

Abstract
Posttraumatic osteoarthritis (PTOA) is the most common form of osteoarthritis (OA) of the ankle joint. PTOA occurs as a result of several factors, including the poor regenerative capacity of hyaline articular cartilage as well as increased contact stresses following trauma. The purpose of this article is to review the epidemiology, pathogenesis, and potential targets for treatment of PTOA in the ankle joint. Previous reviews primarily addressed clinical approaches to ankle PTOA, while the focus of the current article will be specifically on the newly acquired knowledge of the cellular mechanisms that drive PTOA in the ankle joint and means for potential targeted therapeutics that might halt the progression of cartilage degeneration and/or improve the outcome of surgical interventions. Three experimental treatment strategies are discussed in this review: (1) increasing the anabolic potential of chondrocytes through treatment with growth factors such as bone morphogenetic protein-7; (2) limiting chondrocyte cell death either through the protection of cell membrane with poloxamer 188 or inhibiting activity of intracellular proteases, caspases, which are responsible for cell death by apoptosis; and (3) inhibiting catabolic/inflammatory responses of chondrocytes by treating them with anti-inflammatory agents such as tumor necrosis factor-α antagonists. Future studies should focus on identifying the appropriate timing for treatment and an appropriate combination of anti-inflammatory, chondro- and matrix-protective biologics to limit the progression of trauma-induced cartilage degeneration and prevent the development of PTOA in the ankle joint.
Keywords: cartilage, ankle, posttraumatic osteoarthritis, chondroprotection, anti-inflammatory
Introduction
Ankle joint (talocrural) cartilage is unique in anatomy, mechanical load, biochemical composition, metabolism, and disease pattern. While this joint is highly susceptible to physical injury that may lead to the development of posttraumatic osteoarthritis (PTOA), it is simultaneously less prone to idiopathic degenerative disease of the cartilage (i.e., primary osteoarthritis) as compared to other joints.1,2 The prevalence of ankle joint osteoarthritis (OA) is less than 2% in comparison to more than 40% in the knee or hip joints. PTOA occurs as a result of direct injury to articular cartilage after events such as articular fracture, chondral injury, or ankle sprain. Overall, PTOA accounts for more than 12% of symptomatic OA in all articular joints, with the ankle joint among the most commonly affected.3 Because of the ankle’s increased susceptibility to PTOA, a number of experimental studies have been performed in recent years to more clearly define the mechanism of ankle PTOA and to identify targets for biologic interventions to impede the osteoarthritic process following injury to the ankle joint.
The purpose of this article is to review the epidemiology, pathogenesis, and potential targets for treatment of PTOA modeled in cadaveric human cartilage obtained from the ankle joint. The current review literature on the experimental treatment of ankle PTOA is lacking. In 2006, Polyzois et al.4 reviewed new biologic interventions for repair of cartilage damage. However, since this time many new studies have been published that increase our understanding of the mechanism of ankle PTOA and thus provide insight into new treatment strategies, including but not limited to (1) increasing the anabolic activity of chondrocytes through treatment with bone morphogenetic protein-7 (BMP-7); (2) limiting chondrocyte cell death with the use of poloxamer 188 (P188) or caspase inhibitors; and (3) inhibiting catabolic/inflammatory responses of chondrocytes by treating them with anti-inflammatory agents such as tumor necrosis factor-α (TNF-α) antagonists.
Unique Characteristics of Human Ankle Cartilage
Ankle cartilage has been found to be thinner and more uniform than knee cartilage, with tibial and talar surfaces ranging from 1.06 to 1.63 mm and from 0.94 to 1.62 mm, respectively.5 Ankle cartilage is composed of hyaline cartilage. Hyaline articular cartilage is divided into 4 zones: tangential (superficial), transitional (middle), radial (deep), and calcified.6 The superficial zone consists of horizontally aligned chondrocytes, most of which are paired with approximately 25% of chondrocytes found in single-cell patterns.7 This is distinct from the knee joint, in which more than half of the chondrocytes of the femoral condyles and patellar groove are arranged in strings and clusters, respectively. Furthermore, the distance between chondrocytes is significantly greater in the talus compared to the femoral condyles.7
It is thought that ankle cartilage, in comparison to knee cartilage, has a greater capacity for repair.8 Ankle cartilage has increased stiffness and decreased permeability due to an increased amount of proteoglycans and water.8 Furthermore, the extracellular matrix of ankle cartilage has an increased density, which provides an increased resistance to load and decreased sensitivity to mechanical damage.9 In articular cartilage, chondrocytes represent the only cell type and maintain the extracellular matrix through the balance of anabolic and catabolic activities. Chondrocytes found in ankle cartilage synthesize proteoglycans at an increased rate, leading to increased capacity for repair.8 In comparison to knee chondrocytes, ankle chondrocytes are more responsive to anabolic factors, such as osteogenic protein-1.10 Ankle lesions have shown upregulation of C-terminal type II procollagen propeptide and aggrecan turnover (epitope 836), markers of synthesis, as compared to knee lesions.11 Meanwhile, ankle chondrocytes are less responsive to catabolic mediators, including fibronectin fragments and interleukin-1β, both of which have been shown to inhibit proteoglycan synthesis.12,13 Levels of matrix metalloproteinases (MMPs), enzymes responsible for the degradation of extracellular matrix proteins, also differ between the ankle and knee joints. In normal ankle cartilage, mRNA expression of matrix metalloproteinase-8 (MMP-8) has been found to be undetectable, whereas normal knee cartilage showed the presence of this MMP.14
Prevalence of Osteoarthritis and Posttraumatic Osteoarthritis in the Ankle
Osteoarthritis is the leading cause of joint disability in the United States, affecting 27 million people.15 Of the lower limbs, the hips, knees, and first metatarsophalangeal joints are most commonly affected. In comparison, OA of the ankle is relatively rare, affecting as low as 1% of the population.16 In a study looking at 500 patients with limb joint OA, the incidence of ankle OA was 4.4% with a male-to-female ratio of 0.8:1.0.17 Similar trends have been observed in our long-standing collaboration (over 25 years) with the Gift of Hope Organ and Tissue Donor Network (Itasca, IL) using cartilage from donors with no documented history of joint diseases. Cartilage degeneration was less pronounced in the ankle joint than in the knee as defined by joint morphology scale ( Fig. 1A ). Also, with aging, cartilage degeneration was less severe in the ankle joint than that in the knee joint ( Fig. 1B and C ).18
Figure 1.

Changes in morphological appearance of the ankle and knee cartilage from human organ donors with no documented history of joint disease with age and degeneration as defined by the Collins morphology scale8,18.
PTOA is the predominant form of OA in the ankle, accounting for 70% to 78% of cases, followed by secondary arthritis (12% to 13%) and finally primary/idiopathic OA (7% to 9%).19,20 This differs from hip or knee OA, which is largely idiopathic. Furthermore, among patients undergoing total ankle arthroplasty, more than half have a primary diagnosis of PTOA.21
Epidemiologic studies have reported that 13% to 18% of patients undergoing a total ankle joint replacement had an identifiable acute trauma to the joint.22 It has also been shown that early onset of OA can occur within 10 years after injury,23 thereby indicating that patients with PTOA are much younger (18-44 years) than those with idiopathic OA. On average, the latency time between injury and end-stage OA is 20 years, though patients who are younger at the time of initial injury typically have a longer latency period.24 In the United States alone, there are approximately 5.6 million people suffering from PTOA, which translates to an annual financial burden of $3.06 billion on the health care system.25
Mechanism of Posttraumatic Osteoarthritis
The key difference between PTOA and primary or idiopathic OA is the presence of a precipitating insult to the joint, where the extent of cartilage damage depends on the intensity and force of the impact and/or the change in joint biomechanics and integrity due to overuse, overload, or damage of other joint tissues.26 Regardless of the cause of joint injury, PTOA develops as a result of poor intrinsic regenerative ability of hyaline articular cartilage.26 In terms of intraarticular forces, tibiotalar instability has been shown to significantly increase peak contact stresses.27,28 Even in patients with surgically reduced ankle fractures, peak contact stresses are significantly higher compared to normal, contralateral ankles.29 Moreover, an inflammatory response that occurs immediately after fracture has been detected. Synovial fluid from patients who suffered an intra-articular ankle fracture showed elevated inflammatory cytokines at 17 days postinjury compared to controls.30,31
For the most part, current surgical approaches treat the developed pathology. The idea of biological interventions or pharmacological treatment is based on the premise of arresting and/or preventing the onset and progression of the disease. Developing therapies that could reduce the inflammatory and degrading mediators will be key to prevent or slow down PTOA.
Regenerative Capacity Following Cartilage Impact
In 1743, William Hunter famously remarked that “ulcerated cartilage is universally allowed to be a very troublesome disease . . . when destroyed, it is never recovered.”32 Exactly 250 years later, Shapiro et al.33 partially disproved Hunter’s assertion. In 1993, Shapiro showed that experimental PTOA in New Zealand White rabbits resulted in mesenchymal stem cell proliferation leading to regeneration of cartilage that closely resembled normal hyaline cartilage as long as 8 weeks following impaction. However, by 12 weeks postimpaction, degeneration of superficial zone cartilage had begun with formation of vertical fissuring; worsened degeneration took place between 36 and 48 weeks. Perhaps most importantly, Shapiro et al.33 showed that the newly synthesized cartilage failed to integrate with the residual, unharmed chondrocytes. In fact, polarized light microscopy found a gap between areas of new and residual cartilage. Thus, although it was possible for new cartilage cells to grow into the areas affected by trauma, there were no signs of true cartilage repair in which newly synthesized matrix integrated with the remaining host tissue to create a functional cartilage interphase.
Our laboratory simulated ex vivo injury to ankle cartilage that potentially can resemble cartilage degeneration observed in PTOA by subjecting a normal human talus to a single impaction using a 4-mm cylindrical indenter with an impulse of 1 N s as described.34 We found that impaction resulted in chondrocyte death within the area of damage, with only about one third of cells in the superficial layer remaining alive immediately following injury. These results coincided with those of Hembree et al.,35 who showed that chondrocyte death was significantly greater along the edge of fractures and in the superficial zone in traumatically injured human joint fragments.
Our studies showed that, over time, the level of cell death increased both laterally and horizontally beyond the area that experienced impaction directly. Importantly, cells that surrounded the immediate damage zone were viable within the first 48 hours; however, with time they showed evidence of apoptosis and death, suggesting radial progression of cartilage damage. These results were consistent with those of Tochigi et al.,36 who created distal tibial intraarticular fractures in human ankles harvested immediately following amputation. The authors found that chondrocyte death was significantly greater in fracture-edge regions compared to nonfracture regions immediately postfracture, though chondrocyte fractional death continued to increase in both regions over 48 hours following fracture.
Biologic Approaches to PTOA
Currently, a number of biologic approaches are utilized in the clinical setting to treat ankle osteochondral lesions. These include BMAC (bone marrow aspirate concentrate), PRP (platelet-rich plasma), and HA (hyaluronic acid).37 However, their potency as DMOADs (disease-modifying OA drugs) for the treatment of PTOA in the ankle joint has not been explored as of yet.
Our laboratory described immediate cellular responses to injury.26 Three phases have been identified: early phase, intermediate phase, and late phase.26 The early phase is characterized by cell death and the release and activation of intracellular pro-inflammatory mediators that trigger matrix degradation; intermediate phase is characterized by initiation of anabolic responses; and late phase supposedly favors matrix remodeling and anabolic processes. Based on these mechanisms, 4 classes of potential therapies have been considered: (1) chondroprotective, (2) anti-inflammatory, (3) matrix protective, and (4) pro-anabolic.26,38-42
The latest efforts in our laboratory have focused on testing targeted therapeutics that could be easily translated to patient care using the same methodology and the same model of ankle cartilage injury as previously described.34 Among these were: BMP-7, also called osteogenic protein-1 (OP-1)43; anti-TNF-α; N-acetylcysteine (NAC)44; interleukin-receptor antagonist (IRAP)45; P188 surfactant; and inhibitors of caspases.34,46 We found that each of these listed biologics was able to prevent chondrocyte death, although to a different degree, in our acute human ankle injury model ( Fig. 2 ). Application of P188, OP-1, and anti-TNF-α also resulted in a better histological appearance of cartilages subjected to impaction based on Safranin O staining.
Figure 2.
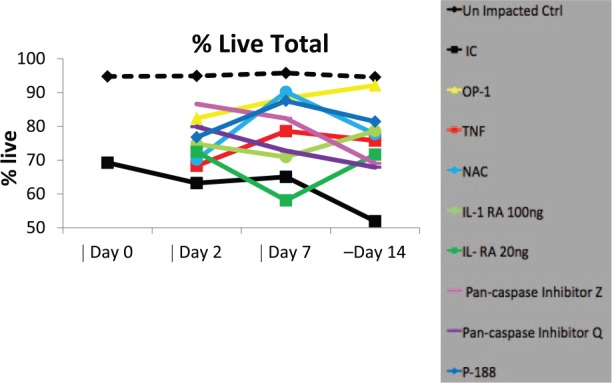
Cell viability. Total refers to all layers of cartilage explant (superficial, middle, deep). All agents were added to parallel cultures immediately after impaction on day 0 and were kept for 48 hours, after which they were replaced with fresh media only. Measurements were made on day 0 immediately following impaction, and then on days 2, 7, and 14. IC = impacted control; OP-1 = osteogenic protein-1 added at 100 ng/mL concentration; TNF = tumor necrosis factor-α (TNF-α) antagonist, 100 ng/mL; NAC = N-acetyl-l-cysteine, 2.5 mM; IL-RA = IL-1 receptor antagonist used at 20 ng/mL and 100 ng/mL; Pan-caspase inhibitor Z = Z-VAD-FMK pan caspase inhibitor, 100 µM; Pan-caspase inhibitor Q = Q-VD-OPh pan caspase inhibitor, 100 µM. Cell viability was assessed with Live/Dead Cell assay.
Based on these data and in line with timing of cellular responses described in Anderson et al.,26 we selected the 3 most effective agents for combined therapy: P188 to protect the cell membrane, anti-TNF-α to inhibit inflammatory responses, and OP-1 to stimulate chondrocyte anabolism. Three treatment schemas were tested over the course of 14 days culture using the same model as in Pascual-Garrido et al.34 ( Fig. 3 ): Combination 1: P188 administered at day 0 for 48 hours + OP-1 administered at day 0 for 48 hours and at days 7 to 14 + anti-TNF-α administered at days 0 to 7; Combination 2: all 3 agents administered at day 0 for 48 hours and anti-TNF-α and BMP-7 administered again at day 7 for 48 hours; Combination 3: all agents administered simultaneously at day 0 for 48 hours ( Fig. 3A ). Tissue and media were collected on days 0, 2, 7, and 14 and analyzed for cell viability, Safranin O staining, and proteoglycan (PG) synthesis. It was found that Combined treatment 1 and to a lesser extent 2 markedly improved cell survival not only in the superficial zone but also throughout the entire cartilage ( Fig. 3B and C ). Over the course of culture, the percentage of viable cells remained higher in these 2 combined groups. The percentage of live chondrocytes increased from 59% in the impacted control (IC) to 84% in Combined schema 1 (P = .006). Both Combinations 1 and 2 also had a stronger effect on Safranin O staining and preservation of matrix integrity than Combination 3 (data not shown). Contrary to this, Combination 3 exhibited the highest effect on PG synthesis, especially at the end of culture on day 14 (1.8-fold increase vs. IC or the other 2 combinations) (P < 0.05; Fig. 3D ), confirming that anabolic/remodeling responses are secondary to cell death and inflammation. To the best of our knowledge, this is the first attempt to combine biologic treatments of various modes of action in a model of experimental PTOA. Presented data generated 2 important findings: (1) the same combination of agents, but administered at various treatment regimens, can induce different effects. Prolonged administration of anti-TNF-α and BMP-7 (Combination 1) had a strong effect on cell survival and matrix preservation, but was less effective in inducing chondrocyte synthetic activity, thereby suggesting that overstimulation/overdosing may have a detrimental effect on chondrocyte anabolism; and (2) a window of opportunity exists to arrest cell death and delay/prevent cartilage degeneration. Reported in vitro data warrant further exploration of combined biologic therapies for PTOA, especially if they can target distinct mechanisms: cell survival, inhibition of catabolism, and stimulation of remodeling processes. In this particular case, the chosen triad may have clinical relevance since all 3 agents have already been approved by the Food and Drug Administration for other use.
Figure 3.

Effect of combined treatment on chondrocyte viability and proteoglycan synthesis in the model of ankle acute injury as described in Pascual-Garrido et al.34 (A) Schematic representation of combo groups. (B) Cell viability as measured by Live/Dead Cell assay. UIC = un-impacted control; IC = impacted control. (C) Representative images of cartilage sections stained with calcein AM (green fluorescence, live cells) and ethidium bromide homodimer-1 (red fluorescence, dead cells) (Molecular Probes, Eugene, OR). (D) Proteoglycan synthesis was measured as described47: the cells were labeled with 5 µCi/mL 35S-sulfate (Perkin Elmer, Boston, MA) for 4 hours. The amount of 35S-labeled PGs was analyzed by the alcian blue (Bio-Rad, Hercules, CA) precipitation method; the values were normalized to the wet weight of each cartilage explant. All measurements were done in triplicate for each donor sample.
Conclusion
Ankle cartilage consists of the same cell type and the same matrix constituents as other articular cartilages, although it differs in thickness, biomechanical and biochemical responses, and vulnerability to catabolic processes. The unique anatomic and cellular characteristics of the articular cartilage within the ankle joint make it more susceptible to PTOA. Recent ex vivo studies have shown promise for therapeutic approaches to ankle PTOA, both by increasing the anabolic characteristics of ankle joint chondrocytes as well as by limiting chondrocyte cell death. Unfortunately, there is a limited number of investigations that have focused on the understanding of human ankle cartilage physiology, pathophysiology, and regenerative capacity. A concentrated effort is therefore critical for further advancement of biologic repair in PTOA. Along with surgical options and cell-based cartilage repair applications, mechanism-based targeted biologic intervention will have its niche if future studies address the most effective formulation, composition (in the case of platelet-rich plasma, for example), dose, and delivery method of the biologic cocktail. In addition, the ideal time and duration of the intervention must be tested as well as possible adverse side effects and the potential for a combination of surgery and biologic therapy.
Footnotes
Acknowledgments and Funding: The authors would like to acknowledge Mrs. Arnavaz Hakimiyan, MS; Mrs. Carol Pacione, MS; and Dr. Lev Rappoport, MD, for technical assistance; and Dr. Arkady Margulis, MD, for procurement of human donor tissue. The authors would like to acknowledge the Gift of Hope Organ & Tissue donor network and donor families. Experiments with combined treatment were generated by Dr. Rylee Olewinski as part of her honor research project. The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Football League Charitable Foundation grant (SC) and Ciba-Geigy Endowed Chair at Rush University Medical Center (SC).
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval: The Deceased Subject Rule applies to this study and as such, there is no need for the Institutional Review Board review and approval.
References
- 1. Nelson AJ, Collins CL, Yard EE, Fields SK, Comstock RD. Ankle injuries among United States high school sports athletes, 2005-2006. J Athl Train. 2007;42(3):381-7. [PMC free article] [PubMed] [Google Scholar]
- 2. Huch K, Kuettner KE, Dieppe P. Osteoarthritis in ankle and knee joints. Semin Arthritis Rheum. 1997;26(4):667-74. [DOI] [PubMed] [Google Scholar]
- 3. Brown TD, Johnston RC, Saltzman CL, Marsh JL, Buckwalter JA. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma. 2006;20(10):739-44. [DOI] [PubMed] [Google Scholar]
- 4. Polyzois VD, Papakostas I, Zgonis T, Polyzois DG, Soucacos PN. Current concepts and techniques in posttraumatic arthritis. Clin Podiatr Med Surg. 2006;23(2):455-65. [DOI] [PubMed] [Google Scholar]
- 5. Shepherd DE, Seedhom BB. Thickness of human articular cartilage in joints of the lower limb. Ann Rheum Dis. 1999;58(1):27-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sophia Fox AJ, Bedi A, Rodeo SA. The basic science of articular cartilage: structure, composition, and function. Sports Health. 2009;1(6):461-8. doi: 10.1177/1941738109350438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rolauffs B, Williams JM, Grodzinsky AJ, Kuettner KE, Cole AA. Distinct horizontal patterns in the spatial organization of superficial zone chondrocytes of human joints. J Struct Biol. 2008;162(2):335-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kuettner KE, Cole AA. Cartilage degeneration in different human joints. Osteoarthritis Cartilage. 2005;13(2):93-103. doi: 10.1016/j.joca.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 9. Treppo S, Koepp H, Quan EC, Cole AA, Kuettner KE, Grodzinsky AJ. Comparison of biomechanical and biochemical properties of cartilage from human knee and ankle pairs. J Orthop Res. 2000;18(5):739-48. [DOI] [PubMed] [Google Scholar]
- 10. Cole AA, Kuettner KE. Molecular basis for differences between human joints. Cell Mol Life Sci. 2002;59(1):19-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aurich M, Squires GR, Reiner A, Mollenhauer JA, Kuettner KE, Poole AR, et al. Differential matrix degradation and turnover in early cartilage lesions of human knee and ankle joints. Arthritis Rheum. 2005;52(1):112-9. [DOI] [PubMed] [Google Scholar]
- 12. Dang Y, Cole AA, Homandberg GA. Comparison of the catabolic effects of fibronectin fragments in human knee and ankle cartilages. Osteoarthritis Cartilage. 2003;11(7):538-47. [DOI] [PubMed] [Google Scholar]
- 13. Eger W, Schumacher BL, Mollenhauer J, Kuettner KE, Cole AA. Human knee and ankle cartilage explants: catabolic differences. J Orthop Res. 2002;20(3):526-34. [DOI] [PubMed] [Google Scholar]
- 14. Chubinskaya S, Kuettner KE, Cole AA. Expression of matrix metalloproteinases in normal and damaged articular cartilage from human knee and ankle joints. Lab Invest. 1999;79(12):1669-77. [PubMed] [Google Scholar]
- 15. Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part II. Arthritis Rheum. 2008;58(1):26-35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Peyron JG. The epidemiology of osteoarthritis. In: Moskowitz RW, Howell DS, Goldberg VM, Mankin HJ. editors. Osteoarthritis: diagnosis and treatment. Philadelphia: Saunders; 1984. p. 9-27. [Google Scholar]
- 17. Cushnaghan J, Dieppe P. Study of 500 patients with limb joint osteoarthritis. I. Analysis by age, sex, and distribution of symptomatic joint sites. Ann Rheum Dis. 1991;50(1):8-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collins DH. Osteoarthritis. In: The pathology of articular and spinal diseases. London: Edward Arnold; 1949. p. 74-115. [Google Scholar]
- 19. Saltzman CL, Salamon ML, Blanchard GM, Huff T, Hayes A, Buckwalter JA, et al. Epidemiology of ankle arthritis: report of a consecutive series of 639 patients from a tertiary orthopaedic center. Iowa Orthop J. 2005;25:44-6. [PMC free article] [PubMed] [Google Scholar]
- 20. Valderrabano V, Horisberger M, Russell I, Dougall H, Hintermann B. Etiology of ankle osteoarthritis. Clin Orthop Relat Res. 2009;467(7):1800-6. doi: 10.1007/s11999-008-0543-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Daniels TR, Mayich DJ, Penner MJ. Intermediate to long-term outcomes of total ankle replacement with the Scandinavian Total Ankle Replacement (STAR). J Bone Joint Surg Am. 2015;97(11):895-903. doi: 10.2106/JBJS.N.01077. [DOI] [PubMed] [Google Scholar]
- 22. Kern D, Zlatkin MB, Dalinka MK. Occupational and post-traumatic arthritis. Radiol Clin North Am. 1988;26(6):1349-58. [PubMed] [Google Scholar]
- 23. Roos H, Adalberth T, Dahlberg L, Lohmander LS. Osteoarthritis of the knee after injury to the anterior cruciate ligament or meniscus: the influence of time and age. Osteoarthritis Cartilage. 1995;3(4):261-7. [DOI] [PubMed] [Google Scholar]
- 24. Horisberger M, Valderrabano V, Hintermann B. Posttraumatic ankle osteoarthritis after ankle-related fractures. J Orthop Trauma. 2009;23(1):60-7. doi: 10.1097/BOT.0b013e31818915d9. [DOI] [PubMed] [Google Scholar]
- 25. March L, Smith EU, Hoy DG, Cross MJ, Sanchez-Riera L, Blyth F, et al. Burden of disability due to musculoskeletal (MSK) disorders. Best Pract Res Clin Rheumatol. 2014;28(3):353-66. doi: 10.1016/j.berh.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 26. Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, et al. Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res. 2011;29(6):802-9. doi: 10.1002/jor.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McKinley TO, McKinley T, Rudert MJ, Koos DC, Pedersen DR, Baer TE, et al. Stance-phase aggregate contact stress and contact stress gradient changes resulting from articular surface stepoffs in human cadaveric ankles. Osteoarthritis Cartilage. 2006;14(2):131-8. [DOI] [PubMed] [Google Scholar]
- 28. McKinley TO, Tochigi Y, Rudert MJ, Brown TD. Instability-associated changes in contact stress and contact stress rates near a step-off incongruity. J Bone Joint Surg Am. 2008;90(2):375-83. doi: 10.2106/JBJS.G.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li W, Anderson DD, Goldsworthy JK, Marsh JL, Brown TD. Patient-specific finite element analysis of chronic contact stress exposure after intraarticular fracture of the tibial plafond. J Orthop Res. 2008;26(8):1039-45. doi: 10.1002/jor.20642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adams SB, Setton LA, Bell RD, Easley ME, Huebner JL, Stabler T, et al. Inflammatory cytokines and matrix metalloproteinases in the synovial fluid after intra-articular ankle fracture. Foot Ankle Int. 2015;36(11):1264-71. doi: 10.1177/1071100715611176. [DOI] [PubMed] [Google Scholar]
- 31. Furman BD, Kimmerling KA, Zura RD, Reilly RM, Zlowodzki MP, Huebner JL, et al. Articular ankle fracture results in increased synovitis, synovial macrophage infiltration, and synovial fluid concentrations of inflammatory cytokines and chemokines. Arthritis Rheumatol. 2015;67(5):1234-9. doi: 10.1002/art.39064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hunter W. Of the structure and disease of articulating cartilages. 1743. Clin Orthop Relat Res. 1995;(317):3-6. [PubMed] [Google Scholar]
- 33. Shapiro F, Koide S, Glimcher MJ. Cell origin and differentiation in the repair of full- thickness defects of articular cartilage. J Bone Joint Surg Am. 1993;75(4):532-53. [DOI] [PubMed] [Google Scholar]
- 34. Pascual-Garrido C, Hakimiyan AA, Rappoport L, Oegema TR, Wimmer MA, Chubinskaya S. Anti-apoptotic treatments prevent cartilage degradation after acute trauma to human ankle cartilage. Osteoarthritis Cartilage. 2009;17(9):1244-51. doi: 10.1016/j.joca.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hembree WC, Ward BD, Furman BD, Zura RD, Nichols LA, Guilak F, et al. Viability and apoptosis of human chondrocytes in osteochondral fragments following joint trauma. J Bone Joint Surg Br. 2007;89(10):1388-95. doi: 10.1302/0301-620X.89B10.18907. [DOI] [PubMed] [Google Scholar]
- 36. Tochigi Y, Buckwalter JA, Martin JA, Hillis SL, Zhang P, Vaseenon T, et al. Distribution and progression of chondrocyte damage in a whole-organ model of human ankle intra-articular fracture. J Bone Joint Surg Am. 2011;93(6):533-9. doi: 10.2106/JBJS.I.01777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mei-Dan O, Carmont MR, Laver L, Mann G, Maffulli N, Nyska M. Platelet-rich plasma or hyaluronate in the management of osteochondral lesions of the talus. Am J Sports Med. 2012;40(3):534-41. doi: 10.1177/0363546511431238. [DOI] [PubMed] [Google Scholar]
- 38. Bodker A, Lattermann C, Chubinskaya S. Biomarkers in early and post-traumatic osteoarthritis. In: Cole BJ, Harris JD. editors. Biologic knee reconstruction: a surgeon’s guide. Thorofare, NJ: Slack Inc.; 2015. p. 41-5. [Google Scholar]
- 39. Chubinskaya S, Wimmer MA. Key pathways to prevent posttraumatic arthritis for future molecule-based therapy. Cartilage. 2013;4(3 Suppl):13S-21S. doi: 10.1177/1947603513487457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lidder S, Chubinskaya S. Post-traumatic OA: Biologic approaches to treatment. In: Rothschild BM. editor. Principles of osteoarthritis—its definition, character, derivation and modality-related recognition. Rijeka, Croatia: InTechWeb.org; 2012. p. 233-60. [Google Scholar]
- 41. Moran CJ, Pascual-Garrido C, Chubinskaya S, Potter HG, Warren RF, Cole BJ, et al. Restoration of articular cartilage. J Bone Joint Surg Am. 2014;96(4):336-44. doi: 10.2106/JBJS.L.01329. [DOI] [PubMed] [Google Scholar]
- 42. Pascual-Garrido C, Chubinskaya S. Potential targets for pharmacologic therapies. In: Olson S, Guilak F. editors. Post-traumatic arthritis: pathogenesis, diagnosis and management. Berlin, Germany: Springer; 2015. p. 331-42. [Google Scholar]
- 43. Chubinskaya S, Otten L, Soeder S, Borgia JA, Aigner T, Rueger DC, et al. Regulation of chondrocyte gene expression by osteogenic protein-1. Arthritis Res Ther. 2011;13(2):R55. doi: 10.1186/ar3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin JA, McCabe D, Walter M, Buckwalter JA, McKinley TO. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91(8):1890-7. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pelletier JP, Caron JP, Evans C, Robbins PD, Georgescu HI, Jovanovic D, et al. In vivo suppression of early experimental osteoarthritis by interleukin-1 receptor antagonist using gene therapy. Arthritis Rheum. 1997;40(6):1012-9. [DOI] [PubMed] [Google Scholar]
- 46. D’Lima DD, Hermida J, Hashimoto S, Colwell C, Lotz M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54(6):1814-21. [DOI] [PubMed] [Google Scholar]
- 47. Masuda K, Shirota H, Thonar EJ. Quantification of 35S-labeled proteoglycans complexed to alcian blue by rapid filtration in multiwell plates. Anal Biochem. 1994;217(2):167-75. [DOI] [PubMed] [Google Scholar]