

Abstract
AIMS/HYPOTHESIS
Ketogenic diets (KDs) increasingly gained attention as effective means for weight loss and potential adjunctive treatment of cancer. Metabolic benefits of KDs are regularly ascribed towards enhanced hepatic secretion of fibroblast growth factor (FGF) 21, and its systemic effects on fatty acid oxidation, energy expenditure and body weight. Ambiguous data from Fgf21 knockout strains and low FGF21 concentrations reported for humans in ketosis have nevertheless cast doubt regarding the endogenous function of FGF21. We here aimed to elucidate the causal role of FGF21 in mediating therapeutic benefits of KDs on metabolism and cancer.
METHODS
We established a dietary model of increased vs. decreased FGF21 by feeding C57BL/6J mice with KDs, either depleted or enriched with protein, respectively. We furthermore used wild type and Fgf21 knockout mice that were subjected to the respective diets, and monitored energy and glucose homeostasis as well as tumor growth after transplantation of Lewis-Lung-Carcinoma cells.
RESULTS
Hepatic and circulating but not adipose tissue FGF21 levels were profoundly increased by protein starvation and independent of the state of ketosis. We demonstrate that endogenous FGF21 is not essential for the maintenance of normoglycemia upon protein and carbohydrate starvation and is dispensable for the effects of KDs on energy expenditure. Furthermore, the tumor-suppressing effects of KDs were independent from FGF21, and rather driven by concomitant protein and carbohydrate starvation.
CONCLUSION/INTERPRETATION
Our data indicate that multiple systemic effects of KDs exposure in mice that were previously ascribed towards increased FGF21 secretion are rather a consequence of protein malnutrition.
Keywords: Fibroblast growth factor 21, ketogenic diets, protein starvation, glucose homeostasis, energy expenditure, tumor suppression
Introduction
Ketogenic diets (KDs) are actively used for weight loss [1–4] and anticonvulsant therapy [5]. More recently KDs have been intensively studied as potential adjunctive treatment for neurodegenerative diseases [6] and malignant brain cancer [7–9]. Metabolic benefits of KD feeding have been ascribed to an increased hepatic expression and higher circulating levels of fibroblast growth factor (FGF) 21 [10–12]. By enhancing fatty acid oxidation and ketogenesis in response to fasting and KD feeding in rodents, FGF21 has been suggested as a critical component of the metabolic adaptations to a state of fasting and ketosis [10–13]. Profound variations in body weight, fat mass and glucose and lipid metabolism in various strains of FGF21 KO mice [12–16] and the wide range of serum FGF21 concentrations in humans challenged the relevance of endogenous FGF21 on metabolism [17–19]. Nevertheless, the pharmacological application of FGF21 holds promise as an effective therapeutic agent for the treatment of obesity and diabetes [20–25], while uncertainty remains regarding its physiological role as endocrine mediator of ketogenic adaptations.
We recently showed that KDs need to be low in carbohydrates and protein to trigger weight loss and ketosis in rats [26]. Accordingly, we hypothesized that metabolic benefits of KDs on energy expenditure, glucose and ketone body metabolism rather result from protein starvation than directly from increased endogenous FGF21. We further hypothesized that protein starvation but not FGF21 drive therapeutic effects of KDs on tumour suppression.
To test these hypotheses, we studied male C57BL/6J mice fed a control low-fat diet (LFD) or ketogenic diets containing regular (RP-KD) or low (LP-KD) amounts of protein. We observed elevated circulating FGF21 levels only in LP-KD-fed mice, while increased ketone body levels as well as enhanced expression and activity of renal gluconeogenic enzymes were found in both LP-KD- and RP-KD-fed mice. Acute effects of KDs on energy expenditure were similar in wild type (WT) and global Fgf21 knockout (KO) mice. Furthermore, FGF21 was dispensable for the maintenance of euglycemia or ketone body synthesis upon KD feeding. Finally, we verified that tumour-suppressing effects of KDs were independent of FGF21 and mainly determined by carbohydrate and protein starvation.
Material and Methods
Mice
Male 8-week-old C57BL/6J mice (Jackson Laboratory, Bar Harbor, MA) had ad libitum access to low fat diet (LFD: 13% calories from protein, 10% from fat, 77% from carbohydrates), a LFD-matched ketogenic diet (KD) with regular protein (RP) content (RP-KD: 13% calories from protein, 87% from fat, 0% from carbohydrates) or a LFD-matched KD with low- protein (LP) content [10, 27, 28] (LP-KD: 5% calories from protein, 95% from fat, 0% from carbohydrates). Diet compositions (Research Diets, New Brunswick, NJ) are shown in Supplemental Table 1. Experimental cohort 1 received LFD (N=9), RP-KD (N=15) or LP-KD (N=15) for 8 weeks. Cohort 2 received LFD (N=10), RP-KD (N=10) or LP-KD (N=10) for 2 weeks. Cohort 3 to 5 consisted of Fgf21 KO mice [15, 29] and WT littermates, which received LFD (N=8), RP-KD (N=8) or LP-KD (N=8) for 2 weeks. All procedures for animal use were approved by the University of Cincinnati Institutional Animal Care and Use Committee.
Body composition
Body composition was analysed using nuclear magnetic resonance (Echo Medical Systems, Houston, TX).
Oral glucose tolerance test
After 6 hours of fasting, mice received an oral bolus of 1.5 g glucose per kilogram lean mass. Blood glucose was measured at 0, 15, 30, 60 and 120 min by using a glucose analyser (Accucheck, Roche, Indianapolis, IN). Plasma insulin from blood taken at 0 and 15 min was assessed by ELISA (CrystalChem, Downers Grove, IL).
Pyruvate tolerance test
After 16 hours of fasting, 1.5 g sodium pyruvate (Sigma Aldrich, St. Louis, MO) per kg body weight was administered intraperitoneally to Fgf21 WT and KO fed mice that were fed for 2 weeks with LFD, RP-KD or LP-KD. Tail blood was taken at 0, 15, 30, 60 and 120 min and glucose levels were determined with an Accucheck glucose analyser.
Energy expenditure
Measurements of energy expenditure, locomotor activity, respiratory quotient as well as food and water intake were performed by combined indirect calorimetry (PhenoMaster, TSE Systems GmbH, Bad Homburg, Germany) as described previously [30]. Experimental details are given in the Supplemental Material and Methods.
Tumor cell injection
Fgf21 WT and KO mice received subcutaneous injections of 4×105 cells from the murine Lewis Lung Cancer cell line (LLC1, from ATCC), dissolved in PBS. Mice were subsequently switched from chow to LFD (N=8), RP-KD (N=8) or LP-KD (N=8). Tumours were excised and weighed 14 days after tumour cell injection.
Plasma analyses
Trunk blood from ad libitum fed mice was collected for plasma preparation. Plasma FGF21 was assessed by FGF21-ELISA (Millipore, Billerica, MA). 3-Hydroxybutyrate (3-HB) was quantified using the colorimetric Autokit 3-HB (Wako, Richmond, VA).
Hepatic and renal PEPCK activity assay
PEPCK activity was measured as described previously [31, 32] with modifications detailed in the Supplemental Material and Methods.
Cell culture experiments
HepG2 cells (ATCC® HB-8065™) were seeded in 6-well plates and cultured in minimal essential medium (MEM, M5650, SIGMA Aldrich) supplemented with 10% (v/v) fetal bovine serum (FBS) until reaching confluence. Cells were subsequently washed with PBS and incubated for 6h with serum-free MEM with or without non-essential amino acids (M5650 and M2279, SIGMA Aldrich).
Gene expression analyses
Quantitative gene expression analyses from 7–12 random samples per group were performed as described [33, 34] using gene-specific TaqMan probes (Supplemental Table 2). The relative expression of each gene was normalized to beta-actin (Actb) or phosphoglycerate kinase 1 (Pgk1) [34].
Statistics
Data are reported as means ± SEM. Statistical differences were assessed by One-Way or Two-Way ANOVA with Tukey’s post-tests. Energy expenditure was analysed by analysis of covariance (ANCOVA) using fat and lean mass as covariates and Bonferroni post-hoc tests. P values ≤ 0.05 were regarded as statistically significant, with P < 0.05 (*), P < 0.01 (**) P < 0.001 (***). All analyses were performed using Prism 6.0 (GraphPad, Inc, La Jolla, CA) or SPSS 20 (IBM, Armonk, NY).
Results
Body weight changes in response to ketogenic diets depend on the relative amounts of dietary fat and protein
Wild type C57BL/6 mice were ad libitum fed with LFD, RP-KD or LP-KD for 8 weeks. While RP-KD feeding led to a significant increase in body weight compared to LFD mice, prolonged feeding with LP-KD significantly decreased body weight (Figure 1a). The increased body weight in RP-KD mice is in accordance to the significantly increased fat mass after 8 weeks of feeding, while LP-KD mice showed a trend towards lower body fat contents compared to LFD-fed mice (Figure 1b). Significant lean mass loss after 4 and 8 weeks of LP-KD feeding accompanies the pronounced body weight loss of this diet group (Figure 1c). Mice consumed less of the more energy dense KDs (Figure 1d), but the cumulative caloric intake was similar in all diet groups (Figure 1e). Body weight loss in LP-KD mice correlated with the significant reduction in food efficiency (Figure 1f). Glucose excursion and area under the curve were significantly lower in LP-KD mice, as expected based on their significantly lower body weight and body fat (Figure 1g–h). Insulin levels tended to be lower in LP-KD mice (Figure 1i) compared to LFD or RP-KD mice.
Figure 1.
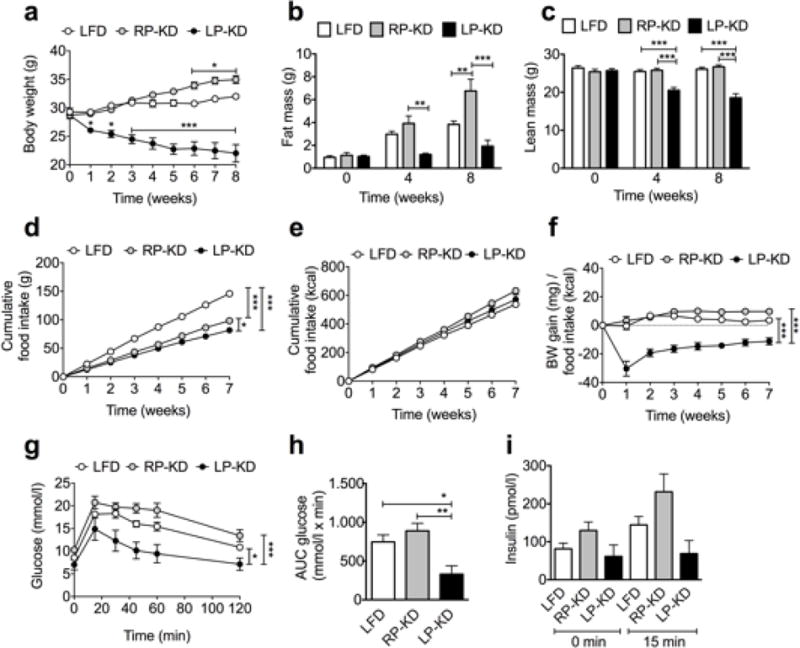
C57BL/6 mice subjected for 8 weeks to RP-KD demonstrated increased body weight (a) and fat mass (b) compared to LFD controls. LP-KD-fed mice revealed decreased body weight (a) and lean mass (c). Mice consumed lower amounts of more energy dense KDs (d), resulting in similar calorie consumption (e). Decreased food efficiency in LP-KD-fed mice (f). LP-KD feeding for 7 weeks decreased glucose excursions (g) and blood glucose area under the curve (AUC) (h), and lowered insulin levels (i) in an oral glucose tolerance test. Mean±SEM, (A–F) N=9 (LFD), N=15 (RP-KD, LP-KD). (G–H) N=7.
Exposure to protein starved ketogenic diet results in muscle wasting in mice
In accordance with the observed lean mass loss, LP-KD feeding resulted in a significant loss of gastrocnemius wet tissue mass after one week (Figure 2a) and two weeks (Figure 2b) of LP-KD feeding. Similarly, soleus wet tissue mass was significantly decreased after one week (Figure 2c) and two weeks (Figure 2d) of LP-KD feeding. Expression of the muscle atrophy marker muscle RING-finger protein-1 Murf1 [35] tended to be increased in the gastrocnemius (Figure 2e) and the soleus (Figure 2f) of 2-weeks LP-KD-fed mice, compared to RP-KD- or LFD-fed mice. Muscle atrophy marker atrogin-1 (Fbxo32) expression [35] was significantly increased in the gastrocnemius (Figure 2g) and the soleus (Figure 2f) after 2 weeks of LP-KD exposure.
Figure 2.
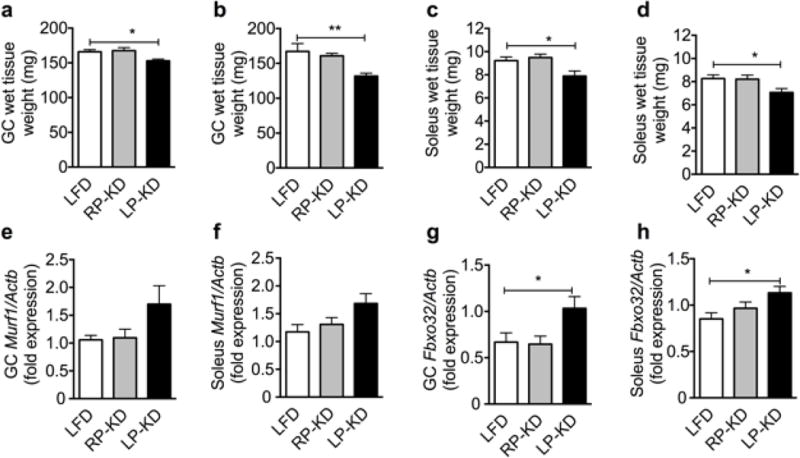
Decreased wet tissue weight of gastrocnemius (GC) after 7 (a) and 14 days (b) of LP-KD exposure, and of soleus after 7 (c) and 14 days (d) of LP-KD exposure. Muscle RING-finger protein-1 (Murf1) expression in the GC (e) and the soleus (f) tends to increase after 14 days of LP-KD exposure. Atrogin-1 (Fbxo31) is significantly increased in GC (g) and soleus (h) of LP-KD-fed mice after 14 days. Mean±SEM, N=8.
Liver but not adipose tissue FGF21 expression upon ketosis depends on the amount of dietary protein
Plasma levels of 3-hydroxybutyrate (3-HB) were increased by 6.9- fold after 8-weeks exposure to LP-KD, and 3.8- fold after 8-weeks exposure to RP-KD, compared to LFD, respectively (Figure 3a), indicating that both diets are causing a state of ketosis. Expression of mitochondrial D-beta-hydroxybutyrate dehydrogenase (Bdh) 1, which is involved in the conversion of acetoacetate to 3-HB, was significantly increased in LP-KD mice, compared to LFD controls (Figure 3b). RP-KD and LP-KD mice displayed a significant reduction of Bdh2 expression, which is involved in the reversion of 3-HB to acetoacetate (Figure 3c). Previous reports suggest an essential role for FGF21 in hepatic ketogenesis during fasting [12]. Accordingly, our finding of elevated ketone body secretion in LP-KD mice should be accompanied by elevated FGF21 expression and secretion. However, only the exposure to LP-KD for 8 weeks, but not to RP-KD resulted in a significant up-regulation of liver Fgf21 gene expression (Figure 3d). Expression differences in liver Fgf21 were reflected by similar differences in plasma FGF21 levels (Figure 3e). A similar increase in 3-HB levels in two weeks KD-fed Fgf21 KO and WT mice further confirmed that FGF21 is not essential for ketogenesis (Figure 3f). To assess whether protein starvation can cause the increase in hepatic FGF21 expression, HepG2 liver cells were starved selectively of non-essential amino acids (NEAS) or non-selectively by deprivation of fetal bovine serum (FBS). The lack of FBS caused a slight but significant increase in FGF21 expression. Additional reduction in NEAS resulted in a pronounced up-regulation of FGF21 expression compared to FBS starvation (Figure 3g). Fgf21 expression was significantly increased in gWAT of both RP-KD and LP-KD-fed mice (Figure 3h) and thus independent of the protein content in the KD.
Figure 3.
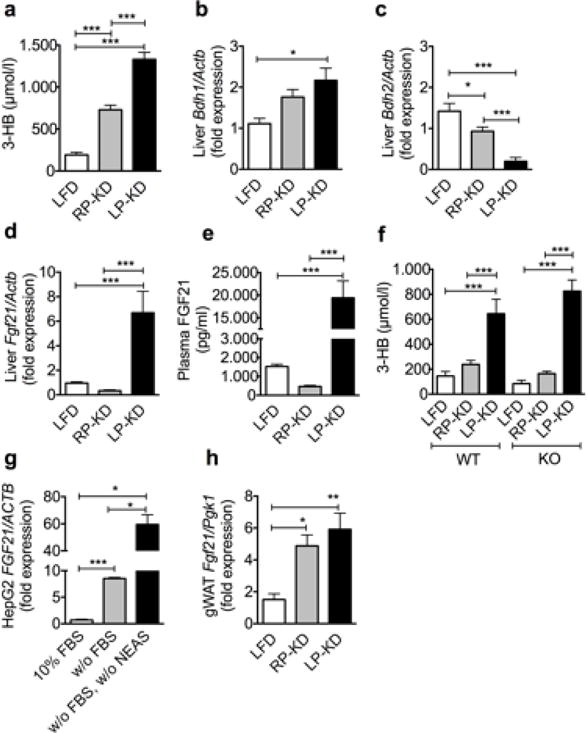
Increased plasma 3-hydroxybutyrate (3-HB) levels following 8 weeks exposure to RP-KD and LP-KD (a). Increased hepatic D-beta-hydroxybutyrate dehydrogenase (Bdh) 1 expression in RP-KD-fed mice (b), reduced hepatic Bdh2 expression in RP-KD- and LP-KD-fed mice (c), compared to LFD-fed controls. Higher hepatic Fgf21 expression (d) and plasma FGF21 levels (e) in LP-KD- compared to LFD- and RP-KD-fed mice. (f) Similar and significant increase of 3-HB in Fgf21 WT and KO mice following two weeks of LP-KD feeding. (g) Increased FGF21 expression in HepG2 cells following concomitant fetal bovine serum (FBS) and non- essential amino acid (NEAS) starvation (w/o FBS, w/o NEAS), compared to FBS starved cells (w/o FBS) and control cells (10% FBS). (h) Significant increase of Fgf21 in the gonadal while adipose tissue (gWAT) of RP-KFD and LP-KD-fed mice. Mean±SEM. N = 7–14. For cell culture experiments 3 independent wells derived from 3 different 6-well plates were analysed.
Short-term dietary effects of ketogenic diets on energy expenditure are independent from endogenous FGF21
Weight-matched WT (Figure 4a) and Fgf21 KO mice (Figure 4b) revealed similar respiratory quotients (RQ) when fed regular chow diet but a drop of RQ when acutely exposed to RP-KD or LP-KD. Chronic exposure of WT mice to chow or KD for 7 weeks resulted in a similar drop in RQ (Figure 4c) and corroborated the switch in nutrient partitioning from carbohydrate to lipid oxidation. WT (Figure 4d) and KO mice (Figure 4e) displayed similar locomotor activities on chow diet, but reduced activities when exposed to RP-KD or LP-KD for 3 days. Exposure of WT mice to KDs for 7 weeks resulted in a similar decrease in activity, compared to chow-fed WT controls (Figure 4f). Further dissection of locomotor patterns revealed similar dark and light phase activities of WT and KO mice on chow diet (Figure 4g), but a drop in dark phase locomotion after acute KD exposure in WT (Figure 4h) and KO mice (Figure 4i), and after 7 weeks of KD exposure in WT mice (Figure 4j).
Figure 4.
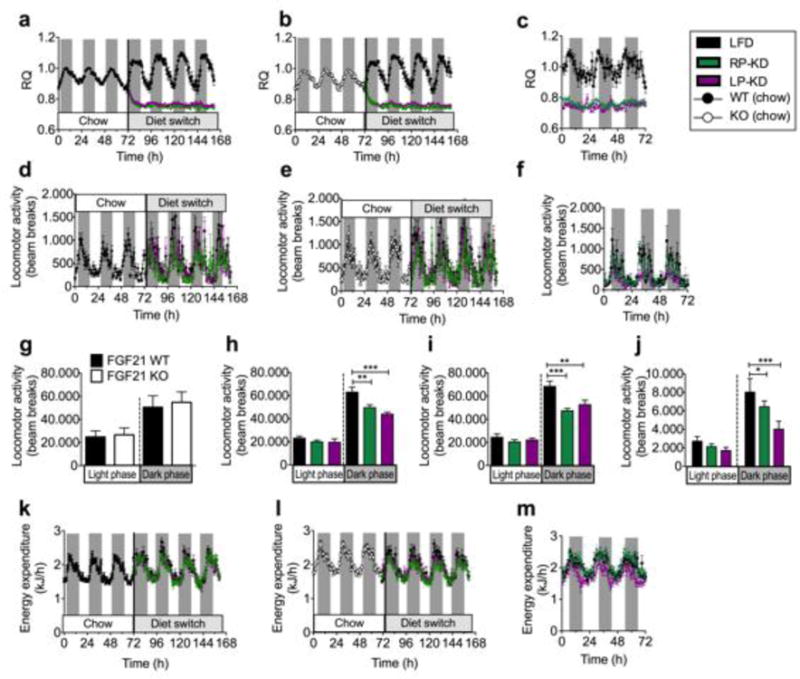
Respiratory quotients (RQ) in (a) Fgf21 WT and (b) KO mice following an acute dietary switch from chow to either LFD, RP-KD or LP-KD, or in WT mice after 7 weeks of chow, RP-KD or LP-KD exposure (c). Locomotor activity in WT (d) and KO (e) mice exposed to chow, and after an acute switch to LFD, RP-KD or LP-KD. In WT mice, locomotor activity was further monitored after 7 weeks of dietary exposure (f). Daily locomotor activity during the dark and light phase in chow-fed WT and KO mice (g) and after the dietary switch in WT (h) and KO mice (i). Daily dark phase locomotor activity of WT mice following 7 weeks of RP-KD or LP-KD exposure (j). Changes in energy expenditure (EE) in WT (k) and KO mice (l) following the dietary switch from chow to LFD, RP-KD or LP-KD, and in WT mice after 7 weeks of LFD, RP-KD or LP-KD (m). Mean±SEM, N=7–9.
Analysis of covariance (ANCOVA) revealed a significant effect of the dietary switch on dark phase energy expenditure (EE) in WT (Figure 4k) and KO mice (Figure 4l) after controlling for the effects of covariates lean mass and fat mass (Supplemental Figure 1). The covariate lean mass, but not fat mass, was significantly related to EE (Supplemental Table 4). Bonferroni post-hoc tests showed a significant decrease in dark phase EE in WT mice fed with LP-KD compared to LFD (p=0.031), and a trend for decreased total EE in WT mice fed with LP-KD compared to LFD-fed mice (p=0.053). Similarly, KO mice fed with LP-KD displayed significantly decreased dark phase EE (p=0.008) compared to LFD-fed mice. We did not observe any significant interaction effects between genotypes and dietary exposure (Genotype*Diet for EE total: F=0.004, p=0.996; EE dark phase: F=0.113, p=0.894; EE light phase: F=0.078, p=0.925), demonstrating that both WT and KO mice respond similarly to the exposure to LFD, RP-KD, or LP-KD, respectively. Interestingly, after 7 weeks of feeding the dietary impact on energy expenditure was lost (Figure 4m); EE continued to be strongly related to lean mass, but LP-KD or RP-KD exposure no longer showed significant effects (Supplementary Table 4).
FGF21 is not required for maintaining glucose levels upon ketosis
Both, RP-KD and LP-KD-fed mice were able to maintain normo-glycemia despite the absence of carbohydrates (Figure 1g). Based on this finding and our findings of muscle catabolism and higher liver Fgf21 expression in LP-KD but not RP-KD-fed mice, we hypothesized that FGF21 could be an important factor for muscle protein catabolism to ultimately provide amino acids as substrates for gluconeogenesis. Following 8 weeks of feeding to LFD or KDs, hepatic expression levels of gluconeogenic enzymes glucose-6-phosphatase catalytic subunit (G6pc, Figure 5a) and phosphoenolpyruvate carboxykinase (Pck1, Figure 5b) as well as phosphoenolpyruvate carboxykinase (PEPCK) activity (Figure 5c) were similar under all dietary conditions. Similarly, liver peroxisome proliferator-activated receptor-gamma co-activator alpha (Pgc1α), a transcriptional co-activator that controls the expression of gluconeogenic genes [36] remained unchanged (Supplemental Figure 2). In contrast, eight weeks of LP-KD feeding resulted in a significant increase of kidney G6pc (Figure 5d) and Pck1 (Figure 5e) expression. A similar increase of kidney Pck1, but not G6pc expression was also detected in RP-KD-fed mice (Figure 5 d–e). The increase of renal Pck1 expression was in accordance with a significant increase of PEPCK activity in the kidney of RP-KD mice and a trend towards an increased PEPCK activity in LP-KD-fed mice (Figure 5f). Notably, the expression of the fibroblast growth factor receptor (Fgfr) 1 and ß-klotho (Klb) was increased in isolated renal cortices from LP-KD fed mice (Supplemental Figure 3). Neither substantial differences in hepatic Fgf21 expression (Supplemental Figure 4), nor genetic ablation of Fgf21 seemed to affect gluconeogenesis, as revealed by similar glucose excursions in pyruvate tolerance tests in WT (Figure 5g) and Fgf21 KO mice (Figure 5h) and similar ad libitum blood glucose levels (Figure 5i) in WT and KO mice after 2 weeks of LFD, RP-KD or LP-KD feeding.
Figure 5.
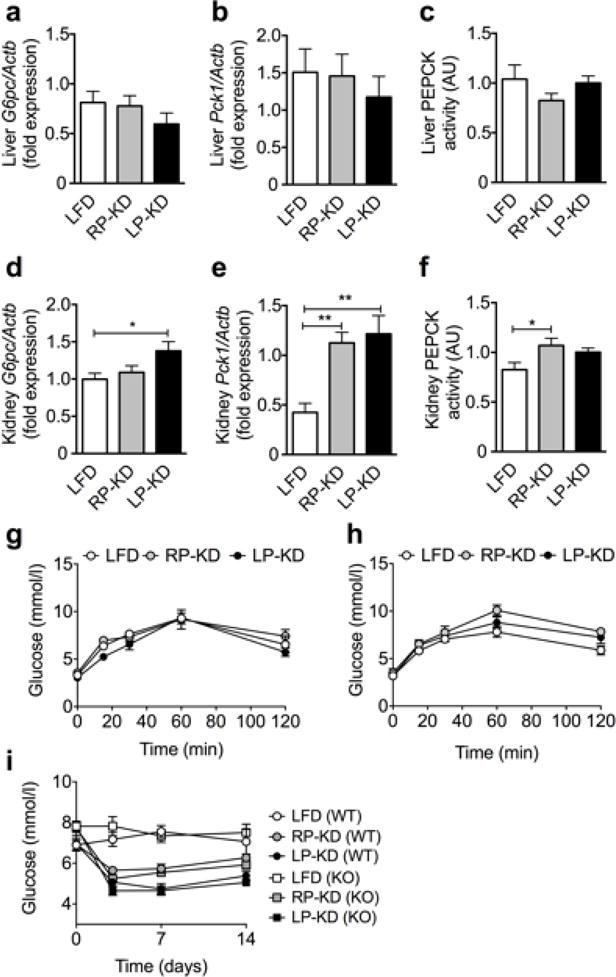
Hepatic expression of glucose-6-phosphatase (G6pc) (a) and phosphoenolpyruvat-carboxykinase (Pck1) (b), and hepatic phosphoenolpyruvat-carboxykinase (PEPCK) activity (c) after 8 weeks of LFD, RP-KD and LP-KD exposure. Renal cortex expression of G6pc (d), Pck1 (e) and renal PEPCK activity (f). Pyruvate tolerance tests in Fgf21 WT (g) and KO mice (f) fed KD for 2 weeks. Ad libitum blood glucose of WT and KO mice before and after 7 and 14 days of dietary exposure (i). Mean±SEM, N=8–10.
Exposure to protein starved ketogenic diet suppresses tumor growth independently of FGF21
Previous data indicate that ketogenoic diets can slow down tumour growth [7, 8]. Using a subcutaneous Lewis Lung Carcinoma allograft cancer model we demonstrate that the beneficial effect of KDs on tumour growth depends on the diet composition. While RP-KD did not significantly affect the tumour weight, LP-KD feeding resulted in a significant reduction of tumour weight in WT mice (Figure 6). The effect was independent from FGF21 signalling, since similar results were obtained in WT and Fgf21 KO mice.
Figure 6.
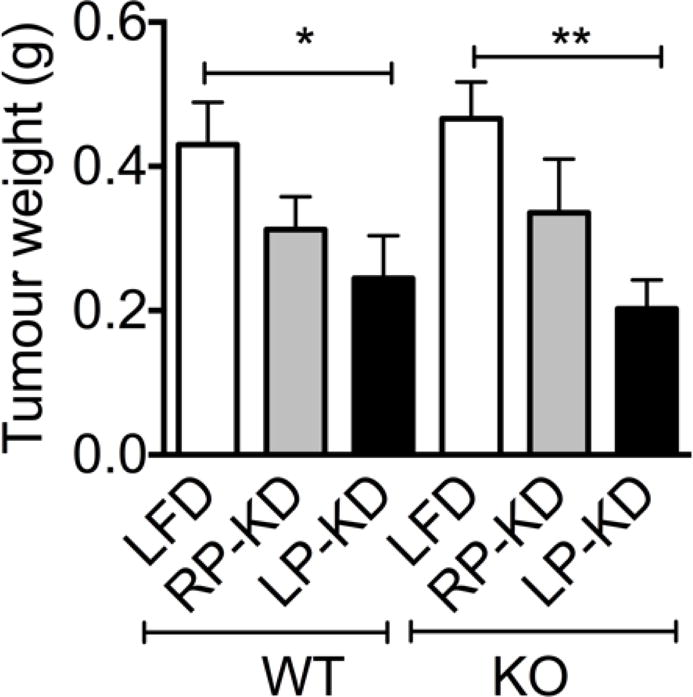
Tumour weights of subcutaneous Lewis Lung Carcinoma allografts were reduced by LP-KD exposure in both WT mice and Fgf21 KO mice. Mean±SEM, N=8.
Discussion
Ketogenic diets (KDs) have garnered attention as an effective treatment of obesity by causing weight loss and improving glucose tolerance and plasma lipid profiles [1–4], potentially via release of FGF21 [10, 12]. Through a series of experiments, we reveal that FGF21 is not an essential mediator of metabolic alterations seen with KD feeding in mice. Our data suggests that the overall protein content in KDs is an important contributor to its effects on body weight, energy and glucose homeostasis.
We reveal that body weight and lean mass are potently decreased by LP-KD feeding. In contrast, a gain in fat mass in RP-KD mice led to significantly higher body weights compared to LFD and LP-KD mice. Both LP-KD and RP-KD feeding induced ketosis, as revealed by increased 3-HB levels in RP-KD and LP-KD mice, compared to LFD controls. However, circulating FGF21 levels were only increased in LP-KD mice compared to RP-KD or LFD mice. Accordingly, our data corroborates previous findings that hepatic and plasma FGF21 levels are indicative of protein starvation [26, 37–39] but not the state of ketosis [15]. In contrast to the protein-dependent regulation of hepatic Fgf21 expression and release, RP-KD and LP-KD fed mice displayed a similar up-regulation of Fgf21 in the gWAT, suggesting alternative regulation of FGF21 in adipose as compared to liver.
Mechanisms underlying the weight lowering effects of KD still remain a matter of debate. In our model, we observed a similar intake of calories in all dietary groups, but severely diminished food efficiency in LP-KD mice. The inability of LP-KD mice to translate ingested calories into weight gain may either be due to malabsorption, urinary loss of ketones, or increased energy expenditure. Indeed, recent evidence suggests KD feeding increases energy expenditure [27, 28]. FGF21 may play a role in this thermogenic effect of KDs, as also suggested by increased energy expenditure in obese rats or mice after central or intraperitoneal infusion of pharmacological FGF21 doses, respectively [40, 41]. Further, hypothalamic ß-klotho (KLB), the obligate co-receptor for FGF21 receptor fibroblast growth factor receptor (FGFR) 1, was required for central FGF21 effects on energy expenditure in tgFgf21 mice [42].
To investigate the impact of endogenous FGF21 on energy expenditure, we performed indirect calorimetry measurements in Fgf21 WT and KO mice following an acute challenge with LFD, RP-KD or LP-KD. The acute feeding paradigm assured that lean mass as predictive covariate for EE was similar for all groups. LP-KD but not RP-KD mice showed a significant decrease of EE during the dark phase compared to LFD mice. This stands in contrast to previous findings by Leager et al. showing increased energy expenditure in mice fed protein-restricted regular diet [39]. Further, in contrast to Laeger et al. who showed that dietary effects on EE were no longer present in an alternative strain of Fgf21 KO mice, we observed a similar decrease in EE in LP-KD-fed KO and WT mice, suggesting that this effect was FGF21-independent [39]. To assess whether these differences were due to a shorter exposure of our mice, we compared C57BL/6 WT mice that were chronically fed with RP-KD and LP-KD and displayed low and high circulating FGF21 levels, respectively. ANCOVA analyses revealed that effects on EE were strongly related to changes in lean mass, but not to LP-KD or RP-KD exposure. Together, our findings do not support a functional role for endogenous FGF21 in energy expenditure, at least not in a model of KD exposure.
Current studies investigating effects of FGF21 on carbohydrate metabolism are controversial. FGF21 has been implicated in the regulation of gluconeogenesis during the progression from fasting to starvation [13]. Administration of recombinant FGF21 induced hepatic expression of key regulators of gluconeogenesis such as Pgc1α and Pck1 but not G6pc [43]. In contrast, therapeutic administration of FGF21 had a potent glucose-lowering effect in ob/ob and db/db mice [44]. Glucose output was suppressed in H4IIE hepatoma cells exposed to FGF21 [45].
In our experimental setup neither the expression of hepatic gluconeogenic enzymes G6pc, Pck1 and Pgc1α nor PEPCK activity were altered by 8-weeks of KD feeding, which is consistent with findings from 4-weeks KD-fed rats [26] and 6-days KD-fed Fgf21 WT and KO mice [46]. In contrast, G6pc and Pck1 expression as well as PEPCK activity were elevated in the kidney cortex of our KD-fed mice. Overall, our data suggest enhanced renal gluconeogenesis as important contributor to systemic glucose control in a state of ketosis. However, similar changes in blood glucose levels during 14 days of KD feeding, and similar glucose excursions following an acute pyruvate challenge in KD-fed WT and KO mice, do not support an essential role of endogenous FGF21 in the overall maintenance of normoglycemia during KD feeding.
We finally aimed to investigate if FGF21 might be involved in anti-tumorigenic effects of KD feeding, and implanted Lewis-Lung Carcinoma cells into Fgf21 WT and KO mice fed with LP-KD, RP-KD and LFD. We observed significantly diminished tumour growth in both WT and KO mice after LP-KD feeding, suggesting that concomitant dietary protein and carbohydrate deprivation but not FGF21 are necessary to reduce tumour growth. This finding may have important clinical implications for cancer patients that are subjected to adjuvant KD therapy. The prospect of increased anti-tumorigenic efficacy by adjuvant LP-KD may be promising for cancer patients and warrants future clinical studies, but adverse clinical symptoms for protein malnutrition should be monitored closely.
In conclusion, our studies demonstrate that endogenous FGF21 is a biomarker for protein starvation, but dispensable for the maintenance of normoglycemia, ketone body synthesis and tumour suppression in mice fed KD. Metabolic alterations on murine energy expenditure and glucose and ketone body metabolism by KD were rather a consequence of concomitant carbohydrate and protein starvation. Future studies will need to address remaining questions such as the correct protein content and composition of KD to induce similar states of ketosis in mice and humans, or the often-discrepant findings obtained by endogenous FGF21 induction vs. pharmacological doses of FGF21. Nevertheless, we believe that our physiological model of increased or decreased FGF21 in response to protein-starved or regular-protein KD provides a useful tool to investigate the impact of endogenous FGF21 on therapeutic effects of KD for metabolism and cancer.
Supplementary Material
Acknowledgments
The authors would like to thank Nobuyuki Itho from the Kyoto University Graduate School of Pharmaceutical Sciences, Sakyo, Kyoto, Japan for providing Fgf21 knockout mice.
Funding:
This work was supported in part by the NIH grant U01 CA141464-02. K.S. was supported by a fellowship from the German Research Foundation (STE 1466/4-1). K.M.H. was supported by the ADA grant 1-13-JF-21.
Abbreviations
- KO
knock out
- WT
wild type
- FGF21
fibroblast growth factor 21
- LFD
low fat diet
- KD
ketogenic diet
- LP-KD
low protein ketogenic diet
- RP-KD
regular protein ketogenic diet
- LLC
Lewis Lung Carcinoma
- EE
energy expenditure
- RQ
respiratory quotient
Footnotes
Duality of interest
R.J.S has received research support from Novo Nordisk, Ethicon Surgical Care, Eisai and Boehringer Ingelheim. R.J.S has served as a paid consultant for Novo Nordisk, Ethicon Surgical Care, Boehringer Ingelheim, Sanofi, Novartis, Takeda and has equity in Zafgen and Endobetix. All other authors declare no conflicts of interest.
Contribution statement
K.S. and R.J.S. designed the study; K.S., F.Z., C.N., P.K., M.B., S.Y., A.Z., M.L., and T.D.M., provided essential data for analysis, K.S. and P.T.P. analysed data and wrote the manuscript. P.J.F.M., K.M.H. and R.J.S. contributed to discussions and reviewed and edited the manuscript. All authors read and approved the final manuscript. K.S. is responsible for the integrity of the work as whole.
References
- 1.Brehm BJ, Seeley RJ, Daniels SR, D’Alessio DA. A randomized trial comparing a very low carbohydrate diet and a calorie-restricted low fat diet on body weight and cardiovascular risk factors in healthy women. J Clin Endocrinol Metab. 2003;88:1617–1623. doi: 10.1210/jc.2002-021480. [DOI] [PubMed] [Google Scholar]
- 2.Foster GD, Wyatt HR, Hill JO, McGuckin BG, Brill C, Mohammed BS, et al. A randomized trial of a low-carbohydrate diet for obesity. N Engl J Med. 2003;348:2082–2090. doi: 10.1056/NEJMoa022207. [DOI] [PubMed] [Google Scholar]
- 3.Dashti HM, Mathew TC, Khadada M, Al-Mousawi M, Talib H, Asfar SK, et al. Beneficial effects of ketogenic diet in obese diabetic subjects. Mol Cell Biochem. 2007;302:249–256. doi: 10.1007/s11010-007-9448-z. [DOI] [PubMed] [Google Scholar]
- 4.Acheson KJ. Carbohydrate for weight and metabolic control: where do we stand? Nutrition. 2010;26:141–145. doi: 10.1016/j.nut.2009.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Kessler SK, Neal EG, Camfield CS, Kossoff EH. Dietary therapies for epilepsy: future research. Epilepsy Behav. 2011;22:17–22. doi: 10.1016/j.yebeh.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paoli A, Bianco A, Damiani E, Bosco G. Ketogenic diet in neuromuscular and neurodegenerative diseases. Biomed Res Int. 2014;2014:474296. doi: 10.1155/2014/474296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nebeling LC, Miraldi F, Shurin SB, Lerner E. Effects of a ketogenic diet on tumor metabolism and nutritional status in pediatric oncology patients: two case reports. J Am Coll Nutr. 1995;14:202–208. doi: 10.1080/07315724.1995.10718495. [DOI] [PubMed] [Google Scholar]
- 8.Seyfried TN, Sanderson TM, El-Abbadi MM, McGowan R, Mukherjee P. Role of glucose and ketone bodies in the metabolic control of experimental brain cancer. Br J Cancer. 2003;89:1375–1382. doi: 10.1038/sj.bjc.6601269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seyfried TN, Kiebish MA, Marsh J, Shelton LM, Huysentruyt LC, Mukherjee P. Metabolic management of brain cancer. Biochim Biophys Acta. 2011;1807:577–594. doi: 10.1016/j.bbabio.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 10.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 11.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 12.Badman MK, Koester A, Flier JS, Kharitonenkov A, Maratos-Flier E. Fibroblast growth factor 21-deficient mice demonstrate impaired adaptation to ketosis. Endocrinology. 2009;150:4931–4940. doi: 10.1210/en.2009-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potthoff MJ, Inagaki T, Satapati S, Ding X, He T, Goetz R, et al. FGF21 induces PGC-1alpha and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc Natl Acad Sci U S A. 2009;106:10853–10858. doi: 10.1073/pnas.0904187106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, et al. Fibroblast growth factor-21 regulates PPARgamma activity and the antidiabetic actions of thiazolidinediones. Cell. 2012;148:556–567. doi: 10.1016/j.cell.2011.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hotta Y, Nakamura H, Konishi M, Murata Y, Takagi H, Matsumura S, et al. Fibroblast growth factor 21 regulates lipolysis in white adipose tissue but is not required for ketogenesis and triglyceride clearance in liver. Endocrinology. 2009;150:4625–4633. doi: 10.1210/en.2009-0119. [DOI] [PubMed] [Google Scholar]
- 16.Adams AC, Coskun T, Cheng CC,LSOF, Dubois SL, Kharitonenkov A. Fibroblast growth factor 21 is not required for the antidiabetic actions of the thiazoladinediones. Mol Metab. 2013;2:205–214. doi: 10.1016/j.molmet.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galman C, Lundasen T, Kharitonenkov A, Bina HA, Eriksson M, Hafstrom I, et al. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008;8:169–174. doi: 10.1016/j.cmet.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 18.Christodoulides C, Dyson P, Sprecher D, Tsintzas K, Karpe F. Circulating fibroblast growth factor 21 is induced by peroxisome proliferator-activated receptor agonists but not ketosis in man. J Clin Endocrinol Metab. 2009;94:3594–3601. doi: 10.1210/jc.2009-0111. [DOI] [PubMed] [Google Scholar]
- 19.Dushay J, Chui PC, Gopalakrishnan GS, Varela-Rey M, Crawley M, Fisher FM, et al. Increased fibroblast growth factor 21 in obesity and nonalcoholic fatty liver disease. Gastroenterology. 2010;139:456–463. doi: 10.1053/j.gastro.2010.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coskun T, Bina HA, Schneider MA, Dunbar JD, Hu CC, Chen Y, et al. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology. 2008;149:6018–6027. doi: 10.1210/en.2008-0816. [DOI] [PubMed] [Google Scholar]
- 21.Muller TD, Sullivan LM, Habegger K, Yi CX, Kabra D, Grant E, et al. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. J Pept Sci. 2012;18:383–393. doi: 10.1002/psc.2408. [DOI] [PubMed] [Google Scholar]
- 22.Kharitonenkov A, Beals JM, Micanovic R, Strifler BA, Rathnachalam R, Wroblewski VJ, et al. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS One. 2013;8:e58575. doi: 10.1371/journal.pone.0058575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adams AC, Halstead CA, Hansen BC, Irizarry AR, Martin JA, Myers SR, et al. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS One. 2013;8:e65763. doi: 10.1371/journal.pone.0065763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gimeno RE, Moller DE. FGF21-based pharmacotherapy–potential utility for metabolic disorders. Trends Endocrinol Metab. 2014;25:303–311. doi: 10.1016/j.tem.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 25.Kharitonenkov A, Adams AC. Inventing new medicines: The FGF21 story. Mol Metab. 2014;3:221–229. doi: 10.1016/j.molmet.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bielohuby M, Menhofer D, Kirchner H, Stoehr BJ, Muller TD, Stock P, et al. Induction of ketosis in rats fed low-carbohydrate, high-fat diets depends on the relative abundance of dietary fat and protein. Am J Physiol Endocrinol Metab. 2011;300:E65–76. doi: 10.1152/ajpendo.00478.2010. [DOI] [PubMed] [Google Scholar]
- 27.Jornayvaz FR, Jurczak MJ, Lee HY, Birkenfeld AL, Frederick DW, Zhang D, et al. A high-fat, ketogenic diet causes hepatic insulin resistance in mice, despite increasing energy expenditure and preventing weight gain. Am J Physiol Endocrinol Metab. 2010;299:E808–815. doi: 10.1152/ajpendo.00361.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kennedy AR, Pissios P, Otu H, Roberson R, Xue B, Asakura K, et al. A high-fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Endocrinol Metab. 2007;292:E1724–1739. doi: 10.1152/ajpendo.00717.2006. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim Biophys Acta. 2000;1492:203–206. doi: 10.1016/s0167-4781(00)00067-1. [DOI] [PubMed] [Google Scholar]
- 30.Stemmer K, Kotzbeck P, Zani F, Bauer M, Neff C, Muller TD, et al. Thermoneutral housing is a critical factor for immune function and diet-induced obesity in C57BL/6 nude mice. Int J Obes (Lond) 2014 doi: 10.1038/ijo.2014.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wiese TJ, Lambeth DO, Ray PD. The intracellular distribution and activities of phosphoenolpyruvate carboxykinase isozymes in various tissues of several mammals and birds. Comp Biochem Physiol B. 1991;100:297–302. doi: 10.1016/0305-0491(91)90378-q. [DOI] [PubMed] [Google Scholar]
- 32.Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, et al. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J Biol Chem. 2009;284:26578–26590. doi: 10.1074/jbc.M109.011775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stemmer K, Bielohuby M, Grayson BE, Begg DP, Chambers AP, Neff C, et al. Roux-en-Y gastric bypass surgery but not vertical sleeve gastrectomy decreases bone mass in male rats. Endocrinology. 2013;154:2015–2024. doi: 10.1210/en.2012-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, et al. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004;18:39–51. doi: 10.1096/fj.03-0610com. [DOI] [PubMed] [Google Scholar]
- 36.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 37.De Sousa-Coelho AL, Marrero PF, Haro D. Activating transcription factor 4-dependent induction of FGF21 during amino acid deprivation. Biochem J. 2012;443:165–171. doi: 10.1042/BJ20111748. [DOI] [PubMed] [Google Scholar]
- 38.Pissios P, Hong S, Kennedy AR, Prasad D, Liu FF, Maratos-Flier E. Methionine and choline regulate the metabolic phenotype of a ketogenic diet. Mol Metab. 2013;2:306–313. doi: 10.1016/j.molmet.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laeger T, Henagan TM, Albarado DC, Redman LM, Bray GA, Noland RC, et al. FGF21 is an endocrine signal of protein restriction. J Clin Invest. 2014;124:3913–3922. doi: 10.1172/JCI74915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarruf DA, Thaler JP, Morton GJ, German J, Fischer JD, Ogimoto K, et al. Fibroblast growth factor 21 action in the brain increases energy expenditure and insulin sensitivity in obese rats. Diabetes. 2010;59:1817–1824. doi: 10.2337/db09-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J, Lloyd DJ, Hale C, Stanislaus S, Chen M, Sivits G, et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes. 2009;58:250–259. doi: 10.2337/db08-0392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owen BM, Ding X, Morgan DA, Coate KC, Bookout AL, Rahmouni K, et al. FGF21 Acts Centrally to Induce Sympathetic Nerve Activity, Energy Expenditure, and Weight Loss. Cell Metab. 2014;20:670–677. doi: 10.1016/j.cmet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fisher FM, Estall JL, Adams AC, Antonellis PJ, Bina HA, Flier JS, et al. Integrated regulation of hepatic metabolism by fibroblast growth factor 21 (FGF21) in vivo. Endocrinology. 2011;152:2996–3004. doi: 10.1210/en.2011-0281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, et al. FGF-21 as a novel metabolic regulator. J Clin Invest. 2005;115:1627–1635. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kong LJ, Feng W, Wright M, Chen Y, Dallas-yang Q, Zhou YP, et al. FGF21 suppresses hepatic glucose production through the activation of atypical protein kinase Ciota/lambda. Eur J Pharmacol. 2013;702:302–308. doi: 10.1016/j.ejphar.2012.11.065. [DOI] [PubMed] [Google Scholar]
- 46.Murata Y, Nishio K, Mochiyama T, Konishi M, Shimada M, Ohta H, et al. Fgf21 impairs adipocyte insulin sensitivity in mice fed a low-carbohydrate, high-fat ketogenic diet. PLoS One. 2013;8:e69330. doi: 10.1371/journal.pone.0069330. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.