

Summary
Memories are believed to be stored in distributed neuronal assemblies through activity-induced changes in synaptic and intrinsic properties. However, the specific mechanisms by which different memories become associated or linked remain a mystery. Here, we develop a simplified, biophysically inspired network model that incorporates multiple plasticity processes and explains linking of information at three different levels: (1) learning of a single associative memory, (2) rescuing of a weak memory when paired with a strong one, and (3) linking of multiple memories across time. By dissecting synaptic from intrinsic plasticity and neuron-wide from dendritically restricted protein capture, the model reveals a simple, unifying principle: linked memories share synaptic clusters within the dendrites of overlapping populations of neurons. The model generates numerous experimentally testable predictions regarding the cellular and sub-cellular properties of memory engrams as well as their spatiotemporal interactions.
Keywords: computational model, simplified neurons, non-linear dendrites, plasticity, synaptic tagging and capture, intrinsic excitability, information binding, synaptic clustering, memory allocation
Graphical Abstract
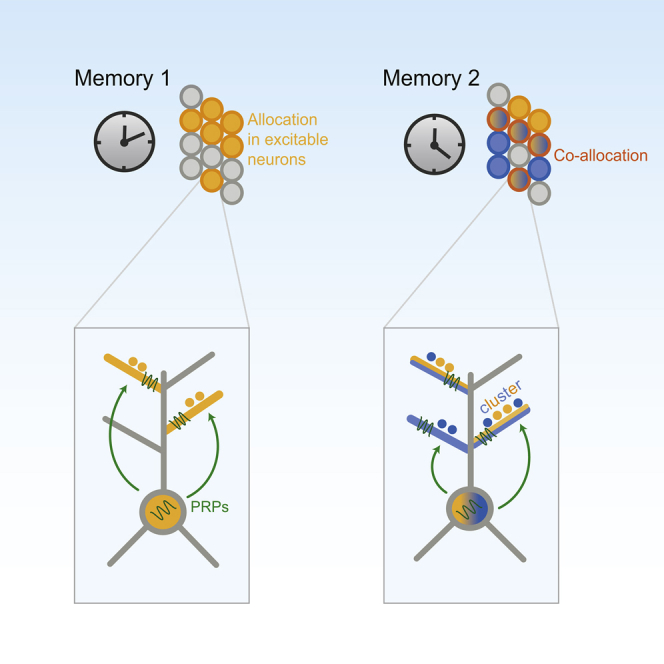
Highlights
-
•
Network model with active dendrites and synaptic, somatic, homeostatic plasticity
-
•
Linked memories are stored in overlapping populations of neurons
-
•
Linked memories share synaptic clusters in common dendritic branches
-
•
The locus of protein synthesis or capture shapes the structure of the memory trace
Kastellakis et al. present a biologically relevant network model of associative memory formation and dissect the subcellular mechanisms underlying the linking of memories of different strengths and numbers across time. In all cases, linked memories exhibit synaptic co-clustering within the dendrites of common neurons.
Introduction
Associative memories are believed to be stored in specific neuronal assemblies (Reijmers et al., 2007) through long-lasting synaptic and excitability modifications (Disterhoft and Oh, 2006, Frick et al., 2004), which can be localized within dendrites (Frick et al., 2004, Losonczy et al., 2008, Zhang and Linden, 2003) or seen throughout the cell (Oh et al., 2010). Activity of the CREB (cAMP response element-binding protein) transcription factor was shown to increase excitability and bias the allocation of associative memories into excitable neuronal ensembles (Han et al., 2007, Restivo et al., 2009, Silva et al., 2009). Based on the dynamics of CREB activation, it was proposed that memories learned within short time intervals will be stored in overlapping neuronal populations (Rogerson et al., 2014, Silva et al., 2009), and thus interact during recall. Indeed, it was recently shown that two associative memories learned within a period of a few hours interact with each other and are allocated to overlapping populations of neurons in both the hippocampus (Cai et al., 2016) and the amygdala (Rashid et al., 2016). These findings show that memory linking across time relies on the overlap of cellular memory engrams. What remains unclear are the sub-cellular mechanisms that enable this type of memory linking and to what extent they are generic, namely, what underlies the linking of memories of different strengths and numbers across different time intervals.
For example, apart from neuronal excitability, the phenomenon of synaptic tagging and capture (STC) provides a mechanistic model for the specificity and co-operativity of synaptic plasticity (Govindarajan et al., 2011, Redondo and Morris, 2011), a prerequisite for long-term memory linking. Moreover, the locus of protein synthesis and capture is critical because it determines the distribution (neuron-wide or spatially restricted) of synapses that get strengthened or weakened during learning (Rogerson et al., 2014). It remains unclear whether the proteins needed to stabilize a synapse are available throughout a neuron or isolated within strongly activated branches, typically equipped with nonlinear mechanisms, or a combination of the two extremes (Redondo and Morris, 2011, Steward and Schuman, 2007). Together with homeostatic mechanisms (Turrigiano, 2008), these multi-level processes shape the structure (neuronal, dendritic, and synaptic features) and interactions of memory engrams in ways that remain largely unexplored.
Previous modeling studies have investigated individual memory processes, such as branch strength potentiation (Legenstein and Maass, 2011), homeostasis (Wu and Mel, 2009), and synaptic capture (Barrett et al., 2009, Clopath et al., 2008, O’Donnell and Sejnowski, 2014), and their role in information binding. However, to date, no models integrate these phenomena into a biophysically constrained network model. Given their differential contributions to memory formation, integration of these phenomena is crucial to extract the rules underlying the allocation and linking of memories. Toward this goal we build a biophysically inspired network model of generic neurons with non-linear dendritic subunits that incorporates plasticity of intrinsic excitability, homeostasis, and STC with somatic, dendritic, or combined protein synthesis or capture.
The model is used to examine the cellular and sub-cellular mechanisms underlying memory formation under three different settings: (1) encoding and recall of a single associative memory, (2) rescuing of a weak memory by pairing with a strong memory, and (3) linking of multiple subsequent memories presented at different time intervals. By varying the expression of individual plasticity processes, we aim to dissect their effects on synaptic and neuronal properties of memory engrams and infer the key mechanisms that enable memory linking across time.
Results
The proposed network model is outlined in the Experimental Procedures and consists of excitatory neurons with independent dendritic subunits along with feedback inhibitory neurons (Figure 1). Simulations were conducted under the following conditions: (1) plasticity-related proteins (PRPs) required for synaptic tagging and capture are synthesized at the soma and made available to all dendritic subunits simultaneously (somatic); (2) PRPs are synthesized and made available only to synapses in a dendritic branch (local); and (3) a combination of somatic and local (S&L) PRP synthesis takes place. The effect of intrinsic neuronal excitability on memory was examined under the following conditions: (4) enhanced excitability, whereby excitatory neurons recruited in a memory had a reduced afterhyperpolarization (AHP) for 12 hr (Zhou et al., 2009); and (5) static excitability, where neuronal excitability did not change (see Supplemental Experimental Procedures).
Figure 1.

Neuronal and Network Model
(A) Schematic of synaptic integration in two-layer excitatory model neurons. Neurons integrate synaptic signals independently in dendritic branches and subsequently in the somatic layer. Encoding events originate from separate excitatory populations and contact random branches. Protein production is required for the consolidation of synaptic tags, and it can be somatic (PRPs available to all branches), local (PRPs available only to strongly activated branches), or combined (S&L).
(B) Plasticity-related protein transients are generated after a strong LTP-inducing event and enable LTP associativity. The time course of the level of PRPs is modeled as an alpha function.
(C) Calcium levels after training (at the level of the synapse) determine the sign and magnitude of synaptic tags.
(D and E) Principle of heterosynaptic late LTP interactions: synaptic weight updates depend on the interaction between synaptic tags and the availability of PRPs (D). When the two processes do not overlap in time, heterosynaptic potentiation does not take place (E).
(F) Connectivity of the network model. Left: interneurons (red) provide feedback inhibition to the excitatory cell population (brown). Right: excitatory neurons receive background excitatory input (blue). Connectivity parameters are listed in Table S2.
Learning a Single Associative Memory
The model was trained to encode a single associative memory composed of two events or stimuli (S1 and S2), via the concurrent activation for 4 s of the inputs representing each stimulus. These events could represent a pair of conditioned and unconditioned stimuli, as in fear conditioning, or any pair of sensory stimuli (e.g., a sound and a visual cue) experienced together as in contextual memories. Successful formation of an associative memory between S1 and S2 was indicated by an enhanced response to the presentation of the first stimulus (S1) during recall, as observed experimentally (Quirk et al., 1995). Recall was assessed 24 hr post-training, to allow for homeostasis to take place.
Validation: Network Responses to S1 Increase after Learning
Model parameters were calibrated so that learning increased the network responsiveness to S1 presentation (Figure 2), to indicate that the two stimuli became associated. Specifically, parameters (plasticity thresholds, dendritic spike threshold, number of afferent connections, and initial synaptic weight) were tuned so that the percentage of coding neurons after learning was approximately 30% (as per Quirk et al., 1995, Reijmers et al., 2007, Rumpel et al., 2005). A sensitivity analysis with respect to model parameters for all PRP conditions is shown in Figures S2 and S4A.
Figure 2.

Neuronal Activity Properties of a Single Associative Memory
(A) Size of the neuronal population encoding an associative memory: percentage of excitatory neurons with average ff > 10 Hz upon S1 presentation, measured before (Pre) and after training (recall), under the three PRP conditions.
(B) Average firing rate of the coding neurons before (Pre) and after training (recall), upon S1 presentation, under the three conditions.
(C) Sparseness of the entire neuronal population measured by the Treves-Rolls metric, before (Pre) and after training (recall), upon S1 presentation, under the three conditions.
(D) Conceptual models of population responses before and after learning for somatic, local, and combined (S&L) PRP conditions. The neuronal memory engram denoted by the coding cells (blue circles filled with yellow-orange colors) is similar in size under conditions of somatic or dendritic (local) PRPs and slightly larger if PRPs are available throughout the neuron (S&L). However, it is highest in contrast (differences in color of coding versus responsive but non-coding cells), under somatic, followed by S&L and local PRP conditions.
Graphs show average ± SEM of 10 trials. ∗∗p < 0.01, one-way ANOVA followed by Bonferroni post-test. See also Figures S2, S4, and S5.
As a result, both the percentage of coding neurons, excitatory neurons firing above 10 Hz in response to S1 presentation, and their average firing rate increased during recall for all PRP conditions. Specifically, the percentage of coding neurons (Figure 2A) increased from 0.7% ± 0.1% before training (Pre) to 29.5% ± 0.9% under somatic, 28.3% ± 0.7% under local, and 35.7% ± 0.8% under S&L PRP conditions, respectively. The average firing rate of these coding neurons (Figure 2B) increased from 10.8 ± 0.4 Hz before training to 15.4 ± 0.2 Hz under somatic, 12.3 ± 0.2 Hz under local, and 14.6 ± 0.3 Hz under S&L PRP conditions. Note that S&L PRPs lead to larger engrams, yet somatic PRPs lead to engrams with largest mean activity. The difference (p < 0.01, one-way ANOVA followed by Bonferroni post-test) between somatic and S&L firing frequencies is due to the steeper sparseness distribution in the former compared with the latter (Figures S1B and S1D).
Population Activity Becomes Sparser after Learning and Depends on PRP Condition
Associative fear conditioning was shown to increase population sparseness, so that fewer neurons respond more vigorously to conditioned stimulus (CS) presentation after learning (Gdalyahu et al., 2012). We assessed the activity sparseness of the excitatory model neurons using the Treves-Rolls metric (Treves and Rolls, 1991), which measures the steepness of the population activity (firing rate) distribution (Figures 2C and S1A–S1D). We found that learning increased the sparseness of the population response under all PRP conditions, albeit to different levels (Figure 2C), without any additional parameter tuning.
An interesting prediction is that sparseness will be greatest under somatic and smallest under local PRP conditions. This difference can be explained as follows (see Figures S1A–S1D): under somatic PRP conditions, neurons that cross the calcium threshold for PRP synthesis will have all their tagged synapses potentiated, leading to a marked increase in their activity compared with other responsive cells, and thus a steep population activity distribution (for the same reason, the average firing rate of the coding population is highest in Figure 2B). In the local PRP case, only synapses tagged within PRP-producing dendrites will be potentiated, thus leading to a broader activity distribution. Under S&L PRP conditions, neurons will exhibit variable activity levels, thus leading to an intermediate steepness of the activity distribution. Activity sparseness represents the signal-to-noise ratio or contrast between coding and non-coding neurons during memory recall, with less sparseness potentially resulting in more interference between different memories and/or high noise levels.
Overall, model predictions regarding the population response characteristics during recall of a single associative memory are graphically summarized in Figure 2D: the neuronal memory engram (coding neurons) is expected to be similar in size under conditions of somatic or dendritic (local) PRPs and slightly larger under S&L conditions. However, it is expected to be highest in contrast under somatic, followed by S&L and local PRP conditions. Differences in mean firing rates would be small (2–3 Hz).
Cellular and Sub-cellular Features of the Learned Memory Differ between PRP Conditions
The model also predicts marked differences in the sub-cellular (synaptic, dendritic) properties of the learned memory among PRP conditions, which can be used to design experiments that tease out the dominant mode of PRP synthesis in vitro and/or in vivo. First, the percentage of excitatory neurons with ≥1 potentiated synapses is much smaller under somatic PRP conditions (29.2% ± 2.5%) compared with local and S&L PRP conditions (80.3% ± 1.3%; Figure 3A). Second, the total number of potentiated synapses after learning is similar in local and somatic conditions, but significantly larger under S&L conditions (Figure 3B). Third, within each neuron, more synapses are potentiated on average under somatic (32.4 ± 4.5) compared with local (10.9 ± 6.5) and S&L (17.4 ± 13.2) PRP conditions. Importantly, the distributions of potentiated synapses per neuron (Figures S1E–S1G) are quite different among the three cases (p < 0.001, Kruskal-Wallis test), with the former (somatic) approximating the sum of the other two. These differences suggest that it is feasible to infer the mode of PRPs experimentally with a reasonable sample size, given the assumptions used to configure our model. Specifically, if sampling from a uniform population of neurons and trying to infer which one of the three distributions best describes the data, having just 50 neurons would be sufficient to discriminate between local versus somatic and local versus S&L conditions, whereas 200 samples would be needed to discriminate between somatic and S&L conditions (alpha: p < 0.01, power: 1 − β = 0.9; see Figure S1).
Figure 3.

Neuronal, Dendritic, and Synaptic Features of a Single Associative Memory
(A) Percentage of excitatory neurons having at least one potentiated synapse after learning under the three PRP conditions.
(B) Total number of potentiated synapses under the three PRP conditions. Potentiated synapses are defined as having a weight > 0.7 (maximum synapse weight = 1.0) during recall.
(C) Boxplots of the number of potentiated synapses per excitatory neuron under the three PRP conditions. Neurons without potentiated synapses are not included. The respective distributions are shown in Figure S1.
(D) Boxplots of the number of potentiated synapses per dendritic branch under the three PRP conditions. Branches without potentiated synapses are not included. The respective distributions are shown in Figure S1.
(E) Conceptual illustration of a single associative memory engram under the three PRP conditions. Left: somatic PRP availability forms memory engrams with high activity sparsity, whose synaptic trace is restricted within a small neuronal population and potentiated synapses form smaller synaptic clusters within dendrites. Middle: local PRPs lead to memory engrams with low activity-sparsity, whose synaptic trace is distributed across the majority of the neuronal population and potentiated synapses form large clusters within few dendritic branches of these neurons. Right: an intermediate of the two cases occurs under conditions of combined PRPs.
Graphs show average ± SEM of 10 trials. ∗∗p < 0.01, one-way ANOVA followed by Bonferroni post-test. See also Figure S1.
A different picture emerges when looking within dendritic branches. The average number of potentiated synapses per branch is largest under local conditions (3.9 ± 0.9 synapses; Figure 3D) and smaller under somatic and S&L conditions (2.8 ± 1.0; Figure 3D). The distributions of potentiated synapses per branch (Figures S1H–S1J) reveal that with local PRPs, synapses are potentiated in groups ≥3, reflecting synapse clustering, whereas with somatic or S&L PRPs, isolated synapses are also potentiated. Again, the distributions of potentiated synapses per branch are different among the three cases (p < 0.001, Kruskal-Wallis test), but distinguishing between PRP modes based on this feature is more challenging. A sample size of 50 branches would be required to discriminate between local versus S&L and local versus somatic PRPs, but discriminating between somatic and S&L conditions requires thousands of branches because the two distributions are very similar.
In summary, model predictions regarding a single associative memory are conceptually depicted in Figure 3E and listed later. First, in line with experimental data, acquisition of a single associative memory increases the activity and sparseness of the population response across all PRP conditions (Figure 2). Second, the locus of PRP synthesis or capture determines the neuronal, dendritic, and synaptic features of the memory engram: somatic PRPs result in engrams with high activity and sparsity/contrast (Figures 2C, 2D, and 3E, left), whose synaptic trace is restricted to a small neuronal population (Figure 3A), containing clusters of two to three potentiated synapses that are widely distributed within dendritic trees (Figure 3D). Local PRPs lead to engrams with lower activity and sparsity/contrast (Figures 2C, 2D, and 3E, middle), whose synaptic trace is distributed across the majority of the neuronal population (Figure 3A), containing clusters of three to five potentiated synapses that are concentrated within a few dendritic branches (Figures 3C and 3D). An intermediate version of these characteristics is observed under conditions of combined S&L PRPs (Figure 3E, right).
These results suggest a number of predictions that could be tested experimentally. For example, plasticity markers such as pHluorin-tagged glutamate receptors (Zhang et al., 2015) and phosphorylated cofilin (Lynch et al., 2015), as well as two-photon in vivo imaging (Fu et al., 2012, Makino and Malinow, 2011), could be used to test the distributions of potentiated synapses after learning. Evidence for widespread plasticity across a majority of neurons, with localized clusters of potentiated synapses within a small subset of their dendritic branches, would indicate that local (as opposed to somatic) PRP mechanisms predominate during associative memory formation.
Dendritic Spikes Facilitate Engram Formation and Diversity
Because a key feature of our model is the incorporation of dendritic non-linearities, to understand their contribution, we examined engram formation in an alternative configuration of the network, in which dendrites do not spike. We found that to have a comparable coding population size, the number of afferent synapses must be increased by a factor of 2.36 to compensate for the loss of calcium influx through dendritic spikes. A sensitivity analysis of the alternative model is shown in Figure S2, whereby the main difference documented is a smaller size of the coding neuronal population, which is more pronounced under local PRP conditions.
Interestingly, eliminating dendritic spikes also eliminated the differences in the characteristics of memory engrams formed under somatic versus S&L PRP conditions, making them indistinguishable (see Figure S5). These simulations suggest important roles for dendritic excitability: dendritic spikes not only facilitate engram formation via the use of substantially lower numbers of synapses (thus saving resources) but also induce memory engrams whose features are distinguishably different depending on the locus of PRPs, thus expanding the dynamical range of engram formation. As a consequence, we predict that changes in the biophysics of dendrites that affect dendritic spiking (e.g., N-methyl-D-aspartate [NMDA] receptor deficits; Magnusson, 2012) would result in smaller, less diverse memory engrams especially if PRPs are restricted in dendrites.
Pairing Weak and Strong Memories
Locus of PRP Synthesis or Capture Affects Weak Memory Rescuing
We next investigated whether the mechanisms that underlie the formation of a single associative memory can also account for the rescue of a weak memory when paired with a strong one (so-called behavioral tagging; Ballarini et al., 2009), which is believed to be a consequence of the synaptic tagging and capture memory consolidation model (Frey and Morris, 1997, Redondo and Morris, 2011).
Weak learning was simulated by reducing the duration of the S1 and S2 stimuli so that only a small percentage of synapses representing these stimuli were consolidated (see Supplemental Experimental Procedures). Because both STC and learning-induced enhancement of intrinsic excitability could affect the two memories, we performed simulations under the three PRP conditions whereby the neuronal excitability of recruited neurons was either enhanced or remained static.
We found that rescuing of the weak memory, indicated by an increased percentage of coding neurons compared with baseline at 24 hr, was achieved in all cases and depended on the PRP condition and neuronal excitability (Figures 4A–4C versus 4D–4F). With enhanced excitability, rescuing was asymmetric and occurred under all PRP conditions for pairing intervals between −5 and +2 hr (Figure 4A). The effect was more pronounced in the somatic condition, followed by the S&L condition, and smallest in the local PRP condition. The latter is due to the lower probability of tagged synapses (representing the weak memory) being co-localized in the same dendritic branches with strong synapses that produce PRPs (representing the strong memory). The earlier-mentioned asymmetry was abolished if neuronal excitability remained unaltered after learning (Figure 4D), in which case the rescuing window was reduced to [−2, 2 hr].
Figure 4.

Rescuing of a Weak Memory When Paired with a Strong One
With enhanced excitability of neurons after learning (A–C) and without enhanced excitability of neurons after learning (D–F) are shown.
(A) Size of neuronal population responding during the recall of the weak memory (percentage of coding neurons, ff > 10 Hz) as a function of the weak-strong pairing time interval (negative intervals indicate that the strong memory precedes the weak one).
(B) Neurons that encode both the weak and the strong memory as a percentage of the sum of the neurons coding for each memory divided by 2.
(C) Dendritic branches containing clusters from both memories (i.e., two synapses of the weak and two synapses of the strong memory) as a percentage of the number of branches containing at least one cluster (i.e., two synapses) from either memory.
(D–F) Same as in (A)–(C), without learning-induced enhanced excitability.
Graphs show average ± SEM of 10 simulation trials. See also Figures S3 and S4.
These simulations make several predictions. First, an asymmetry in the pairing window for behavioral tagging is suggestive of learning-induced enhanced neuronal excitability. Second, such asymmetry could act as a mechanism for encoding the order of events: a weak memory learned before a strong one would be smaller (in size) than a weak memory learned after a strong one. Third, manipulations that attenuate or block the excitability enhancement, like drugs that enhance the slow afterhyperpolarization, should either blunt or eliminate the ordered encoding of events.
Weak Memory Rescuing Is Achieved through Overlapping Memory Engrams
According to the STC model, synapses coding for the weak memory can be strengthened through the sharing of PRP products. Our model predicts that this mechanism also leads to the overlapped storage (or co-allocation) of the weak and strong memories to many common neurons (Figures 4B and 4E) and many common dendrites of these neurons (Figures 4C and 4F). We find that the degree of co-allocation of the strong and weak memories follows the size of the ensemble encoding the weak memory. Similar to neuronal coding populations (Figures 4A and 4D), neuronal (Figures 4B and 4E) as well as dendritic overlaps (at least two potentiated synapses from each memory; Figures 4C and 4F) are asymmetric under conditions of enhanced, but not static, neuronal excitability. The asymmetry is more pronounced under conditions of somatic and S&L rather than local PRPs. Moreover, co-allocation within 2 hr is largest under somatic and S&L PRP conditions and is significant in the local PRP condition in the case of enhanced excitability (p < 0.01, one-way ANOVA), but not under static excitability (p > 0.1). Finally, rescuing of the weak memory by the strong memory is associated with increased dendritic co-localization of the two memories (co-clustering) in all PRP and both excitability conditions (p < 0.01, one-way ANOVA; Figures 4C and 4F).
The earlier simulations highlight the interactions and dissect the contributions of two distinct plasticity mechanisms on weak-strong memory rescuing: intrinsic excitability and STC, with the latter also depending on the mode of PRP synthesis or capture. The following predictions are generated by the model. First, irrespective of the mode of PRP synthesis or capture and neuronal excitability changes, weak-strong memory pairing within a window of 2 hr can lead to rescuing of the weak memory, as shown experimentally (Ballarini et al., 2009, Moncada and Viola, 2007), with the exception of local PRPs and static excitability, pairing at −2 hr. Second, this rescue is expected to be more effective if learning of a strong memory leads to neuronal excitability increases and if PRPs are available throughout the neuron (somatic). Third, the rescue could be extended to the [−5, 2 hr] window if the strong memory precedes the weak and leads to increases in somatic excitability. Fourth, under all conditions tested, rescuing of the weak memory is mediated by neuronal co-allocation of the two memories (Figures 4B and 4E), as well as respective co-clustering of their synaptic contacts in common dendritic branches (Figures 4C and 4F).
Neuronal and Dendritic Co-allocation Link Two Strong Associative Memories
It has been proposed that co-allocation of two memories to overlapping populations of neurons links the two memories by increasing the probability of co-recall (Silva et al., 2009). Indeed, two associative memories learned within a time period of a few hours were shown to be allocated to overlapping populations of neurons and interact during recall, in both the hippocampus (Cai et al., 2016) and the amygdala (Rashid et al., 2016). The main hypothesis for co-allocation entails that CREB-dependent transcription in the neurons encoding the first memory results in temporary increases in excitability (Stanciu et al., 2001) that, for a time, bias the allocation of subsequent memories to many of the same neurons that encoded the first memory (Viosca et al., 2009, Zhou et al., 2009). We used our model to test this hypothesis by presenting two strong (capable of producing PRPs) learning events separated by an inter-stimulus interval (ISI) of 1, 2, 5, or 24 hr. Recall was tested 24 hr later by measuring responses to one of the two stimuli associated with each of the memories. Simulations were performed under all three PRP conditions, with and without enhanced neuronal excitability.
We found that under conditions of enhanced excitability, the two memories interacted for ISIs of 1–5 hr (Figures 5A–5D), whereas under conditions of static excitability, this interaction was limited to ISIs of 1–2 hr (Figures 5E–5H). Specifically, under all conditions, the coding population size of both memories increased substantially for ISIs of 1–2 hr compared with baseline at 24 hr (20%–60% increase at 1 hr; Figures 5A, 5B, 5E, and 5F). The enhancement of the first memory was independent of the PRP condition and neuronal excitability (Figures 5A and 5E) and was not significant at 5 hr (p > 0.1, t test). In contrast, the neuronal size of the second memory was larger under conditions of enhanced excitability and depended on the mode of PRPs (somatic PRP conditions resulted in the biggest traces, followed by S&L and local conditions; Figures 5B and 5F). Moreover, increases in memory size were extended to ISIs of 5 hr under enhanced excitability conditions (p < 0.001; Figure 5B), indicating that learning-induced increases in neuronal excitability prime the allocation of the second memory in more neurons, in accordance with experimental studies (Cohen et al., 1999).
Figure 5.

Linking of Strong Memories across Time
With enhanced excitability of neurons after learning (A–D) and without enhanced excitability of neurons after learning (E–H) are shown.
(A) Increase in the size (percentage of coding neurons) of the first strong memory as a function of the ISI, compared with the 24-hr interval (baseline), under the three PRP conditions.
(B) As in (A), for the second strong memory.
(C) Percentage of neurons coding for both memories, indicating population overlaps (as in Figure 4B).
(D) Percentage of branches containing clusters from both memories (≥2 synapses from each memory), indicating dendritic overlaps and co-clustering (as in Figure 4C).
(E–H) As in (A)–(D), without learning-induced enhanced excitability.
Dashed lines indicate baseline at 24-hr interval. Graphs show average ± SEM of 10 trials.
Increases in the size of the two memories were accompanied by respective increases in the overlap between the populations coding for the two memories under both enhanced and static excitability conditions (Figures 5C and 5G), except for the case of local PRPs with static excitability, in which the neuronal co-allocation was not significantly increased beyond an ISI of 1 hr (p > 0.1, one-way ANOVA followed by Bonferroni post-test; Figure 5G). As with the case of weak-strong memory pairing, the pairing of strong memories led to co-clustering of synaptic contacts from the two memories in dendritic branches for ISIs of 1–2 hr for all cases tested. Co-clustering extended to 5 hr under conditions of enhanced excitability and somatic or S&L but not local, PRPs (p < 0.01, one-way ANOVA followed by Bonferroni post-test; Figures 5D and 5H). Our results agree with recent experimental data on the overlap of time-linked memory ensembles in CA1 in vivo (Cai et al., 2016), which indicate an ∼50% increase of overlap at 5 hr compared with 7 days.
In all cases, the restriction of PRPs to dendritic branches led to a smaller degree of neuronal co-allocation between the two memories for ISIs of 1 and 2 hr (p < 0.01, one-way ANOVA). The effect on dendritic co-clustering, however, was not significant (p > 0.1), indicating that dendritic co-allocation is not heavily dependent on the levels of neuronal co-allocation.
Overall, our model makes the following predictions regarding the interactions between two strong memories and the underlying mechanisms. First, our simulations support the hypothesis that changes in excitability lead to co-allocation of memories separated by several hours (Silva et al., 2009, Rogerson et al., 2014) and are in line with experimental evidence that linked memories are stored in overlapping neuronal populations (Cai et al., 2016, Rashid et al., 2016). Second, our model also supports the hypothesis that STC mechanisms will lead to synapse clustering, in agreement with prior work (Govindarajan et al., 2011, Govindarajan et al., 2006, O’Donnell and Sejnowski, 2014). Third, pairing of two strong memories will affect both memories if learned within a couple of hours, but just the second one for longer ISIs (i.e., 5 hr). Fourth, the asymmetric enhancement of the second memory is due to increases in neuronal excitability after the acquisition of the first memory and predicts that blocking this increase in excitability should eliminate this asymmetry and possibly restrict memory enhancement to a 2-hr interval. This prediction is in line with experimental evidence from aged mice (where neuronal excitability after learning is decreased) showing impaired memory linking and reduced neuronal overlaps (Cai et al., 2016). Fifth, for ISIs shorter than 2 hr, the high overlaps between the neuronal ensembles representing both memories under somatic and S&L PRP conditions may lead to memory interference (Robertson, 2012). Restricting PRPs to dendritic branches (e.g., by reducing transcription) should reduce neuronal co-allocation and, therefore, decrease interference. Finally, we predict that interactions between strong memories are mediated by neuronal co-allocation and dendritic co-clustering of synapses representing the two memories.
Creating Memory Episodes by Binding Memories via Population Overlaps
The linking of strong memories suggests that overlapping allocation may be a general mechanism for binding together sequences of events to create memory episodes. The interplay between excitability and STC is likely to favor the creation of memory episodes for the following reasons: (1) STC links memories according to their temporal proximity, that is, the closer the memories, the higher their co-allocation; and (2) learning-induced enhancement of neuronal excitability influences prior and subsequent memories in an asymmetric manner, thus enabling ordered linking of events. To test this hypothesis, we simulated the encoding of 10 sequentially presented memories separated by inter-stimulus intervals (ISIs) of 1, 5, and 24 hr, under conditions of enhanced versus static excitability and somatic versus local PRP synthesis and capture.
As shown in Figure 6A, sequential encoding with an ISI of 1 hr leads to high population overlaps between memories that are close in time (near the diagonal). These overlaps extend to more distant memories in conditions of somatic PRPs and enhanced neuronal excitability. As shown in Figure 6B (left), when memories are presented with 1-hr intervals, neuronal overlaps are largest under conditions of somatic PRPs and enhanced neuronal excitability compared with the other cases (p < 0.01, one-way ANOVA followed by Bonferroni post-test), and are non-significant under conditions of local PRPs and static neuronal excitability, for all ISIs tested. When memories are separated by 5 hr (Figure 6B, middle), the neuronal overlap is significantly smaller compared with 1 hr for all cases (p < 0.01) and remains significantly above baseline only for memories separated by 5–10 hr under conditions of enhanced (p < 0.01), but not static, neuronal excitability. This phenomenon can be explained by considering the effect of homeostatic mechanisms, which operate over time periods beyond 24 hr and reduce the overall excitability of the entire neuronal population, as well as the time window of enhanced neuronal excitability that does not extend beyond 12 hr. This effect is most pronounced when memories are separated by 24 hr, where neuronal overlaps are not different from baseline for both enhanced and static excitability cases and both PRP conditions (Figure 6B, right).
Figure 6.

Overlapping Allocation of Sequences of Memories
(A) Percentage of common coding neurons between 10 memories encoded sequentially separated by 1 hr. Top: enhanced excitability. Bottom: static excitability. Left: somatic PRPs. Right: local PRPs.
(B) Average percentage of neuronal overlaps as a function of temporal proximity (distance in hours) between the memories (i.e., average of the diagonals in [A]). Left: all four combinations of enhanced/static excitability and somatic/local PRPs for ISI = 1 hr. Middle: neuronal overlaps for ISI = 5 hr. Right: neuronal overlaps for ISI = 24 hr between memories.
(C) Average percentage of branches containing clusters of both memories (co-clustering) as a function of temporal proximity (distance in hours) between the memories (A). Left: all four combinations of enhanced/static excitability and somatic/local PRPs for ISI = 1 hr between memories. Middle: co-clustering for ISI = 5 hr between memories. Right: co-clustering for ISI = 24 hr between memories.
Graphs show average ± SEM of 10 trials.
Overall, these simulations predict the overlapping allocation of strings of memories when subsequent events are interleaved with ISIs up to 5 hr. As with all prior experiments, the overlapping allocation at the neuronal level is accompanied by an increased co-clustering of synapses from interacting memories (Figure 6C), which follows the pattern of population overlaps: higher under somatic PRPs and enhanced excitability, and diminished when memories are separated by large intervals. These simulations suggest that overlapping memory allocation mediated by STC and neuronal excitability mechanisms can be a natural candidate for binding together subsequent events over large timescales to create coherent memory episodes. Our model predicts that manipulations or conditions that disrupt these mechanisms, as, for example, in aging, would lead to diminished ability to bind events, leading to dissociation and partial forgetting of details in memory episodes.
Discussion
The notion that dendrites may act as semi-independent computational and storage units (Mel, 1992, Poirazi and Mel, 2001), together with evidence for dendritically localized synaptic plasticity (Govindarajan et al., 2011, Hardie and Spruston, 2009, Kang and Schuman, 1996), has led to multiple hypotheses regarding the role of dendritic plasticity in the formation of memory engrams (Branco and Häusser, 2010, Govindarajan et al., 2006, Kastellakis et al., 2015, Rogerson et al., 2014, Zhou et al., 2009). Modeling studies have only recently begun to investigate this issue, albeit using simplified models and/or focusing on single plasticity rules (Legenstein and Maass, 2011, O’Donnell and Sejnowski, 2014, Wu and Mel, 2009). The model presented here incorporates biologically constrained dendritic compartments, depolarization-dependent plasticity mechanisms, synaptic tagging and capture, and changes in neuronal excitability in neurons embedded in a cortical microcircuit to study memory engram formation. Importantly, we show that each of these mechanisms plays a role in the formation and interaction of memory engrams that would be missed if using simplified models. Thus, the key contribution of this work is the systematic investigation of the biologically relevant parameter space that allows memory linking so as to characterize the underlying sub-cellular mechanisms and the extent to which they are generic, namely underlie information binding across the levels of a single, a pair, or multiple associative memories.
We find that the locus of protein synthesis or capture leads to marked differences in both the neuronal (e.g., size/sparsity of the coding population) and synaptic features (e.g., distributions of potentiated synapses within neurons and branches) of associative memories in our model. The predicted differences can, through targeted experiments, help identify whether dendritic or somatic protein synthesis is dominant. For example, experimental evidence for widespread plasticity across a majority of neurons, with localized clusters of potentiated synapses within a small subset of their dendritic branches, would indicate that local (as opposed to somatic) PRP mechanisms predominate during associative memory formation. Distinguishable characteristics endowed by the locus of PRPs are also seen in the interaction of associative memories (weak or strong) across time. In addition, we predict a role for intrinsic excitability in weak-strong memory rescuing, which has thus far been attributed to STC mechanisms alone (Ballarini et al., 2009). Our model also predicts roles for dendritic spikes: they serve as a boosting mechanism for storing and linking memories via the use of fewer synaptic resources, whereas also enhancing the diversity of engram features (e.g., sparsity and distributions of potentiated synapses), thus increasing their dynamic range.
Importantly, our model is in agreement with recent experimental studies, which confirm that there is excitability-dependent overlap of memory engrams learned within time intervals of a few hours. Memory engrams in CA1 are overlapping when they are separated by 5-hr interval, but not at 7 days, and the overlap is linked with behavioral expression (Cai et al., 2016). Similarly, in the lateral amygdala, memory engrams were allocated in overlapping populations for memories separated up to 6 hr (Rashid et al., 2016) and engrams competed for recruitment of neurons. Our model indicates that this overlapping allocation is also affected by the locus of PRP synthesis. For example, we predict that shorter intervals (1–2 hr) would lead to higher and qualitatively different co-allocation, primarily because of synaptic tagging and capture mechanisms rather than neuronal excitability (Figure 5). Moreover, we propose that the neuronal overlap will be significantly higher if one of the memories is weak (Figure 4).
This work capitalizes on prior experimental and modeling findings (Barrett et al., 2009, Clopath et al., 2008, Govindarajan et al., 2011, Legenstein and Maass, 2011, Losonczy et al., 2008, O’Donnell and Sejnowski, 2014, Smolen et al., 2006) to put together many pieces of a large puzzle. This is done by identifying a unifying principle for linking information across time: through neuronal and dendritic co-allocation realized via synapse clustering (schematically illustrated in Figure 7). This simple rule explains memory interactions at the cellular and sub-cellular levels, and is robust under all of the different scenarios tested here, including variations in the locus of PRP synthesis and the plasticity of neuronal excitability, different numbers, and strengths of memory, variable time intervals, among others. In all cases tested, the strength of interactions between memories was proportional to the degree of neuronal and dendritic overlap between those memories. Importantly, our findings are in line with recent studies showing that learning is accompanied by increased synaptic clustering (Fu et al., 2012, McBride et al., 2008), although evidence is currently missing for or against the co-clustering of afferents representing distinct memories.
Figure 7.

Proposed Conceptual Model of Memory Linking through Neuronal and Dendritic Overlaps
(A) Temporally close memories are stored in overlapping neuronal populations because of the enhancement of neuronal excitability that follows learning and the sharing of PRP products.
(B) Within a single neuron of this population, axons coding for both memories converge to the same branches, where the probability for PRP capture is highest.
(C) Allocation to the same branches leads to composite clusters that contain strengthened synapses encoding for both memory events. Thus, activation of one memory facilitates the activation of the second memory by engaging local nonlinearities.
Prior work has also examined memory associations and contributed important insights with respect to the mechanisms underlying information binding and weak-strong memory interactions (Legenstein and Maass, 2011, O’Donnell and Sejnowski, 2014), several of which are in agreement with this study. Through a systematic investigation of a much larger parameter space, our model predicts that synapse clustering is a universal mechanism that links both weak-strong and strong-strong memories, in the absence of high axonal overlap, irrespective of the location of protein synthesis and for time intervals much longer than the ones explained by prior studies or models.
Although most studies have treated memories as independent entities, they are almost always linked to other memories in ways that help us to organize our world into predictive patterns. Our biologically realistic model captured this critical facet of memory and yielded a number of experimental predictions that could guide research on how weak and strong memories are linked across time. Disruptions of the processes that link and organize memories are likely to affect cognitive function and result in psychopathology. For example, it has been proposed that molecular and cellular disruptions leading to over- or under-clustering may be related to the cognitive symptoms with a number of psychiatric problems, such as schizophrenia (memory interference caused by over-clustering) or autism (difficulty in contextual processing caused by under-clustering) (Kastellakis et al., 2015). Future experiments that test predictions proposed here will address the mechanisms underlying these pathologies in animal models, and therefore open new avenues for the understanding and treatment of memory deficits.
Experimental Procedures
The model’s complexity lies between biophysical and abstract mathematical models. Dendritic structure (a branch is a single compartment) and nonlinearities are incorporated in a simplified manner and the plasticity rules are modeled in a phenomenological way, without modeling the biophysics of membrane mechanisms or the elaborated morphology of dendritic trees. The model thus provides a general framework for dissecting the contributions of each plasticity mechanism to different memory phenomena.
The proposed model (Figure 1) consists of 400 excitatory and 100 inhibitory neurons. Excitatory neurons are modeled as two-stage integrators (Poirazi et al., 2003), consisting of independent, nonlinear dendritic subunits capable of compartmentalized plasticity (Poirazi and Mel, 2001). The somatic spike response was modeled as an adaptive integrate-and-fire unit responsive to the summed dendritic input. The neurons received feedback inhibition from inhibitory point neurons, as well as excitation from background synapses.
Synaptic plasticity conforms to the STC model, which requires both synaptic tagging and the availability of plasticity-related proteins (PRPs) for stable strengthening and weakening of synapses (Redondo and Morris, 2011). Calcium acts as the main trigger for the induction of synaptic tags and for the synthesis of PRPs. The calcium influx after a presynaptic spike depends on the voltage of the postsynaptic dendritic subunit in an NMDA-dependent manner. The total calcium influx to a synapse during a learning event determines the sign (long-term potentiation [LTP] or long-term depression [LTD]) of synaptic tags according to the calcium control model (Shouval et al., 2002) and triggers the production of PRPs, which are modeled by alpha functions. The calcium level also determines whether neuronal excitability increases after a learning event. This effect is mediated by the reduction of the spike adaptation at the soma, mimicking a decreased afterhyperpolarization current (Oh et al., 2009, Zhou et al., 2009). The effect of homeostasis on synaptic plasticity is modeled via a synaptic scaling homeostatic rule (Turrigiano, 2008). The parameter values of our neuron models are listed in Table S1.
The network model consists of a population comprising of the following: (1) excitatory neurons of a target region, (2) inhibitory neurons in the same region, (3) the stimulus-carrying input neurons, and (4) background noise input neurons. Each memory encoding is performed by the activation of a set of stimulus-carrying neurons, which carry the conditioned and unconditioned stimulus information that represents the memory to be encoded (Figure 1A). Stimulus-carrying neurons are grouped in sets of six (three representing stimulus 1 [S1] and three representing stimulus 2 [S2]), which fire with an average firing frequency of 30 Hz during the encoding of each memory (S1+S2 neurons), as well as during the recall of each individual event (S1 neurons only). Stimulus-carrying neurons create potential synapses by randomly targeting the dendritic branches of the excitatory neurons. These stimulus-to-excitatory synapses are the only plastic synapses in the network and are initialized to the same low initial weight. The background inputs create a number of synaptic contacts to random dendrites and fire with an average frequency of 0.5 Hz throughout the simulations. Connectivity parameters among the neuronal populations in the network model are listed in Table S2.
When encoding multiple memories, inter-stimulus intervals (ISIs) of ≥1 hr are introduced between consecutive memories. During ISI periods, spiking activity is not simulated, but slow consolidation processes (PRP production, synaptic tag consolidation, and homeostasis) take place. A consolidation period of 36 hr follows the end of all encoding events. Recall takes places after this consolidation period, via the activation of S1 input neurons. Population responses during recall are used to assess the properties of memory engrams. The significance of differences in engram properties was measured using ANOVA models. A detailed description of the model and experimental procedures is provided in the Supplemental Experimental Procedures. The source code of the model is provided under the open source GPLv2 license at http://dendrites.gr/ and under ModelDB: 206249.
Author Contributions
G.K. and P.P. conceived the study and designed the model. G.K. implemented and performed the simulations and analyzed the data. G.K., P.P., and A.J.S. wrote the paper.
Acknowledgments
We thank D.J. Cai, A. Papoutsi, and P. Petrantonakis for critical reading and useful discussions. This work was supported by the European Research Council Starting Grant dEMORY (GA 311435) to P.P., the National Institute on Aging (grant R37 AG013622), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (A.J.S.).
Published: November 1, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.10.015.
Accession Numbers
The accession number for the computational model reported in this paper is ModelDB: 206249.
Supplemental Information
References
- Ballarini F., Moncada D., Martinez M.C., Alen N., Viola H. Behavioral tagging is a general mechanism of long-term memory formation. Proc. Natl. Acad. Sci. USA. 2009;106:14599–14604. doi: 10.1073/pnas.0907078106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett A.B., Billings G.O., Morris R.G.M., van Rossum M.C.W. State based model of long-term potentiation and synaptic tagging and capture. PLoS Comput. Biol. 2009;5:e1000259. doi: 10.1371/journal.pcbi.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco T., Häusser M. The single dendritic branch as a fundamental functional unit in the nervous system. Curr. Opin. Neurobiol. 2010;20:494–502. doi: 10.1016/j.conb.2010.07.009. [DOI] [PubMed] [Google Scholar]
- Cai D.J., Aharoni D., Shuman T., Shobe J., Biane J., Song W., Wei B., Veshkini M., La-Vu M., Lou J. A shared neural ensemble links distinct contextual memories encoded close in time. Nature. 2016;534:115–118. doi: 10.1038/nature17955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clopath C., Ziegler L., Vasilaki E., Büsing L., Gerstner W. Tag-trigger-consolidation: a model of early and late long-term-potentiation and depression. PLoS Comput. Biol. 2008;4:e1000248. doi: 10.1371/journal.pcbi.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen A.S., Coussens C.M., Raymond C.R., Abraham W.C. Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J. Neurophysiol. 1999;82:3139–3148. doi: 10.1152/jn.1999.82.6.3139. [DOI] [PubMed] [Google Scholar]
- Disterhoft J.F., Oh M.M. Learning, aging and intrinsic neuronal plasticity. Trends Neurosci. 2006;29:587–599. doi: 10.1016/j.tins.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Frey U., Morris R.G.M.G. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Frick A., Magee J., Johnston D. LTP is accompanied by an enhanced local excitability of pyramidal neuron dendrites. Nat. Neurosci. 2004;7:126–135. doi: 10.1038/nn1178. [DOI] [PubMed] [Google Scholar]
- Fu M., Yu X., Lu J., Zuo Y. Repetitive motor learning induces coordinated formation of clustered dendritic spines in vivo. Nature. 2012;483:92–95. doi: 10.1038/nature10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gdalyahu A., Tring E., Polack P.-O., Gruver R., Golshani P., Fanselow M.S., Silva A.J., Trachtenberg J.T. Associative fear learning enhances sparse network coding in primary sensory cortex. Neuron. 2012;75:121–132. doi: 10.1016/j.neuron.2012.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan A., Kelleher R.J., Tonegawa S. A clustered plasticity model of long-term memory engrams. Nat. Rev. Neurosci. 2006;7:575–583. doi: 10.1038/nrn1937. [DOI] [PubMed] [Google Scholar]
- Govindarajan A., Israely I., Huang S.-Y., Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis-dependent LTP. Neuron. 2011;69:132–146. doi: 10.1016/j.neuron.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.-H., Kushner S.A., Yiu A.P., Cole C.J., Matynia A., Brown R.A., Neve R.L., Guzowski J.F., Silva A.J., Josselyn S.A. Neuronal competition and selection during memory formation. Science. 2007;316:457–460. doi: 10.1126/science.1139438. [DOI] [PubMed] [Google Scholar]
- Hardie J., Spruston N. Synaptic depolarization is more effective than back-propagating action potentials during induction of associative long-term potentiation in hippocampal pyramidal neurons. J. Neurosci. 2009;29:3233–3241. doi: 10.1523/JNEUROSCI.6000-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H., Schuman E.M. A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science. 1996;273:1402–1406. doi: 10.1126/science.273.5280.1402. [DOI] [PubMed] [Google Scholar]
- Kastellakis G., Cai D.J., Mednick S.C., Silva A.J., Poirazi P. Synaptic clustering within dendrites: an emerging theory of memory formation. Prog. Neurobiol. 2015;126:19–35. doi: 10.1016/j.pneurobio.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legenstein R., Maass W. Branch-specific plasticity enables self-organization of nonlinear computation in single neurons. J. Neurosci. 2011;31:10787–10802. doi: 10.1523/JNEUROSCI.5684-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A., Makara J.K., Magee J.C. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature. 2008;452:436–441. doi: 10.1038/nature06725. [DOI] [PubMed] [Google Scholar]
- Lynch G., Kramár E.A., Gall C.M. Protein synthesis and consolidation of memory-related synaptic changes. Brain Res. 2015;1621:62–72. doi: 10.1016/j.brainres.2014.11.060. [DOI] [PubMed] [Google Scholar]
- Magnusson K.R. Aging of the NMDA receptor: from a mouse’s point of view. Future Neurol. 2012;7:627–637. doi: 10.2217/fnl.12.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H., Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride T.J., Rodriguez-Contreras A., Trinh A., Bailey R., Debello W.M. Learning drives differential clustering of axodendritic contacts in the barn owl auditory system. J. Neurosci. 2008;28:6960–6973. doi: 10.1523/JNEUROSCI.1352-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mel B.W. NMDA-based pattern discrimination in a modeled cortical neuron. Neural Comput. 1992;4:502–517. [Google Scholar]
- Moncada D., Viola H. Induction of long-term memory by exposure to novelty requires protein synthesis: evidence for a behavioral tagging. J. Neurosci. 2007;27:7476–7481. doi: 10.1523/JNEUROSCI.1083-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell C., Sejnowski T.J. Selective memory generalization by spatial patterning of protein synthesis. Neuron. 2014;82:398–412. doi: 10.1016/j.neuron.2014.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M.M., McKay B.M., Power J.M., Disterhoft J.F. Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A. Proc. Natl. Acad. Sci. USA. 2009;106:1620–1625. doi: 10.1073/pnas.0807708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh M.M., Oliveira F.A., Disterhoft J.F. Learning and aging related changes in intrinsic neuronal excitability. Front. Aging Neurosci. 2010;2:2. doi: 10.3389/neuro.24.002.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirazi P., Mel B.W. Impact of active dendrites and structural plasticity on the memory capacity of neural tissue. Neuron. 2001;29:779–796. doi: 10.1016/s0896-6273(01)00252-5. [DOI] [PubMed] [Google Scholar]
- Poirazi P., Brannon T., Mel B.W. Arithmetic of subthreshold synaptic summation in a model CA1 pyramidal cell. Neuron. 2003;37:977–987. doi: 10.1016/s0896-6273(03)00148-x. [DOI] [PubMed] [Google Scholar]
- Quirk G.J., Repa C., LeDoux J.E. Fear conditioning enhances short-latency auditory responses of lateral amygdala neurons: parallel recordings in the freely behaving rat. Neuron. 1995;15:1029–1039. doi: 10.1016/0896-6273(95)90092-6. [DOI] [PubMed] [Google Scholar]
- Rashid A.J., Yan C., Mercaldo V., Hsiang H.-L., Park S., Cole C.J., De Cristofaro A., Yu J., Ramakrishnan C., Lee S.Y. Competition between engrams influences fear memory formation and recall. Science. 2016;353:383–387. doi: 10.1126/science.aaf0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo R.L., Morris R.G.M. Making memories last: the synaptic tagging and capture hypothesis. Nat. Rev. Neurosci. 2011;12:17–30. doi: 10.1038/nrn2963. [DOI] [PubMed] [Google Scholar]
- Reijmers L.G., Perkins B.L., Matsuo N., Mayford M. Localization of a stable neural correlate of associative memory. Science. 2007;317:1230–1233. doi: 10.1126/science.1143839. [DOI] [PubMed] [Google Scholar]
- Restivo L., Tafi E., Ammassari-Teule M., Marie H. Viral-mediated expression of a constitutively active form of CREB in hippocampal neurons increases memory. Hippocampus. 2009;19:228–234. doi: 10.1002/hipo.20527. [DOI] [PubMed] [Google Scholar]
- Robertson E.M. New insights in human memory interference and consolidation. Curr. Biol. 2012;22:R66–R71. doi: 10.1016/j.cub.2011.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogerson T., Cai D.J., Frank A., Sano Y., Shobe J., Lopez-Aranda M.F., Silva A.J. Synaptic tagging during memory allocation. Nat. Rev. Neurosci. 2014;15:157–169. doi: 10.1038/nrn3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S., LeDoux J., Zador A., Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Shouval H.Z., Bear M.F., Cooper L.N. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl. Acad. Sci. USA. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva A.J., Zhou Y., Rogerson T., Shobe J., Balaji J. Molecular and cellular approaches to memory allocation in neural circuits. Science. 2009;326:391–395. doi: 10.1126/science.1174519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolen P., Baxter D.A., Byrne J.H. A model of the roles of essential kinases in the induction and expression of late long-term potentiation. Biophys. J. 2006;90:2760–2775. doi: 10.1529/biophysj.105.072470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanciu M., Radulovic J., Spiess J. Phosphorylated cAMP response element binding protein in the mouse brain after fear conditioning: relationship to Fos production. Brain Res. Mol. Brain Res. 2001;94:15–24. doi: 10.1016/s0169-328x(01)00174-7. [DOI] [PubMed] [Google Scholar]
- Steward O., Schuman E.M. Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci. 2007;24:299–325. doi: 10.1146/annurev.neuro.24.1.299. [DOI] [PubMed] [Google Scholar]
- Treves A., Rolls E. What determines the capacity of autoassociative memories in the brain? Network. 1991;2:371–397. [Google Scholar]
- Turrigiano G.G. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viosca J., Lopez de Armentia M., Jancic D., Barco A. Enhanced CREB-dependent gene expression increases the excitability of neurons in the basal amygdala and primes the consolidation of contextual and cued fear memory. Learn. Mem. 2009;16:193–197. doi: 10.1101/lm.1254209. [DOI] [PubMed] [Google Scholar]
- Wu X.E., Mel B.W. Capacity-enhancing synaptic learning rules in a medial temporal lobe online learning model. Neuron. 2009;62:31–41. doi: 10.1016/j.neuron.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Linden D.J. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat. Rev. Neurosci. 2003;4:885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Cudmore R.H., Lin D.-T., Linden D.J., Huganir R.L. Visualization of NMDA receptor-dependent AMPA receptor synaptic plasticity in vivo. Nat. Neurosci. 2015;18:402–407. doi: 10.1038/nn.3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Won J., Karlsson M.G., Zhou M., Rogerson T., Balaji J., Neve R., Poirazi P., Silva A.J. CREB regulates excitability and the allocation of memory to subsets of neurons in the amygdala. Nat. Neurosci. 2009;12:1438–1443. doi: 10.1038/nn.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.