fields of 0.15, 0.17, and 0.19 V/nm in the bulk phase, and 0.56, 0.83, and 1.09 V/nm inside the nanotube membrane at external fields of E0 = 0.3, 0.4, and 0.5 V/nm, respectively. The electric field in the bulk water region calculated from the slope of Φ(z) and the orientation of water dipoles are consistent with the previously determined relationship between the polarization and the electric field of TIP3P bulk water (32).
The electric field in typical nucleic acid translocation experiments with membrane-inserted α-hemolysin channels is in the range of 0.02–0.04 V/nm (120 mV across 3- to 5-nm membrane thickness) (2, 5), which is ≈20 times smaller than the smallest estimated electric field in our nanotube membrane simulations. Use of relatively high electric fields was necessary to induce at least nanometer-scale motion on the nanosecond simulation time scales. We recently (32) showed that the hydrodynamic and electrophoretic properties (33) obtained from simulations of single-stranded RNA homopolymers in free solution at similar electric fields are in accord with experiment. Also, in a recent microsecond-electrophoresis experiment with micrometer-separation paths, electric fields exceeding 0.01 V/nm were achieved without any significant alteration of mobilities because of Joule heating (34). Solid-state nanopore systems like the nanotubes used in our simulations are expected to be more stable than biological membranes (9, 35). Indeed, in all our simulations under high electric fields, the carbon nanotube membrane models were found to remain intact, held together by strong hydrophobic interactions between neighboring nanotubes, even though the nanotubes were flexible and could move relative to each other and the solvent (12, 13).
RNA Translocation Kinetics. In the analysis of RNA translocation kinetics, the duration of entry events is defined as the time between the first RNA atom entering into the membrane region (within 1.1 nm of the membrane center), and either the last RNA atom entering or the first atom exiting the membrane region. The duration of exit events is defined as the time between the completion of the entry event and the last atom leaving the membrane region. To correct for incomplete entry or exit events (i.e., the simulation was terminated before the RNA molecule finished entering or exiting the membrane region, respectively), we calculated average entry and exit times by using a maximum-likelihood estimate for randomly censored data, 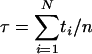 , where N is the total number of attempts, n is the number of successful events, and ti is the duration of an event, if completed, and otherwise the time between the start of an event and the end of the simulation. For cumulative distributions, we used the Kaplan–Meier nonparametric estimator (36).
, where N is the total number of attempts, n is the number of successful events, and ti is the duration of an event, if completed, and otherwise the time between the start of an event and the end of the simulation. For cumulative distributions, we used the Kaplan–Meier nonparametric estimator (36).
Trap-Diffusion Model. To describe the exit-time statistics, we consider a model in which RNA molecules can become transiently trapped as they pass through the pore. Similar models have been used previously in modeling molecular motors (37) and polymer translocation (21). The exit-time probability distribution can be obtained by solving the coupled equations describing the exchange between diffusively drifting and bound RNA populations,
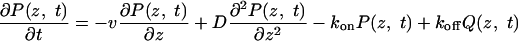 |
[1] |
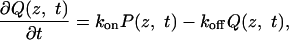 |
[2] |
where P(z, t) and Q(z, t) are the probability densities of free and trapped RNA, respectively, at position z and time t. kon and koff are the position-independent rate coefficients for RNA to become trapped in the pore and to dissociate from the pore walls, respectively. v is the drift velocity of the untrapped RNA, given approximately by the Nernst–Einstein relation, v ≈ qDE/kBT, with D the diffusion coefficient, q the effective RNA charge (32), E the electric field, and kB Boltzmann's constant.
In the trap-diffusion model, the exit time is given by the arrival time of molecules that start at a fixed distance Δz (here, 2.2 nm) upstream from the exit. We obtain the Laplace transform 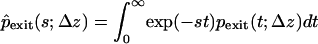 of the exit-time distribution by calculating the outgoing flux from the Green's functions for Eqs. 1 and 2 on a semiinfinite interval with absorbing boundary conditions at the exit:
of the exit-time distribution by calculating the outgoing flux from the Green's functions for Eqs. 1 and 2 on a semiinfinite interval with absorbing boundary conditions at the exit:
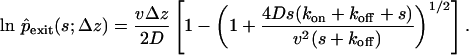 |
[3] |
The average exit time is then given by
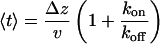 |
[4] |
with a variance of
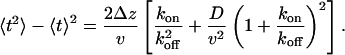 |
[5] |
Within this model, the probability of exiting without becoming trapped is given by
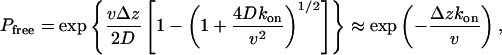 |
[6] |
where the approximate expression at the end is valid for slow diffusion, Dkon ≪ v2. Without trapping (kon = 0), we can invert the Laplace transform to obtain the exit-time distribution as (38)
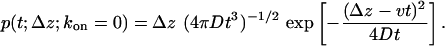 |
[7] |
This distribution is non-Gaussian and skewed to slow exit times. Weighted with the fraction of untrapped molecules, Pfree, Eq. 7 accounts for an initial burst in the exit-time distribution. The subsequent slow phase of trapped molecules in Eq. 3 has an amplitude of 1 - Pfree and a characteristic time of
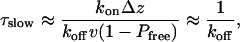 |
[8] |
where the last relation holds in the limit of small diffusion coefficients (D ≪ v2/kon) and trapping rates (kon ≪ v/Δz). If the drift induced by an external field is fast compared with diffusion, we can invert the Laplace transform in Eq. 3. In that limit, the exit-time distribution consists of a δ-function contribution from molecules traversing the pore without attaching to the pore wall, followed by a tail of transiently trapped molecules,
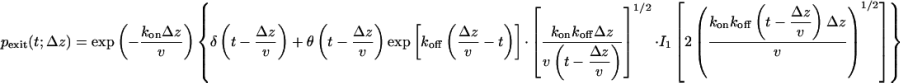 |
[9] |
where θ(x) is the Heaviside step function [i.e., θ(x) = 1 for x > 0 and 0 otherwise], and I1(x) is the modified Bessel function of the first kind of order 1.
Results and Discussion
RNA Translocation. During the combined simulation time of over 800 ns, we observe 173 translocations of RNA and 14,435 translocations of K+ counterions across the nanotube membranes. RNA transport across the membranes is driven by electric fields acting on the charged phosphate groups, but hydrophobic interactions between the bases of the RNA and the carbon nanotubes strongly affect the translocation process, as shown in Figs. 1B and 2 A–C.
Fig. 2.

Representative configurations of RNA during translocation through the nanotube membrane. (A) Head-on entry. (B) Translocation. (C) Exit with single trapped base. The close-up shows a tight hydrophobic interaction between an unstacked base and the nanotube wall, together with surrounding water molecules. (D) Sideways entry. (E) Five trapped bases (side and top view). (F) RNA trapped inside two nanotubes (without electric field, E0 = 0).
Fig. 2 shows snapshots of typical translocation events. Rapid entry occurs if a terminal phosphate group enters first into a nanotube pore (Fig. 2 A). Less frequently, one of the central phosphate groups is pulled into a pore by the electric field (Fig. 2D). The resulting confinement distorts the RNA structure, forcing it into a tightly packed U-shape. Nevertheless, despite the severe geometric restraints, we find that the small RNA molecules studied here translocate through the membrane, driven by the high electric fields inside the pore, without disrupting the nanotube membrane.
To determine the orientation of RNA before, during, and after translocations, we calculate the cosine of the angle θ between the long axis of RNA, as determined from the inertia tensor, and the z axis as a function of the z position of RNA (Fig. 3A). A decrease of cosθ near z ≈ 1.1 nm indicates that RNA molecules initially dock onto the membrane surface and, typically, spread out on it under the combined influence of hydrophobic and electrostatic forces. A large increase of cosθ around z = 0 indicates that RNAs are oriented along the z axis during the translocation events. The drop in cosθ just outside the membrane (z ≈ -1.0 nm) is caused by RNA molecules that remain hydrophobically attached to the membrane upon exit and spread out on it. The subsequent increase of cosθ for z < -1.0 nm is caused in part by the elongation of RNA during the exit process if one end remains in hydrophobic contact with the nanotube while the other end is pulled away from the nanotube by the electric field (Fig. 2C). The orientation of the RNA in the presence of electric fields is also evident in the distributions of base and phosphate groups along the z direction normal to the membrane plane (Fig. 3B). On the entrance side (z > 0), phosphate groups penetrate into the nanotube membranes, pulled by the high electric fields inside the pores, whereas base atoms are built up along the water/nanotube interface. On the exit side (z < 0), we find a high density of base atoms inside the carbon nanotubes, originating from bases forming long-lived hydrophobic interactions with the nanotube walls. In contrast, phosphate groups concentrate outside the nanotube membrane near the exit. The results shown in Fig. 3 are for an average over A6 RNA trajectories, but the main features are present also in data from individual runs. Similar results are obtained for U6 (data not shown).
Fig. 3.

Orientational and density profiles of RNA. (A) Cosine of the angle θ between the long axis of A6 RNA, as determined from the inertia tensor, and the z axis as a function of RNA position, averaged over all simulations with applied electric fields. (B) Density profiles of bases (blue) and phosphates (red) of A6 RNA along the z direction.
Kinetics of RNA Exit from Pores. During RNA exit from the membrane, the charged phosphate groups usually proceed rapidly into the high-dielectric, low-field solvent, as shown in Fig. 3. However, attractive hydrophobic interactions between individual bases and the nanotube channel can delay the release of RNA into the solvent (Fig. 2C). We observe structures with up to five RNA bases simultaneously attached to the nanotube pore wall (Fig. 2E). At a field of 0.3 V/nm, hydrophobic trapping prevents the release of RNA from the nanotube in a significant fraction of the ≈20-ns simulations. In the absence of an external electric field driving translocation, RNA slides into the nanotube pores but remains trapped by strong hydrophobic interactions between the bases and the channel walls. In some cases, RNA can become trapped simultaneously in two neighboring pores. Such trapping results in tight hydrophobic contacts between the bases and the nanotube channels but leaves much of the charged sugarphosphate backbone fully exposed to the solvent outside the membrane (Fig. 2F).
Results for the translocation kinetics of RNA through carbon nanotube membranes are shown in Figs. 4 and 5. The exit-time distribution of A6 RNA at 0.5 V/nm (Fig. 4A) has a lag time, followed by a sharp peak of fast exit events at t ≈ 150 ps, and then a long tail extending to ≈3 ns. This distribution of exit times is described well by the trap-diffusion model of Eqs. 1 and 2, which combines free RNA diffusion with intermittent random trapping inside the pore. In Fig. 4A, we have included results calculated with a drift distance of Δz = 2.2 nm across the membrane, an RNA diffusion coefficient of D = 0.7 × 10-5 cm2·s-1, and a drift velocity v = 14 nm·ns-1 for untrapped RNA at the highest field. These values are similar to those determined previously for free diffusion (32). The estimated rate coefficients for attaching to and dissociating from the pore wall are kon = 7 ns-1 and koff = 2.5 ns-1, respectively. With those values, the roughly 30% of RNA molecules that exit the tube without becoming trapped produce the sharp peak in the exit times. Subsequently, arriving RNA molecules were trapped in the pore and result in the slow tail found in the distribution. The amplitude and decay of the exit-time tail are accurately reproduced by the approximate analytic expression for slow diffusion, Eq. 9.
Fig. 4.

Kinetics of RNA exit from the membrane pores. (A) Cumulative probability distributions Pexit(t) of exit times t of A6 RNA (blue line) at an electric field of 0.5 V/nm compared with the trap-diffusion model (solid black line). (Inset) The corresponding histogram pexit(t) of exit times, with the first peak being dominated by freely diffusing RNA, and the slow tail by RNA that was trapped in the pore. The black dotted lines show the results from the approximate analytic expression (Eq. 9), which are almost indistinguishable from the full distributions at times beyond the free-drift peak. (B) Mean RNA exit times of A6 (blue circles) and U6 (red triangles) RNA depending on electric field. Error bars correspond to one estimated standard deviation.
Fig. 5.

Kinetics of RNA entry into the membrane pores. (A) Cumulative probability distributions Pentry(t) for the duration t of entry events of A6 (blue; right-hand scale) and U6 (red; left-hand scale) RNA at electric fields of 0.3 V/nm (long dashed lines), 0.4 V/nm (dotted lines), and 0.5 V/nm (solid lines). Plotted is the survival probability 1 - Pentry(t) on a logarithmic scale. Exponential distributions with time constants of 1 (U6) and 1.3 (A6) ns are shown as solid black lines. (B) Mean RNA entry times of A6 and U6 RNA depending on electric field. For further details, see Fig. 4.
As expected, exit slows down with decreasing electric field. This slow down is the result of a reduced drift velocity and, more importantly, a slower dissociation of the hydrophobically trapped RNA molecules from the pore walls. With some uncertainty, caused by the small number of events at low fields, we find that the characteristic time of the slow decay in the exit-time distributions seems to decrease exponentially with the applied electric field. This result is consistent with the experimental observations of Bates et al. (5). Based on Eq. 8, for slow diffusion, the characteristic time for exit is dominated by the rate of dissociation from the pore wall, koff. An exponential field dependence of koff is, indeed, expected from phenomenological theories of force-induced ligand dissociation (39, 40).
Exit rates (Fig. 4B) depend more strongly on electric fields than rates of entry into the pores (Fig. 5B), and drop to <1/(10 ns) at a field of E0 = 0.3 V/nm, below which exit is limiting the rate of translocation. This sharp reduction in exit rates with decreasing electric fields (Fig. 4B) explains why we have not observed any exit of RNA from the nanotubes without external electric field on the simulation time scale. Indeed, if we assume an exponential dependence of the exit times on the electric field inside the pores, we estimate exit times of roughly 100 μs to 1 ms for a 120-mV voltage drop across the membrane. This result is on a time scale similar to that measured for DNA translocation through α-hemolysin (5).
Strong hydrophobic interactions between RNA bases and the nonpolar nanotubes (Fig. 2C) trap RNA inside the membrane pores, despite the cost of partially desolvating the phosphatebackbone charges and the entropic cost of confining a flexible polymer. At all electric fields, we find longer average exit times of A6 compared with U6 RNA (Fig. 4B). Based on their respective exit-time distributions, the difference in exit rates seems to be caused by a slightly higher probability of A6 to become trapped, which we attribute to stronger hydrophobic interactions of the larger A6 purine bases with the nanotubes, compared with the U6 pyrimidine bases.
Kinetics of RNA Entry into Pores. Fig. 5A shows the cumulative distributions of entry times of RNA into the membrane pores for different electric fields and RNA sequences. The entry-time distributions show two distinct phases. A fast exponential phase is independent of the external electric field but depends on the RNA sequence, with time constants of ≈1 ns for U6 and 1.3 ns for A6 RNA, respectively. A second slow phase, although negligible at 0.5 V/nm, has an amplitude that grows strongly with decreasing electric field. We associate this slow phase with RNA molecules that became transiently trapped upon entry because of unfavorable geometry (e.g., sideways entry; Fig. 2D). To explain the fast, field-independent phase, we point out that, in addition to electrostatic and hydrophobic effects, the conformational dynamics of RNA molecules will affect their rate of translocation through the carbon nanotube membranes. Conformational dynamics should be particularly relevant during RNA entry into the pores. To explore the hypothesis that the initial fast phase in the entry-time distributions of Fig. 5A is caused by conformational dynamics on a nanosecond timescale, we measure the rate at which RNA conformations evolve in time. We quantify the RNA structural change by calculating a time-dependent rms distance (rmsd). In Fig. 7, which is published as supporting information on the PNAS web site, we plot the average rmsd between two RNA conformations separated by fixed time differences Δt along trajectories in free solution (32) as a function of Δt. A simple linear scaling of time by 1.3 and amplitude by 0.9 brings the rmsd-vs.-time curve of U6 RNA to overlap with A6. The ≈30% faster configurational dynamics of U6 compared with A6 RNA is consistent with the 30% difference in the time constants of the fast phase for RNA entry into the pores observed in Fig. 5A. This result suggests that nanosecond configurational dynamics is indeed a dominant factor in the fast phase of RNA entry that becomes rate limiting at high electric fields and accounts for shorter average entry times of U6 RNA into the nanotube membrane (Fig. 5B). Although the entry rates in Fig. 5B are almost identical at electric fields of 0.4 and 0.5 V/nm, trapping of RNA at the membrane interface becomes relevant at a lower electric field of 0.3 V/nm, resulting in a slow tail in the entry-time distributions (Fig. 5A) and a sharp increase in the average duration of entry events.
Concluding Remarks. Exit-time distributions similar to those obtained here in the simulations and predicted by the trap-diffusion model of Eqs. 1 and 2 were observed in recent single-molecule measurements of single-stranded DNA exiting from membrane-inserted α-hemolysin channels (5). The approximately exponential tail after a rapid initial burst in the measured exit times was attributed to pore-binding DNA populations, consistent with our observations and model. In Fig. 6, we show that the trap-diffusion model suggested by our simulations can indeed account for the experimental translocation data. The model accurately captures the multiphasic kinetics measured for DNA pore blockage both with and without applied electric field. In particular, we recover the slow “tails” in the translocation and exit-time distributions without invoking more elaborate models (38).
Fig. 6.

DNA translocation experiment of Bates et al. (5) modeled by the trap-diffusion model. (A) Probability distributions of poly(dA)60 translocation time t across α-hemolysin channels at 120 mV. The experimental results (bars and open circles for probability density and cumulative distribution, respectively; data taken from figure 2 of ref. 5) are compared with Brownian dynamics simulations of the trap-diffusion model [Eqs. 1 and 2 (lines)]. In the simulations, t was estimated as the first-passage time to drift downstream by a distance L (where L is an arbitrary scaling length corresponding here to the extension of the DNA) with parameters D = 1.35 × 10-4 L2/μs, v = 2.5 × 10-3 L/μs, kon = 1/(1,750 μs), and koff = 1/(600 μs). (B) Cumulative probability distributions of exit times. Experimental results, shown as open circles, are taken from figure 5 of ref. 5. In the simulations (line), as in the experiments, the “polymer” was drawn into the pore for 200 μs at 120 mV with the parameters of A, before the electric field was turned off and the cumulative exit-time distribution was collected (with DNA exit at both ends of the pore). In the field-free period, we used D = 3.6 × 10-4 L2/μs, v = 0, kon = 1/(450 μs), and koff = 1/(1,900 μs).
In our molecular dynamics simulations, we found that the translocation kinetics of RNA through the nanotube membranes is sequence-dependent. At first glance, this finding may seem surprising, considering that the nanotube pores are atomically smooth and featureless, compared with more structured protein pores (2, 4–6). However, we showed that the RNA molecules differ in their affinity for the pore wall and their conformational dynamics. These differences affect the rates of exit and entry, respectively, and thus the overall rate of RNA translocation, and highlight that RNA is not just a simple polyanion. Our results suggest that sequence information can be extracted from synthetic-pore translocation data (35, 41, 42) and that simulations could prove useful in the experimental analysis.
The number of U6 translocations through the nanotube membranes is significantly larger than that of A6 RNA (52, 42, and 7 vs. 45, 25, and 2 at electric fields of 0.5, 0.4, and 0.3 V/nm, respectively, during simulations covering ≈110 ns for each RNA and field). By averaging over multiple trajectories of individual RNA molecules through the periodic nanotube membranes, we effectively recreate an ensemble measurement from our single-molecule simulation data. Differences in entry and exit rates result in sequence-dependent electrophoretic mobilities of RNA, and, as shown in Fig. 1B, the “ensemble” of U6 RNA gradually separates from that of A6. On a time scale of multiple translocations, the simulation system of periodically spaced nanotube membranes thus acts similarly to a porous gel in an electrophoretic measurement. We expect the underlying differences in RNA conformational flexibility and affinity for the pore walls to result in sequence-dependent rates of membrane translocation also for longer polymers in larger pores (10, 11), and in nanometer-size pores of other materials (9, 35, 41), as long as frequent biopolymer-pore interactions occur during translocation. Functionalization of the carbon nanotube rims, for instance by attaching complementary nucleic acids (43) for sequence-dependent trapping (6), should further increase the specificity of carbon nanotube membranes in separation devices.
Supplementary Material
Acknowledgments
We thank Drs. Attila Szabo, Alexander Berezhkovskii, and Christine Peter for many helpful discussions and suggestions. This study used the biowulf pc/linux cluster at the National Institutes of Health.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Huang, S. H., Ratliff, K. S. & Matouschek, A. (2002) Nat. Struct. Biol. 9, 301-307. [DOI] [PubMed] [Google Scholar]
- 2.Kasianowicz, J. J., Brandin, E., Branton, D. & Deamer, D. W. (1996) Proc. Natl. Acad. Sci. USA 93, 13770-13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deamer, D. W. & Branton, D. (2002) Acc. Chem. Res. 35, 817-825. [DOI] [PubMed] [Google Scholar]
- 4.Bezrukov, S. M., Vodyanoy, I. & Parsegian, V. A. (1994) Nature 370, 279-281. [DOI] [PubMed] [Google Scholar]
- 5.Bates, M., Burns, M. & Meller, A. (2003) Biophys. J. 84, 2366-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howorka, S., Cheley, S. & Bayley, H. (2001) Nat. Biotechnol. 19, 636-639. [DOI] [PubMed] [Google Scholar]
- 7.Song, L. Z., Hobaugh, M. R., Shustak, C., Cheley, S., Bayley, H. & Gouaux, J. E. (1996) Science 274, 1859-1866. [DOI] [PubMed] [Google Scholar]
- 8.Volkmuth, W. D. & Austin, R. H. (1992) Nature 358, 600-602. [DOI] [PubMed] [Google Scholar]
- 9.Li, J., Stein, D., McMullan, C., Branton, D., Aziz, M. J. & Golovchenko, J. A. (2001) Nature 412, 166-169. [DOI] [PubMed] [Google Scholar]
- 10.Fan, S. S., Chapline, M. G., Franklin, N. R., Tombler, T. W., Cassell, A. M. & Dai, H. J. (1999) Science 283, 512-514. [DOI] [PubMed] [Google Scholar]
- 11.Miller, S. A., Young, V. Y. & Martin, C. R. (2001) J. Am. Chem. Soc. 123, 12335-12342. [DOI] [PubMed] [Google Scholar]
- 12.Hummer, G., Rasaiah, J. C. & Noworyta, J. P. (2001) Nature 414, 188-190. [DOI] [PubMed] [Google Scholar]
- 13.Kalra, A., Garde, S. & Hummer, G. (2003) Proc. Natl. Acad. Sci. USA 100, 10175-10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu, F. & Schulten, K. (2003) Biophys. J. 85, 236-244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dellago, C., Naor, M. M. & Hummer, G. (2003) Phys. Rev. Lett. 90, 105902. [DOI] [PubMed] [Google Scholar]
- 16.Gao, H. J., Kong, Y., Cui, D. X. & Ozkan, C. S. (2003) Nano Lett. 3, 471-473. [Google Scholar]
- 17.Gogotsi, Y., Libera, J. A., Guvenc-Yazicioglu, A. & Megaridis, C. M. (2001) Appl. Phys. Lett. 79, 1021-1023. [Google Scholar]
- 18.Maniwa, Y., Kataura, H., Abe, M., Suzuki, S., Achiba, Y., Kira, H. & Matsuda, K. (2002) J. Phys. Soc. Jpn. 71, 2863-2866. [Google Scholar]
- 19.Ito, T., Sun, L. & Crooks, R. M. (2003) Chem. Commun., 1482-1483. [DOI] [PubMed]
- 20.de Gennes, G. P. (1999) Proc. Natl. Acad. Sci. USA 96, 7262-7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lubensky, D. K. & Nelson, D. R. (1999) Biophys. J. 77, 1824-1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berezhkovskii, A. M. & Gopich, I. V. (2003) Biophys. J. 84, 787-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong, C. Y. & Muthukumar, M. (2002) Electrophoresis 23, 2697-2703. [DOI] [PubMed] [Google Scholar]
- 24.Kalé, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Phys. 151, 283-312. [Google Scholar]
- 25.Cornell, W. D., Cieplak, P., Bayly, C. I., Gould, I. R., Merz, K. M., Ferguson, D. M., Spellmeyer, D. C., Fox, T., Caldwell, J. W. & Kollman, P. A. (1995) J. Am. Chem. Soc. 117, 5179-5197. [Google Scholar]
- 26.Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. (1983) J. Chem. Phys. 79, 926-935. [Google Scholar]
- 27.Ghosh, S., Sood, A. K. & Kumar, N. (2003) Science 299, 1042-1044. [DOI] [PubMed] [Google Scholar]
- 28.Nowakowski, J. & Tinoco, I., Jr. (1999) in Oxford Handbook of Nucleic Acid Structure, ed. Neidle, S. (Oxford Univ. Press, New York), pp. 567-602.
- 29.Åqvist, J. (1990) J. Phys. Chem. 94, 8021-8024. [Google Scholar]
- 30.Essmann, U., Perera, L., Berkowitz, M. L., Darden, T., Lee, H. & Pedersen, L. G. (1995) J. Chem. Phys. 103, 8577-8593. [Google Scholar]
- 31.Yeh, I.-C. & Berkowitz, M. L. (1999) J. Chem. Phys. 110, 7935-7942. [Google Scholar]
- 32.Yeh, I.-C. & Hummer, G. (2004) Biophys. J. 86, 618-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Berne, B. J. & Pecora, R. (1976) Dynamic Light Scattering (Wiley, New York).
- 34.Plenert, M. L. & Shear, J. B. (2003) Proc. Natl. Acad. Sci. USA 100, 3853-3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li, J. L., Gershow, M., Stein, D., Brandin, E. & Golovchenko, J. A. (2003) Nat. Mater. 2, 611-615. [DOI] [PubMed] [Google Scholar]
- 36.Kaplan, E. L. & Meier, P. (1958) J. Am. Stat. Assoc. 53, 457-481. [Google Scholar]
- 37.Jülicher, F., Ajdari, A. & Prost, J. (1997) Rev. Mod. Phys. 69, 1269-1281. [Google Scholar]
- 38.Metzler, R. & Klafter, J. (2003) Biophys. J. 85, 2776-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bell, G. I. (1978) Science 200, 618-627. [DOI] [PubMed] [Google Scholar]
- 40.Hummer, G. & Szabo, A. (2003) Biophys. J. 85, 5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storm, A. J., Chen, J. H., Ling, X. S., Zandbergen, H. W. & Dekker, C. (2003) Nat. Mater. 2, 537-540. [DOI] [PubMed] [Google Scholar]
- 42.Austin, R. H. (2003) Nat. Mater. 2, 567-568. [DOI] [PubMed] [Google Scholar]
- 43.Williams, K. A., Veenhuizen, P. T. M., de la Torre, B. G., Eritja, R. & Dekker, C. (2002) Nature 420, 761-761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}