

Abstract
The chemical speciation of arsenic in sediments and porewaters of aquifers is the critical factor that determines whether dissolved arsenic accumulates to potentially toxic levels. Sequestration of arsenic in solid phases, which may occur by adsorption or precipitation processes, controls dissolved concentrations. We present synchrotron x-ray absorption spectra of arsenic in shallow aquifer sediments that indicate the local structure of realgar (AsS) as the primary arsenic-bearing phase in sulfate-reducing conditions at concentrations of 1–3 mmol·kg–1, which has not previously been verified in sediments at low temperature. Spectroscopic evidence shows that arsenic does not substitute for iron or sulfur in iron sulfide minerals at the molecular scale. A general geochemical model derived from our field and spectroscopic observations show that the ratio of reactive iron to sulfur in the system controls the distribution of solid phases capable of removing arsenic from solution when conditions change from oxidized to reduced, the rate of which is influenced by microbial processes. Because of the difference in solubility of iron versus arsenic sulfides, precipitation of iron sulfide may remove sulfide from solution but not arsenic if precipitation rates are fast. The lack of incorporation of arsenic into iron sulfides may result in the accumulation of dissolved As(III) if adsorption is weak or inhibited. Aquifers particularly at risk for such geochemical conditions are those in which oxidized and reduced waters mix, and where the amount of sulfate available for microbial reduction is limited.
Despite intensive study in recent years, quantitative biogeochemical models that explain and predict conditions controlling the uptake or release of dissolved arsenic in groundwaters remain incomplete, due partly to a lack of knowledge of arsenic speciation in subsurface sediments. Worldwide, elevated concentrations of arsenic (>10 μg·liter–1, the World Health Organization and new U.S. Environmental Protection Agency drinking-water standard) in groundwater have the potential to have an adverse impact on ≈90 million people (1, 2), including 13 million in the United States (3, 4). Arsenic is a known carcinogen and mutagen that can lead to both acute and chronic adverse health effects (5, 6). Arsenic in ground- and surface waters results from both natural and anthropogenic sources, but its mobility and attenuation, and thus impact on humans and other organisms, is directly tied to its chemical speciation (3, 7, 8).
Recent studies using direct spectroscopic characterizations have begun to verify differing chemical states of arsenic in natural samples and their influence on arsenic mobility (9–12). Under oxic conditions, it is widely accepted that arsenic in sediments is removed from solution by adsorption to or coprecipitation with ferric oxyhydroxide and possibly by adsorption to manganese oxide and phyllosilicate minerals (7, 13). As subsurface conditions change from oxidized to progressively more reduced, As(V) is reduced to As(III), probably mediated by microbial activity that includes metabolic and detoxification mechanisms (8, 14). Reduced arsenic may be released to solution by the reductive dissolution of host iron phases (15). This mechanism is thought to be a primary cause of elevated arsenic in groundwaters in the Bengal basin and elsewhere, although factors that drive reduction in a particular system remain obscure (2, 16–18).
Accumulation of arsenic at redox boundaries in sediments has been documented and association with sulfide has been observed under very reduced conditions (10, 19–21). At conditions where sulfide is stable, the concentration of dissolved arsenic at equilibrium may be controlled by the solubility of sulfide phases and therefore depends on the rate of microbially mediated sulfate reduction (8, 22). Previous studies have assumed or postulated that arsenic associated with sulfide in aquifer sediments under reduced conditions is present either as a minor substituent in fine-grained biogenic pyrite (FeS2) or arsenopyrite (FeAsS) (23–25), as a sorption complex on sulfide minerals, or as the mineral orpiment (As2S3) (21, 26, 27). Under sulfate-reduced conditions, orpiment is often assumed as the arsenic sulfide phase. The formation of arsenic sulfide phases in aquifer sediments as a mechanism for removal from solution, as opposed to adsorption or substitution mechanisms, requires that the solubility of the sulfide mineral is exceeded (at least locally) and that mineral precipitation rates are fast compared with the rate of groundwater flow.
To our knowledge, the formation of realgar or its polymorphs pararealgar and alacranite (written as AsS or As4S4) at ambient temperatures in natural sediments, perhaps as a consequence of microbiological activity, has not been directly verified (28). Realgar polymorphs (all monoclinic) contain covalently bonded clusters of four arsenic and four sulfur atoms; differences in their crystal structures arise from subtle differences in the arrangement of arsenic and sulfur clusters (29–31). Huber et al. (32) have described a thermophyllic archaeon that respires arsenate and precipitates realgar in its growth range of 68–100°C in laboratory cultures. Newman et al. (33, 34) reported a bacterium (Desulfotomaculum auripigmentum) capable of reducing As(V) to As(III) and precipitating orpiment or amorphous As2S3 in laboratory culture. Orpiment, realgar, and other arsenic sulfide minerals are typical high-temperature phases found together in hydrothermal deposits, but few studies discuss the occurrence of realgar polymorphs at lower temperatures. Pararealgar has been observed as an alteration product of realgar at ambient conditions (31, 35). In another study, an arsenic sulfide phase identified as the realgar polymorph alacranite (As4S4) was precipitated from solution after acid extraction from sediments (27). Identification of arsenic sulfide phases in reduced sediments is difficult because they are often fine-grained, in low abundance, and unstable in air.
We used synchrotron x-ray absorption spectroscopy (XAS), an element-specific, nonvacuum technique that is sensitive to low levels of arsenic, to directly examine its chemical speciation in shallow, arsenic-impacted aquifer sediments. The study site serves as an accessible model system where changes in sediment and porewater arsenic concentrations can be examined in response to variations in subsurface oxidation state over small vertical and horizontal spatial scales (cm to m) (36). Samples were carefully handled to preclude exposure to air and preserve the reduced states of arsenic, iron, and sulfur for spectroscopic characterization. We combined spectral and chemical analysis of arsenic and iron in natural sediments with thermodynamic reaction-progress calculations to develop a general model for arsenic, iron, and sulfur speciation as a function of the oxidation state of the system. Changes in the chemical speciation of these elements are used to explain the geochemical behavior of arsenic as subsurface conditions change between oxidized and reduced and to identify conditions that maximize dissolved arsenic concentrations.
Methods
At a site adjacent to San Francisco Bay (in East Palo Alto, CA), arsenic was released to surface sediments from a former pesticide-manufacturing facility that operated from 1926 to 1970. Arsenic migrated into the subsurface and infiltrated a shallow aquifer at ≈2 m depth, where sediment arsenic concentrations range from <0.1 to 5 mmol·kg–1 and aqueous concentrations are <0.07 to 200 μM (pH ≈ 6–7). A 3.5-m sediment core was collected at this site and transported within hours to the Stanford Synchrotron Radiation Laboratory. Samples were immediately loaded into Teflon sample holders in a N2 atmosphere, sealed with Kapton tape, and quenched in liquid N2. Samples were kept frozen until XAS data collection, which occurred within 3 days. Data were collected at Stanford Synchrotron Radiation Laboratory on wiggler beamline 4-3 [3 GeV (1 eV = 1.602 × 10–19 J), 70–90 mA] by using an unfocused beam. Arsenic and iron K-edge x-ray absorption near-edge structure (XANES) and extended x-ray absorption fine-structure (EXAFS) fluorescence spectra were collected by using Si(220) monochromator crystals (vertical beam size = 1 mm) with a 13-element solid-state Ge-array fluorescence detector. Samples were held 15–20 K with a He cryostat and 6–40 scans were collected and averaged. Bulk absorption spectra were collected on the same samples for both the arsenic (calibrated at 11,867 eV by using arsenic foil) and iron (calibrated at 7,112 eV by using iron foil) absorption edges. No changes in spectral features were noted during the course of data collection that would indicate oxidation or reduction. Solid arsenic reference compounds were diluted with inert B(OH)3 and collected at 15–20 K in transmission mode by using gas-filled ion chambers. XAS data were analyzed with the computer packages exafspak (Stanford Synchrotron Radiation Laboratory) and feff6 (37) (see Supporting Text, which is published as supporting information on the PNAS web site, for details). In our EXAFS analysis, we rely on differences in near-neighbor atom identities and interatomic distances, which are accurate to within ±0.02 Å (12, 38), to deduce the arsenic-bonding environment. A significant advantage to our approach is that XAS data were collected on samples that did not experience any extraction or processing methods that might alter the chemical speciation of arsenic, iron, and sulfur in the sediments. A total of 11 spectra were collected from this core and an additional 21 spectra were collected from two other cores at this site. All samples were characterized by bulk chemical methods reported elsewhere (36). This suite of samples shows changes in arsenic and iron oxidation states and speciation as a function of depth from the surface to ≈3.5 m. Here, we discuss XAS results from two samples in the upper 1.5 m of core where sulfate reduction is occurring in fine-grained, organic-matter-rich sediments (36).
Reaction path models were used to examine changes in the thermodynamic state of a model subsurface system constrained by field-site observations through a series of irreversible equilibrium reaction steps. This approach is commonly used in the analysis of complex natural systems in the absence of kinetic data. Reaction path models were calculated with the react program of the geochemist's workbench modeling package by using a modified version of the Lawrence Livermore National Laboratory thermodynamic database augmented with thermodynamic data from ref. 28 for arsenic species and ref. 39 for green rusts (see Table 1, which is published as supporting information on the PNAS web site, for thermodynamic data and details of the modeling).
Results
XAS. Arsenic XANES and EXAFS spectra of the sediments are compared to those of reference arsenic sulfide and oxide compounds in Fig. 1. In XANES spectra, the energy of maximum absorption (the absorption edge) is sensitive to the oxidation state of the target element. The XANES spectra (Fig. 1a) show that the absorption edge for the sediment samples coincides with that of the mineral realgar. The absorption edge for the sediment samples and realgar are shifted to slightly lower energy (≈0.5–1 eV) than that of orpiment. The absorption edge position for arsenopyrite (FeAsS) is slightly lower than that of realgar (0.3–0.5 eV), but the spectrum differs in the XANES above the edge and the EXAFS is significantly different (see below). Note that the absorption edge for As(III) oxide (As2O3) is at higher energy (≈1.5 eV) than that of the corresponding As(III) sulfide orpiment (Fig. 1a).
Fig. 1.

Arsenic K-edge x-ray absorption spectra. (a) XANES spectra for arsenic sulfide and oxide reference compounds compared with two sediment samples A and B. Total arsenic concentrations were 1.76 and 2.73 mmol·kg–1 for sediment A and B, respectively. (b) EXAFS spectra and corresponding Fourier transforms for reference compounds and sediment sample B. Solid lines are data; dashed lines are nonlinear least-squares fits. (See Table 2, which is published as supporting information on the PNAS web site, for numerical fit results. Fit results for arsenopyrite and arsenic substituted into pyrite are published in ref. 12.)
Quantitative analysis of EXAFS spectra provides structural data about the near-neighbor atomic environment (interatomic distances and the identity and number of backscattering atoms) to ≈4 Å around the target element. The EXAFS spectrum of sediment sample B and realgar are similar, indicating a similar local atomic environment around arsenic, but they are distinctly different from the spectra of the other compounds (Fig. 1b). Least-squares fits to the sediment data produced interatomic distances identical (within error) with average interatomic distances in crystalline realgar (see Table 2 for comparison of EXAFS and x-ray diffraction interatomic distances). The local atomic coordination of arsenic in the realgar polymorphs pararealgar (31) and alacrinite (29) is similar to that of arsenic in realgar. Because the EXAFS spectrum is an average of all arsenic sites in the sample, differences in interatomic distances resolvable with x-ray diffraction (<≈0.1 Å) are averaged into a single backscattering distance in the EXAFS. Thus, these polymorphs are not distinguishable on the basis of their EXAFS spectrum alone. Minor differences in the spectral features of realgar and sediment B (Fig. 1b) are attributed to a greater degree of atomic disorder, small particle size, and/or chemical impurities in the sediments (40). In the realgar local atomic structure, arsenic forms dimers such that arsenic shares a direct bond with another arsenic atom at a relatively short interatomic distance of ≈2.58 Å; more distant arsenic atoms are found at 3.27–3.63 Å. In orpiment, arsenic is bonded directly to three sulfur atoms, and the nearest arsenic atoms are found at ≈3.19 Å. Because of the difference in atomic mass between arsenic and sulfur, the presence of arsenic atoms at a close interatomic distance gives rise to the strong beat pattern seen in the EXAFS of realgar, which corresponds to the large peaks in the Fourier transform between 2 and 3.5 Å (uncorrected for phase shift) (Fig. 1b). These strong backscattering features are characteristic of realgar and isomorphic compounds and are absent in orpiment and its polymorphs, as seen in the weak scattering beyond the first shell of sulfur atoms in the orpiment spectrum (Fig. 1b).
The sediment sample EXAFS also shows that arsenic is not precipitating as arsenopyrite, nor is arsenic substituting into pyrite at the molecular scale. The spectral signature of sediment B is distinctly different from that of crystalline arsenopyrite or a natural pyrite sample containing on average ≈1.2 wt % arsenic (detailed analyses of arsenopyrite and the arsenic-bearing pyrite sample are given in ref. 12) (Fig. 1b). If arsenic was primarily associated with pyrite or arsenopyrite, the local coordination would differ considerably from that of the arsenic sulfide compounds and the EXAFS would be dominated by iron-backscattering atoms, which are easily distinguished from arsenic backscatterers.
Analysis of the iron EXAFS spectra on the same samples supports the formation of pyrite in the sediments. We compare the sediment data with the reference compounds pyrite and an iron-bearing phyllosilicate (illite), and with a mechanical mixture of these two phases (Fig. 2; see Table 3, which is published as supporting information on the PNAS web site, and ref. 41 for detailed analysis). The spectral signal for iron in sediments is dominated by iron substitution in detrital phyllosilicate minerals (probably mica). The nonsilicate iron fraction is primarily pyrite (not amorphous FeS; see Fig. 2), which comprises ≈10–40% of the iron (by atomic mass) present in the sediments (total iron concentrations are 2.8–5.4 wt %). Other supporting evidence suggests that realgar is present in the sediments as very small, poorly crystalline particles, which may form as surface coatings or particulates in association with pyrite. We were unable to detect the presence of any arsenic with scanning electron microscopy and energy dispersive spectrometry, suggesting that particle size is small (<2 μm). No sulfide phases, either arsenic or iron, were detectable in bulk sediments with powder XRD (36). Sequential extraction of arsenic and iron from sediments using an extraction scheme optimized for arsenic (42) showed that arsenic is not dominantly associated with easily exchanged sorption sites or with reducible iron (36). This finding indicates that much of the arsenic is strongly sequestered in the sediments rather than surface-adsorbed or associated with easily dissolved phases, which is consistent with the presence of small particles.
Fig. 2.

Iron K-edge EXAFS spectra. EXAFS spectra and corresponding Fourier transforms for sediment samples A and B compared with reference pyrite, freshly precipitated FeS and illite (see ref. 41 for quantitative fits), and a mechanical mixture of pyrite plus illite (50:50 wt % Fe). Solid lines are data, dashed lines are nonlinear least-squares fits, and the shaded areas indicate the fraction of the fit that is composed of pyrite (sulfide component). The nonsulfide component of the sediments is a composite of iron-bearing phyllosilicate and oxide phases (see Table 3 for numerical fit results).
Geochemical Modeling. Reaction-path modeling was performed to provide insight into the overall thermodynamic controls on arsenic speciation and concentration under the dynamic redox environment of a shallow, oxidized freshwater aquifer infiltrated by reducing marine waters (San Francisco Bay water). We used observations at the field site as model constraints for the general analysis of subsurface changes from oxidized to sulfate reducing in arsenic-bearing sediments. The model system consisted of an arsenic-contaminated sediment initially containing arsenic adsorbed onto Fe(III) oxyhydroxide (represented by goethite or ferrihydrite) and organic matter (represented by CH2O). The reaction path simulated microbially mediated organic matter degradation through a sequence of terminal electron-accepting processes, including aerobic (dissolved oxygen) and arsenate, ferric iron, and sulfate reduction, but does not include any specific kinetic reactions. Porewater was initially set at saturation with atmospheric oxygen and contained sulfate at seawater concentration (28 mM). Arsenic, at a total concentration of 100 μM, was initially distributed between the porewater and sorption sites on the Fe(III) oxyhydroxide phase in the sediment and was allowed to reequilibrate in response to the irreversible oxidation of organic matter. Porewater and adsorbed concentrations and mineral abundances were tracked as a function of the amount of organic matter oxidized at constant pH (= 7), typical of many subsurface environments and close to measurements at the study site. A summary of the primary adsorption and precipitation reactions controlling arsenic distribution are shown in Fig. 3. Reaction-path results from quantitative modeling are plotted as a function of system oxidation state (pe) and the activity of total sulfide (H2S) to emphasize the role of sulfide in controlling arsenic speciation (Fig. 4).
Fig. 3.

Biogeochemical model. Shown is a summary of adsorption and precipitation reactions that control arsenic uptake and release in the As-Fe-S system. Open arrows indicate adsorption reactions, filled arrows indicate dissolution/precipitation reactions, and curved arrows indicate reactions that may couple with microbial organic matter oxidation. Diagram shows the primary reactions for a system transitioning from oxidized to reducing conditions; reactions for oxidation pathways may differ.
Fig. 4.

Reaction path model. Quantitative model for the schematic reactions shown in Fig. 3 illustrated as pe-log aH2S stability diagram for the As-Fe-S-O-H system at 25°C and pH = 7. Log activity (S)total = –2; iron mineral stability fields are shown for log activity (Fe)total = –5 (stability fields shaded gray) and –7 (stability fields stippled); and arsenic mineral stability domains are calculated for log activity (As)total = –3 to –6, as labeled. Reaction paths are shown for evolution of iron-rich (path A, solid red arrow) and iron-poor (path B, dotted blue arrow) sediments during oxidation of organic matter. Equilibrium reaction paths were calculated for evolution of groundwater containing 28 mmol·kg–1 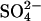 and 100 μmol·kg–1 as during progressive oxidation of sedimentary organic matter in iron-rich (100 mmol·kg–1 reactive Fe) and iron-poor (1 mmol·kg–1 reactive Fe) sediment. At log activity (Fe)total =–7, the stability field for green rust is absent and iron exists as Fe2+(aq).
and 100 μmol·kg–1 as during progressive oxidation of sedimentary organic matter in iron-rich (100 mmol·kg–1 reactive Fe) and iron-poor (1 mmol·kg–1 reactive Fe) sediment. At log activity (Fe)total =–7, the stability field for green rust is absent and iron exists as Fe2+(aq).
By varying the amount of Fe(III) oxyhydroxide initially present in the sediment, a series of reaction-path calculations were performed over a range of reactive Fe/S ratios. These results showed that, depending on the initial Fe/S molar ratio in the system, two distinct reaction paths determined the fate of arsenic (Fig. 4). In other words, aquifer systems can be classified as either iron-controlled or sulfur-controlled with respect to the behavior of arsenic. The model predicts that maximum dissolved arsenic concentrations are attained in the redox transition between conditions where arsenate sorbed on Fe(III) oxyhydroxide is stable and the stability region where sufficient reduction of arsenate and sulfate leads to the formation of realgar or other arsenic sulfide phases. In this intermediate region, concentrations of dissolved As(III), Fe(II), and sulfide are controlled by the rate of reductive dissolution of Fe(III) oxyhydroxide (with sorbed arsenic) and the rate of sulfate reduction, both of which are influenced by microbial activity (Fig. 3). A limitation of the model calculation is that it does not include specific reaction rates because of the difficulty in obtaining reliable kinetic data for all reactions in complex natural systems. Realgar (or a polymorph of similar thermodynamic stability) is predicted as the initial arsenic sulfide to precipitate in high-iron, reducing environments where H2S activity is buffered by the coexistence of iron sulfide with ferric (hydr)oxide [either goethite or ferrihydrite (not shown)], or with Fe(II/III) (hydr)oxide (either green rust or magnetite) at circumneutral pH (Fig. 4, path A). Our model predicts that initial precipitation and subsequent stabilization of realgar also depends on dissolved arsenic concentration. A threshold arsenic concentration is required to exceed realgar solubility during the course of the reaction path. Although the threshold concentration depends on several factors such as iron and sulfide concentrations and pH, our simulations indicate that it is in the range of 10 to 100 μM. At lower total arsenic concentrations and reduced conditions, the only mechanism available for arsenic removal is adsorption to mineral surfaces. In contrast, iron-poor environments do not effectively buffer dissolved H2S (because FeS or pyrite precipitation is limited) and allow it to increase to concentrations high enough to stabilize orpiment (Fig. 4, path B). Orpiment becomes increasingly soluble with further increases in dissolved H2S at >1 mM because of formation of arsenic-sulfide aqueous complexes (43).
Discussion
Under sulfate-reducing conditions, our field data and geochemical models show that dissolved arsenic concentrations can be limited by the low-temperature formation of a realgar-like arsenic sulfide if groundwater arsenic concentrations are sufficiently high (i.e., μM levels), which has not been previously recognized. Under these circumstances, precipitation of realgar (relatively high-iron/low-sulfur environments) or orpiment (relatively low-iron/high-sulfur environments) will remove dissolved arsenic from solution. Our spectroscopic data confirm the lack of a kinetic barrier to realgar formation within the time-frame of contamination (the pesticide facility began operations ≈78 years ago). Our spectroscopic data also show that, at the molecular scale, coprecipitation of arsenic with sedimentary iron sulfide minerals is not the dominant mechanism of arsenic uptake in these sediments. This finding is somewhat surprising because the substitution of arsenic in pyrite is well known in hydrothermal and metamorphic systems. For example, Savage et al. (12) documented arsenic concentrations up to 5 wt % in individual pyrite grains and showed that arsenic substituted for sulfur in the pyrite crystal structure rather than forming inclusions or small grains of arsenopyrite. In reduced sediments, iron sulfide probably precipitates initially as amorphous FeS and converts to pyrite (44). We find no evidence for FeS in these sediments, which should be detectable by XAS at concentrations of ≈5% of the total iron present (40). Although a small amount of substitution of arsenic into the pyrite structure may occur at low temperatures, coprecipitation of arsenic with diagenetic iron sulfide minerals is not the dominant removal mechanism here.
In porewaters where arsenic concentrations never exceed the solubility for realgar, orpiment, or their metastable polymorphs (i.e., sub-μM levels) under reduced conditions, arsenic attenuation is limited to adsorption processes. Laboratory investigations of arsenite sorption on sulfide minerals indicate weak adsorption on FeS and pyrite, in particular, below pH ≈6 (45, 46). Under mildly reducing conditions, where iron sulfide phases are undersaturated because of low sulfide activity, our model suggests that arsenite may coexist with Fe(II, III) oxides or hydroxides, such as magnetite (Fe3O4) or metastable “green rust” {in general, 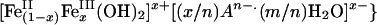 , if dissolved iron concentrations are high enough to exceed their solubility (Fig. 4). If such phases are present, then arsenic adsorption onto their surfaces and surface redox reactions may be important (47, 48). Current understanding of arsenic adsorption and reaction with mixed-valence iron oxides is incomplete, and more research is needed to determine the role these phases play in the environmental fate of arsenic. The efficiency and reversibility of arsenite adsorption on iron sulfide, iron oxide, iron hydroxide, and phyllosilicate minerals in natural systems depend on many factors, including pH, dissolved arsenic concentration, type of mineral substrate, and presence of competing species, and are thus difficult to predict quantitatively. In general, however, precipitation is a more complete and stable mechanism for removal of solutes from solution than adsorption (49, 50).
, if dissolved iron concentrations are high enough to exceed their solubility (Fig. 4). If such phases are present, then arsenic adsorption onto their surfaces and surface redox reactions may be important (47, 48). Current understanding of arsenic adsorption and reaction with mixed-valence iron oxides is incomplete, and more research is needed to determine the role these phases play in the environmental fate of arsenic. The efficiency and reversibility of arsenite adsorption on iron sulfide, iron oxide, iron hydroxide, and phyllosilicate minerals in natural systems depend on many factors, including pH, dissolved arsenic concentration, type of mineral substrate, and presence of competing species, and are thus difficult to predict quantitatively. In general, however, precipitation is a more complete and stable mechanism for removal of solutes from solution than adsorption (49, 50).
High rates of microbial arsenate, ferric iron, and sulfate reduction may increase dissolved concentrations locally in porewaters, where precipitation of iron and arsenic sulfides may occur heterogeneously in sediment pore spaces or oscillate as phases are locally over- and undersaturated. Bacterial cell walls or biofilms may also serve as nucleation sites for sulfide mineral precipitation, which may be nanometer-sized (51). These processes would result in a close spatial association of iron and arsenic sulfides (around millimeters to centimeters) as coatings, inclusions, or small particles. Our spectroscopic evidence shows that the arsenic and iron sulfides are present as distinct phases at the molecular scale, which validates our thermodynamic calculations as a baseline for quantitative analysis given that field reaction rates are unknown.
The present findings have implications for arsenic contamination and migration in sedimentary aquifers. In a number of drinking-water aquifers in east Asia, including those in Bangladesh, West Bengal, Vietnam, and Taiwan, groundwater is affected by the release of arsenic that occurs at natural abundance levels in response to the reductive dissolution of sedimentary Fe(III) oxyhydroxides in aquifer sediments (1, 16–18). In general, total sediment and dissolved arsenic concentrations are lower in these examples than in our study site, typically <10–600 μmol·kg–1 for sediments and <1–5 μM for groundwater, but higher in some cases (2). In general, groundwaters are reduced as a result of high rates of microbiological activity but tend to be low in sulfate (1, 16). If iron concentrations are relatively high but the amount of sulfate available for reduction is limited, the solubility of arsenic sulfide phases may never be exceeded, even in some cases where the solubility of iron sulfide is exceeded. In this case, adsorption on mineral substrates is a possible removal mechanism but one that is strongly influenced by pH. When pH decreases below circumneutral, arsenite adsorption tends to decrease, particularly in the presence of competing anions such as phosphate (15, 52). Under these aquifer conditions, no strong geochemical attenuation mechanism for arsenite removal exists because dissolved H2S is scavenged from solution by FeS/pyrite precipitation whereas arsenic is not, allowing dissolved arsenite concentrations to increase. In aquifers where agricultural runoff has an impact, competition for adsorption sites among arsenic and other species, such as phosphate and silica, would inhibit adsorption and promote elevated dissolved arsenic concentrations.
Concluding Remarks
A striking feature of arsenic occurrence in groundwater world-wide is its variability over hydrologically small spatial intervals (centimeters to meters) (1, 2). Temporal variations may be similarly erratic but are not known for most aquifers. This variability is a reflection of the interplay among changes in the chemical composition and redox state of groundwater, microbiological activity, and adsorption and precipitation processes in the subsurface that establish and evolve within the overall hydrologic framework. Our spectroscopic evidence and modeling point out the importance of the geochemical regime where oxidation potential is intermediate between the stability fields for oxidized iron(III) phases and arsenate, and the region where arsenic sulfide phases are stable (Fig. 4). In this intermediate redox state at circumneutral pH, conditions generally favor partitioning of arsenic to solution. Dissolved arsenic concentrations may remain difficult to predict quantitatively in specific cases because they are controlled by rates of dissolution and precipitation of iron and sulfide phases and their solubilities, and by competing pH-dependent adsorption reactions. Within this general framework, however, we can predict hydrogeochemical states most at risk for contamination by naturally occurring arsenic. These conditions include aquifers that are rich in organic matter, nitrogen, and phosphorus with high rates of microbial reduction creating anoxic conditions and that are limited in available (i.e., labile or reactive) sulfur and/or iron. In areas with large seasonal fluctuations in recharge, nutrients are continually resupplied, but oxygen is rapidly depleted in the subsurface. If sulfate is limited, other electron acceptors such as nitrate, arsenate, and ferric iron become important for microbial respiration. The release of arsenic to solution and its accumulation to hazardous levels in the transition from oxidized to reducing, which may vary seasonally, depend on the amount of available iron and sulfur in the aquifer system, on the rate of reductive dissolution of iron(III) phases, and on the rate of precipitation of iron sulfide, which removes dissolved sulfide. In horizons of relatively low permeability, reduced arsenic may accumulate in solution if a precipitation or adsorption removal process does not keep pace with vertical or lateral groundwater flow.
Supplementary Material
Acknowledgments
We thank Mr. Robert Ferguson and SLLI, Inc. for access to the field site, Michael Rafferty and Ken Chiang (S. S. Papadopulos & Associates), and Geomatrix Consultants for assistance with sample collection. The Stanford Synchrotron Radiation Laboratory is operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. This work was supported by National Science Foundation Grant EAR-0073984.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: XAS, x-ray absorption spectroscopy; XANES, x-ray absorption near-edge structure; EXAFS, extended x-ray absorption fine structure.
References
- 1.Smedley, P. L. (2003) in Arsenic in Goundwater, eds. Welch, A. H. & Stollenwerk, K. G. (Kluwer, Boston), pp. 179–209.
- 2.Smedley, P. L. & Kinniburgh, D. G. (2002) Appl. Geochem. 17, 517–568. [Google Scholar]
- 3.Welch, A. H., Westjohn, D. B., Helsel, D. R. & Wanty, R. B. (2000) Ground Water 38, 589–604. [Google Scholar]
- 4.Ryker, S. J. (2003) in Arsenic in Goundwater, eds. Welch, A. H. & Stollenwerk, K. G. (Kluwer, Boston), pp. 165–178.
- 5.Hindmarsh, J. T. & McCurdy, R. F. (1986) CRC Crit. Rev. 23, 315–347. [DOI] [PubMed] [Google Scholar]
- 6.Morton, W. E. & Dunnette, D. A. (1994) in Arsenic in the Environment, ed. Nriagu, J. O. (Wiley, New York), Part 2, pp. 17–34.
- 7.Hering, J. G. & Kneebone, P. E. (2002) in Environmental Chemistry of Arsenic, ed. Frankenberger, W. T., Jr. (Dekker, New York), pp. 155–181.
- 8.Inskeep, W. P., McDermott, T. R. & Fendorf, S. (2002) in Environmental Chemistry of Arsenic, ed. Frankenberger, W. T., Jr. (Dekker, New York), pp. 183–215.
- 9.Foster, A. L., Brown, G. E., Jr., Tingle, T. N. & Parks, G. A. (1998) Am. Mineral. 83, 553–568. [Google Scholar]
- 10.La Force, M. J., Hansel, C. M. & Fendorf, S. (2000) Environ. Sci. Technol. 34, 3937–3943. [DOI] [PubMed] [Google Scholar]
- 11.Kneebone, P. E., O'Day, P. A., Jones, N. & Hering, J. G. (2002) Environ. Sci. Technol. 36, 381–386. [DOI] [PubMed] [Google Scholar]
- 12.Savage, K. E., Tingle, T. N., O'Day, P. A., Waychunas, G. A. & Bird, D. K. (2000) Appl. Geochem. 15, 1219–1244. [Google Scholar]
- 13.Stollenwerk, K. G. (2003) in Arsenic in Goundwater, eds. Welch, A. H. & Stollenwerk, K. G. (Kluwer, Boston), pp. 67–100.
- 14.Oremland, R. S., Newman, D. K., Kail, B. W. & Stolz, J. F. (2002) in Environmental Chemistry of Arsenic, ed. Frankenberger, W. T., Jr. (Dekker, New York), pp. 273–295.
- 15.Dixit, S. & Hering, J. G. (2003) Environ. Sci. Technol. 37, 4182–4189. [DOI] [PubMed] [Google Scholar]
- 16.Harvey, C. F., Swartz, C. H., Badruzzaman, A. B. M., Keon-Blute, N., Yu, W., Ali, M. A., Jay, J., Beckie, R., Niedan, V., Brabander, D., et al. (2002) Science 298, 1602–1606. [DOI] [PubMed] [Google Scholar]
- 17.McArthur, J. M., Banerjee, D. M., Hudson-Edwards, K. A., Mishra, R., Purohit, R., Ravenscroft, P., Cronin, A., Howarth, R. J., Chatterjee, A., Talukder, T., et al. (2004) Appl. Geochem. 19, 1255–1293. [Google Scholar]
- 18.Nickson, R. T., McArthur, J. M., Ravenscroft, P., Burgess, W. G. & Ahmed, K. M. (2000) Appl. Geochem. 15, 403–413. [Google Scholar]
- 19.Harrington, J. M., Fendorf, S. E. & Rosenzweig, R. F. (1998) Environ. Sci. Technol. 32, 2425–2430. [Google Scholar]
- 20.Brannon, J. M. & Patrick, W. H., Jr. (1987) Environ. Sci. Technol. 21, 450–459. [DOI] [PubMed] [Google Scholar]
- 21.Moore, J. N., Ficklin, W. H. & Johns, C. (1988) Environ. Sci. Technol. 22, 432–437. [Google Scholar]
- 22.Stolz, J. F. & Oremland, R. S. (1999) FEMS Microbiol. Rev. 23, 615–627. [DOI] [PubMed] [Google Scholar]
- 23.Rittle, K. A., Drever, J. I. & Colberg, P. J. S. (1995) Geomicrobiol. J. 13, 1–11. [Google Scholar]
- 24.Saunders, J. A., Pritchett, M. A. & Cook, R. B. (1997) Geomicrobiol. J. 14, 203–217. [Google Scholar]
- 25.Kim, M. J., Nriagu, J. & Haack, S. (2002) Environ. Pollut. 120, 379–390. [DOI] [PubMed] [Google Scholar]
- 26.Aggett, J. & O'Brian, G. A. (1985) Environ. Sci. Technol. 19, 231–238. [DOI] [PubMed] [Google Scholar]
- 27.Wilkin, R. T. & Ford, R. G. (2002) Environ. Sci. Technol. 36, 4921–4927. [DOI] [PubMed] [Google Scholar]
- 28.Nordstrom, D. K. & Archer, D. G. (2003) in Arsenic in Goundwater, eds. Welch, A. H. & Stollenwerk, K. G. (Kluwer, Boston), pp. 1–25.
- 29.Burns, P. C. & Percival, J. B. (2001) Can. Mineral. 39, 809–818. [Google Scholar]
- 30.Mullen, D. J. E. & Nowacki, W. (1972) Z. Kristallogr. 136, 48–65. [Google Scholar]
- 31.Bonazzi, P., Menchetti, S. & Pratesi, G. (1995) Am. Mineral. 80, 400–403. [Google Scholar]
- 32.Huber, R., Sacher, M., Vollmann, A., Huber, H. & Rose, D. (2000) Syst. Appl. Microbiol. 23, 305–314. [DOI] [PubMed] [Google Scholar]
- 33.Newman, D. K., Kennedy, E. K., Coates, J. D., Ahmann, D., Ellis, D. J., Lovely, D. R. & Morel, F. M. M. (1997) Arch. Microbiol. 168, 380–388. [DOI] [PubMed] [Google Scholar]
- 34.Newman, D. K., Beveridge, T. J. & Morel, F. M. M. (1997) Appl. Environ. Microbiol. 63, 2022–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douglass, D. L., Shing, C. C. & Wang, G. (1992) Am. Mineral. 77, 1266–1274. [Google Scholar]
- 36.Root, R. A. (2003) M.S. thesis (Arizona State University, Tempe).
- 37.Rehr, J. J., Albers, R. C. & Zabinsky, S. I. (1992) Phys. Rev. Lett. 69, 3397–3400. [DOI] [PubMed] [Google Scholar]
- 38.O'Day, P. A., Rehr, J. J., Zabinsky, S. I. & Brown, G. E., Jr. (1994) J. Am. Chem. Soc. 116, 2938–2949. [Google Scholar]
- 39.Bourrie, G., Trolard, F., Genin, J. M. R., Jaffrezic, A., Maitre, V. & Abdelmoula, M. (1999) Geochim. Cosmochim. Acta 63, 3417–3427. [Google Scholar]
- 40.Carroll, S. A., O'Day, P. A., Esser, B. & Randall, S. (2002) Geochem. Trans. 3, 81–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Day, P. A., Rivera, N., Root, R. & Carroll, S. A. (2004) Am. Mineral. 89, 572–585. [Google Scholar]
- 42.Keon, N. E., Swartz, C. H., Brabander, D. J., Harvey, C. & Hemond, H. F. (2001) Environ. Sci. Technol. 35, 2778–2784. [DOI] [PubMed] [Google Scholar]
- 43.Wilkin, R. T., Wallschlager, D. & Ford, R. G. (2003) Geochem. Trans. 4, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lennie, A. R. & Vaughan, D. J. (1996) in Mineral Spectroscopy: A Tribute to Roger G. Burns, eds. Dyar, M. D., McCammon, C. & Schaefer, M. W. (Geochem. Soc., St. Louis, MO), Special Publication no. 5, pp. 117–131.
- 45.Farquhar, M. L., Charnock, J. M., Livens, F. R. & Vaughan, D. J. (2002) Environ. Sci. Technol. 36, 1757–1762. [DOI] [PubMed] [Google Scholar]
- 46.Bostick, B. C. & Fendorf, S. (2003) Geochim. Cosmochim. Acta 67, 909–921. [Google Scholar]
- 47.Randall, S. R., Sherman, D. M. & Ragnarsdottir, K. V. (2001) Geochim. Cosmochim. Acta 65, 1015–1023. [Google Scholar]
- 48.Bowell, R. J. (1994) Appl. Geochem. 9, 279–286. [Google Scholar]
- 49.O'Day, P. A. (1999) Rev. Geophys. 37, 249–274. [Google Scholar]
- 50.Brown, G. E., Jr., & Parks, G. A. (2001) Int. Geol. Rev. 43, 963–1073. [Google Scholar]
- 51.Labrenz, M., Druschel, G. K., Thomsen-Ebert, T., Gilbert, B., Welch, S. A., Kemner, K. M., Logan, G. A., Summons, R. E., De Stasio, G., Bond, P. L., et al. (2000) Science 290, 1744–1747. [DOI] [PubMed] [Google Scholar]
- 52.Raven, K. P., Jain, A. & Loeppert, R. H. (1998) Environ. Sci. Technol. 32, 344–349. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}