

Abstract
This work presents a general method for determining single-molecule intramolecular dynamics in biomolecules by using a reporter fluorophore, whose fluorescence is quenched or partially quenched as a result of intramolecular motion, and a remote observer fluorophore. These fluorophores were excited independently with two different lasers, and the ratio of the two fluorophores' fluorescence was calculated. The time-varying ratio was then filtered to reduce contributions from molecules outside the overlapped laser volume and then correlated. The rates of opening and closing of a DNA hairpin were measured by using both fluorescence correlation spectroscopy and this method for comparison. We found at 50 pM, where molecules were studied one by one as they diffused through the probe volume, we obtained accurate opening and closing rates and could also measure dynamic heterogeneity. To demonstrate applicability to a more complex biological molecule we then probed intramolecular motions in the dimer of a human telomerase RNA fragment (hTR380-444), in the presence of an excess of monomer. The motion was found to occur on the time scale of 180-750 μs and slowed with increasing magnesium ion concentration. Blocking experiments using complementary oligonucleotides suggested that the motion involves substantial changes in dimer tertiary structure. This method appears to be a general method for selectively studying intramolecular motion in large biomolecules or complexes.
Many biological molecules perform intramolecular dynamics to convert between different conformations and perform their biological functions. These motions can be revealed by fluorescence studies based on fluorescence resonance energy transfer (FRET) or quenching. These studies are of two general types: single-molecule experiments, where molecules are analyzed one at a time, and fluctuation spectroscopy, where a few molecules (typically <10) are analyzed. Single-molecule experiments have the advantage of directly determining heterogeneity without signal averaging, but they have reduced signal-to-noise ratio, limiting the possible time resolution. In contrast, fluctuation-based methods have improved single-to-noise ratio, allowing the measurement of faster processes, but they may average out some of the dynamical heterogeneity.
Single-molecule fluorescence spectroscopy (1-8) at room temperature has been performed both on surface-attached molecules and in solution. Studies on immobilized molecules offer longer observation times but there are issues about the surface perturbing the dynamics of interest, particularly for small molecules (9). For solution-phase studies confocal microscopy is used to define a small open probe volume, and one or two lasers are focused to a diffraction-limited spot to make the measurements as the molecules diffuse across the probe volume. This process eliminates the problem with the perturbation of dynamics by the surface. However, it creates new constraints such as reduced signal-to-noise ratio and limited observation time, and for these reasons there have been a very limited number of studies of the intramolecular dynamics of biomolecules in solution at the single-molecule level (7, 10, 11).
Fluctuation spectroscopy is based on analyzing the fluctuations of physical observables around equilibrium from a few molecules at a time. There are two possible sources of information: (i) fluctuations in time and (ii) fluctuations in amplitude. For fluorescence intensity measurements, these fluctuations gave rise to the techniques of fluorescence correlation spectroscopy (FCS) (12-16) and the photon counting histogram, respectively (17-19). DNA hairpin dynamics has been studied in solution by both FRET and fluorophore quenching with FCS-based methods. Bonnet et al. (20) measured the autocorrelation function of a DNA hairpin labeled with fluorophore and quencher, divided by the autocorrelation function of a reference hairpin labeled only with the fluorophore. We will hereafter refer to this general approach as the reference method. By doing ratios, the contribution to the autocorrelation function from diffusion is eliminated, leaving the contribution exclusively caused by intramolecular motion. In an alternative approach, Wallace and coworkers (21, 22) used FRET between a donor and an acceptor fluorophore on the DNA hairpin, where autocorrelation of the FRET signal provided measurement of intramolecular motion. Both of these studies were performed at 10 nM DNA hairpin concentration, where there are ≈10 molecules in the probe volume on average, so some degree of population-based averaging of the dynamics was observed.
In this work we present a general method for measuring the dynamics of biomolecules based on determining ratios for the fluorescence between an observer fluorophore, at some convenient position on the molecule, and a reporter fluorophore whose fluorescence is quenched or partially quenched as a result of motion. The quenching can be caused by a change in the microenvironment or proximity to an extrinsic quencher. Ratiometric single-molecule fluorescence measurements were originally introduced by Weiss and coworkers (13, 23, 24) to remove the effect of excitation beam heterogeneity. Recently, alternating excitation of donor and acceptor fluorophores has been introduced (25). Here, we extend ratiometric measurements to dynamics at the single-molecule level. We used a model DNA hairpin system so we could make measurements of the opening and closing rates by using the method of Bonnet et al. (20) and comparing those results to our method. We found excellent agreement, validating our method.
To demonstrate that the method is applicable to larger biological molecules we then probed local motion in a region of the RNA component of the enzyme human telomerase (hTR) (26). We studied the fragment hTR380-444 containing the CR7 domain and the J7b/8a loop, which we have previously shown to comprise an RNA-RNA interaction site that promotes hTR dimerization. We reasoned that dimerization of hTR380-444 would alter both the structural and dynamic properties of the fragment, making it an important system suitable for fundamental study. We found that the hTR380-444 dimer showed submillisecond intramolecular dynamics.
Experimental Procedures
The HPLC-purified oligonucleotides I, II, III, IV, V, VI, and VII used in these experiments are summarized in Table 1 (for details see Supporting Text, which is published as supporting information on the PNAS web site). The hTR380-444 was made and labeled at the 5′ end with either Alexa 488 or Alexa 647 as described (26).
Table 1. Oligonucleotide sequence of samples used in this study.
| DNA samples | Sequence |
|---|---|
| I | 5′-X1ACG CCC CAA AAA AAA AAA AAA AAC CGG CGT GCC ATC CGC ACC GTG GCC ATT ATC CTT TTA CCT CA-3′ |
| II | 5′-X2TGA GGT AAA AGG ATA ATG GCC ACG GTG CGG ATG GCX3-3′ |
| III | 5′-TGA GGT AAA AGG ATA ATG GCC ACG GTG CGG ATG GC-3′ |
| IV | 5′-X1TAC CGA TTA CTG GCT CTT ATC CTA GGC TTA AGT TAC ACT A-3′ |
| V | 5′-X2TAG TGT AAC TTA AGC CTA GGA TAA GAG CCA GTA ATC GGT A-3′ |
| VI | 5′-TCG CTC C-3′ |
| VII | 5′-TCG CTC CTT TTT GAG CCG A-3′ |
The fluorophores are coupled to DNA with a six-carbon linkage. X1, rhodamine green; X2, Alexa 647; X3, dabcyl.
The apparatus used to achieve dual-color single-molecule fluorescence coincidence detection has been described (27). Briefly, an argon ion laser and helium neon laser were used for simultaneous excitation, with a Nikon PlanApo ×60 numerical aperture 1.4 oil-immersion objective. The overlap volume was carefully optimized so that it was 30% of the total excited volume. The fluorescence was collected by using the same objective, separated by using a dichroic, and detected with two avalanche photodiodes. All experiments were carried out on a temperature stage controlled within ±0.1°C (Linkman Scientific, Surrey, U.K.). The excitation laser powers were adjusted to 10 μW for the red laser and 30 μW for the blue laser to give comparable counts on both channels. For ratio autocorrelation experiments a bin time of 20 μs was used, and the total time for one experiment was 3 min. For measurement of the single-molecule histogram the red laser power was 100 μW, and the blue laser was 300 μW with a time bin of 1 ms. The experiment took 90 min.
The DNA hairpin sample in Fig. 1A was prepared by mixing two equivalents of the 65-base DNA I with its complementary 35-base DNA II for the ratio experiments or III for the reference experiments. The mixture was heated to 90°C for 10 min and slowly cooled down to room temperature. Bulk fluorescence measurements showed no FRET. DNA samples IV and V were used to form a 40-bp duplex as a control experiment, shown in Fig. 2A. For the single DNA hairpin molecule experiments, the hairpin DNA was diluted to 50 pM in 100 mM KCl and 10 mM Tris·HCl, pH 7.4 plus 0.01% Tween 20 to prevent surface adhesion of the DNA molecules.
Fig. 1.

Samples used in these experiments. (A) The DNA hairpin labeled with a red-excited dye (Alexa 647), a blue-excited dye (rhodamine green), and a quencher (dabcyl). The molecule was designed to fluctuate between open and closed states in solution at ambient temperature as shown by the dotted arrow. (B) The hTR380-444 dimer labeled with red-excited dye (Alexa 647) and blue-excited dye (Alexa 488). The gray bases on the top and bottom monomers can be blocked by DNA VI or VII, respectively.
Fig. 2.

Filtered ratiometric autocorrelation. (A) Two-color ratio autocorrelation curves for the 50 pM DNA hairpin sample. ○, the unfiltered two-color ratio autocorrelation at 50 pM; □, the filtered two-color ratio autocorrelation (ratio range 0.3-0.7); and ▵, the control sample of 40-bp duplex DNA in which there is no hairpin loop. The lines are fits using a stretched exponential function. (B) Schematic of the filtered ratio autocorrelation method. (C) Single-molecule histograms of the ratio, R. The AND model requires coincident events above a threshold of 15 counts per ms on both channels, whereas the OR model requires 15 counts per ms on either channel. The dashed lines indicate the subunit ranges between 0.3 and 0.7. The number of events in the AND histogram is 3% of the OR histogram.
For the hTR experiments, hTR380-444 solution containing equimolar amounts (2 nM) of both Alexa 488-labeled and Alexa 647-labeled samples was prepared in TKM buffer (10 mM Tris·HCl, pH 7.4/100 mM KCl/10 mM MgCl2). The stock solution was incubated at 37°C for 1 h and then diluted to 100 pM total hTR concentration in TKM buffer containing an additional 0.01% Tween 20. Using a similar method to that used previously for DNA blocking experiments (26), the hTR380-444 stock sample was mixed with a 100-fold molar excess of complementary DNA (VI or VII) (Table 1) and incubated at 37°C for 1 h before diluting to 100 pM total hTR concentration for experiments. A 395UUUU398 mutant monomer hTR380-444 labeled with Alexa 647 at its 5′ end was also used in a control experiment since this mutant cannot dimerize (26). It was mixed with DNA VI labeled with rhodamine green at its 5′ end, at 10 nM in equimolar ratio, and further diluted to 50 pM for a control experiment.
Reference Method. We ran fluorescence correlation spectroscopy experiments for the hairpin sample (DNA I plus DNA II) and the control sample (DNA I plus DNA III) at 10 nM. The laser powers were kept the same as the ratiometric autocorrelation experiments with a 20-μs bin time.
Filtered Ratiometric Autocorrelation (FRAC) Spectroscopy. The ratiometric method for determination of the relaxation time for the hairpin is based on simultaneously recording the fluorescence of the reporter and observer as shown in Fig. 2B. The fluorescence is divided by the total fluorescence intensity to determine the ratio, R:
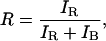 |
1 |
where IR and IB are the red excited and blue excited fluorescence intensities, respectively. R can take a value between 0 and 1 and vary with time due to molecules diffusing into and out of the probe volumes and intramolecular motion. R is therefore a function of time, R(t). The autocorrelation function of R provides the time scale of these dynamics and can be calculated by
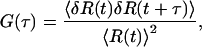 |
2 |
where 〈R(t)〉 is the time average of R(t) and δR(t) is the difference of R(t) from 〈R(t)〉, the fluctuation in R. 〈δR(t) δR(t + τ)〉 is the time average of the product of the fluctuation in R at time t and after a delay t + τ and will only be nonzero within the time scale of the dynamics.
In principle, just like autocorrelation of FRET (21, 22, 28), autocorrelation of R should eliminate or greatly reduce the contribution of diffusion to the autocorrelation function, although this needs to be checked by control experiments. However, there is an additional complication for ratio autocorrelation since there is imperfect overlap of the red and blue laser focal volumes. This complication means it is necessary to remove the contribution to the ratio autocorrelation function from molecules that pass through the volumes excited by the red laser or blue laser only. These molecules will have R values of 0 and 1. To reduce this background, we filtered the data and analyzed only ratio values of 0.3-0.7, as shown in Fig. 2C, to extract the real intramolecular dynamics. This range was optimized to select >90% of all coincidence fluorescence bursts and minimize the contribution from noncoincident events. All values outside this range were set to zero to perform autocorrelation. A smaller range of R led to poorer signal-to-noise ratio, and a larger range led to some diffusional contribution to the autocorrelation function.
Both the filtered and unfiltered two-color ratio autocorrelation functions shown in Fig. 2 A could be fitted by a stretched exponential (21, 22),
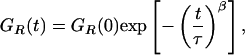 |
3 |
where τ corresponds to the effective relaxation time associated with the correlated motion, and β is a stretch parameter, describing the heterogeneity of the system. β can vary between 1 (where the system displays normal two-state Arrhenius kinetics, with one discrete energy barrier) and 0 (where there is a continuum of equal energy barriers and the system shows power-law kinetics). The mean relaxation time 〈τ〉 can be related to τ and β by
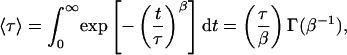 |
4 |
where Γ(β-1) is a gamma function.
Results and Discussion
DNA Hairpin. We first studied the opening and closing of a DNA hairpin (Fig. 1 A) to validate our method. The DNA hairpin has the reporter fluorophore quenched in the closed state and has the same length loop and stem sequence as the hairpin studied previously by FRET (28). The duplex region enables attachment of the observer fluorophore to a part of the structure that is not expected to change during hairpin opening and closing. The single-molecule fluorescence melting curve was measured (Fig. 6, which is published as supporting information on the PNAS web site). As in our previous work, we used a two-state model to fit the fluorescence-melting curve (21),
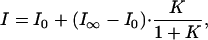 |
5 |
where I0, I∞ denote the fluorescence intensity of all-open and all-closed states (high and low temperature limits), and K is the equilibrium constant. Assuming that the enthalpy and entropy changes are temperature-independent, then the equilibrium constant K = exp(-ΔH/RT + ΔS/R). This function fitted the melting curves well. The standard enthalpy change for hairpin opening, ΔH, was determined to be -140 ± 20 kJ/mol, and the standard entropy change, ΔS, was 450 ± 70 J/mol, in reasonable agreement with ensemble measurements of -120 kJ/mol and 390 J/mol, respectively (see Supporting Text for experimental details).
We then went on to measure the dynamics of hairpin opening and closing by making measurements at a concentration of DNA hairpin of 50 pM. Typical unfiltered and filtered two-color ratio GR(t) autocorrelation functions are shown in Fig. 2 A. A control experiment with a 40-bp DNA duplex (DNA IV and DNA V) at 20°C was also performed, and the filtered two-color ratio autocorrelation was determined (Fig. 2 A). The control is flat with amplitude close to zero. No dynamics is observable, showing that there is no contribution of local f luorophore motion to the filtered ratio autocorrelation function.
For comparison the reference method was also used on the same DNA hairpin sample. Experiments were performed at 10 nM and also fitted with a stretched exponential function. The results of these experiments are shown in Table 2. The mean relaxation time using the filtered ratio method was in good agreement with the reference method. It was also clear that without filtering there was a difference in relaxation times of a factor of two because of the diffusional contribution to the autocorrelation function.
Table 2. DNA hairpin relaxation times at 20°C.
| Methods | GR(τ) | τ(μs) | β | <τ>(μs) |
|---|---|---|---|---|
| Filtered ratio correlation at 50 pM | 0.091 ± 0.002 | 143 ± 18 | 0.443 ± 0.005 | 326 ± 17 |
| Ratio correlation at 50 pM | 0.049 ± 0.001 | 327 ± 6 | 0.452 ± 0.004 | 726 ± 12 |
| Reference | 0.253 ± 0.007 | 340 ± 7 | 0.943 ± 0.110 | 360 ± 9 |
Times were measured in this work by three different methods. See text for details.
To determine the opening and closing rates for the hairpin at different temperatures, we followed the approach described in our previous work (23). We assumed a two-state model (open and closed) and measured the apparent relaxation time and equilibrium constant from 20°C to 50°C. The results are presented as an Arrhenius plot (Fig. 3). Non-Arrhenius kinetics for the closing rate of hairpin sample was observed. This behavior was reported previously for the same hairpin but without the overhang (21). The activation energies for the opening and closing processes were calculated based on four points with a function k(T) = Cexp(-Ea/RT) at low and high temperatures, respectively. The opening activation energies were 33.4 ± 3.4 kJ/mol calculated by the filtered ratio autocorrelation method in comparison to 29.8 ± 3.6 kJ/mol from the reference method. The closing activation energies were -45.9 ± 8.3 and -44.8 ± 3.1 kJ/mol, respectively. The results of these experiments show that the filtered ratio method can accurately determine the rates and activation energies for DNA hairpin opening and closing.
Fig. 3.

Arrhenius plots of the closing and opening kinetics for the DNA hairpin sample. Squares represent the results from the filtered two-color ratio autocorrelation at 50 pM. Triangles represent the results of the reference method.
hTR380-444 Dimer. We then applied the same method to the hTR380-444 dimer (Fig. 1B). Under the experimental conditions, 8% of the molecules entering the laser-excited volumes were dimers (26). We obtained a filtered ratio autocorrelation for dimer molecules, which could be fitted to a stretched exponential as shown in Fig. 7, which is published as supporting information on the PNAS web site. The beta parameter was 0.5 ± 0.1 in this case, indicating dynamical heterogeneity. A control experiment with the 395UUUU398 mutant hTR380-444, in which there was no detectable dimer formed (26), gave no ratio autocorrelation function. It also was not possible to obtain a cross-correlation function in this experiment since only 8% of the molecules were dimers (data not shown). To elucidate the nature of the motion we then determined the activation energy at different magnesium ion concentrations by measuring the apparent rate constant over a range of temperatures from 20°C to 50°C (Fig. 4). The presence of magnesium ions is known to influence the structure and folding of RNA (29-34). We confirmed that the addition of magnesium ions did not reduce the fluorescence intensity of individual molecules or the average count rates for the experiment. The apparent activation energy for the dynamics increased with magnesium ion and was 15.5 ± 1.2, 23.7 ± 1.3, and 27.5 ± 1.9 kJ/mol-1 in 0, 10, and 200 mM magnesium ion, respectively.
Fig. 4.

Arrhenius plot of the apparent rate constant for hTR380-444 dimer intramolecular dynamics at different magnesium ion concentrations.
Further DNA “blocking” experiments were carried out at 20°C, using complementary oligonucleotides VI and VII, to elucidate which part of the hTR dimer is responsible for the observed intramolecular motion. First, DNA VI was used to block one side of the 5′ end of hTR380-444 at 20°C (Fig. 5). The amplitude of the autocorrelation function was similar to the unblocked hTR dimer but the relaxation time increased from 320 ± 45 to 629 ± 135 μs. When 19-base DNA VII was used to “tie together” the 5′ and 3′ ends of hTR380-444, the relaxation time was further increased to 886 ± 200 μs without a reduction in amplitude. An experiment was performed on only the monomer by using the 395UUUU398 mutant monomer hTR380-444 that cannot form dimer, blocked with DNA VII labeled with Alexa 647. This complex has both fluorophores on the monomer for coincidence experiments. The relaxation time decreased to 200 ± 18 μs, but more importantly we found that the correlation amplitude was reduced to 15% of that for hTR380-444 dimer.
Fig. 5.

Relaxation times for blocking experiments on the hTR380-444 dimer at 20°C. See details in the text.
Comparison of Methods. The filtered ratio method gave values for the mean relaxation time, the opening and closing rates, and activation energies in good agreement with the reference method. The main difference was that the FRAC measurements were performed in the single-molecule regime. This process removed averaging and gave a β parameter value of 0.45 ± 0.02 in good agreement with the value of 0.46 measured previously on a similar hairpin by using FRET. In contrast to the reference method, there were on average 10 molecules in the probe volume at one time, which averaged out intramolecular motion and gave a beta value close to 1.
It is clear that filtering of the ratio autocorrelation is needed. Without filtering, the signal caused by molecules not passing through the overlapped lasers dominates the autocorrelation function, leading to incorrect measurement of the relaxation time and hence intramolecular dynamics. In the hairpin experiments we used filtering to predominantly correlate fluorescence from molecules that passed through the overlapped laser volume. However, in the hTR study the filtering served a second purpose and selected only dimerized RNA molecules passing through the overlapped laser probe volume from the excess of monomers.
Intramolecular Motion of the hTR380-444 Dimer. After validating the method on a DNA hairpin sample we have applied it to a more complex and biologically relevant system, the hTR380-444 dimer. This dimer consists of 128 bases and is believed to be formed by hydrogen-bonding of up to eight complementary bases (26). The tertiary structure of the dimer is not known. We put a fluorophore at the 5′ end of the monomer to measure intramolecular motion. Here, the experimental system differed from the hairpin, since no quencher was needed, and motion of the RNA or fluorophore linkage resulted in reduced fluorescence of the fluorophore. Unlike FRET this quenching presumably requires local contact and the reduced fluorescence state will last only for a short duration compared to the overall time of any intramolecular motion. Thus each fluorophore can act as an observer for the other for most of the time. It is also possible in principle that the two fluorophores undergo motion in phase, which would result in a zero autocorrelation function. However, we found that the amplitude of the FRAC for the hTR380-444 dimer was comparable to the DNA hairpin, showing that the motions are not synchronized and each fluorophore could act as an observer for the other.
The stabilization effect of magnesium ion on RNA structure is well documented, and, generally, it stabilizes tertiary structure and slows down intramolecular motion (31, 34, 35). This effect has been clearly measured in the experiment. The activation energies measured are small and without Mg2+, the activation energy (15.5 ± 1.2 kJ/mol) is almost the same as the activation energy caused by the water viscosity, 14.4 kJ/mol. This finding means that the motion is predominantly the intramolecular movement of the RNA structural units, since rearrangement of these units would be expected to have a higher activation energy (36). The time scale of the motion is also longer than that for the formation of secondary structure for RNA in folding experiments, on a 10- to 100-μs time scale (37-39), supporting this conclusion.
For a DNA duplex, which is rigid, the FRAC spectra were flat with an amplitude close to zero, indicating that local fluorophore motion does not contribute to the measured motion. A similar flat FRAC spectra would be expected for the blocking oligonucleotide experiments if the regions to which the blocking DNA hybridized were directly responsible for the motion. In contrast, we observed that the amplitudes of the FRAC spectra were comparable but the DNA blocks slowed down the intramolecular dynamics. This finding suggests that the motion arises from parts of the dimer that are not blocked. It appears that increasing the rigidity of the RNA, by formation of rigid duplex regions, slows down the motion. This conclusion is strongly supported by the experiment on the 395UUUU398 mutant hTR380-444 monomer that cannot dimerize (26), with the fluorophore-labeled DNA VII block. This experiment on the monomer gave a large reduction in the FRAC amplitude compared with the experiment performed on the hTR380-444 dimer with the same blocking oligonulceotide, but lacking a fluorophore.
Studies of RNA dynamics have been performed at the single-molecule level of large RNA molecules by FRET (8, 29, 30, 40) and optical tweezers (41) at the millisecond time scale. Small RNA molecules, <30 bases, have been studied at the ensemble level on the submillisecond time scale by NMR, EPR, or fluorescence (42-48). Therefore, there are little data to compare with the results of this study where we have studied a large RNA molecule intramolecular dynamics on the submillisecond time scale. The most relevant studies we can find are those on the tRNA early melting transition (46, 49), which show a similar low activation energy and submillisecond time scale to that observed here. This finding suggests that other RNA structures can make similar time-scale motions involving changes in tertiary structure.
Conclusions
In summary, we have shown that the filtered ratio autocorrelation method applied in the single-molecule limit provides accurate measurement of the intramolecular dynamics, allowing the determination of the opening and closing rates of a DNA hairpin and the dynamical heterogeneity. The use of two-color excitation enables more straightforward labeling of the sample without prior knowledge of its tertiary structure. The requirement to control the distance between the two fluorophores, as for FRET, are removed. The observer fluorophore can be placed at any position on the molecule, provided it is not undergoing intramolecular motion, and only the reporter fluorophore needs to be placed at the desired position in the molecule to probe motion. Filtering of the data allows selection of molecules that pass through the overlapped laser volumes and also the complex or species of interest. The method is applicable to a large RNA molecule, providing equilibrium measurement of intramolecular dynamics in solution and could be extended to other large biological molecules such as proteins and RNA and DNA protein complexes.
Supplementary Material
Acknowledgments
This work was funded by the Biotechnology and Biological Sciences Research Council.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: FRET, fluorescence resonance energy transfer; hTR, RNA component of the enzyme human telomerase; FRAC, filtered ratiometric autocorrelation; I, intensity.
References
- 1.Xie, X. S. & Trautman, J. K. (1998) Annu. Rev. Phys. Chem. 49, 441-480. [DOI] [PubMed] [Google Scholar]
- 2.Weiss, S. (1999) Science 283, 1676-1683. [DOI] [PubMed] [Google Scholar]
- 3.Schuler, B., Lipman, E. A. & Eaton, W. A. (2002) Nature 419, 743-747. [DOI] [PubMed] [Google Scholar]
- 4.Plakhotnik, T., Donley, E. A. & Wild, U. P. (1997) Annu. Rev. Phys. Chem. 48, 181-212. [DOI] [PubMed] [Google Scholar]
- 5.Nie, S. M. & Zare, R. N. (1997) Annu. Rev. Biophys. Biomol. Struct. 26, 567-596. [DOI] [PubMed] [Google Scholar]
- 6.Ha, T. J. (2004) Biochemistry 43, 4055-4063. [DOI] [PubMed] [Google Scholar]
- 7.Eggeling, C., Fries, J. R., Brand, L., Gunther, R. & Seidel, C. A. M. (1998) Proc. Natl. Acad. Sci. USA 95, 1556-1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhuang, X. W., Kim, H., Pereira, M. J. B., Babcock, H. P., Walter, N. G. & Chu, S. (2002) Science 296, 1473-1476. [DOI] [PubMed] [Google Scholar]
- 9.Grunwell, J. R., Glass, J. L., Lacoste, T. D., Deniz, A. A., Chemla, D. S. & Schultz, P. G. (2001) J. Am. Chem. Soc. 123, 4295-4303. [DOI] [PubMed] [Google Scholar]
- 10.Edman, L., Mets, U. & Rigler, R. (1996) Proc. Natl. Acad. Sci. USA 93, 6710-6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neuweiler, H., Schulz, A., Bohmer, M., Enderlein, J. & Sauer, M. (2003) J. Am. Chem. Soc. 125, 5324-5330. [DOI] [PubMed] [Google Scholar]
- 12.Magde, D., Elson, E. & Webb, W. W. (1972) Phys. Rev. Lett. 29, 705-708. [Google Scholar]
- 13.Ha, T. J., Ting, A. Y., Liang, J., Caldwell, W. B., Deniz, A. A., Chemla, D. S., Schultz, P. G. & Weiss, S. (1999) Proc. Natl. Acad. Sci. USA 96, 893-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hess, S. T., Huang, S. H., Heikal, A. A. & Webb, W. W. (2002) Biochemistry 41, 697-705. [DOI] [PubMed] [Google Scholar]
- 15.Haustein, E. & Schwille, P. (2003) Methods 29, 153-166. [DOI] [PubMed] [Google Scholar]
- 16.Ying, L. M., Green, J. J., Li, H. T., Klenerman, D. & Balasubramanian, S. (2003) Proc. Natl. Acad. Sci. USA 100, 14629-14634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen, Y., Muller, J. D., So, P. T. C. & Gratton, E. (1999) Biophys. J. 77, 553-567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller, J. D., Chen, Y. & Gratton, E. (2000) Biophys. J. 78, 474-486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perroud, T. D., Bo, H. A., Wallace, M. I. & Zare, R. N. (2003) ChemPhysChem 4, 1121-1123. [DOI] [PubMed] [Google Scholar]
- 20.Bonnet, G., Krichevsky, O. & Libchaber, A. (1998) Proc. Natl. Acad. Sci. USA 95, 8602-8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wallace, M. I., Ying, L. M., Balasubramanian, S. & Klenerman, D. (2001) Proc. Natl. Acad. Sci. USA 98, 5584-5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ying, L. M., Wallace, M. I., Balasubramanian, S. & Klenerman, D. (2000) J. Phys. Chem. B 104, 5171-5178. [Google Scholar]
- 23.Dahan, M., Deniz, A. A., Ha, T. J., Chemla, D. S., Schultz, P. G. & Weiss, S. (1999) Chem. Phys. 247, 85-106. [Google Scholar]
- 24.Deniz, A. A., Dahan, M., Grunwell, J. R., Ha, T. J., Faulhaber, A. E., Chemla, D. S., Weiss, S. & Schultz, P. G. (1999) Proc. Natl. Acad. Sci. USA 96, 3670-3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kapanidis, A., Lee, N. K., Laurence, T., Doose, S., Margeat, E. & Weiss, S. (2004) Proc. Natl. Acad. Sci. USA 101, 8936-8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ren, X. J., Gavory, G., Li, H. T., Ying, L. M., Klenerman, D. & Balasubramanian, S. (2003) Nucleic Acids Res. 31, 6509-6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, H. T., Ying, L. M., Green, J. J., Balasubramanian, S. & Klenerman, D. (2003) Anal. Chem. 75, 1664-1670. [DOI] [PubMed] [Google Scholar]
- 28.Wallace, M. I., Ying, L. M., Balasubramanian, S. & Klenerman, D. (2000) J. Phys. Chem. B 104, 11551-11555. [Google Scholar]
- 29.Kim, H. D., Nienhaus, G. U., Ha, T., Orr, J. W., Williamson, J. R. & Chu, S. (2002) Proc. Natl. Acad. Sci. USA 99, 4284-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie, Z., Srividya, N., Sosnick, T. R., Pan, T. & Scherer, N. F. (2004) Proc. Natl. Acad. Sci. USA 101, 534-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Draper, D. E. (1996) Nat. Struct. Biol. 3, 397-400. [DOI] [PubMed] [Google Scholar]
- 32.Al-Hashimi, H. M., Gosser, Y., Gorin, A., Hu, W. D., Majumdar, A. & Patel, D. J. (2002) J. Mol. Biol. 315, 95-102. [DOI] [PubMed] [Google Scholar]
- 33.Al-Hashimi, H. M., Gorin, A., Majumdar, A., Gosser, Y. & Patel, D. J. (2002) J. Mol. Biol. 318, 637-649. [DOI] [PubMed] [Google Scholar]
- 34.Furtig, B., Richter, C., Wohnert, J. & Schwalbe, H. (2003) Chembiochem 4, 936-962. [DOI] [PubMed] [Google Scholar]
- 35.Al-Hashimi, H. M., Pitt, S. W., Majumdar, A., Xu, W. J. & Patel, D. J. (2003) J. Mol. Biol. 329, 867-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li, Y., Bevilacqua, P. C., Mathews, D. & Turner, D. H. (1995) Biochemistry 34, 14394-14399. [DOI] [PubMed] [Google Scholar]
- 37.Batey, R. T. & Doudna, J. A. (1998) Nat. Struct. Biol. 5, 337-340. [DOI] [PubMed] [Google Scholar]
- 38.Cohen, S. B. & Cech, T. R. (1997) J. Am. Chem. Soc. 119, 6259-6268. [Google Scholar]
- 39.Szewczak, A. A. & Cech, T. R. (1997) RNA Publ. RNA Soc. 3, 838-849. [PMC free article] [PubMed] [Google Scholar]
- 40.Zhuang, X. W. & Rief, M. (2003) Curr. Opin. Struct. Biol. 13, 88-97. [DOI] [PubMed] [Google Scholar]
- 41.Liphardt, J., Onoa, B., Smith, S. B., Tinoco, I. & Bustamante, C. (2001) Science 292, 733-737. [DOI] [PubMed] [Google Scholar]
- 42.Hoogstraten, C. G., Wank, J. R. & Pardi, A. (2000) Biochemistry 39, 9951-9958. [DOI] [PubMed] [Google Scholar]
- 43.Menger, M., Eckstein, F. & Porschke, D. (2000) Biochemistry 39, 4500-4507. [DOI] [PubMed] [Google Scholar]
- 44.Menger, M., Eckstein, F. & Porschke, D. (2000) Nucleic Acids Res. 28, 4428-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Craig, M. E., Crothers, D. M. & Doty, P. (1971) J. Mol. Biol. 62, 383-401. [DOI] [PubMed] [Google Scholar]
- 46.Crotheres, D. M., Cole, P., Hilberts, C. W. & Shulman, R. G. (1974) J. Mol. Biol. 87, 63-88. [DOI] [PubMed] [Google Scholar]
- 47.Qin, P. Z., Hideg, K., Feigon, J. & Hubbell, W. L. (2003) Biochemistry 42, 6772-6783. [DOI] [PubMed] [Google Scholar]
- 48.Dayie, K. T., Brodsky, A. S. & Williamson, J. R. (2002) J. Mol. Biol. 317, 263-278. [DOI] [PubMed] [Google Scholar]
- 49.Cole, P. E. & Crothers, D. M. (1972) Biochemistry 11, 4368-4374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}