

Abstract
Congenital anemias comprise a group of blood disorders characterized by a reduction in the number of peripherally circulating erythrocytes. Various genetic etiologies have been identified that affect diverse aspects of erythroid physiology and broadly fall into two main categories: impaired production or increased destruction of mature erythrocytes. Current therapies are largely focused on symptomatic treatment and are often based on transfusion of donor-derived erythrocytes and management of complications. Hematopoietic stem cell transplantation represents the only curative option currently available for the majority of congenital anemias. Recent advances in gene therapy and genome editing hold promise for the development of additional curative strategies for these blood disorders. The relative ease of access to the hematopoietic stem cell compartment, as well as the possibility of genetic manipulation ex vivo and subsequent transplantation in an autologous manner, make blood disorders among the most amenable to cellular therapies. Here we review cell-based and gene therapy approaches, and discuss the limitations and prospects of emerging avenues, including genome editing tools and the use of pluripotent stem cells, for the treatment of congenital forms of anemia.
Keywords: anemia, red blood cells, cellular therapy, gene therapy, genome editing, hematopoiesis
Introduction
Hematopoietic stem cells (HSCs) are rare cells in the bone marrow that give rise to progressively more lineage-restricted progenitor cells that ultimately form all of the cellular components of the blood [Orkin and Zon, 2008]. Erythropoiesis is the coordinated process of differentiation by which these HSCs and progenitor cells (HSPCs) are able to produce erythrocytes responsible for the transport of oxygen [Palis, 2014]. This process is characterized by the massive accumulation of hemoglobin, distinct changes in morphology, and enucleation of the cells prior to entry into the circulation (Fig 1A). Human erythrocytes have a life span of 120 days and under homeostatic conditions healthy adults produce approximately two million erythrocytes every second to balance production and turnover [Palis, 2014]. The major cytokine that regulates erythropoiesis, erythropoietin (EPO), is produced by interstitial fibroblasts in the kidney that sense tissue oxygenation [Bunn, 2013].
Figure 1.
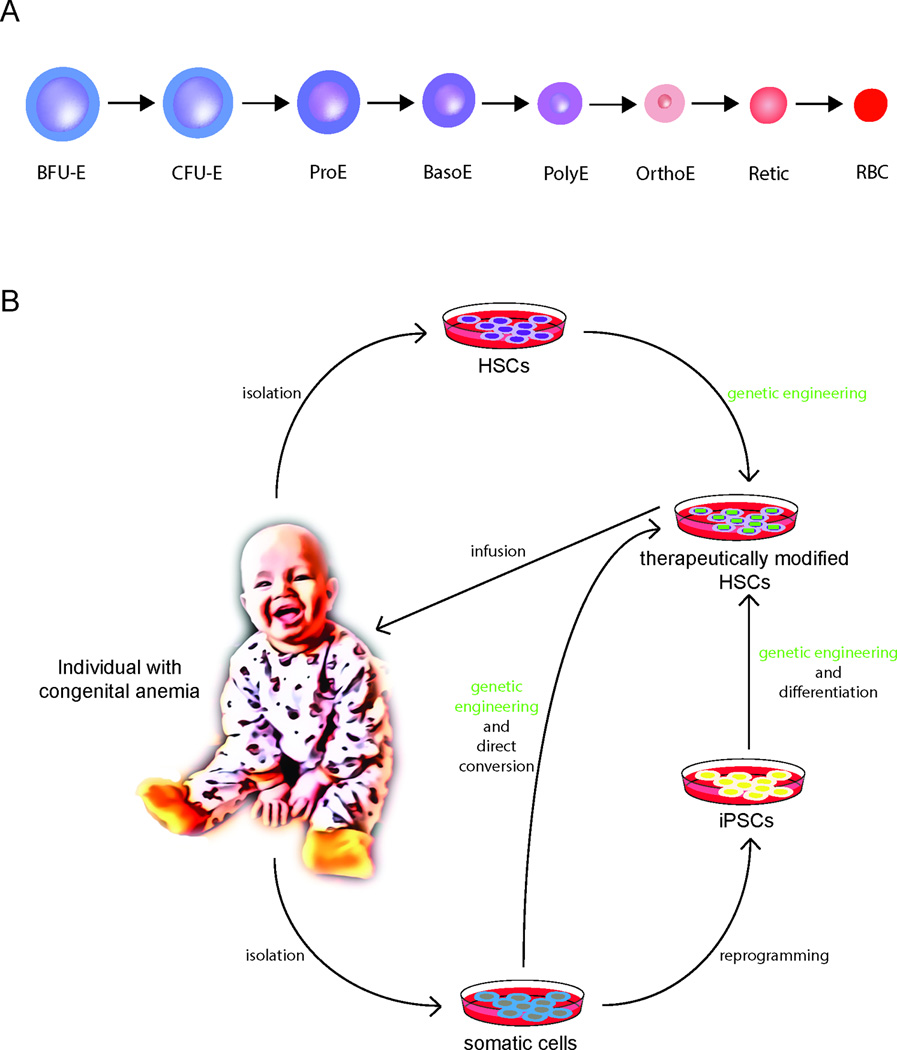
(A) A model for erythropoiesis. Early progenitor cells undergo progressive maturation through successive cellular stages that are accompanied by distinct changes in morphology. Burst forming unit – erythroid (BFU-E); colony forming unit – erythroid (CFU-E); proerythroblast (ProE); basophilic erythroblast (BasoE); polychromatic erythroblast (PolyE); orthochromatic erythroblast (OrthoE); reticulocyte (Retic); red blood cell (RBC). (B) Therapeutic avenues for the treatment of congenital anemias. Gene correction and editing of patients’ autologous cells presents a highly desired strategy to overcome limitations presented by allogeneic HSC transplantation and limited availability of HLA-matched allogeneic donors. Generally, this involves the isolation of the patient’s HSPCs from the bone marrow and subsequent exposure to gene modifying agents using viral or other transient delivery methods. Depending on the genetic entity of the disease, correction may be achieved by the introduction of a functional gene, repair of the mutated gene at its endogenous locus by homology directed repair (HDR) (e.g. the SCD mutation), or by targeting a modifier gene or its regulators known to ameliorate disease (e.g. knockdown or disruption of BCL11A in the hemoglobinopathies). The corrected cells would subsequently be re-infused into the patient to allow repopulation of the bone marrow and generation of healthy cells. Patient-specific induced pluripotent stem cells (iPSCs) derived through reprogramming of somatic cells present another possible approach. They have an unlimited propagation capacity allowing for more faithful genetic modification as they can be more extensively screened for adequate correction compared to manipulated HSPCs. Subsequently, they may be directed to differentiate into hematopoietic cells. Alternatively, a patient’s somatic cells could be directly converted into hematopoietic cells. However, in both instances the derivation of HSPCs with multi-lineage potential and engraftment capability remain key challenges to be overcome.
Anemia is defined as a decrease in the number of circulating erythrocytes. While anemia is often acquired, monogenic disorders leading to anemia are among the most prevalent genetic conditions, particularly among children [Sankaran and Weiss, 2015; Weatherall, 2010]. In these congenital forms of anemia, numerous distinct defects have been identified, affecting diverse aspects of erythroid physiology, including hemoglobin production, membrane stability, metabolism, vesicular trafficking, and ribosome biogenesis [Sankaran and Weiss, 2015]. These defects lead to reduced or ineffective production and maturation of erythroid cells in the bone marrow, or a shortened life span due to increased destruction of mature erythrocytes. Some forms of anemia involve a combination of these defects. Treatment remains largely supportive and symptomatic, consisting of transfusion with healthy donor erythrocytes and management of specific complications. Many patients with such disorders have significant morbidity and mortality, despite receiving the best available therapies [Marsella and Borgna-Pignatti, 2014; Yazdanbakhsh et al., 2012]. Numerous studies have provided important insight into the molecular pathophysiology of these disorders and have led to the development of a variety of compounds, with many under pre-clinical and clinical development [Archer et al., 2015; Ataga and Stocker, 2015]. However, allogeneic transplantation of HSPCs from a healthy donor (ideally a matched sibling) into a conditioned and myeloablated recipient is currently the only curative option available in a clinical setting for these disorders [Lucarelli et al., 2012]. Nevertheless, such transplants can be limited by human leukocyte antigen (HLA)-matched donor availability and transplant-associated complications, such as graft-versus-host disease and graft failure, which can cause considerable morbidity and mortality [Kekre and Antin, 2014; Mattsson et al., 2008; Petersdorf, 2013]. Given these limitations, the development of approaches to genetically manipulate autologous HSPCs holds considerable promise for improved therapies for congenital anemias. Advances in the field of gene therapy and genome editing now raise the prospect of correcting the genetic defect as a curative and more broadly available option. This review will focus on these and other emerging approaches for cellular and gene therapies for congenital forms of anemia.
Gene therapy approaches in congenital anemias
The frequent occurrence of monogenic congenital anemias and the relative ease of isolation of human HSPCs, as well as the possibility of gene modification or correction of HSPCs ex vivo and subsequent reinfusion into an affected patient, make such disorders prime candidates for these emerging therapeutic approaches (Fig 1B) [Sankaran and Weiss, 2015]. Significant advances in gene therapy methods and recent progress in the field of genome editing, such as use of the clustered regularly-interspaced short palindromic repeats (CRISPR)/Cas9 and other approaches, suggest that genetic correction of specific mutations may be feasible in the future [Gupta and Musunuru, 2014; Hsu et al., 2014]. In classical gene therapy, various approaches have been pursued to meet the challenges posed by the diverse set of congenital anemias, which can be caused by a single mutation of a single gene (e.g. sickle cell disease), a variety of different mutations at a single gene locus (e.g. β-thalassemia), or a number of different genes. Potential gene therapy or editing strategies include the introduction of a functional gene, the repair of a gene at its endogenous locus, or targeting a modifier gene known to ameliorate disease severity. While individually tailored approaches are possible in principle, there are practical limitations in applying these advances to disorders caused by diverse mutations that occur in multiple genes.
The addition of an intact gene by means of gene transfer is a strategy long pursued, particularly for the treatment of sickle cell disease (SCD) and the thalassemia syndromes, collectively known as the hemoglobinopathies. They represent the most prevalent forms of congenital anemia in the world and are caused by mutations in the hemoglobin genes [Sankaran et al., 2010]. Thus, the discussion of gene therapy will focus largely on the hemoglobinopathies, with discussion of other conditions at the end of this section.
The human genome has two genes for α-globin (HBA1 and HBA2) on each copy of chromosome 16 and a single gene for β-globin on each copy of chromosome 11 (HBB). The adult form of human hemoglobin (HbA, α2β2) consists of two α- and two β-globin chains forming a heterotetramer with each subunit bound to the iron-containing heme prosthetic group responsible for reversible oxygen binding. Thalassemia is caused by mutations that inhibit the production of either the α- or β-globin chains, which results in an imbalance of both chains required for the formation of the functional HbA tetramer. In β-thalassemia, reduced production of β-globin chains leads to aggregation of excessive free α-globin molecules that damage and destroy erythroid precursors, leading to ineffective erythropoiesis. The resulting anemia and compensatory mechanisms lead to organomegaly and erythroid hyperplasia with cortical thinning resulting in bone abnormalities. In α-thalassemia, there is an excess of free β-globin subunits, which are more stable than free α-globin molecules, and the disorder primarily manifests with hemolysis of mature erythrocytes in the circulation. As there are a total of four α-globin genes, the deletion of one or two copies results in no or mild anemia, while the loss of three α-globin genes leads to hemolytic anemia of variable severity, and deletion of all four α-globin genes is incompatible with life [Galanello and Origa, 2010; Harteveld and Higgs, 2010; Marengo-Rowe, 2007; Peters et al., 2012; Piel and Weatherall, 2014]. In SCD, the substitution of glutamate by valine at position 6 in the β-globin chain leads to the formation of a qualitatively defective hemoglobin S (HbS). The replacement of a hydrophilic amino acid with a hydrophobic residue creates rigid linear HbS polymers upon deoxygenation, deforming red blood cells (RBCs) into the characteristic sickle cell morphology. The sickled RBCs adhere to each other and the endothelium of small blood vessels, predisposing the patient to vaso-occlusive events characterized by tissue hypoxia / ischemia and resultant organ damage. Consequently, SCD is a multi-systemic disorder with numerous clinical manifestations that arise from impaired oxygen delivery to tissues [Frenette and Atweh, 2007; Rees et al., 2010; Steinberg, 1999].
Accordingly, the delivery of a functional β-globin gene has long been a primary strategy to treat SCD and β-thalassemia. However, attempts to use integrating γ-retroviral vectors initially failed to achieve sufficient and erythroid-specific transgene expression when gene expression was driven by viral long terminal repeats (LTRs) [Karlsson et al., 1988]. Appropriate expression was later enabled by the discovery of essential endogenous regulatory elements, such as the locus control region (LCR), a critical aggregate of upstream enhancers of the β-globin gene cluster [Talbot et al., 1989]. Incorporation of components of the LCR into gene transfer vectors significantly enhanced the transcription of the delivered globin genes [Plavec et al., 1993; Sadelain et al., 1995]. An additional obstacle in successful gene therapy was the silencing of transgenes randomly integrated into the genome and the altered expression of genes adjacent to the site of integration due to newly inserted regulatory elements. These problems were overcome through the identification and use of chromatin insulator elements to shield repressive and enhancing effects of endogenous and exogenous chromatin elements, respectively (Figure 2) [Evans-Galea et al., 2007; Rivella and Sadelain, 1998]. Deregulated gene expression due to a tendency of γ-retroviral vectors to insert near transcriptional start sites, frequently resulting in insertional oncogenesis due to integration near proto-oncogenes, led to the more frequent use of lentiviral vectors that exhibit a safer integration profile [Biffi et al., 2011; Cattoglio et al., 2007; Kvaratskhelia et al., 2014]. Nonetheless, lentiviruses have a relatively random integration pattern and tend to insert in actively transcribed genes [Schroder et al., 2002]. Furthermore, lentiviral vectors are more efficient at transducing the primary target cells for gene therapy, the quiescent HSCs. A sufficient level of modification in such HSCs presents a significant bottleneck for gene therapy approaches to be successful. To further enhance safety, modern self-inactivating (SIN) vectors carry deletions in their 3’ LTR to further minimize transactivation of nearby genes [Miyoshi et al., 1998]. These findings highlight the principal challenges for all approaches of gene therapy relying on gene integration to enable high-level and lineage-specific expression without compromising safety.
Figure 2.
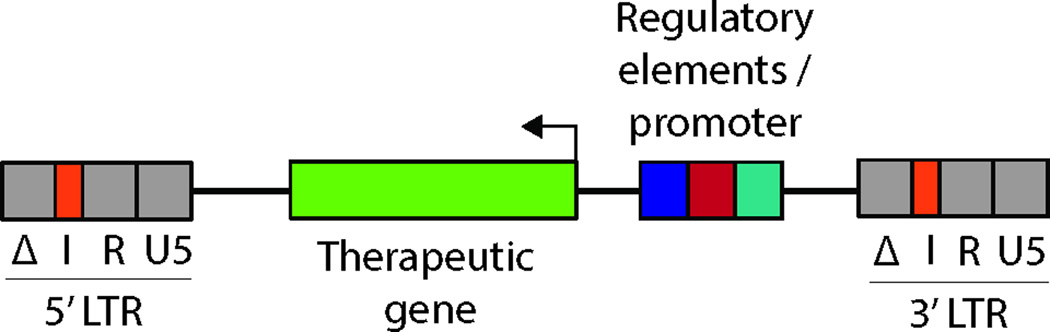
Simplified vector scheme illustrating a commonly used organization of a lentiviral vector for gene therapy. The vector contains the therapeutic gene of interest and transcriptional regulatory elements driving its expression. These elements are in reverse orientation with respect to viral transcription to avoid interference with viral processing, as is often the case in such vectors. The long terminal repeat region (LTR) is segmented into U3, R and U5 regions. For safety, the U3 repeat region is in a self-inactivating (SIN) configuration (Δ). An insulator element (I) has been introduced into the LTR to shield transcriptional effects of endogenous and exogenous chromatin elements after integration.
A number of β-globin gene-containing lentiviral vectors were subsequently shown to correct SCD and β-thalassemia in mouse models [Levasseur et al., 2003; May et al., 2000; Pawliuk et al., 2001] and in human HSPCs transplanted into immunodeficient mice [Breda et al., 2012; Puthenveetil et al., 2004]. A recent study reported the lentiviral transduction of human bone marrow (BM) derived HSPCs from SCD donors to achieve exogenous expression of the human β-globin gene, which prevented sickling of differentiated erythroid cells [Romero et al., 2013]. The anti-sickling β-globin gene activity was retained for at least three months following transplantation into immunodeficient mice, demonstrating a sustained therapeutic effect. Several groups have also evaluated the utility of synthetic β-globins or the use of γ-globin for therapeutic purposes. β-globin variants carrying substitutions at E22A and T87Q have potent anti-sickling properties by affecting axial and lateral contacts essential for the formation of linear HbS polymers [McCune et al., 1994; Negre et al., 2016]. The application of γ-globin vectors stems from the observation of potent anti-sickling effects of fetal hemoglobin (HbF, α2γ2), which ameliorates disease morbidity and mortality in SCD and β-thalassemia syndromes [Musallam et al., 2012; Perumbeti et al., 2009; Pestina et al., 2009; Platt et al., 1994; Wilber et al., 2011]. In fact, patients with either SCD or β-thalassemia and hereditary persistence of fetal hemoglobin exhibit reduced disease severity and in some cases are entirely asymptomatic [Musallam et al., 2012; Sankaran, 2011; Sankaran and Orkin, 2013]. In addition to a direct benefit of replacing defective or dysfunctional β-globin chains in the hemoglobinopathies, HbF also allows for increased RBC survival, so such replacement strategies result in additional alleviation of pathogenesis.
The expression of fetal hemoglobin is primarily confined to the gestational period, after which it is replaced by adult hemoglobin during the first several months of life [Sankaran and Nathan, 2010]. Consequently, identifying the genetic factors contributing to persistent fetal hemoglobin levels and the mechanisms related to the developmental switch of the hemoglobin genes have been areas of intense investigation over many decades. A major discovery came from follow up of genome-wide association studies (GWAS) that identified common polymorphisms associated with fetal hemoglobin levels in the BCL11A gene [Lettre et al., 2008; Menzel et al., 2007; Uda et al., 2008]. Although expressed in the brain and in lymphocytes, there was no known role for BCL11A in erythroid development. Seminal work showed that BCL11A was a major transcriptional regulator of the switch from fetal γ- to adult β-globin expression, and knockdown of BCL11A significantly increased HbF expression without altering erythroid development [Sankaran et al., 2008; Sankaran et al., 2009]. As expected, deletion of Bcl11a in a transgenic mouse model of SCD corrected the associated hematologic and pathologic defects through induction of HbF [Xu et al., 2011]. Importantly, three patients with heterozygous microdeletions of chromosome 2p15p16 encompassing BCL11A presented with developmental delay and autism spectrum disorder, but they had no signs of compromised hematologic and immunologic function and exhibited persistence of HbF expression with levels ranging from 16–30% [Basak et al., 2015]. Subsequent follow-up studies of additional patients with BCL11A deletions or mutations have supported these findings [Dias et al., 2016; Funnell et al., 2015]. These findings support the rationale for partial suppression and hematopoietic-specific therapeutic targeting of BCL11A to treat the hemoglobinopathies, as complete deletion of Bcl11a has been shown to severely affect murine lymphoid development [Yu et al., 2012]. To avoid potential side effects in non-erythroid cells by short hairpin RNA (shRNA) mediated knockdown of BCL11A, erythroid-specific expression cassettes have been developed for use in potential gene therapy vectors [Guda et al., 2015]. The delivery of such shRNAs using SIN lentiviral vectors resulted in HbF induction to levels more than 50% of total hemoglobin in human erythroid cultures, although further studies of such vectors are needed. Furthermore, RNA polymerase II-driven shRNAs appeared to be less cytotoxic than RNA polymerase III-based vectors – perhaps due to lower overall expression levels of the shRNAs – implying possible preferences for using polymerase II-based strategies in clinical settings more generally [Brendel et al., 2016; Guda et al., 2015]. Further strategies may involve the targeting of erythroid-specific regulatory elements of BCL11A instead of coding sequences, an approach discussed in more detail below. Currently, a number of clinical trials utilizing various vector systems aiming to introduce globin variants, as well as approaches to induce HbF for the treatment of SCD and β-thalassemia by targeting BCL11A, are open or in preparation and have recently been reviewed in detail [Archer et al., 2015; Hoban et al., 2016b].
Recently, several additional potential targets for HbF induction have emerged. The leukemia/lymphoma-related factor (LRF), a transcription factor that is encoded by the ZBTB7A gene, has emerged as another potential target. Disruption of ZBTB7A induces HbF in primary human erythroblasts [Masuda et al., 2016]. LRF and BCL11A suppress γ-globin via potentially distinct mechanisms, and concomitant knockout of both genes results in higher fetal/adult globin ratios in erythroid cell lines than knockout of each gene alone [Masuda et al., 2016]. However, LRF is necessary for normal erythropoiesis as well as lymphocyte production, so it is unlikely to be an ideal therapeutic target [Maeda et al., 2007; Sakurai et al., 2011]. An additional potential target is the hematopoietic transcription factor MYB, whose suppression in erythroid progenitors is associated with elevated HbF [Galarneau et al., 2010; Lettre et al., 2008; Sankaran et al., 2011; Stadhouders et al., 2014]. Furthermore, common variants in the hematopoietic enhancers of this gene significantly affect not only on HbF levels, but also the clinical course of SCD and β-thalassemia, suggesting potential beneficial roles of these variants, which warrant more detailed study [Sankaran et al., 2013; Sankaran and Orkin, 2013; Stadhouders et al., 2014]. Rare mutations in the erythroid transcription factor KLF1 have also been identified in individuals with elevated HbF. However, KLF1 mutations in humans can cause variable phenotypes, including severe forms of anemia, and are not always associated with elevated HbF levels [Arnaud et al., 2010; Borg et al., 2010; Giardine et al., 2011; Sankaran and Orkin, 2013; Satta et al., 2011]. Another strategy for HbF induction has been the forced chromatin looping by tethering the self-association domain of Ldb1 to an artificial zinc finger protein targeting the γ-globin promoter [Deng et al., 2014]. In primary adult human erythroblasts, this resulted in increased production of γ-globin mRNA and HbF with a concomitant reduction in adult β-globin expression. It remains to be investigated whether this strategy and its therapeutic effect may be applicable in the long-term in vivo.
Modulation of the expression of the endogenous δ-globin gene may present an alternative strategy to increase the level of hemoglobin A2 consisting of two α- and two δ-globin chains (HbA-2, α2δ2). HbA2 is physiologically expressed at low levels in adults and could be a potential substitute for HbA in the hemoglobinopathies. In a proof-of-principle experiment, the activation of δ-globin in transgenic mice led to a robust improvement of hematologic parameters in β-thalassemic mice [Manchinu et al., 2014]. Therefore, it is possible that this may be an alternative strategy to replace either defective or dysfunctional adult β-globin production.
An alternative approach for the β-thalassemia syndromes may be the targeted reduction of α-globin expression to restore the quantitative balance in globin chain production, thereby reducing pathologic effects of excess α-globin aggregates [Mettananda et al., 2015]. This rationale is supported by the observation that the number of functional α-globin genes has direct clinical consequences as exemplified by the beneficial effects of co-inherited α-thalassemia mutations in β-thalassemia patients [Kan and Nathan, 1970; Thein, 2008] and a more severe clinical phenotype upon increased copy number of the α-globin gene cluster [Premawardhena et al., 2005; Sollaino et al., 2009]. As anticipated, in proof-of-principle experiments, RNA interference strategies that reduce the amount of α-globin mRNAs have resulted in phenotypic improvement in murine models of β-thalassemia [Voon et al., 2008; Xie et al., 2011; Xie et al., 2007].
The efficacy of gene therapy approaches has also been evaluated in murine models for other congenital anemias, including Diamond-Blackfan anemia (DBA). DBA is characterized by an absence of erythroid precursors in the bone marrow and various developmental anomalies [Vlachos et al., 2008]. In 50–60% of cases, DBA is caused by heterozygous loss of function mutations in one of sixteen ribosomal proteins, with ribosomal protein S19 (RPS19) being mutated in ~25% of affected individuals [Danilova and Gazda, 2015]. Using lentiviral vectors, Jaako and colleagues restored ribosomal protein S19 (Rps19) expression, rescuing the anemia in an Rps19 RNA interference knockdown mouse model of DBA [Jaako et al., 2014]. However, some ribosomal protein gene lesions have only been identified in a fraction of patients, and while the development of appropriate gene therapy vectors is possible, their testing of efficacy and safety is laborious and may not be suitable for a very limited number of patients. Recently, the altered translation of GATA1 mRNA has been identified as a common defect downstream of the knockdown of various ribosomal protein genes mutated in DBA [Ludwig et al., 2014]. Although it has also been suggested that the erythroid defect may occur as a result of excessive p53 activation [Ball, 2011; Boultwood et al., 2012; Ellis, 2014], the reduced production of the erythroid-essential transcription factor GATA1 appears to play a fundamental role with respect to the specificity of the observed defect, and rarely, GATA1 mutations are sufficient to cause DBA [Sankaran et al., 2012; Weiss et al., 2012]. Accordingly, lentiviral mediated expression of GATA1 in patient cells with ribosomal protein haploinsufficiency partially restored erythroid differentiation [Ludwig et al., 2014]. Thus, gene therapy approaches aimed at increasing the expression of GATA1 may provide a promising alternative and would be amenable to a large proportion of patients with DBA, independent of their genetic lesion. Erythroid-specific expression of GATA1 is particularly important as ubiquitous expression can promote the differentiation of HSCs with concomitant loss of self-renewal activity [Gentner et al., 2010; Iwasaki et al., 2003]. Gene expression within the HSC compartment may be prevented by the incorporation of microRNA-126 target sequences into GATA1 gene expression vectors. MicroRNAs present a class of short noncoding RNAs that bind to mRNAs and control their gene expression at the post-transcriptional level. MicroRNA-126 is highly expressed in HSCs but quickly downregulated during the early stages of hematopoietic differentiation and this strategy has successfully suppressed ectopic gene expression in HSCs, while maintaining expression in differentiated cells [Gentner et al., 2010]. Furthermore, erythroid-specific regulatory elements for GATA1 expression have already been identified in mice and provide a framework for lineage-restricted expression of GATA1 in hematopoietic cells [Shimizu et al., 2013; Takai et al., 2013]. In the future, unbiased screening approaches, such as massively parallel reporter assays, may have value for identifying regulatory elements with desired expression properties to allow for regulated delivery of such transgenes [Ulirsch et al., 2016].
Fanconi anemia is a rare bone marrow failure disorder (affecting all blood lineages) with patients developing varying degrees of hematologic disease, including anemia, other cytopenias, myelodysplastic syndrome, and acute myeloid leukemia [Kee and D'Andrea, 2012]. Mutations in twenty-one different genes have been identified, with FANCA being the most commonly mutated (in up to 60% of patients) [Bluteau et al., 2016; Rio et al., 2014]. All gene products appear to function in a common pathway, the Fanconi anemia pathway, which is primarily involved in double stranded DNA repair by homologous recombination. Consequently, patients have an increasing predisposition to cancer with age. Lentiviral-mediated gene therapy in Fanca deficient mice efficiently corrected the phenotype without any sign of toxicity [Molina-Estevez et al., 2015] and human gene therapy trials are currently underway [EuroFancolen, 2015; Tolar et al., 2012].
Numerous metabolic defects in erythroid cells have been described and gene replacement strategies have proven to be an effective means of permanent correction in models of these disorders. Glucose-6-phosphate dehydrogenase (G6PD) is a housekeeping gene expressed in all cells and deficiency of its activity is a highly prevalent condition in humans [Luzzatto et al., 2016]. G6PD is part of the pentose phosphate pathway and plays a major role in NADPH production and resistance to oxidative stress in erythrocytes, which lack mitochondria to regenerate antioxidants. Most mutations in G6PD are mild and only become clinically apparent upon challenge of the patient with oxidant drugs or foods. However, some mutations cause severe instability or reduction in gene expression, which therefore lead to lifelong chronic hemolytic anemia [Mason et al., 1995]. Early proof-of-principle experiments have shown stable retroviral transduction of murine and human HSCs with human G6PD in a mouse transplant model and phenotypic correction in G6PD deficient embryonic stem cells [Rovira et al., 2000]. Pyruvate kinase (PK) deficiency is a chronic hemolytic anemia due to mutations in the PKLR gene. PKLR catalyzes the last ATP-generating reaction in the glycolytic pathway [Zanella et al., 2005]. Mature erythrocytes only express the R-type-specific isoform, so any loss of PKLR activity results in reduced ATP formation and increased metabolic and mechanical vulnerability. The resulting hemolytic anemia may range from mild to life threatening in severe cases. Successful gene replacement therapy has recently been demonstrated in a mouse model of PK deficiency [Garcia-Gomez et al., 2016]. Using a lentiviral vector expressing the human PKLR cDNA, Garcia-Gomez and colleagues transduced murine PK-deficient HSCs and showed functional correction of the glycolytic pathway and normalization of the hematologic phenotype.
Congenital erythroid porphyria results from the deficiency in uroporphyrinogen III synthase (UROS), the fourth enzyme of the heme biosynthetic pathway [Di Pierro et al., 2016]. The enzyme defect results in the accumulation of toxic porphyrin metabolites, which cause cutaneous photosensitivity, photo-mutilation, and chronic hemolytic anemia. Mice carrying homozygous mutations in Uros closely mimic the symptoms observed in patients, and lentiviral-mediated transfer of UROS cDNA into mouse HSCs resulted in a complete and long-term enzymatic, metabolic, and phenotypic correction of the disease, favored by a survival advantage of corrected red blood cells [Robert-Richard et al., 2008]. Together, these reports underscore the utility and feasibility of gene therapy approaches in the treatment of various congenital forms of anemia.
Genome editing tools and applications in congenital anemias
The correction of a gene at its endogenous locus presents an appealing solution for the treatment of blood disorders and there have been considerable advances in the field of genome editing over the past years. However, precise editing while completely avoiding the creation of unintended mutations, such as insertions or deletions (indels), remains a major challenge. Current genome editing approaches rely on the expression of a nuclease to induce specific double-stranded DNA breaks (DSBs) that trigger homologous recombination and/or non-homologous end-joining (NHEJ) that leads to the formation of indels [Kim and Kim, 2014] (Fig 3). The process of homologous recombination may be directed by the introduction of a donor template to insert a desired sequence leading to homology-directed repair (HDR) at the endogenous locus, or at a so-called safe harbor locus in a specific region of the genome [Cox et al., 2015; Genovese et al., 2014; Hsu et al., 2014; Kim and Kim, 2014; Sadelain et al., 2012; Sankaran and Weiss, 2015]. While NHEJ occurs throughout the cell cycle, the activity of the HDR machinery is primarily confined to the S and G2 phases [Mao et al., 2008; Orthwein et al., 2015; Saleh-Gohari and Helleday, 2004]. Considering that the most primitive repopulating HSCs with multi-lineage and long-term engraftment potential are largely quiescent, this presents a major hurdle for achieving therapeutically beneficial HDR-mediated editing efficiently, as these cells appear to preferentially repair DSBs by the error-prone NHEJ pathway [Genovese et al., 2014; Mohrin et al., 2010]. Some marginal improvements in homology-directed genome editing can be achieved through the use of rationally designed asymmetric donor DNA templates [Richardson et al., 2016], and modulation of the balance between the activation of HDR vs. NHEJ may also slightly increase precise editing efficiency [Chu et al., 2015; Maruyama et al., 2015; Pinder et al., 2015; Yu et al., 2015], but it remains unclear whether such manipulation of HSPCs may affect engraftment or repopulating capability. Nevertheless, NHEJ can be utilized to induce deletions or other disruptions in coding or regulatory regions, although nuclease off-targets effects must still be considered. Delivery of the nuclease and donor template to HSPCs can be enabled by the use of integrase-defective non-integrating lentiviral vectors (IDLV) or these products can be introduced via transient delivery of plasmids or nucleoprotein complexes [Cai et al., 2016; Giani et al., 2016; Mandal et al., 2014; Nightingale et al., 2006; Schumann et al., 2015; Shaw and Cornetta, 2014]. Consequently, the effect of altered gene expression and insertional oncogenesis due to the integration of foreign DNA would be minimized, as this remains a major concern for classical gene therapy viral-based approaches even with the use of safer SIN vectors [Aiuti et al., 2013; Hacein-Bey-Abina et al., 2008].
Figure 3.
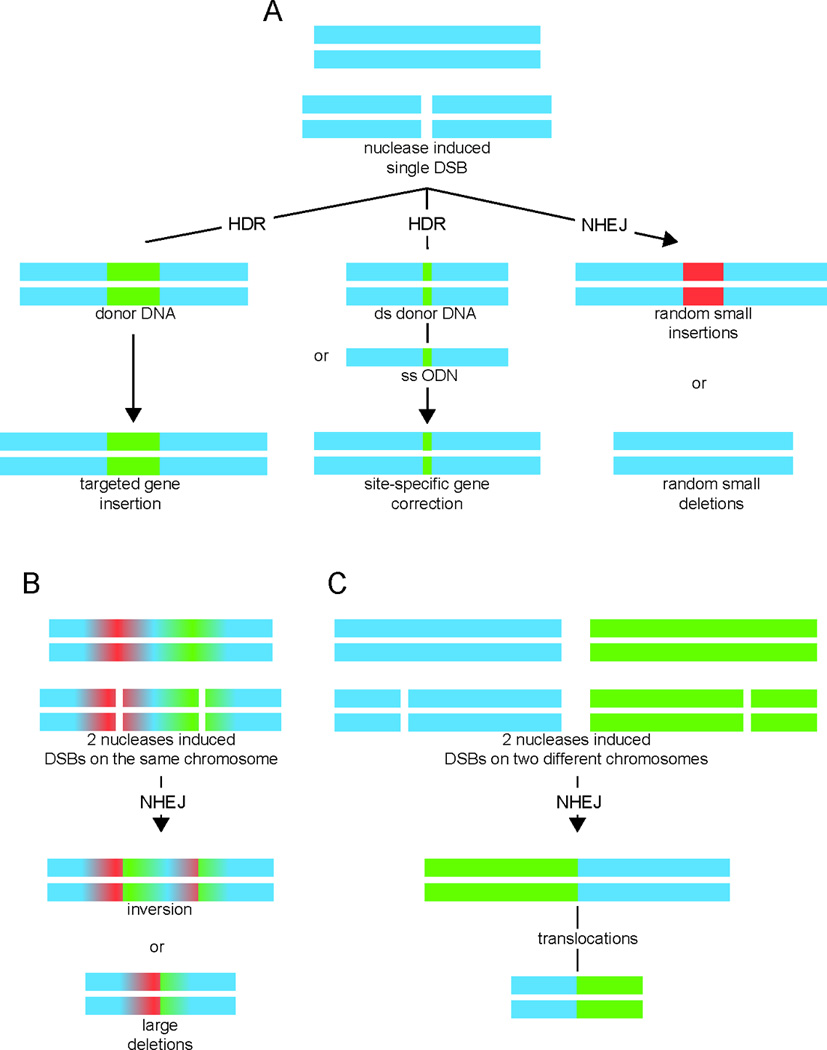
Nuclease-induced genome-editing. (A) Induction of a double strand break (DSB) by a nuclease either leads to non-homologous end joining (NHEJ) with indel formation (small random deletions or small random insertions), or results in homology-directed repair (HDR) after introduction of an either single-stranded oligodeoxynucleotide (ssODN) or a double-stranded donor DNA repair template containing the corrected sequence and flanking sequences that are homologous to the target locus. Alternatively, HDR may be utilized for targeted insertion of a gene, either at the endogenous locus or at a so-called safe harbor locus. (B) Induction of two DSBs by two nucleases on the same chromosome can result in inversion or large deletion of the flanking region. (C) Induction of two DSBs by two nucleases on different chromosomes may result in translocations. Figure has been modified from Kim and Kim [Kim and Kim, 2014].
Several classes of targeted nucleases are available in the genome editing toolkit and have recently been reviewed in detail [Cox et al., 2015; Gaj et al., 2013; Gupta and Musunuru, 2014; Kim and Kim, 2014]. These include meganucleases, zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and most recently the CRISPR/Cas systems (Table 1). Meganucleases are sequence-specific endonucleases that have relatively long target sites, 14–40 base pairs (bp), limiting their application due to the rare presence of target sequences at the majority of human genes [Cox et al., 2015; Hafez and Hausner, 2012; Paques and Duchateau, 2007]. Nonetheless, despite challenges, it is possible to engineer these meganucleases to target diverse sequences within the genome [Silva et al., 2011; Smith et al., 2006]. There are only limited examples of the use of meganucleases for gene correction related to congenital anemias or other blood disorders. For example, one study reported the correction of DCLRE1C deficiency in murine HSCs, albeit in a rather inefficient manner [Riviere et al., 2014].
Table 1.
Key features of the major classes of programmable nucleases
| Meganucleases | ZFNs | TALENs | CRISPR/Cas | |
|---|---|---|---|---|
| Nuclease | Homing endonucleases |
FokI | FokI | Cas9 |
|
Type of recognition |
Protein-DNA | Protein-DNA | Protein-DNA | RNA-DNA |
|
Recognition site length |
14–40 bp | 18–36 bp | 30–40 bp | ~20 bp |
|
Engineering complexity |
high | high | moderate | low |
|
Off-target effects |
low | high | low | variable |
| Size | ~1 kb | ~1 kb/ monomer |
~3 kb/ monomer |
~3.3 kb – ~4.2 kb |
ZFNs are site-specific endonucleases composed of a DNA-binding domain consisting of three to six tandem arrays of Cys2His2 zinc fingers, each able to specifically bind 3 bp of DNA, and a catalytic DNA-cleavage domain, typically derived from the endonuclease FokI [Scott, 2005]. Multiple zinc fingers can be incorporated to recognize a DNA target sequence up to 36 bp in length to increase specificity. DNA cleavage is then induced by hetero-dimerization of the nuclease domains of two independent ZFNs, each binding to adjacent sequences [Gaj et al., 2013; Perez-Pinera et al., 2012]. To avoid homo-dimerization of individual ZFNs resulting in potential non-specific cleavage, FokI variants have been designed that efficiently cleave DNA only when paired as a heterodimer with a second ZFN leading to improved specificity [Doyon et al., 2011; Miller et al., 2007; Szczepek et al., 2007]. Design, construction, and validation present a challenging process that prohibits the wider application of ZFNs [Ramirez et al., 2008]. Nevertheless, ZFNs have been successfully applied to target the CCR5 locus in HSPCs [Holt et al., 2010; Wang et al., 2015] and to correct genetic lesions in patient-specific induced pluripotent stem cells (iPSCs) with SCD and Fanconi anemia mutations [Rio et al., 2014; Sebastiano et al., 2011]. Correction in primary HSPCs from patients with SCD has also recently been reported [Hoban et al., 2015], although this process is rare and inefficient. Therefore, the low rate of successful gene editing presents a significant bottleneck for therapeutic purposes. For example, in SCD samples, the ZFN-mediated correction of HbS to HbA did not exceed 10%. This is consistent with a study that demonstrated site-specific gene correction in human HSPCs derived from patients with severe combined immune deficiency and showed particularly low efficiency of precise editing in quiescent HSCs [Genovese et al., 2014]. This presents a major obstacle for clinical applications, as HSPCs are the primary target for editing strategies.
TALENs comprise a customizable DNA-binding domain that allows for targeting any sequence of interest and a restriction endonuclease, FokI, to introduce DSBs [Cox et al., 2015; Joung and Sander, 2013; Kim and Kim, 2014]. The DNA-binding domain consists of highly conserved TALE repeats arranged in a tandem array. Each repeat is composed of 33–35 amino acids binding to a single base of DNA that is recognized by two hypervariable amino acid residues, usually at positions 12 and 13 of the repeat [Joung and Sander, 2013; Perez-Pinera et al., 2012]. Multiple TALE repeats can be designed in any order of interest and their targeted DNA sequences are typically 15–20 bp in length. Similar to ZFNs, the target DNA is cleaved after dimerization of the nuclease domains of two individual TALENs in the 12–21 bp spacer region between their respective recognition sites [Gaj et al., 2013; Joung and Sander, 2013]. Hybrid nucleases known as megaTAL combining a TALE DNA binding domain with an engineered, sequence-specific meganuclease may promote even more efficient cleavage with increased sequence specificity and improved delivery [Boissel et al., 2014]. Several groups have utilized TALENs in erythroid cell lines and in SCD and β-thalassemia PSC lines to mediate site-specific correction or targeted integration of β-globin at its endogenous locus [Sun and Zhao, 2014; Voit et al., 2014; Xu et al., 2015]. TALEN-mediated homologous recombination has also been used for therapeutic editing of pyruvate kinase deficiency in PSCs with subsequent generation of a high number of erythroid cells from these edited cells [Garate et al., 2015].
TALENs and ZFNs have been utilized successfully to target erythroid-specific regulatory elements of BCL11A to induce fetal globin expression. Follow-up of GWAS signals led to the identification of an erythroid-specific enhancer of BCL11A [Bauer et al., 2013]. TALEN directed targeting of this enhancer resulted in disruption of BCL11A expression in a mouse erythroleukemia cell line, with robust induction of embryonic globin expression. A subsequent study reported the deletion of the +55 kb and +58 kb DNase I–hypersensitive sites of the erythroid-specific BCL11A enhancer in primary human HSPCs using TALENs and ZFNs resulting in elevation of γ-globin mRNA and HbF levels [Vierstra et al., 2015]. Similarly, CRISPR/Cas9 mediated ablation of BCL11A expression has been achieved by targeting its erythroid-specific enhancer in human hematopoietic cells, including primary HSPCs, which robustly induced HbF expression [Canver et al., 2015].
The CRISPR/Cas endonuclease systems rely on the guidance of a small RNA to their target DNA site, which distinguishes it from meganucleases, ZFNs, and TALENs, all of which rely on protein-DNA interactions (Table 1) [Cox et al., 2015; Doudna and Charpentier, 2014]. In the CRISPR/Cas systems a 20 bp single guide RNA (sgRNA) complementary to the target DNA sequence and the presence of a downstream protospacer adjacent motif (PAM) sequence, NGG, are sufficient to allow cleavage by the Cas9 nuclease. This feature of RNA-guided genome targeting allows CRISPR/Cas systems to be incredibly versatile in targeting numerous regions of the genome without the need to engineer DNA binding proteins [Cox et al., 2015; Doudna and Charpentier, 2014; Hsu et al., 2014; Sander and Joung, 2014]. Modification of this system by using Cas9 nickase mutants to introduce single strand DNA breaks (nicks), in combination with using paired sgRNAs to introduce multiple targeted breaks in the desired locus, results in particularly high specificity and significant reduction of off-target effects [Ran et al., 2013b]. Over the past few years, multiple CRISPR/Cas systems with various characteristics have been described and will inevitably further advance genome editing applications [Cong et al., 2013; Ran et al., 2015; Zetsche et al., 2015].
An initial effort to assess the editing efficiency of the CRISPR/Cas9 system in HSPCs reported highly efficient CCR5 ablation with the use of a single guide RNA, which could be improved further with a dual sgRNA approach, achieving biallelic gene deletion rates of ~27% with minimal detectable off-target mutations [Mandal et al., 2014]. The edited HSPCs retained multi-lineage potential in vitro and in vivo upon transplantation in immunodeficient mice. Similarly, CRISPR/Cas9 has also been successfully applied for gene correction by HDR in congenital anemias, including β-thalassemia and SCD patient-derived primary hematopoietic cells, iPSCs and in fibroblasts from patient with Fanconi anemia [Hoban et al., 2016a; Huang et al., 2015; Niu et al., 2016; Osborn et al., 2015; Xie et al., 2014; Xu et al., 2015]. Recently, CRISPR/Cas9-mediated genome editing of a promoter region in the γ-globin gene recapitulated a naturally occurring mutation in people with hereditary persistence of fetal hemoglobin and lead to the production of RBCs with modest elevation of HbF [Traxler et al., 2016]. However, the authors of this work did not comprehensively ascertain the range of indels that may form, particularly if larger deletions occurred, and additionally how effective this strategy is in engraftable HSCs was not tested.
These advances emphasize the promise of genome editing approaches utilizing autologous cells to overcome limitations in allogeneic transplantation for the treatment of congenital anemias. Currently, the efficiency of precise genome editing remains the major bottleneck for site-specific gene correction in HSPC. Furthermore, DNA breaks themselves, such as those created by the vast majority of genome editing approaches, may compromise HSC function and have prompted the development of new nucleotide editing tools that may allow more precise genome modifications without the creation of DNA breaks [Komor et al., 2016]. It is important to note that in many instances, complete correction of all HSPCs will not be required. Even at a smaller frequency, gene-corrected cells would exhibit a selective survival advantage to fill the previously defective erythropoietic niche and increase RBC lifespan to allow for a clinically relevant benefit [Sankaran and Weiss, 2015]. However, at this time HSPCs cannot be expanded indefinitely in vitro while maintaining long-term repopulation and multi-lineage capabilities [Boitano et al., 2010; Fares et al., 2014]. Analogous to classical gene therapy approaches, this significantly hampers the identification and enrichment of site-specific corrected cells and prohibits reliable verification of editing and screening of adverse off-target effects. These limitations of HSPCs prompted the exploration of other potential target cell types for genetic manipulation and correction.
The use of pluripotent stem cells for cell therapies
The ability of pluripotent stem cells (PSCs) to differentiate into cell types derived from any of the three germ layers has made them an attractive cellular target for the purposes of regenerative medicine. Recent advances in the derivation of PSCs, including human embryonic stem cells and induced PSCs (iPSCs), have opened up many new possibilities for the study of human disease and potential cellular therapies [Chen et al., 2014; Takahashi et al., 2007]. Their capacity to expand and maintain pluripotency in vitro offers a potentially unlimited supply for screening and selecting gene-edited clones and has successfully been demonstrated for various congenital anemias, including SCD, β-thalassemia, and PK deficiency [Garate et al., 2015; Sebastiano et al., 2011; Xu et al., 2015; Zou et al., 2011]. Furthermore, mature erythroid cells have been successfully derived from PSCs and may provide a means to overcome deficits in the blood supply for transfusion purposes [Giani et al., 2016]. Despite the potential of PSCs, their application for curative cellular therapies remains restricted by the insufficient generation of fully functional HSCs from PSCs (Fig 1B) [Daniel et al., 2016; Kim and Sankaran, 2016; Vo and Daley, 2015]. The generation of HSCs by mimicking the natural differentiation pathway has long been considered the optimal route. However, although hematopoietic differentiation of PSCs has been found to mirror aspects of in vivo embryonic development, current efforts have been unable to produce the third hematopoietic wave of ontogeny that gives rise to HSCs [Nandakumar et al., 2016; Yoder, 2014]. The first hematopoietic wave, commonly known as the primitive wave, arises in the yolk sac at embryonic day 7.5 (E7.5) of murine development and transiently produces primitive erythroblasts and primitive macrophages and megakaryocytes. A second wave of yolk sac hematopoiesis is characterized by the emergence of erythro-myeloid progenitors (EMPs) at approximately E8.25 that also colonize the fetal liver. EMPs arise from vascular endothelial cells that are present in blood island capillaries of the yolk sac and give rise to erythro-myeloid and lymphocyte progenitor cells. The second wave is considered “definitive” because the produced blood cells display adult-type features and functions, but this wave does not produce HSCs or other progenitors capable of long-term engraftment. The third wave begins at E10.5 and is characterized by the emergence of HSCs from a unique population of endothelial cells, termed the hemogenic endothelium (HE) that can be found in dorsal aorta of the aorta-gonad-mesonephros region and within the arteries found in the umbilical, vitelline, cranial, yolk sac, and placental regions [Yoder, 2014]. These HSCs migrate to the fetal liver before ultimately seeding the bone marrow and giving rise to all blood lineages; they are capable of long-term engraftment after transplantation. To date, protocols for PSC differentiation have only been able to produce the first two waves of hematopoiesis successfully and do not fully recapitulate certain blood disorders, such as the hemoglobinopathies, since the adult β-globin gene is generally poorly expressed [Yang et al., 2014].
Initial experiments successfully generated hematopoietic progenitors by directed differentiation from embryoid bodies, three-dimensional aggregates of PSCs, but their multi-lineage potential and engraftment capacities were limited as these cells primarily exhibited characteristics of the first two hematopoietic waves, as we note above [Kaufman et al., 2001; Keller et al., 1993; Vodyanik et al., 2005]. Further research led to significant progress in the identification of guiding factors and redefined PSC differentiation protocols through HE to derive hematopoietic cells with multi-lineage-potential, but yet have failed to generate genuine HSCs [Boiers et al., 2013; Ditadi et al., 2015; Dzierzak and Speck, 2008; Gori et al., 2015; Kennedy et al., 2012; Ramos-Mejia et al., 2010; Sturgeon et al., 2014].
Recently, the generation of HSCs from human PSCs has been reported through the formation of teratomas in immunodeficient mice [Amabile et al., 2013; Suzuki et al., 2013]. Teratoma-derived CD34+ cells were capable of reconstitution and multi-lineage engraftment upon xenotransplantation. These studies provide a proof-of-principle that PSC-derived human hematopoietic cells with repopulating and reconstituting potential can possibly be obtained, although the described tumor-forming approaches are not suitable for potential clinical applications and certainly cannot be efficiently scaled to generate HSCs reliably from PSCs. The identification of intrinsic and extrinsic factors necessary for the production of HSCs presents a significant challenge in the use of PSCs for cellular therapeutic applications in hematopoietic disorders.
Direct conversion of cellular identities by transcription factor mediated reprogramming without going trough a pluripotent stage has been demonstrated for various somatic cell types and presents an alternative source for the generation of gene-corrected cells for therapeutic purposes [Vo and Daley, 2015] (Fig 1B). Several studies have shown the instructive roles of lineage-specific transcription factors like GATA1, CEBPA, CEBPB, and SPI1 in the direct conversion between hematopoietic cell types [Iwasaki et al., 2003; Laiosa et al., 2006; Xie et al., 2004] and from non-hematopoietic to hematopoietic cell types [Capellera-Garcia et al., 2016]. These approaches may be improved further to increase cellular yield when combined with additional methods of manipulating hematopoietic cytokine signaling [Giani et al., 2016]. The generation of hematopoietic progenitor cells from human and murine fibroblasts and endothelial cells has been demonstrated, but to date show limited self-renewal capacity, engraftment potential, or multi-lineage differentiation potential [Batta et al., 2014; Sandler et al., 2014; Szabo et al., 2010]. However, a similar reprogramming approach aided in the identification of a murine hemogenic precursor cell in vivo capable of maturation into potential bona fide HSCs [Pereira et al., 2016; Pereira et al., 2013]. The first study to successfully generate murine HSCs screened the conversion capability of 36 hematopoietic factors in mature hematopoietic cells to generate engraftable hematopoietic cells [Riddell et al., 2014]. The authors identified a cocktail of 8 transcription factors (Run1t1, Hlf, Lmo2, Prdm5, Pbx1, Zfp37, Mycn and Meis1) to endow lymphoid and myeloid cells with the capacity for multi-lineage engraftment and reconstitute hematopoiesis in serial transplantation. However, it remains unclear to what extent successful reprogramming and maintenance of induced HSCs relied on in vivo cues after the transplantation of transduced cells, currently limiting this approach for potential clinical translation. Whether the same factors can similarly reprogram human hematopoietic cells also remains to be investigated.
Direct conversions from PSCs have also been attempted using a variety of transcription factors, and these manipulations have generated HE and hematopoietic progenitor cell populations, but not yet functional HSCs [Doulatov et al., 2013; Elcheva et al., 2014; Elcheva et al., 2015; Ran et al., 2013a]. While significant progress has been made in the use of PSCs, further studies will be required to optimize transcription factor combinations and to identify additional (extrinsic) factors to refine the process of directed differentiation and direct conversion from pluripotent and readily obtainable somatic cells to generate autologous HSCs. The comprehensive assessment of their functionality, measured by their ability to fully repopulate a recipient upon transplantation and direct molecular comparison of their transcriptional and epigenomic profiles to native HSCs, present fundamental hurdles before their clinical use can be considered. However, if such cells could be obtained, and given that PSCs can readily be manipulated with genome editing tools to enable precise modifications and clonally selected [Giani et al., 2016], this would create an incredibly valuable source of autologous cell therapies to treat congenital forms of anemia.
Concluding remarks
Congenital forms of anemia are a genetically heterogeneous group of disorders that result in significant morbidity and mortality worldwide. At the current time, allogeneic transplantation of HSPCs from a healthy donor is the only cure available. Numerous advances in the fields of gene therapy and genome editing have paved the way to genetically correct patient cells and enable an autologous form of cell therapy with numerous pre-clinical and clinical studies in progress. Further refinement in genome editing tools with improved specificity paired with more elaborate cell culture protocols for HSPCs and PSCs may ultimately enable new curative options for patients with a variety of blood disorders. Despite these promising developments, many congenital forms of anemia are particularly common in resource-poor countries, and most cell and gene therapy approaches may therefore benefit only a minority of patients worldwide. Unless technical advances allow these therapies to be more readily applied, there remains a substantial need to pursue other strategies, such as the development of small molecules. Nonetheless, the insight gained from clinical studies of gene and cell therapy may provide insight into the development of other more readily distributable therapeutics. Thus, major challenges remain and will require the combined efforts of basic scientists and clinical investigators to safely advance these strategies to the clinic.
Acknowledgments
We apologize to all the authors whose work we were unable to specifically discuss due to space limitations. We thank all Sankaran laboratory members for their valuable feedback. This work was funded by NIH grants R01 DK103794, R33 HL120791, and U01 HL117720 (to V.G.S.).
Biographies
Leif S. Ludwig completed his medical studies at the Charité Universitätsmedizin Berlin, Germany and graduated with his Ph.D. in Biochemistry at the Freie Universität Berlin, Germany. He is currently working as a postdoctoral researcher studying hematology and genetics at Boston Children’s Hospital and the Broad Institute.
Rajiv K. Khajuria is an M.D. Ph.D. student at the Charité Universitätsmedizin Berlin, Germany. He is currently conducting his thesis research studying congenital blood disorders at Boston Children’s Hospital and the Broad Institute.
Vijay G. Sankaran is an Assistant Professor of Pediatrics at Boston Children’s Hospital and Harvard Medical School, an Attending Physician in Hematology/ Oncology, and an Associate Member of the Broad Institute. His laboratory utilizes human genetics to understand normal blood production and how this process is perturbed in disease states.
References
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC, Bosticardo M, Evangelio C, Assanelli A, Casiraghi M, Di Nunzio S, Callegaro L, Benati C, Rizzardi P, Pellin D, Di Serio C, Schmidt M, Von Kalle C, Gardner J, Mehta N, Neduva V, Dow DJ, Galy A, Miniero R, Finocchi A, Metin A, Banerjee PP, Orange JS, Galimberti S, Valsecchi MG, Biffi A, Montini E, Villa A, Ciceri F, Roncarolo MG, Naldini L. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile G, Welner RS, Nombela-Arrieta C, D'Alise AM, Di Ruscio A, Ebralidze AK, Kraytsberg Y, Ye M, Kocher O, Neuberg DS, Khrapko K, Silberstein LE, Tenen DG. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121:1255–1264. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer N, Galacteros F, Brugnara C. 2015 Clinical trials update in sickle cell anemia. Am J Hematol. 2015;90:934–950. doi: 10.1002/ajh.24116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud L, Saison C, Helias V, Lucien N, Steschenko D, Giarratana MC, Prehu C, Foliguet B, Montout L, de Brevern AG, Francina A, Ripoche P, Fenneteau O, Da Costa L, Peyrard T, Coghlan G, Illum N, Birgens H, Tamary H, Iolascon A, Delaunay J, Tchernia G, Cartron JP. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet. 2010;87:721–727. doi: 10.1016/j.ajhg.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Stocker J. The trials and hopes for drug development in sickle cell disease. Br J Haematol. 2015;170:768–780. doi: 10.1111/bjh.13548. [DOI] [PubMed] [Google Scholar]
- Ball S. Diamond Blackfan anemia. Hematology Am Soc Hematol Educ Program. 2011;2011:487–491. doi: 10.1182/asheducation-2011.1.487. [DOI] [PubMed] [Google Scholar]
- Basak A, Hancarova M, Ulirsch JC, Balci TB, Trkova M, Pelisek M, Vlckova M, Muzikova K, Cermak J, Trka J, Dyment DA, Orkin SH, Daly MJ, Sedlacek Z, Sankaran VG. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. J Clin Invest. 2015;125:2363–2368. doi: 10.1172/JCI81163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batta K, Florkowska M, Kouskoff V, Lacaud G. Direct reprogramming of murine fibroblasts to hematopoietic progenitor cells. Cell Rep. 2014;9:1871–1884. doi: 10.1016/j.celrep.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Bartolomae CC, Cesana D, Cartier N, Aubourg P, Ranzani M, Cesani M, Benedicenti F, Plati T, Rubagotti E, Merella S, Capotondo A, Sgualdino J, Zanetti G, von Kalle C, Schmidt M, Naldini L, Montini E. Lentiviral vector common integration sites in preclinical models and a clinical trial reflect a benign integration bias and not oncogenic selection. Blood. 2011;117:5332–5339. doi: 10.1182/blood-2010-09-306761. [DOI] [PubMed] [Google Scholar]
- Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, Dubois d'Enghien C, Bluteau O, Cuccuini W, Gachet S, Peffault de Latour R, Leblanc T, Socie G, Baruchel A, Stoppa-Lyonnet D, D'Andrea AD, Soulier J. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126:3580–3584. doi: 10.1172/JCI88010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiers C, Carrelha J, Lutteropp M, Luc S, Green JC, Azzoni E, Woll PS, Mead AJ, Hultquist A, Swiers G, Perdiguero EG, Macaulay IC, Melchiori L, Luis TC, Kharazi S, Bouriez-Jones T, Deng Q, Ponten A, Atkinson D, Jensen CT, Sitnicka E, Geissmann F, Godin I, Sandberg R, de Bruijn MF, Jacobsen SE. Lymphomyeloid contribution of an immune-restricted progenitor emerging prior to definitive hematopoietic stem cells. Cell Stem Cell. 2013;13:535–548. doi: 10.1016/j.stem.2013.08.012. [DOI] [PubMed] [Google Scholar]
- Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, Shendure J, Stoddard BL, Certo MT, Baker D, Scharenberg AM. megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering. Nucleic Acids Res. 2014;42:2591–2601. doi: 10.1093/nar/gkt1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, Schultz PG, Cooke MP. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329:1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg J, Papadopoulos P, Georgitsi M, Gutierrez L, Grech G, Fanis P, Phylactides M, Verkerk AJ, van der Spek PJ, Scerri CA, Cassar W, Galdies R, van Ijcken W, Ozgur Z, Gillemans N, Hou J, Bugeja M, Grosveld FG, von Lindern M, Felice AE, Patrinos GP, Philipsen S. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boultwood J, Pellagatti A, Wainscoat JS. Haploinsufficiency of ribosomal proteins and p53 activation in anemia: Diamond-Blackfan anemia and the 5q- syndrome. Adv Biol Regul. 2012;52:196–203. doi: 10.1016/j.advenzreg.2011.09.008. [DOI] [PubMed] [Google Scholar]
- Breda L, Casu C, Gardenghi S, Bianchi N, Cartegni L, Narla M, Yazdanbakhsh K, Musso M, Manwani D, Little J, Gardner LB, Kleinert DA, Prus E, Fibach E, Grady RW, Giardina PJ, Gambari R, Rivella S. Therapeutic hemoglobin levels after gene transfer in beta-thalassemia mice and in hematopoietic cells of beta-thalassemia and sickle cells disease patients. PLoS One. 2012;7:e32345. doi: 10.1371/journal.pone.0032345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel C, Guda S, Renella R, Bauer DE, Canver MC, Kim YJ, Heeney MM, Klatt D, Fogel J, Milsom MD, Orkin SH, Gregory RI, Williams DA. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. J Clin Invest. 2016 doi: 10.1172/JCI87885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunn HF. Erythropoietin. Cold Spring Harb Perspect Med. 2013;3:a011619. doi: 10.1101/cshperspect.a011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Laustsen A, Zhou Y, Sun C, Anderson MV, Li S, Uldbjerg N, Luo Y, Jakobsen MR, Mikkelsen JG. Targeted, homology-driven gene insertion in stem cells by ZFN-loaded 'all-in-one' lentiviral vectors. Elife. 2016;5 doi: 10.7554/eLife.12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, Luc S, Kurita R, Nakamura Y, Fujiwara Y, Maeda T, Yuan GC, Zhang F, Orkin SH, Bauer DE. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature. 2015;527:192–197. doi: 10.1038/nature15521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellera-Garcia S, Pulecio J, Dhulipala K, Siva K, Rayon-Estrada V, Singbrant S, Sommarin MN, Walkley CR, Soneji S, Karlsson G, Raya A, Sankaran VG, Flygare J. Defining the Minimal Factors Required for Erythropoiesis through Direct Lineage Conversion. Cell Rep. 2016;15:2550–2562. doi: 10.1016/j.celrep.2016.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio A, Cassani B, Schmidt M, von Kalle C, Howe S, Thrasher AJ, Aiuti A, Ferrari G, Recchia A, Mavilio F. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood. 2007;110:1770–1778. doi: 10.1182/blood-2007-01-068759. [DOI] [PubMed] [Google Scholar]
- Chen KG, Mallon BS, McKay RD, Robey PG. Human pluripotent stem cell culture: considerations for maintenance, expansion, and therapeutics. Cell Stem Cell. 2014;14:13–26. doi: 10.1016/j.stem.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu VT, Weber T, Wefers B, Wurst W, Sander S, Rajewsky K, Kuhn R. Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol. 2015;33:543–548. doi: 10.1038/nbt.3198. [DOI] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21:121–131. doi: 10.1038/nm.3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel MG, Pereira CF, Lemischka IR, Moore KA. Making a Hematopoietic Stem Cell. Trends Cell Biol. 2016;26:202–214. doi: 10.1016/j.tcb.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015;8:1013–1026. doi: 10.1242/dmm.020529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rupon JW, Krivega I, Breda L, Motta I, Jahn KS, Reik A, Gregory PD, Rivella S, Dean A, Blobel GA. Reactivation of developmentally silenced globin genes by forced chromatin looping. Cell. 2014;158:849–860. doi: 10.1016/j.cell.2014.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Pierro E, Brancaleoni V, Granata F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br J Haematol. 2016;173:365–379. doi: 10.1111/bjh.13978. [DOI] [PubMed] [Google Scholar]
- Dias C, Estruch SB, Graham SA, McRae J, Sawiak SJ, Hurst JA, Joss SK, Holder SE, Morton JE, Turner C, Thevenon J, Mellul K, Sanchez-Andrade G, Ibarra-Soria X, Deriziotis P, Santos RF, Lee SC, Faivre L, Kleefstra T, Liu P, Hurles ME, Study DDD, Fisher SE, Logan DW. BCL11A Haploinsufficiency Causes an Intellectual Disability Syndrome and Dysregulates Transcription. Am J Hum Genet. 2016;99:253–274. doi: 10.1016/j.ajhg.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditadi A, Sturgeon CM, Tober J, Awong G, Kennedy M, Yzaguirre AD, Azzola L, Ng ES, Stanley EG, French DL, Cheng X, Gadue P, Speck NA, Elefanty AG, Keller G. Human definitive haemogenic endothelium and arterial vascular endothelium represent distinct lineages. Nat Cell Biol. 2015;17:580–591. doi: 10.1038/ncb3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Vo LT, Chou SS, Kim PG, Arora N, Li H, Hadland BK, Bernstein ID, Collins JJ, Zon LI, Daley GQ. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Vo TD, Mendel MC, Greenberg SG, Wang J, Xia DF, Miller JC, Urnov FD, Gregory PD, Holmes MC. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8:74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008;9:129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcheva I, Brok-Volchanskaya V, Kumar A, Liu P, Lee JH, Tong L, Vodyanik M, Swanson S, Stewart R, Kyba M, Yakubov E, Cooke J, Thomson JA, Slukvin I. Direct induction of haematoendothelial programs in human pluripotent stem cells by transcriptional regulators. Nat Commun. 2014;5:4372. doi: 10.1038/ncomms5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elcheva I, Brok-Volchanskaya V, Slukvin I. Direct Induction of Hemogenic Endothelium and Blood by Overexpression of Transcription Factors in Human Pluripotent Stem Cells. J Vis Exp. 2015:e52910. doi: 10.3791/52910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta. 2014;1842:765–768. doi: 10.1016/j.bbadis.2013.12.013. [DOI] [PubMed] [Google Scholar]
- EuroFancolen. Phase I/II Gene Therapy Trial of Fanconi Anemia Patients with a New Orphan Drug Consisting of a Lentiviral Vector Carrying the FANCA Gene: A Coordinated International Action (EuroFancolen) Hum Gene Ther Clin Dev. 2015;26:81–82. doi: 10.1089/humc.2015.2522. [DOI] [PubMed] [Google Scholar]
- Evans-Galea MV, Wielgosz MM, Hanawa H, Srivastava DK, Nienhuis AW. Suppression of clonal dominance in cultured human lymphoid cells by addition of the cHS4 insulator to a lentiviral vector. Mol Ther. 2007;15:801–809. doi: 10.1038/sj.mt.6300103. [DOI] [PubMed] [Google Scholar]
- Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, Csaszar E, Knapp DJ, Miller P, Ngom M, Imren S, Roy DC, Watts KL, Kiem HP, Herrington R, Iscove NN, Humphries RK, Eaves CJ, Cohen S, Marinier A, Zandstra PW, Sauvageau G. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014;345:1509–1512. doi: 10.1126/science.1256337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850–858. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funnell AP, Prontera P, Ottaviani V, Piccione M, Giambona A, Maggio A, Ciaffoni F, Stehling-Sun S, Marra M, Masiello F, Varricchio L, Stamatoyannopoulos JA, Migliaccio AR, Papayannopoulou T. 2p15-p16.1 microdeletions encompassing and proximal to BCL11A are associated with elevated HbF in addition to neurologic impairment. Blood. 2015;126:89–93. doi: 10.1182/blood-2015-04-638528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049–1051. doi: 10.1038/ng.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garate Z, Quintana-Bustamante O, Crane AM, Olivier E, Poirot L, Galetto R, Kosinski P, Hill C, Kung C, Agirre X, Orman I, Cerrato L, Alberquilla O, Rodriguez-Fornes F, Fusaki N, Garcia-Sanchez F, Maia TM, Ribeiro ML, Sevilla J, Prosper F, Jin S, Mountford J, Guenechea G, Gouble A, Bueren JA, Davis BR, Segovia JC. Generation of a High Number of Healthy Erythroid Cells from Gene-Edited Pyruvate Kinase Deficiency Patient-Specific Induced Pluripotent Stem Cells. Stem Cell Reports. 2015;5:1053–1066. doi: 10.1016/j.stemcr.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Gomez M, Calabria A, Garcia-Bravo M, Benedicenti F, Kosinski P, Lopez-Manzaneda S, Hill C, Del Mar Manu-Pereira M, Martin MA, Orman I, Vives-Corrons JL, Kung C, Schambach A, Jin S, Bueren JA, Montini E, Navarro S, Segovia JC. Safe and Efficient Gene Therapy for Pyruvate Kinase Deficiency. Mol Ther. 2016;24:1187–1198. doi: 10.1038/mt.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese P, Schiroli G, Escobar G, Di Tomaso T, Firrito C, Calabria A, Moi D, Mazzieri R, Bonini C, Holmes MC, Gregory PD, van der Burg M, Gentner B, Montini E, Lombardo A, Naldini L. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A, Schira G, Amendola M, Quattrini A, Martino S, Orlacchio A, Dick JE, Biffi A, Naldini L. Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy. Sci Transl Med. 2010;2:58ra84. doi: 10.1126/scitranslmed.3001522. [DOI] [PubMed] [Google Scholar]
- Giani FC, Fiorini C, Wakabayashi A, Ludwig LS, Salem RM, Jobaliya CD, Regan SN, Ulirsch JC, Liang G, Steinberg-Shemer O, Guo MH, Esko T, Tong W, Brugnara C, Hirschhorn JN, Weiss MJ, Zon LI, Chou ST, French DL, Musunuru K, Sankaran VG. Targeted Application of Human Genetic Variation Can Improve Red Blood Cell Production from Stem Cells. Cell Stem Cell. 2016;18:73–78. doi: 10.1016/j.stem.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardine B, Borg J, Higgs DR, Peterson KR, Philipsen S, Maglott D, Singleton BK, Anstee DJ, Basak AN, Clark B, Costa FC, Faustino P, Fedosyuk H, Felice AE, Francina A, Galanello R, Gallivan MV, Georgitsi M, Gibbons RJ, Giordano PC, Harteveld CL, Hoyer JD, Jarvis M, Joly P, Kanavakis E, Kollia P, Menzel S, Miller W, Moradkhani K, Old J, Papachatzopoulou A, Papadakis MN, Papadopoulos P, Pavlovic S, Perseu L, Radmilovic M, Riemer C, Satta S, Schrijver I, Stojiljkovic M, Thein SL, Traeger-Synodinos J, Tully R, Wada T, Waye JS, Wiemann C, Zukic B, Chui DH, Wajcman H, Hardison RC, Patrinos GP. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat Genet. 2011;43:295–301. doi: 10.1038/ng.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gori JL, Butler JM, Chan YY, Chandrasekaran D, Poulos MG, Ginsberg M, Nolan DJ, Elemento O, Wood BL, Adair JE, Rafii S, Kiem HP. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J Clin Invest. 2015;125:1243–1254. doi: 10.1172/JCI79328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guda S, Brendel C, Renella R, Du P, Bauer DE, Canver MC, Grenier JK, Grimson AW, Kamran SC, Thornton J, de Boer H, Root DE, Milsom MD, Orkin SH, Gregory RI, Williams DA. miRNA-embedded shRNAs for Lineage-specific BCL11A Knockdown and Hemoglobin F Induction. Mol Ther. 2015;23:1465–1474. doi: 10.1038/mt.2015.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RM, Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J Clin Invest. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, Clappier E, Caccavelli L, Delabesse E, Beldjord K, Asnafi V, MacIntyre E, Dal Cortivo L, Radford I, Brousse N, Sigaux F, Moshous D, Hauer J, Borkhardt A, Belohradsky BH, Wintergerst U, Velez MC, Leiva L, Sorensen R, Wulffraat N, Blanche S, Bushman FD, Fischer A, Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafez M, Hausner G. Homing endonucleases: DNA scissors on a mission. Genome. 2012;55:553–569. doi: 10.1139/g2012-049. [DOI] [PubMed] [Google Scholar]
- Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010;5:13. doi: 10.1186/1750-1172-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, Cost GJ, Mendel MC, Romero Z, Kaufman ML, Joglekar AV, Ho M, Lumaquin D, Gray D, Lill GR, Cooper AR, Urbinati F, Senadheera S, Zhu A, Liu PQ, Paschon DE, Zhang L, Rebar EJ, Wilber A, Wang X, Gregory PD, Holmes MC, Reik A, Hollis RP, Kohn DB. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood. 2015;125:2597–2604. doi: 10.1182/blood-2014-12-615948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, Lumaquin D, Kuo CY, Romero Z, Long J, Ho M, Young CS, Mojadidi M, Fitz-Gibbon S, Cooper AR, Lill GR, Urbinati F, Campo-Fernandez B, Bjurstrom CF, Pellegrini M, Hollis RP, Kohn DB. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol Ther. 2016a doi: 10.1038/mt.2016.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016b;127:839–848. doi: 10.1182/blood-2015-09-618587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, Crooks GM, Kohn DB, Gregory PD, Holmes MC, Cannon PM. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wang Y, Yan W, Smith C, Ye Z, Wang J, Gao Y, Mendelsohn L, Cheng L. Production of Gene-Corrected Adult Beta Globin Protein in Human Erythrocytes Differentiated from Patient iPSCs After Genome Editing of the Sickle Point Mutation. Stem Cells. 2015;33:1470–1479. doi: 10.1002/stem.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H, Mizuno S, Wells RA, Cantor AB, Watanabe S, Akashi K. GATA-1 converts lymphoid and myelomonocytic progenitors into the megakaryocyte/erythrocyte lineages. Immunity. 2003;19:451–462. doi: 10.1016/s1074-7613(03)00242-5. [DOI] [PubMed] [Google Scholar]
- Jaako P, Debnath S, Olsson K, Modlich U, Rothe M, Schambach A, Flygare J, Karlsson S. Gene therapy cures the anemia and lethal bone marrow failure in a mouse model of RPS19-deficient Diamond-Blackfan anemia. Haematologica. 2014;99:1792–1798. doi: 10.3324/haematol.2014.111195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan YW, Nathan DG. Mild thalassemia: the result of interactions of alpha and beta thalassemia genes. J Clin Invest. 1970;49:635–642. doi: 10.1172/JCI106274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson S, Bodine DM, Perry L, Papayannopoulou T, Nienhuis AW. Expression of the human beta-globin gene following retroviral-mediated transfer into multipotential hematopoietic progenitors of mice. Proc Natl Acad Sci U S A. 1988;85:6062–6066. doi: 10.1073/pnas.85.16.6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DS, Hanson ET, Lewis RL, Auerbach R, Thomson JA. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A. 2001;98:10716–10721. doi: 10.1073/pnas.191362598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee Y, D'Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest. 2012;122:3799–3806. doi: 10.1172/JCI58321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekre N, Antin JH. Hematopoietic stem cell transplantation donor sources in the 21st century: choosing the ideal donor when a perfect match does not exist. Blood. 2014;124:334–343. doi: 10.1182/blood-2014-02-514760. [DOI] [PubMed] [Google Scholar]
- Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy M, Awong G, Sturgeon CM, Ditadi A, LaMotte-Mohs R, Zuniga-Pflucker JC, Keller G. T lymphocyte potential marks the emergence of definitive hematopoietic progenitors in human pluripotent stem cell differentiation cultures. Cell Rep. 2012;2:1722–1735. doi: 10.1016/j.celrep.2012.11.003. [DOI] [PubMed] [Google Scholar]
- Kim AR, Sankaran VG. Development of autologous blood cell therapies. Exp Hematol. 2016 doi: 10.1016/j.exphem.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15:321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvaratskhelia M, Sharma A, Larue RC, Serrao E, Engelman A. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014;42:10209–10225. doi: 10.1093/nar/gku769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBP alpha and PU.1 transcription factors. Immunity. 2006;25:731–744. doi: 10.1016/j.immuni.2006.09.011. [DOI] [PubMed] [Google Scholar]
- Lettre G, Sankaran VG, Bezerra MA, Araujo AS, Uda M, Sanna S, Cao A, Schlessinger D, Costa FF, Hirschhorn JN, Orkin SH. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105:11869–11874. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur DN, Ryan TM, Pawlik KM, Townes TM. Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood. 2003;102:4312–4319. doi: 10.1182/blood-2003-04-1251. [DOI] [PubMed] [Google Scholar]
- Lucarelli G, Isgro A, Sodani P, Gaziev J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb Perspect Med. 2012;2:a011825. doi: 10.1101/cshperspect.a011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig LS, Gazda HT, Eng JC, Eichhorn SW, Thiru P, Ghazvinian R, George TI, Gotlib JR, Beggs AH, Sieff CA, Lodish HF, Lander ES, Sankaran VG. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat Med. 2014;20:748–753. doi: 10.1038/nm.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzatto L, Nannelli C, Notaro R. Glucose-6-Phosphate Dehydrogenase Deficiency. Hematol Oncol Clin North Am. 2016;30:373–393. doi: 10.1016/j.hoc.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Maeda T, Merghoub T, Hobbs RM, Dong L, Maeda M, Zakrzewski J, van den Brink MR, Zelent A, Shigematsu H, Akashi K, Teruya-Feldstein J, Cattoretti G, Pandolfi PP. Regulation of B versus T lymphoid lineage fate decision by the proto-oncogene LRF. Science. 2007;316:860–866. doi: 10.1126/science.1140881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchinu MF, Marongiu MF, Poddie D, Casu C, Latini V, Simbula M, Galanello R, Moi P, Cao A, Porcu S, Ristaldi MS. In vivo activation of the human delta-globin gene: the therapeutic potential in beta-thalassemic mice. Haematologica. 2014;99:76–84. doi: 10.3324/haematol.2012.082768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal PK, Ferreira LM, Collins R, Meissner TB, Boutwell CL, Friesen M, Vrbanac V, Garrison BS, Stortchevoi A, Bryder D, Musunuru K, Brand H, Tager AM, Allen TM, Talkowski ME, Rossi DJ, Cowan CA. Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells using CRISPR/Cas9. Cell Stem Cell. 2014;15:643–652. doi: 10.1016/j.stem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo-Rowe AJ. The thalassemias and related disorders. Proc (Bayl Univ Med Cent) 2007;20:27–31. doi: 10.1080/08998280.2007.11928230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsella M, Borgna-Pignatti C. Transfusional iron overload and iron chelation therapy in thalassemia major and sickle cell disease. Hematol Oncol Clin North Am. 2014;28:703–727. vi. doi: 10.1016/j.hoc.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33:538–542. doi: 10.1038/nbt.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PJ, Sonati MF, MacDonald D, Lanza C, Busutil D, Town M, Corcoran CM, Kaeda JS, Stevens DJ, al-Ismail S, et al. New glucose-6-phosphate dehydrogenase mutations associated with chronic anemia. Blood. 1995;85:1377–1380. [PubMed] [Google Scholar]
- Masuda T, Wang X, Maeda M, Canver MC, Sher F, Funnell AP, Fisher C, Suciu M, Martyn GE, Norton LJ, Zhu C, Kurita R, Nakamura Y, Xu J, Higgs DR, Crossley M, Bauer DE, Orkin SH, Kharchenko PV, Maeda T. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science. 2016;351:285–289. doi: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson J, Ringden O, Storb R. Graft Failure after Allogeneic Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant. 2008;14:165–170. doi: 10.1016/j.bbmt.2007.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May C, Rivella S, Callegari J, Heller G, Gaensler KM, Luzzatto L, Sadelain M. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- McCune SL, Reilly MP, Chomo MJ, Asakura T, Townes TM. Recombinant human hemoglobins designed for gene therapy of sickle cell disease. Proc Natl Acad Sci U S A. 1994;91:9852–9856. doi: 10.1073/pnas.91.21.9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, Zelenika D, Boland A, Rooks H, Best S, Spector TD, Farrall M, Lathrop M, Thein SL. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- Mettananda S, Gibbons RJ, Higgs DR. alpha-Globin as a molecular target in the treatment of beta-thalassemia. Blood. 2015;125:3694–3701. doi: 10.1182/blood-2015-03-633594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, Gregory PD, Pabo CO, Rebar EJ. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. J Virol. 1998;72:8150–8157. doi: 10.1128/jvi.72.10.8150-8157.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M, Bourke E, Alexander D, Warr MR, Barry-Holson K, Le Beau MM, Morrison CG, Passegue E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Estevez FJ, Nowrouzi A, Lozano ML, Galy A, Charrier S, von Kalle C, Guenechea G, Bueren JA, Schmidt M. Lentiviral-Mediated Gene Therapy in Fanconi Anemia-A Mice Reveals Long-Term Engraftment and Continuous Turnover of Corrected HSCs. Curr Gene Ther. 2015;15:550–562. doi: 10.2174/1566523215666150929110903. [DOI] [PubMed] [Google Scholar]
- Musallam KM, Sankaran VG, Cappellini MD, Duca L, Nathan DG, Taher AT. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood. 2012;119:364–367. doi: 10.1182/blood-2011-09-382408. [DOI] [PubMed] [Google Scholar]
- Nandakumar SK, Ulirsch JC, Sankaran VG. Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol. 2016;173:206–218. doi: 10.1111/bjh.13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre O, Eggimann AV, Beuzard Y, Ribeil JA, Bourget P, Borwornpinyo S, Hongeng S, Hacein-Bey S, Cavazzana M, Leboulch P, Payen E. Gene Therapy of the beta-Hemoglobinopathies by Lentiviral Transfer of the beta(A(T87Q))-Globin Gene. Hum Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale SJ, Hollis RP, Pepper KA, Petersen D, Yu XJ, Yang C, Bahner I, Kohn DB. Transient gene expression by nonintegrating lentiviral vectors. Mol Ther. 2006;13:1121–1132. doi: 10.1016/j.ymthe.2006.01.008. [DOI] [PubMed] [Google Scholar]
- Niu X, He W, Song B, Ou Z, Fan D, Chen Y, Fan Y, Sun X. Combining single-strand oligodeoxynucleotides and CRISPR/Cas9 to correct gene mutations in Beta-thalassemia-induced Pluripotent Stem Cells. J Biol Chem. 2016 doi: 10.1074/jbc.M116.719237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, Xia B, Peter M, Durocher D. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015;528:422–426. doi: 10.1038/nature16142. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Osborn MJ, Gabriel R, Webber BR, DeFeo AP, McElroy AN, Jarjour J, Starker CG, Wagner JE, Joung JK, Voytas DF, von Kalle C, Schmidt M, Blazar BR, Tolar J. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum Gene Ther. 2015;26:114–126. doi: 10.1089/hum.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J. Primitive and definitive erythropoiesis in mammals. Front Physiol. 2014;5:3. doi: 10.3389/fphys.2014.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F, Duchateau P. Meganucleases and DNA double-strand break-induced recombination: perspectives for gene therapy. Curr Gene Ther. 2007;7:49–66. doi: 10.2174/156652307779940216. [DOI] [PubMed] [Google Scholar]
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, Humphries RK, Beuzard Y, Nagel RL, Leboulch P. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Gomes A, Bernitz J, Papatsenko D, Niu X, Swiers G, Azzoni E, de Bruijn MF, Schaniel C, Lemischka IR, Moore KA. Hematopoietic Reprogramming In Vitro Informs In Vivo Identification of Hemogenic Precursors to Definitive Hematopoietic Stem Cells. Dev Cell. 2016;36:525–539. doi: 10.1016/j.devcel.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira CF, Chang B, Qiu J, Niu X, Papatsenko D, Hendry CE, Clark NR, Nomura-Kitabayashi A, Kovacic JC, Ma'ayan A, Schaniel C, Lemischka IR, Moore K. Induction of a hemogenic program in mouse fibroblasts. Cell Stem Cell. 2013;13:205–218. doi: 10.1016/j.stem.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Ousterout DG, Gersbach CA. Advances in targeted genome editing. Curr Opin Chem Biol. 2012;16:268–277. doi: 10.1016/j.cbpa.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perumbeti A, Higashimoto T, Urbinati F, Franco R, Meiselman HJ, Witte D, Malik P. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114:1174–1185. doi: 10.1182/blood-2009-01-201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestina TI, Hargrove PW, Jay D, Gray JT, Boyd KM, Persons DA. Correction of murine sickle cell disease using gamma-globin lentiviral vectors to mediate high-level expression of fetal hemoglobin. Mol Ther. 2009;17:245–252. doi: 10.1038/mt.2008.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M, Heijboer H, Smiers F, Giordano PC. Diagnosis and management of thalassaemia. BMJ. 2012;344:e228. doi: 10.1136/bmj.e228. [DOI] [PubMed] [Google Scholar]
- Petersdorf EW. The major histocompatibility complex: a model for understanding graft-versus-host disease. Blood. 2013;122:1863–1872. doi: 10.1182/blood-2013-05-355982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Weatherall DJ. The alpha-thalassemias. N Engl J Med. 2014;371:1908–1916. doi: 10.1056/NEJMra1404415. [DOI] [PubMed] [Google Scholar]
- Pinder J, Salsman J, Dellaire G. Nuclear domain 'knock-in' screen for the evaluation and identification of small molecule enhancers of CRISPR-based genome editing. Nucleic Acids Res. 2015;43:9379–9392. doi: 10.1093/nar/gkv993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- Plavec I, Papayannopoulou T, Maury C, Meyer F. A human beta-globin gene fused to the human beta-globin locus control region is expressed at high levels in erythroid cells of mice engrafted with retrovirus-transduced hematopoietic stem cells. Blood. 1993;81:1384–1392. [PubMed] [Google Scholar]
- Premawardhena A, Fisher CA, Olivieri NF, de Silva S, Sloane-Stanley J, Wood WG, Weatherall DJ. A novel molecular basis for beta thalassemia intermedia poses new questions about its pathophysiology. Blood. 2005;106:3251–3255. doi: 10.1182/blood-2005-02-0593. [DOI] [PubMed] [Google Scholar]
- Puthenveetil G, Scholes J, Carbonell D, Qureshi N, Xia P, Zeng L, Li S, Yu Y, Hiti AL, Yee JK, Malik P. Successful correction of the human beta-thalassemia major phenotype using a lentiviral vector. Blood. 2004;104:3445–3453. doi: 10.1182/blood-2004-04-1427. [DOI] [PubMed] [Google Scholar]
- Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, Cathomen T, Voytas DF, Joung JK. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Mejia V, Melen GJ, Sanchez L, Gutierrez-Aranda I, Ligero G, Cortes JL, Real PJ, Bueno C, Menendez P. Nodal/Activin signaling predicts human pluripotent stem cell lines prone to differentiate toward the hematopoietic lineage. Mol Ther. 2010;18:2173–2181. doi: 10.1038/mt.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran D, Shia WJ, Lo MC, Fan JB, Knorr DA, Ferrell PI, Ye Z, Yan M, Cheng L, Kaufman DS, Zhang DE. RUNX1a enhances hematopoietic lineage commitment from human embryonic stem cells and inducible pluripotent stem cells. Blood. 2013a;121:2882–2890. doi: 10.1182/blood-2012-08-451641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013b;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- Riddell J, Gazit R, Garrison BS, Guo G, Saadatpour A, Mandal PK, Ebina W, Volchkov P, Yuan GC, Orkin SH, Rossi DJ. Reprogramming committed murine blood cells to induced hematopoietic stem cells with defined factors. Cell. 2014;157:549–564. doi: 10.1016/j.cell.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio P, Banos R, Lombardo A, Quintana-Bustamante O, Alvarez L, Garate Z, Genovese P, Almarza E, Valeri A, Diez B, Navarro S, Torres Y, Trujillo JP, Murillas R, Segovia JC, Samper E, Surralles J, Gregory PD, Holmes MC, Naldini L, Bueren JA. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med. 2014;6:835–848. doi: 10.15252/emmm.201303374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivella S, Sadelain M. Genetic treatment of severe hemoglobinopathies: the combat against transgene variegation and transgene silencing. Semin Hematol. 1998;35:112–125. [PubMed] [Google Scholar]
- Riviere J, Hauer J, Poirot L, Brochet J, Souque P, Mollier K, Gouble A, Charneau P, Fischer A, Paques F, de Villartay JP, Cavazzana M. Variable correction of Artemis deficiency by I-Sce1-meganuclease-assisted homologous recombination in murine hematopoietic stem cells. Gene Ther. 2014;21:529–532. doi: 10.1038/gt.2014.20. [DOI] [PubMed] [Google Scholar]
- Robert-Richard E, Moreau-Gaudry F, Lalanne M, Lamrissi-Garcia I, Cario-Andre M, Guyonnet-Duperat V, Taine L, Ged C, de Verneuil H. Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells. Am J Hum Genet. 2008;82:113–124. doi: 10.1016/j.ajhg.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero Z, Urbinati F, Geiger S, Cooper AR, Wherley J, Kaufman ML, Hollis RP, de Assin RR, Senadheera S, Sahagian A, Jin X, Gellis A, Wang X, Gjertson D, Deoliveira S, Kempert P, Shupien S, Abdel-Azim H, Walters MC, Meiselman HJ, Wenby RB, Gruber T, Marder V, Coates TD, Kohn DB. beta-globin gene transfer to human bone marrow for sickle cell disease. J Clin Invest. 2013;123:3317–3330. doi: 10.1172/JCI67930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovira A, De Angioletti M, Camacho-Vanegas O, Liu D, Rosti V, Gallardo HF, Notaro R, Sadelain M, Luzzatto L. Stable in vivo expression of glucose-6-phosphate dehydrogenase (G6PD) and rescue of G6PD deficiency in stem cells by gene transfer. Blood. 2000;96:4111–4117. [PubMed] [Google Scholar]
- Sadelain M, Papapetrou EP, Bushman FD. Safe harbours for the integration of new DNA in the human genome. Nat Rev Cancer. 2012;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
- Sadelain M, Wang CH, Antoniou M, Grosveld F, Mulligan RC. Generation of a high-titer retroviral vector capable of expressing high levels of the human beta-globin gene. Proc Natl Acad Sci U S A. 1995;92:6728–6732. doi: 10.1073/pnas.92.15.6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai N, Maeda M, Lee SU, Ishikawa Y, Li M, Williams JC, Wang L, Su L, Suzuki M, Saito TI, Chiba S, Casola S, Yagita H, Teruya-Feldstein J, Tsuzuki S, Bhatia R, Maeda T. The LRF transcription factor regulates mature B cell development and the germinal center response in mice. J Clin Invest. 2011;121:2583–2598. doi: 10.1172/JCI45682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004;32:3683–3688. doi: 10.1093/nar/gkh703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler VM, Lis R, Liu Y, Kedem A, James D, Elemento O, Butler JM, Scandura JM, Rafii S. Reprogramming human endothelial cells to haematopoietic cells requires vascular induction. Nature. 2014;511:312–318. doi: 10.1038/nature13547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG. Targeted therapeutic strategies for fetal hemoglobin induction. Hematology Am Soc Hematol Educ Program. 2011;2011:459–465. doi: 10.1182/asheducation-2011.1.459. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, Gazda HT. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122:2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Joshi M, Agrawal A, Schmitz-Abe K, Towne MC, Marinakis N, Markianos K, Berry GT, Agrawal PB. Rare complete loss of function provides insight into a pleiotropic genome-wide association study locus. Blood. 2013;122:3845–3847. doi: 10.1182/blood-2013-09-528315. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Lettre G, Orkin SH, Hirschhorn JN. Modifier genes in Mendelian disorders: the example of hemoglobin disorders. Ann N Y Acad Sci. 2010;1214:47–56. doi: 10.1111/j.1749-6632.2010.05821.x. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Scepanovic D, Vergilio JA, Ji P, Kim J, Thiru P, Orkin SH, Lander ES, Lodish HF. MicroRNA-15a and-16-1 act via MYB to elevate fetal hemoglobin expression in human trisomy 13. Proc Natl Acad Sci U S A. 2011;108:1519–1524. doi: 10.1073/pnas.1018384108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Nathan DG. Reversing the hemoglobin switch. N Engl J Med. 2010;363:2258–2260. doi: 10.1056/NEJMcibr1010767. [DOI] [PubMed] [Google Scholar]
- Sankaran VG, Orkin SH. The switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3:a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21:221–230. doi: 10.1038/nm.3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran VG, Xu J, Ragoczy T, Ippolito GC, Walkley CR, Maika SD, Fujiwara Y, Ito M, Groudine M, Bender MA, Tucker PW, Orkin SH. Developmental and species-divergent globin switching are driven by BCL11A. Nature. 2009;460:1093–1097. doi: 10.1038/nature08243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satta S, Perseu L, Moi P, Asunis I, Cabriolu A, Maccioni L, Demartis FR, Manunza L, Cao A, Galanello R. Compound heterozygosity for KLF1 mutations associated with remarkable increase of fetal hemoglobin and red cell protoporphyrin. Haematologica. 2011;96:767–770. doi: 10.3324/haematol.2010.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, Haliburton GE, Ye CJ, Bluestone JA, Doudna JA, Marson A. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A. 2015;112:10437–10442. doi: 10.1073/pnas.1512503112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CT. The zinc finger nuclease monopoly. Nat Biotechnol. 2005;23:915–918. doi: 10.1038/nbt0805-915. [DOI] [PubMed] [Google Scholar]
- Sebastiano V, Maeder ML, Angstman JF, Haddad B, Khayter C, Yeo DT, Goodwin MJ, Hawkins JS, Ramirez CL, Batista LF, Artandi SE, Wernig M, Joung JK. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A, Cornetta K. Design and potential of non-integrating lentiviral vectors. Biomedicines. 2014:14–35. doi: 10.3390/biomedicines2010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu R, Hasegawa A, Ottolenghi S, Ronchi A, Yamamoto M. Verification of the in vivo activity of three distinct cis-acting elements within the Gata1 gene promoter-proximal enhancer in mice. Genes Cells. 2013;18:1032–1041. doi: 10.1111/gtc.12096. [DOI] [PubMed] [Google Scholar]
- Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Paques F. Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy. Curr Gene Ther. 2011;11:11–27. doi: 10.2174/156652311794520111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J, Grizot S, Arnould S, Duclert A, Epinat JC, Chames P, Prieto J, Redondo P, Blanco FJ, Bravo J, Montoya G, Paques F, Duchateau P. A combinatorial approach to create artificial homing endonucleases cleaving chosen sequences. Nucleic Acids Res. 2006;34:e149. doi: 10.1093/nar/gkl720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollaino MC, Paglietti ME, Perseu L, Giagu N, Loi D, Galanello R. Association of alpha globin gene quadruplication and heterozygous beta thalassemia in patients with thalassemia intermedia. Haematologica. 2009;94:1445–1448. doi: 10.3324/haematol.2009.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Aktuna S, Thongjuea S, Aghajanirefah A, Pourfarzad F, van Ijcken W, Lenhard B, Rooks H, Best S, Menzel S, Grosveld F, Thein SL, Soler E. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J Clin Invest. 2014;124:1699–1710. doi: 10.1172/JCI71520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–1030. doi: 10.1056/NEJM199904013401307. [DOI] [PubMed] [Google Scholar]
- Sturgeon CM, Ditadi A, Awong G, Kennedy M, Keller G. Wnt signaling controls the specification of definitive and primitive hematopoiesis from human pluripotent stem cells. Nat Biotechnol. 2014;32:554–561. doi: 10.1038/nbt.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun N, Zhao H. Seamless correction of the sickle cell disease mutation of the HBB gene in human induced pluripotent stem cells using TALENs. Biotechnol Bioeng. 2014;111:1048–1053. doi: 10.1002/bit.25018. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Yamazaki S, Yamaguchi T, Okabe M, Masaki H, Takaki S, Otsu M, Nakauchi H. Generation of engraftable hematopoietic stem cells from induced pluripotent stem cells by way of teratoma formation. Mol Ther. 2013;21:1424–1431. doi: 10.1038/mt.2013.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo E, Rampalli S, Risueno RM, Schnerch A, Mitchell R, Fiebig-Comyn A, Levadoux-Martin M, Bhatia M. Direct conversion of human fibroblasts to multilineage blood progenitors. Nature. 2010;468:521–526. doi: 10.1038/nature09591. [DOI] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takai J, Moriguchi T, Suzuki M, Yu L, Ohneda K, Yamamoto M. The Gata1 5' region harbors distinct cis-regulatory modules that direct gene activation in erythroid cells and gene inactivation in HSCs. Blood. 2013;122:3450–3460. doi: 10.1182/blood-2013-01-476911. [DOI] [PubMed] [Google Scholar]
- Talbot D, Collis P, Antoniou M, Vidal M, Grosveld F, Greaves DR. A dominant control region from the human beta-globin locus conferring integration site-independent gene expression. Nature. 1989;338:352–355. doi: 10.1038/338352a0. [DOI] [PubMed] [Google Scholar]
- Thein SL. Genetic modifiers of the beta-haemoglobinopathies. Br J Haematol. 2008;141:357–366. doi: 10.1111/j.1365-2141.2008.07084.x. [DOI] [PubMed] [Google Scholar]
- Tolar J, Becker PS, Clapp DW, Hanenberg H, de Heredia CD, Kiem HP, Navarro S, Qasba P, Rio P, Schmidt M, Sevilla J, Verhoeyen E, Thrasher AJ, Bueren J. Gene therapy for Fanconi anemia: one step closer to the clinic. Hum Gene Ther. 2012;23:141–144. doi: 10.1089/hum.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler EA, Yao Y, Wang YD, Woodard KJ, Kurita R, Nakamura Y, Hughes JR, Hardison RC, Blobel GA, Li C, Weiss MJ. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med. 2016 doi: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, Usala G, Busonero F, Maschio A, Albai G, Piras MG, Sestu N, Lai S, Dei M, Mulas A, Crisponi L, Naitza S, Asunis I, Deiana M, Nagaraja R, Perseu L, Satta S, Cipollina MD, Sollaino C, Moi P, Hirschhorn JN, Orkin SH, Abecasis GR, Schlessinger D, Cao A. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105:1620–1625. doi: 10.1073/pnas.0711566105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulirsch JC, Nandakumar SK, Wang L, Giani FC, Zhang X, Rogov P, Melnikov A, McDonel P, Do R, Mikkelsen TS, Sankaran VG. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell. 2016;165:1530–1545. doi: 10.1016/j.cell.2016.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Reik A, Chang KH, Stehling-Sun S, Zhou Y, Hinkley SJ, Paschon DE, Zhang L, Psatha N, Bendana YR, O'Neil CM, Song AH, Mich AK, Liu PQ, Lee G, Bauer DE, Holmes MC, Orkin SH, Papayannopoulou T, Stamatoyannopoulos G, Rebar EJ, Gregory PD, Urnov FD, Stamatoyannopoulos JA. Functional footprinting of regulatory DNA. Nat Methods. 2015;12:927–930. doi: 10.1038/nmeth.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, Meerpohl J, Karlsson S, Liu JM, Leblanc T, Paley C, Kang EM, Leder EJ, Atsidaftos E, Shimamura A, Bessler M, Glader B, Lipton JM, Participants of Sixth Annual Daniella Maria Arturi International Consensus C. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo LT, Daley GQ. De novo generation of HSCs from somatic and pluripotent stem cell sources. Blood. 2015;125:2641–2648. doi: 10.1182/blood-2014-10-570234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodyanik MA, Bork JA, Thomson JA, Slukvin II. Human embryonic stem cell-derived CD34+ cells: efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood. 2005;105:617–626. doi: 10.1182/blood-2004-04-1649. [DOI] [PubMed] [Google Scholar]
- Voit RA, Hendel A, Pruett-Miller SM, Porteus MH. Nuclease-mediated gene editing by homologous recombination of the human globin locus. Nucleic Acids Res. 2014;42:1365–1378. doi: 10.1093/nar/gkt947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon HP, Wardan H, Vadolas J. siRNA-mediated reduction of alpha-globin results in phenotypic improvements in beta-thalassemic cells. Haematologica. 2008;93:1238–1242. doi: 10.3324/haematol.12555. [DOI] [PubMed] [Google Scholar]
- Wang J, Exline CM, DeClercq JJ, Llewellyn GN, Hayward SB, Li PW, Shivak DA, Surosky RT, Gregory PD, Holmes MC, Cannon PM. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115:4331–4336. doi: 10.1182/blood-2010-01-251348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MJ, Mason PJ, Bessler M. What's in a name? J Clin Invest. 2012;122:2346–2349. doi: 10.1172/JCI63989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber A, Hargrove PW, Kim YS, Riberdy JM, Sankaran VG, Papanikolaou E, Georgomanoli M, Anagnou NP, Orkin SH, Nienhuis AW, Persons DA. Therapeutic levels of fetal hemoglobin in erythroid progeny of beta-thalassemic CD34+ cells after lentiviral vector-mediated gene transfer. Blood. 2011;117:2817–2826. doi: 10.1182/blood-2010-08-300723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Xie SY, Li W, Ren ZR, Huang SZ, Zeng F, Zeng YT. Correction of beta654-thalassaemia mice using direct intravenous injection of siRNA and antisense RNA vectors. Int J Hematol. 2011;93:301–310. doi: 10.1007/s12185-010-0727-1. [DOI] [PubMed] [Google Scholar]
- Xie SY, Ren ZR, Zhang JZ, Guo XB, Wang QX, Wang S, Lin D, Gong XL, Li W, Huang SZ, Zeng F, Zeng YT. Restoration of the balanced alpha/beta-globin gene expression in beta654-thalassemia mice using combined RNAi and antisense RNA approach. Hum Mol Genet. 2007;16:2616–2625. doi: 10.1093/hmg/ddm218. [DOI] [PubMed] [Google Scholar]
- Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Chong BG, Ippolito GC, Fujiwara Y, Ebert BL, Tucker PW, Orkin SH. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334:993–996. doi: 10.1126/science.1211053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Tong Y, Liu XZ, Wang TT, Cheng L, Wang BY, Lv X, Huang Y, Liu DP. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in beta-thalassemia-derived iPSCs. Sci Rep. 2015;5:12065. doi: 10.1038/srep12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CT, French A, Goh PA, Pagnamenta A, Mettananda S, Taylor J, Knight S, Nathwani A, Roberts DJ, Watt SM, Carpenter L. Human induced pluripotent stem cell derived erythroblasts can undergo definitive erythropoiesis and co-express gamma and beta globins. Br J Haematol. 2014;166:435–448. doi: 10.1111/bjh.12910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanbakhsh K, Ware RE, Noizat-Pirenne F. Red blood cell alloimmunization in sickle cell disease: pathophysiology, risk factors, and transfusion management. Blood. 2012;120:528–537. doi: 10.1182/blood-2011-11-327361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder MC. Inducing definitive hematopoiesis in a dish. Nat Biotechnol. 2014;32:539–541. doi: 10.1038/nbt.2929. [DOI] [PubMed] [Google Scholar]
- Yu C, Liu Y, Ma T, Liu K, Xu S, Zhang Y, Liu H, La Russa M, Xie M, Ding S, Qi LS. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell. 2015;16:142–147. doi: 10.1016/j.stem.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y, Wang J, Khaled W, Burke S, Li P, Chen X, Yang W, Jenkins NA, Copeland NG, Zhang S, Liu P. Bcl11a is essential for lymphoid development and negatively regulates p53. J Exp Med. 2012;209:2467–2483. doi: 10.1084/jem.20121846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanella A, Fermo E, Bianchi P, Valentini G. Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br J Haematol. 2005;130:11–25. doi: 10.1111/j.1365-2141.2005.05527.x. [DOI] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Mali P, Huang X, Dowey SN, Cheng L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood. 2011;118:4599–4608. doi: 10.1182/blood-2011-02-335554. [DOI] [PMC free article] [PubMed] [Google Scholar]