

Abstract
SUZ12 is a recently identified Polycomb group (PcG) protein, which together with EZH2 and EED forms different Polycomb repressive complexes (PRC2/3). These complexes contain histone H3 lysine (K) 27/9 and histone H1 K26 methyltransferase activity specified by the EZH2 SET domain. Here we show that mice lacking Suz12, like Ezh2 and Eed mutant mice, are not viable and die during early postimplantation stages displaying severe developmental and proliferative defects. Consistent with this, we demonstrate that SUZ12 is required for proliferation of cells in tissue culture. Furthermore, we demonstrate that SUZ12 is essential for the activity and stability of the PRC2/3 complexes in mouse embryos, in tissue culture cells and in vitro. Strikingly, Suz12-deficient embryos show a specific loss of di- and trimethylated H3K27, demonstrating that Suz12 is indeed essential for EZH2 activity in vivo. In conclusion, our data demonstrate an essential role of SUZ12 in regulating the activity of the PRC2/3 complexes, which are required for regulating proliferation and embryogenesis.
Keywords: embryonic development, EZH2, histone methylation, polycomb, SUZ12
Introduction
Polycomb group proteins (PcG) and Trithorax proteins regulate the correct expression of HOX genes by maintaining their repressive or active transcriptional state, respectively. Both classes of proteins are essential for Drosophila development and their mutations lead to homeotic transformation and fly lethality (Brock and van Lohuizen, 2001; Jacobs and van Lohuizen, 2002). PcG proteins are conserved throughout evolution, forming multiprotein complexes that regulate transcription through the induction of chromatin changes (Jenuwein and Allis, 2001).
In mammals, the best-characterized PcG complexes are the Polycomb repressive complexes (PRC), PRC1 and PRC2. PRC1 is a large complex consisting of more than 10 subunits, including the oncoprotein BMI1 as well as other PcG proteins, such as HPC, HPH and SCML (Jacobs and van Lohuizen, 2002). The PRC2 complex is a smaller complex containing at least four different subunits, including the three PcG proteins EZH2, EED and SUZ12 and the histone-binding proteins RbAp48/46 (reviewed in Cao and Zhang, 2004; Pasini et al, 2004). Recent findings have also reported the existence of an alternative larger form of the PRC2 complex named PRC3 containing EZH2, SUZ12, RbAp48/46 and two isoforms of EED (Kuzmichev et al, 2004). The EZH2-containing complexes (PRC2/3) have intrinsic histone methyltransferase (HMT) activities, by methylating lysine (K) residues 9 and 27 of histone H3 and K26 of histone H1. These modifications are dependent on the integrity of the EZH2 SET domain, and are believed to result in transcriptional repression of target genes (Cao and Zhang, 2004; Kuzmichev et al, 2004).
Recent results have shown that the PcG genes of the mammalian PRC2/3 complexes, EZH2, EED and SUZ12, are downstream targets of the pRB/E2F pathway (Weinmann et al, 2001; Bracken et al, 2003). Moreover, we and others have shown that both EZH2 and EED are essential for proliferation of primary and tumor cells (Varambally et al, 2002; Bracken et al, 2003). Consistent with the fact that deregulation of the pRB pathway is a hallmark of human cancer, recent data have demonstrated that EZH2 is overexpressed in a wide spectrum of human tumors, and its overexpression can be due to amplification of the EZH2 locus. Furthermore, EZH2 is a marker of the metastatic state of prostate and breast tumors and may have a causal role in development of cancer (Varambally et al, 2002; Bracken et al, 2003; Kleer et al, 2003). Indeed, ectopic expression of EZH2 can promote growth advantage in mouse embryo fibroblasts (MEFs) and can induce cellular transformation and invasive capacity to immortalized human breast epithelial cells (Bracken et al, 2003; Kleer et al, 2003).
The SUZ12 protein is the most recently identified protein of the EZH2-containing complexes. It contains two stretches of conserved amino acids (aa) containing a zinc-finger binding motif and a VEFS box, which is required for the interaction between SUZ12 and EZH2 (Yamamoto et al, 2004). To date, the physiological and functional role of SUZ12 in mammals has not been addressed, although the SUZ12 locus is part of a frequent translocation identified in endometrial stromal sarcomas (ESSs) (Koontz et al, 2001). The predicted fusion protein contains 88% of the C-terminal part of SUZ12, fused at its N-terminus to an uncharacterized zinc-finger-containing protein. These data suggest that overexpression of SUZ12 and/or the generation of a fusion protein with additional features to SUZ12 has a causal role in the genesis of ESSs.
Studies in Drosophila showed that consistent with being a PcG protein, su(z)12 mutations lead to strong homeotic transformation and fly lethality (Birve et al, 2001). Su(z)12 is conserved from plants to humans (Birve et al, 2001). Interestingly, even though Caenorhabditis elegans has EZH2 and EED homologs in a complex that retains H3 K27 HMT activity, this complex does not contain a SUZ12 homolog and a SUZ12 C. elegans homolog has not been identified (Cao and Zhang, 2004). This finding raises the question whether SUZ12 is required for PRC2/3 HMT activities, and if the biological function of SUZ12 is exerted through the binding to EZH2 and EED.
In this work we have addressed the biological and functional role of SUZ12 in mammals. We show that mice lacking Suz12 are not viable and die during embryogenesis at early postimplantation stages. We demonstrate that Suz12 is required for cellular proliferation, and that it is essential for the HMT activity of the PRC2/3 complexes both in vivo and in tissue culture. Furthermore, we show that SUZ12 is essential for the integrity of the PRC2/3 complexes and for the stability of EZH2. Taken together with the fact that both Ezh2 and Eed, as the Suz12 knockout mice, die during the postimplantation period of embryogenesis (Faust et al, 1995; O'Carroll et al, 2001), our data demonstrate that SUZ12 is an essential regulator of the activity of the EZH2-containing complexes.
Results
Analysis of Suz12 genetrap embryonic stem cells
To characterize the role of Suz12 in mouse development, we generated a mouse model lacking Suz12. An embryonic stem (ES) cell line containing a genetrap vector inserted in the Suz12 locus on mouse chromosome 11 was identified in the BayGenomics database (http://www.baygenomics.ucsf.edu). In this cell line, the genetrap cassette is inserted in the intron between exons 7 and 8 (Figure 1A). This insertion is predicted to lead to a C-terminal truncation of Suz12, resulting in the production of 276 N-terminal amino acids of Suz12 fused to 1323 aa of the β-galactosidase-neomycin (β-GEO) protein with a molecular weight of 179 kDa (Figure 1B). The truncated form of Suz12 does not contain the two conserved regions of the wild-type (WT) protein, including the domain required for binding to EZH2 (Yamamoto et al, 2004). This suggests that the truncated form of Suz12 would be a functionally inactive mutant. This is supported by the demonstration that production of a similar mutant form of Drosophila su(z)12 leads to strong homeotic transformations and fly lethality (Birve et al, 2001).
Figure 1.
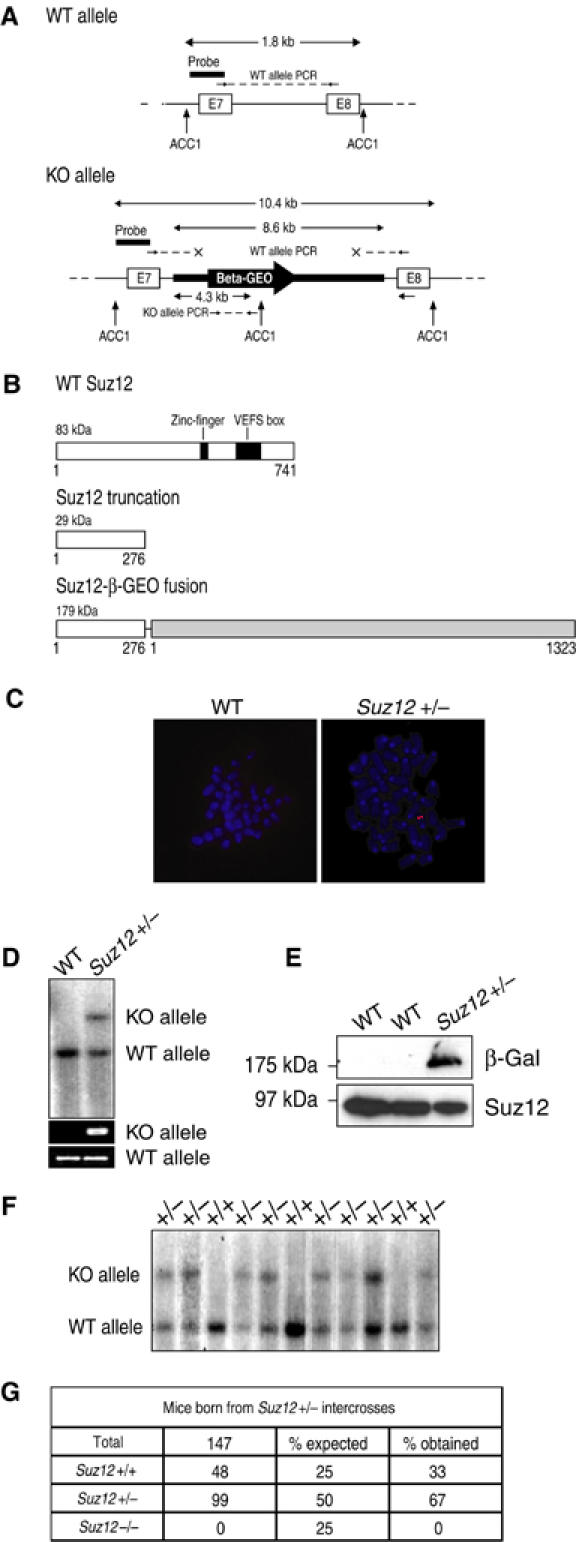
Suz12 is essential for mouse viability. (A) Close-up between exons 7 and 8 of the Suz12 locus is presented for both the WT (top panel) and the genetrap (KO) clones (bottom part). The strategies for genotyping the mice are presented. The probe is indicated (black horizontal bar) as well as the restriction sites (ACC1) used for Southern blot analysis. The PCR primers used are also indicated. The WT allele is detected by amplification of the entire intron 7. The presence of the KO allele was detected by PCR as part of the LacZ gene contained in the genetrap cassette. All fragments and amplified product sizes are indicated in kilobases. (B) Schematical representation of the WT Suz12 protein with the two conserved regions indicated, and the truncation products obtained in the KO mice. (C) FISH analysis of metaphase spreads of WT and Suz12 genetrap ES cells showing normal karyotype of the ES cells and a single insertion of the genetrap cassette in Suz12 targeted cells. (D) Southern blot analysis of WT and Suz12 genetrap ES cells showing the presence of the KO cassette in the Suz12 locus. (E) WB analysis showing the expression of the Suz12 WT protein in ES cells and the expression of the Suz12-β-GEO fusion only in the targeted ES cells. (F) Southern blot analysis showing the absence of Suz12 −/− mice from Suz12+/− intercrosses. (G) Summary table of the genotype of mice born from Suz12+/− intercrosses demonstrating that Suz12 KO mice are not born.
We analyzed the Suz12 genetrap ES cells to confirm the correct positioning and functionality of the insertion cassette. We designed a probe that specifically recognizes the genetrap cassette, and performed fluorescence in situ hybridization (FISH) analysis on metaphase spreads of Suz12 genetrap ES cells, which showed that the ES cells present a normal number of chromosomes and one single insertion of the genetrap cassette (Figure 1C). To confirm that the insertion cassette was placed in the correct locus, we performed Southern blotting and polymerase chain reaction (PCR) to screen for the presence of both the WT and the targeted (KO) Suz12 alleles (Figure 1A). Southern blot analysis on WT and Suz12 genetrap ES cells showed that only the WT allele was present in WT ES cells, whereas the Suz12+/− ES cells contain both the WT and KO alleles (Figure 1D). As a further control, we showed that while WT Suz12 is expressed in all the analyzed ES cells, a genetrap fusion protein of the predicted molecular weight was only detectable in the Suz12 heterozygous ES cells (Figure 1E). Thus, we conclude that the ES Suz12 genetrap cells contain one insertion cassette, leading to the C-terminal truncation of the SUZ12 protein.
Generation of Suz12 KO mice
Several highly chimeric mice (>90%) were generated upon injection of the Suz12 genetrap ES cells. Three chimeric males were crossed with WT C57BL/6 females and we obtained germline transmission of the Suz12 KO allele. We did not observe any phenotypic differences between WT and Suz12 heterozygous mice, when analyzing size, morphology and fertility (data not shown).
To generate Suz12 KO mice, we crossed Suz12+/− mice and genotyped 147 pups either by Southern blotting or PCR analysis (Figure 1F). We did not identify any Suz12 KO mice among the 147 pups analyzed, suggesting that Suz12 depletion results in embryonic lethality. This is supported by the correct Mendelian ratio between Suz12+/− and Suz12+/+ mice (Figure 1G).
Developmental defects in Suz12 KO embryos
To understand when loss of Suz12 induces embryonic lethality, we analyzed embryos generated from Suz12 heterozygous crosses, at different stages of development. While we did not detect the presence of Suz12 KO embryos at later stages, at 10.5 days post coitus (dpc) we found embryonic tissues in smaller deciduae that were nearly completely reabsorbed. PCR analysis on genomic DNA demonstrated that Suz12−/− embryos are nearly completely reabsorbed, suggesting that embryonic lethality occurs at earlier embryonic stages (Figure 2A).
Figure 2.
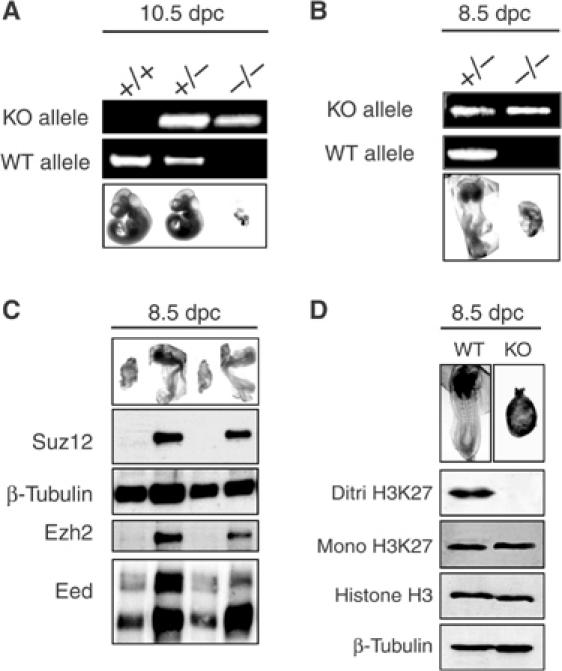
Suz12 is required for early embryonic development. (A) Picture and PCR analysis of 10.5 dpc embryos from Suz12+/− intercrosses showing nearly complete reabsorption of Suz12 KO embryos. (B) Picture and PCR analysis of 8.5 dpc embryos from Suz12+/− intercrosses showing the developmental defects of Suz12 KO embryos. (C) Picture and WB analysis of 8.5 dpc embryos from Suz12+/− intercrosses showing the complete absence of the Suz12 WT protein and the strong reduction of the Ezh2 protein levels in the KO embryos. β-Tubulin was used as loading control. (D) Picture and WB analysis of 8.5 dpc embryos from Suz12+/− intercrosses showing loss of H3K27 di/trimethylation. Each lane corresponds to a pool of four WT and four SUZ12 KO embryos. A representative picture of the pooled embryos is presented on top of each lane. Histone H3 and β-tubulin were used as loading control.
At 8.5 dpc, we found normal as well as much smaller embryos with morphology similar to that of a 7.5 dpc embryo. PCR analysis of these embryos showed that they were Suz12 KO, suggesting that the effects of Suz12 loss arise at around 7.5–8.5 dpc (Figure 2B). To confirm that the Suz12 protein was not expressed in the KO embryos, we prepared protein lysates from whole 8.5 dpc embryos, and analyzed the Suz12 protein levels using an antibody generated against a 179 aa C-terminal portion of Suz12. Western blot analysis (WB) demonstrated that Suz12 is expressed in normal 8.5 dpc embryos while, consistent with their genotype, none of the mutant embryos had detectable levels of WT Suz12 (Figure 2C). Interestingly, the Ezh2 levels were strongly reduced (Figure 2C), suggesting that Suz12 could be required for the stability of the Ezh2 protein. In contrast, Eed levels were only decreased to a minor extent in mutant embryos.
Our results suggest that Suz12 loss results in improper development around 7.5 dpc and that this phenotype could depend directly on the loss of the PRC2/3 complex activity. To better understand these observations, we analyzed embryos from Suz12+/− intercrosses at 7.5 dpc. Hematoxylin and eosin (H&E) staining at 7.5 dpc (Figure 3A, top panels) showed that KO embryos were significantly reduced in size and had several developmental defects. Immunohistochemical (IHC) staining demonstrated that Suz12 was strongly expressed in WT but completely lost in KO embryos (Figure 3A, middle panels). As a confirmation for the specificity of the Suz12 antibody, we showed that Suz12 expression was detected in the maternal tissues proximal to the uterus cavity in both the WT and mutant embryos (Figure 3A, bottom panels). Mutant embryos present severe defects in the development of most of the common structures of a 7.5 dpc embryo. The amniotic, exocoelomic and ectoplacental cavities appear not to form correctly and neither amnion nor chorion was distinguishable. The distinction between the three embryonic sheets was also difficult to appreciate. In addition, an aberrant presence of vacuolated cells was found in the region of the ectoplacental cavity. Very similar results were reported for the Ezh2- and Eed-deficient mice (Faust et al, 1995, 1998; O'Carroll et al, 2001). Furthermore, Faust et al have carried out an extensive and detailed characterization of the Eed mutant mice, using different markers of gastrulation, which confirmed the morphological defects observed in these mutant embryos. Taken together with the previous observations, we conclude that like Ezh2 and Eed, Suz12 KO embryos present strong defects in gastrulation.
Figure 3.
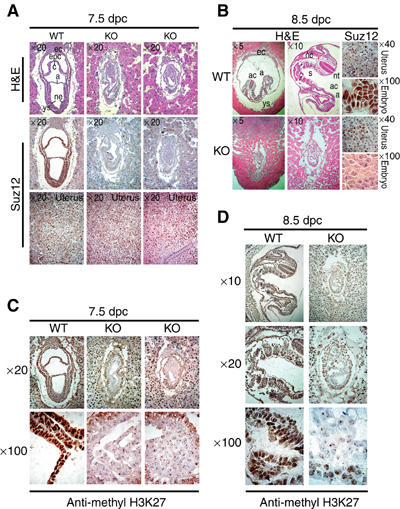
Developmental defects and lack of H3K27 methylation in Suz12 KO embryos. (A) (Top panels) H&E staining of 7.5 dpc in utero embryo sections from Suz12+/− intercrosses showing normal morphology of WT and developmental defects of Suz12 KO embryos. (Middle panels) IHC staining showing the presence or absence of the WT Suz12 protein in consecutive sections of the embryos shown in the top panels. (Lower panels) IHC staining for Suz12 in the uterus of each section is presented as a control for proper staining of the analyzed sections. ec: ectoplacental cone; epc: ectoplacental cavity; c: chorion; a: amnion; ne: neural ectoderm; ys: yolk sac. (B) H&E (left and middle panels) and Suz12 IHC staining (right panels) in the embryos and in the uterus of 8.5 dpc in utero embryo sections from Suz12+/− intercrosses. Different magnifications of the same embryos are presented to better evaluate the differences in size and morphology between WT and KO embryos. ac: amniotic cavity; s: somites; nt: neural tube; nc: notochord. (C) Different magnifications of IHC staining of H3K27 di/trimethylation in WT (left panels) and Suz12 KO (middle and right panels) 7.5 dpc embryos. (D) As in panel (C), but for 8.5 dpc embryos. WT (left panels) and Suz12 KO (right panels) embryos. Suz12 IHC staining of maternal cells in (C, D) was used as antibody staining control.
To monitor the ability of the mutant embryos to develop further, we analyzed embryos at 8.5 dpc. Embryo sections were stained to visualize morphological changes (H&E) and for the expression of Suz12 (Figure 3B). At 8.5 dpc, the difference in size between normal and mutant embryos was greater than at 7.5 dpc. Mutant embryos appear unable to develop, and KO embryos have a similar morphology to the 7.5 Suz12−/− embryos. The amnion does not extend and the amniotic cavity is not folding around the embryo. In addition, the neural ectoderm does not develop resulting in the complete absence of organogenesis.
In summary, our results demonstrate that Suz12 KO embryos are able to implant in the uterus, but that the postimplantation development is strongly compromised most likely due to gastrulating defects that induce a developmental block around 7.5 dpc.
Di- and trimethylation of H3K27 is lost in Suz12 KO embryos
Considering the high similarity of Suz12, Eed and Ezh2 KO embryos and the strong reduction in the levels of Ezh2 (Figure 2C) in Suz12 KO embryos, it is reasonable to speculate that since Suz12 is a subunit of the PRC2 complex, Suz12 could have an essential role in the PRC2 complex activity. To address this, we tested whether Suz12 loss changed the methylation pattern of histone H3 K27 during embryogenesis. As demonstrated in Figures 3C and D, maternal and WT embryonic cells stained strongly for H3K27 methylation, using an antibody specific for the di- and trimethylated forms. In contrast, loss of Suz12 led to a striking absence of both di- and trimethylated H3K27 in the embryo, whereas the surrounding maternal tissue was not affected. WB analysis performed on WT and SUZ12 mutant 8.5 dpc embryos confirmed the reduction in di- and trimethylation of H3K27 (Figure 2D), and interestingly showed that monomethylation of H3K27 was not affected. These results demonstrate that Suz12 is essential for di- and trimethylation of H3K27 during embryogenesis.
Proliferation is strongly impaired in Suz12 KO embryos
To understand whether Suz12 is required for embryonic proliferation, we analyzed the replicative and mitotic indices of WT and KO embryos. As shown in Figures 4A–D, loss of Suz12 leads to a significant reduction in cells positive for both BrdU and the serine 10 phosphorylated form of histone H3 (H3-P(S10), a marker of mitosis) at 7.5 dpc. Similar results were also obtained at 8.5 dpc (see Supplementary information). Thus, Suz12 KO embryos fail to proliferate providing a likely explanation for the difference in sizes between WT and KO embryos.
Figure 4.
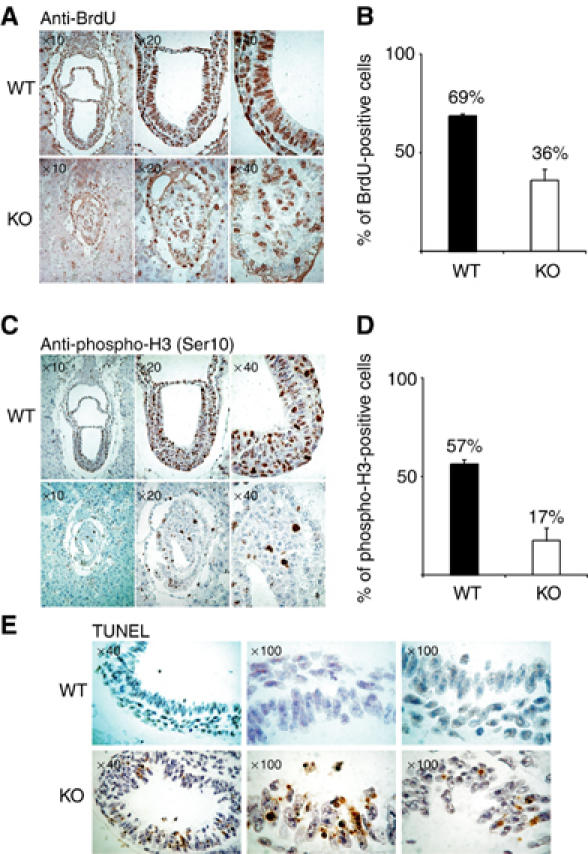
SUZ12 KO embryos are compromised in proliferation. (A) Different magnifications of IHC staining of BrdU in WT (top panels) and KO (bottom panels) 7.5 dpc embryos. The mice were injected with BrdU (150 mg/kg) for 60 min before they were killed. (B) Average of the percentage of BrdU-positive cells in both WT and KO embryos. The standard deviation (s.d.) of the mean is indicated. A total of 1086 cells in WT embryos and 936 cells in KO embryos were counted. (C) Different magnifications of IHC staining of phosphorylation of Ser10 of histone H3 in WT (top panels) and KO (bottom panels) 7.5 dpc embryos. (D) Average of the percentage of H3 Ser10 phosphorylated cells in both WT and KO embryos. s.d. is indicated. A total of 501 cells in WT embryos and 309 cells in KO embryos were counted. (E) Different magnifications of TUNEL staining in WT (top panels) and KO (bottom panels) 7.5 dpc embryos.
Interestingly, we observed that the reduction of BrdU-positive cells in KO embryos was not as strong as the reduction in H3-P(S10)-positive cells. This may suggest that some BrdU-positive cells failed to enter mitosis and we analyzed whether a checkpoint was induced in Suz12 KO embryos. However, both the analysis of p53 levels and the phosphorylation of H2AX (as a measure for an S-phase checkpoint) showed no difference between WT and KO embryos (Supplementary information). In contrast, we observed that a large number of cells in the Suz12 KO embryos underwent cell death as revealed by TUNEL staining (Figure 4E and Supplementary information). These results suggest that the developmental defects induced by loss of PRC2/3 HMT activities lead to a strong decrease in proliferation and survival.
SUZ12 is required for proliferation in differentiated tissue culture cells
Two subunits of the PRC2/3 complexes, EZH2 and EED, are known to be required for the proliferation of both primary and tumor cell lines (Varambally et al, 2002; Bracken et al, 2003). We observed that in vivo, KO embryos for Suz12 present proliferative defects suggesting that SUZ12 is also required for proliferation of human differentiated cells.
To characterize the role of the human SUZ12 protein in cellular proliferation, we tested seven different siRNA oligonucleotides that specifically target the SUZ12 transcript and identified one that efficiently reduced SUZ12 expression to nearly undetectable levels (data not shown). This sequence works efficiently in both transient transfection as double-stranded RNA and in the retroviral vector pRetroSuper (pRS) (Brummelkamp et al, 2002). To determine the effect of SUZ12 loss in human tumor cells, we infected the osteosarcoma tumor cell line U2OS with both pRS and pRS-SUZ12 vectors. The infected cells were plated at low density and stained with crystal violet 2 weeks after plating. The SUZ12 levels were efficiently reduced as shown by WB (Figure 5A). Reduction of SUZ12 levels resulted in a significant decrease in the colony forming ability of U2OS cells, demonstrating that SUZ12 is required for proliferation (Figure 5A).
Figure 5.
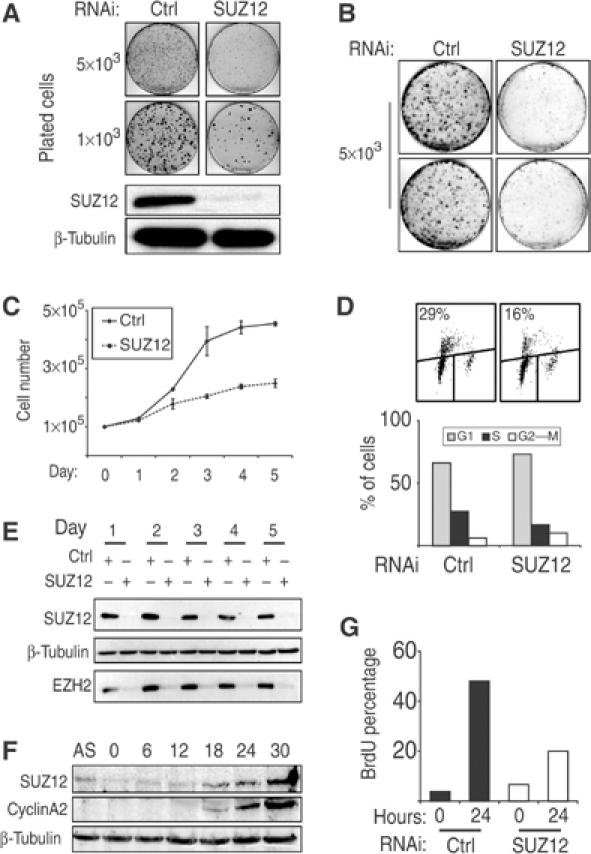
SUZ12 is required for proliferation of both normal and tumor cells. (A) Colony formation of U2OS cells stably interfered with empty (CTRL) and SUZ12 shRNA (SUZ12) retroviral vectors showing the proliferative impairment of SUZ12 interfered U2OS cells. In the bottom panels, WB analyses of SUZ12 levels of the same cells are presented. β-Tubulin was used as loading control. (B–D) Colony, growth curves and BrdU FACS analysis of primary human TIG3-T cells stably interfered with empty (CTRL) and SUZ12 shRNA (SUZ12) retroviral vectors, showing the requirement of SUZ12 for proliferation of diploid human cells. (E) WB analysis of TIG3-T cells at each day of the experiment presented in (C), showing the efficiency of the SUZ12 interference and the reduction of the EZH2 protein levels. β-Tubulin served as loading control. (F) Serum induction of serum-starved human diploid fibroblasts. WB analyses show the accumulation of both SUZ12 and cyclin A2 at the G1–S transition. β-Tubulin was used as loading control. (G) SUZ12 is required for S-phase entry. BrdU incorporation was measured with (24 h) and without (0 h) serum induction of serum-starved TIG3-T cells stably interfered with empty (CTRL) and SUZ12 shRNA (SUZ12) retroviral vectors.
To test whether SUZ12 is also required for proliferation of human diploid cells, we infected TERT immortalized human diploid lung fibroblasts (TIG3-T) with both pRS-empty and pRS-SUZ12. Colony formation assays (Figure 5B) and growth curves (Figure 5C) showed that the proliferation of the SUZ12 interfered cells was strongly compromised. Consistent with this, FACS analysis demonstrated that BrdU incorporation of SUZ12 TIG3-T interfered cells was significantly reduced compared to control cells, leading to an increase in the number of cells in G1 (Figure 5D). WB analysis confirmed that SUZ12 levels were efficiently reduced throughout the experiment (Figure 5E, top panel) and, consistent with the results obtained in mouse embryos, loss of SUZ12 led to a strong reduction of EZH2 levels in human cells (Figure 5E, bottom panel) further suggesting a role of Suz12 in the activity and/or stability of the PRC2/3 complex.
We and others have identified SUZ12 as a target of the E2F transcription factors (Muller et al, 2001; Weinmann et al, 2001; Bracken et al, 2003). In agreement with this, the SUZ12 protein levels are strongly reduced in quiescent cells and subsequently accumulate at the G1–S transition with kinetics similar to cyclin A2 (Figure 5F). To understand whether SUZ12 is required for cells to re-enter the cell cycle, we serum-starved TIG3-T cells stably interfered with both empty and pRS-SUZ12 vectors. Upon cell cycle re-entry, the absence of SUZ12 strongly impaired the ability of the cells to re-enter the cell cycle (Figure 5G). In summary, these results demonstrate that SUZ12 is essential for the proliferation of both tumor and diploid human cells, and for the expression of EZH2.
SUZ12 is required for the integrity of the PRC2/3 complexes
Loss of SUZ12 expression both in vivo and in tissue culture cells strongly reduced the levels of EZH2, suggesting that SUZ12 may be required for the stability of the PRC2/3 complexes. To address this, we transiently interfered with the expression of SUZ12 in HeLa cells by using siRNA oligos. The interference efficiency was controlled by WB analysis (Figure 6A). Protein lysates were prepared from control and SUZ12 interfered cells and separated on a Superose 6 gel filtration column. The elution profile of the different components of the PRC2 complex was monitored by WB analysis. The elution profile in cells interfered with control siRNA (Figure 6B) showed that SUZ12 is found in a 700 kDa complex, and consistent with being part of the PRC2/3 complexes, EZH2 and EED also coeluted in these fractions (Figure 6B). Strikingly, when cells were transfected with SUZ12 RNAi, the EZH2 and EED 700 kDa forms disappeared, and the two proteins accumulated in smaller fractions (Figure 6B). These results demonstrate that SUZ12 is required for the integrity of the PRC2/3 complexes.
Figure 6.
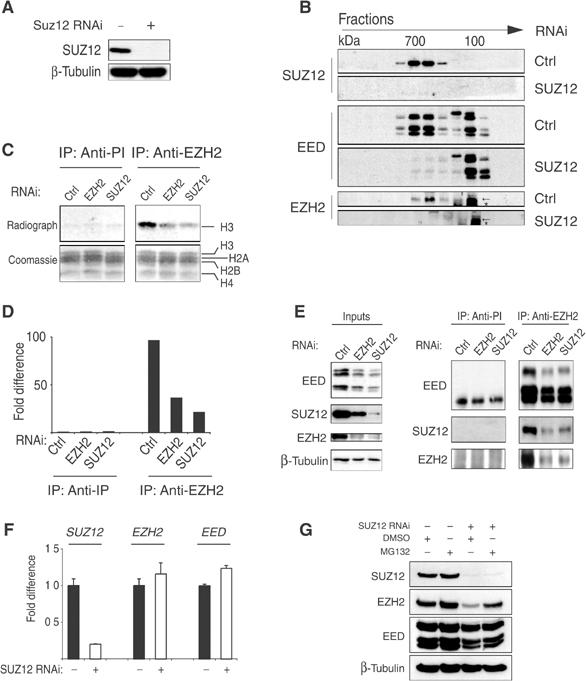
SUZ12 is required for the formation and the HMT activity of EZH2-containing complexes. (A) WB analysis of transiently interfered HeLa cells with control (Ctrl) and SUZ12 siRNA oligos. (B) WB analysis on gel filtration fractions of transiently interfered HeLa cells presented in (A) showing the elution profile of SUZ12, EED and EZH2 in the presence or absence of SUZ12. The arrows indicate EZH2. The asterisk indicates a crossreacting protein recognized by the antibody. (C) HMT assays of IP products of preimmune (anti-PI) and anti-EZH2 serum of transiently interfered HeLa cells with Ctrl, EZH2 and SUZ12 oligos. The bottom panels show the Coomassie staining of the histones used as substrates in the reaction. In the top panel is presented the autoradiograph of the same histones showing the specific H3 HMT of the PRC2 complex and the reduction in activity upon loss of EZH2 and SUZ12. (D) Quantification of the HMT activity presented in (C) by in silico analysis. (E) WB analysis of SUZ12, EZH2 and EED proteins of the experiment presented in (C, D). (F) Expression levels of SUZ12, EZH2 and EED mRNA showing that loss of SUZ12 has no effect on EZH2 and EED expression. (G) WB analysis of transiently interfered HeLa cells with Ctrl and SUZ12 siRNA oligos treated with carrier (DMSO) or 5 μM MG132 proteosome inhibitor for 10 h before harvesting, showing that SUZ12 loss induces a proteosome-dependent degradation of EZH2.
To determine whether disruption of the complexes affects the enzymatic activities, we transfected HeLa cells with control and EZH2- and SUZ12-specific siRNAs. Lysates were immunoprecipitated with preimmune or anti-EZH2 serum and the immunoprecipitation (IP) products were incubated in an in vitro HMT assay. As shown in Figures 6C–E, the EZH2 antibody precipitated a strong HMT activity specific for histone H3. This activity was EZH2 specific since it was strongly reduced in cells transfected with siRNA to EZH2. Significantly, however, together with the demonstration that SUZ12 is essential for the integrity of the PRC2/3 complexes, EZH2 enzymatic activity was also strongly compromised upon SUZ12 downregulation, demonstrating that SUZ12 is required for EZH2 HMT activity.
To address the role of SUZ12 in the stability of the PRC2/3 complexes, we tested whether short-term SUZ12 downregulation affected the transcription of EZH2 or stability of the EZH2 protein. To address this, we analyzed by real-time quantitative PCR the transcription of the three core components of the PRC2/3 complex after transient downregulation of SUZ12. We did not observe any significant changes in the expression of EZH2 and EED mRNAs, suggesting that short-term downregulation of SUZ12 affects the stability of EZH2 rather than its gene transcription (Figure 6F). To test this directly, we incubated HeLa cells transiently transfected with SUZ12-specific siRNA with the proteosome inhibitor MG132 for 10 h before harvesting. As shown in Figure 6G, treatment of HeLa cells with MG132, but not carrier, resulted in stabilization of EZH2. These results demonstrate that SUZ12 prevents the proteolytic degradation of EZH2 by regulating the stability and the enzymatic activity of EZH2-containing complexes.
To understand if the loss of HMT activity is due to EZH2 degradation or whether SUZ12 directly contributes to the enzymatic activity of the complex, we reconstituted the PRC2 complex in vitro using a baculovirus expression system. As shown in Figure 7A, in the absence of Suz12, the recruitment of the subunit RbAp48 is prevented but not the binding between EZH2 and EED. The absence of SUZ12 and the loss of RbAp48 recruitment result in the loss of EZH2 HMT activity on both histones H3 and H1 (Figure 7B). In contrast, the absence of RbAp48 allows the formation of an EZH2–SUZ12–Eed trimeric complex active on both histones H3 and H1 (Figure 7B). In order to obtain an equal amount of stoichiometric complexes, we further purified the different PRC2 complexes by gel filtration (Figure 7C). Fractions containing the different complexes were assayed for HMT activity (Figure 7D, top panel). In the absence of RbAp48, the PRC2 complex is functional but is less efficient than when RbAp48 is present, suggesting that RbAp48 enhances the EZH2 HMT activity. These results demonstrate that SUZ12 is essential for EZH2 HMT activity on both histones H3 and H1 and that SUZ12 enhances this activity by mediating the recruitment of the histone-binding subunit RbAp48 to the PRC2/3 complexes.
Figure 7.
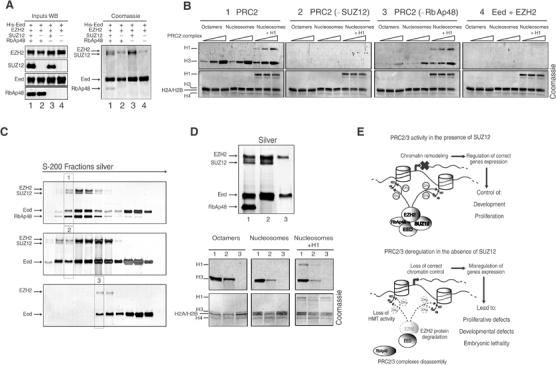
Dual role of SUZ12 in regulating the PRC2 complex activity. (A) SUZ12 is required for the recruitment of RbAp48 to the EZH2 complex. Coomassie staining (right panel) of recombinant proteins bound and eluted from a nickel column. The indicated proteins were coexpressed in insect cells. Eed was produced as a His-tagged protein, whereas the other proteins were produced as nontagged proteins. The left panel shows the expression (input) of the various proteins before His-tag purification. (B) SUZ12 is required for PRC2 HMT activity in vitro on both histones H3 and H1. Increasing amounts (0.5, 1 and 5 μl of 0.1 μg/μl preparation) of the purified proteins shown in (A) were used in an in vitro HMT assay using octamers, oligonucleosomes and oligonucleosomes assembled with histone H1 as substrates as described in Materials and methods. (C) Purification of different PRC2 complex forms. Silver staining of S-200 gel filtration fractions of His purification: PRC2 complex (top panel), PRC2 complex in the absence of RbAp48 (middle panel) and PRC2 complex in the absence of SUZ12 (bottom panel). Highly pure complex fractions (highlighted lanes) were used in (D). (D) Silver staining (top panel) and HMT assays (bottom panels) of different forms of the PRC2 complex (labeled 1–3) showing that EZH2–SUZ12–Eed is the minimal enzymatically active PRC2 complex form. (E) Schematic model of the role of PRC2/3 complexes in transcription and a representation of the effects of SUZ12 loss on the PRC2/3 complexes highlighting the impaired recruitment of RbAp48 and the loss of HMT activity, which induce instability and degradation of EZH2.
Discussion
We have investigated the role of the PcG protein Suz12 in vivo and in tissue culture and have demonstrated that Suz12 is an essential regulator of the activity and stability of the PRC2/3 complexes in both mouse embryogenesis and in human differentiated cells. We have shown that mice lacking functional Suz12 are not viable and die around day 7.5 of embryogenesis. Moreover, we have shown that Suz12 mutant embryos are impaired in proliferation and contain an increased number of apoptotic cells. Proliferative defects were also observed in human tissue culture cells, demonstrating that SUZ12 is required for proliferation and for S-phase entry of serum-stimulated quiescent cells. We have shown that SUZ12 has an essential role in stabilizing the PRC2/3 complexes and maintaining their HMT activities. Disruption of these complexes results in the specific loss of di- and trimethylation of H3K27 in vivo and in the loss of EZH2 HMT activity in vitro. Moreover, we have demonstrated that SUZ12 is required for the enzymatic activity of EZH2 on both histones H3 and H1 and that SUZ12 enhances this activity.
Suz12 and mouse development
SUZ12, together with EZH2, EED and RbAp46/48, forms a complex, which has been shown to contain intrinsic K9/K27 HMT activity (Cao et al, 2002; Czermin et al, 2002; Kuzmichev et al, 2002; Muller et al, 2002). This complex has also been shown recently to contain histone H1K26 methyl transferase activity (Kuzmichev et al, 2004). Ezh2 and Eed are both essential for mouse development and their loss results in embryonic lethality. The mutant embryos are able to go beyond the preimplantation stage and to implant in the uterus, but serious developmental defects arise between 6.5 and 8.5 dpc (Faust et al, 1995; O'Carroll et al, 2001). Gastrulation of the mutant embryos initiates, but fails to correctly develop. Proper embryonic tissues do not form and aberrant outgrowth of mesoderm cells in extraembryonic tissues is observed. In this study, we demonstrate that Suz12 mutant embryos have defects similar to those observed in Ezh2 and Eed KO embryos. Suz12-deficient embryos are able to implant, but their development is blocked around day 7.5. Embryonic and extraembryonic structures in Suz12 KOs do not seem to develop and, like Ezh2 and Eed KOs, organogenesis does not initiate. The difference between WT and mutant embryos is even more evident at 8.5 dpc, when mutant embryos are developmentally blocked and remain morphologically similar to 7.5 dpc embryos.
Based on the significant phenotypic similarities between the KO embryos of Suz12, Eed and Ezh2, we propose that these three genes contribute to the same activities. Interestingly, SUZ12 is part of all EZH2-containing complexes purified so far, suggesting a tight relationship between SUZ12 and EZH2 activities. Moreover, in Drosophila, mutants for the EZH2 and SUZ12 homologs E(z) and Su(z)12 display a similar pattern of homeotic genes deregulation (Jones and Gelbart, 1990; Simon et al, 1992; Birve et al, 2001), suggesting that the transcriptional effects imposed by E(z) and Su(z)12 act in parallel if not identical pathways. In strong support of this notion, we have demonstrated that SUZ12 is required for EZH2 activity and that SUZ12 enhances the PRC2 complex activity by mediating the recruitment of the histone-binding protein RbAp48 to the complex in vitro. Consistent with these in vitro data, we have shown that SUZ12 is required for EZH2 HMT activity in human cells and moreover that H3K27 di- and trimethylation is lost in mouse embryos lacking the Suz12 protein. In the last decade, an overwhelming amount of data has demonstrated the crucial role of histone tail modifications in the regulation of gene transcription. The role of H3K27 methylation is not fully characterized, but it has been linked to transcriptional repression of euchromatic regions (Peters et al, 2003). While in vitro data link EZH2 activity to different histone modifications, in vivo data suggest that the main HMT activity of EZH2 is toward H3K27 (Erhardt et al, 2003). Due to its recent discovery, no data have so far been published regarding H1K26 methylation in vivo. In this work, we have demonstrated that, at least in vitro, SUZ12 is required for EZH2 histone H1K26 methylation, but it will be interesting to understand whether EZH2 complexes are indeed responsible for this modification in vivo.
Taking our and others' data into consideration, K27 methylation is, at present, the most likely enzymatic activity by which the EZH2 complex imposes its control on transcription. Furthermore, since K27 methylation is widely spread throughout the chromatin during embryogenesis and the absence of Suz12 induces a global loss of K27 methylation in vivo, our results suggest that the PRC2/3 complexes are directly involved in controlling the expression of a large number of genes. This result does not exclude the possibility that independently of EZH2, SUZ12 could regulate the activity of other H3K27 HMT enzymes during embryogenesis. To understand this, it would be interesting to investigate if H3K27 methylation is lost in the implanted 7.5 dpc Ezh2 KO embryos, as reported in Ezh2−/− blastocysts (Erhardt et al, 2003). Alternatively, the PRC2/3 complexes might be required for the activity of other H3K27 methyltransferases, and the global loss of H3K27 methylation observed in the Suz12-deficient embryos could therefore reflect the lack of activity of several H3K27 methyltransferases. Another possibility is that the EZH2-containing complexes are involved in the general control of chromatin folding and only indirectly influence transcription. In support of this, E(z), the Drosophila homolog of EZH2, is required for the proper condensation of Drosophila polytene chromosomes (Rastelli et al, 1993). A functional link between EZH2-dependent H1K26 methylation and chromosome condensation has been recently proposed by Kuzmichev et al (2004). Future work involving the identification of target genes for the EZH2-containing complexes, the identification of other H3K27 methyltransferases and more studies on the function of H1K26 methylation will most certainly provide more insights into these questions.
SUZ12 and cellular proliferation
The expression of Suz12 is linked to proliferation and the protein levels are strongly reduced in noncycling compared to cycling cells. We have demonstrated that Suz12 KO embryos, in addition to strong developmental defects, are also severely impaired in proliferation. Since development is strictly linked to proliferation during embryogenesis, it is not possible to determine whether the proliferative defects of the Suz12 KO embryos are due to developmental failures or, conversely, whether the developmental failures are due to lack of proliferation. However, we have also demonstrated that SUZ12 is essential for proliferation of human cells in tissue culture. This is consistent with previous reports demonstrating that EZH2 and EED are also required for proliferation of both tumor and primary cells (Varambally et al, 2002; Bracken et al, 2003). Supporting this notion is the fact that the three PcG genes are transcriptionally regulated by the pRB/E2F pathway, and that they are required for S-phase entry. Although proliferation in tissue culture is not directly comparable with proliferation during embryogenesis, these results suggest that the EZH2-containing complexes are involved in the regulation of genes essential for proliferation. Interestingly, Suz12, Ezh2 and Eed KO mice die during development before the transcription of the Hox genes (reviewed in van Lohuizen, 1998), strongly suggesting that the EZH2-containing complexes have targets other than the Hox genes that are essential for proper development and/or proliferation. Future studies will be aimed at identifying novel PRC2/3 target genes.
Stability and functionality of the PRC2 complex
The data that we have presented on the role of SUZ12 in regulating PRC2/3 function(s) highlight SUZ12 as an essential regulator of these complexes. Monomeric EZH2 does not possess enzymatic activity (Cao et al, 2002; Czermin et al, 2002; Kuzmichev et al, 2002; Muller et al, 2002), and in agreement with this we have demonstrated that abrogation of SUZ12 expression resulted in loss of EZH2 HMT activity in vivo and in vitro. This is explained by the fact that SUZ12 is required for the activity of the PRC2/3 complexes and for the stability of the EZH2 protein. Since deregulation of EZH2 can have a causal role in cancer, it is tempting to speculate that stabilization of EZH2 by mutations in genes required for its degradation could be involved in cancer as well.
In conclusion, we have shown that SUZ12 is essential for the integrity of the PRC2/3 complexes, the stability of EZH2 and hence the HMT activity of these complexes (Figure 7E). We propose that loss of SUZ12 induces changes in the chromatin structure of PRC2/3 target genes. These changes will result in the misregulation of the expression of PRC2/3 target genes, subsequently leading to severe developmental and proliferative defects.
Materials and methods
Generation and genotyping of Suz12 KO mice
Suz12 genetrapped ES cells (XG122) (http://www.baygenomics.ucsf.edu) were used to generate chimeric mice. Suz12 heterozygous mice were interbred to generate Suz12-deficient embryos. ES cells and mice were genotyped either by Southern blotting or PCR analysis. Southern blots were probed with a 432 bp fragment generated by PCR from normal C57BL/6 genomic DNA using the primers Fwd 5′-CTCCATTCCCAAGCCATTTATTC-3′ and Rev 5′-AACAGCAACGAGTAGGACTTCACC-3′ and confirmed by sequencing. Genomic DNA was digested with ACC I (Roche, catalog no. 728-420), separated on a 1.2% agarose gel and transferred to a Hybond N+ membrane (Amersham). The membrane was prehybridized at 65°C for 60 min with hybridization buffer (PerfectHyb™ Sigma). The probe was labeled with [α-32P]dCTP using Rediprime-II DNA labeling system (Amersham), purified with Sephadex G-50 column (Roche) and added to the hybridization buffer for 3 h. The membrane was washed twice with 2 × SSC/0.2% SDS at room temperature (RT) for 15 min and for two times with 0.2 × SSC/0.2% SDS at 65°C for 20 min.
PCR genotyping was performed using the following primers:
WT allele, Fwd 5′-GAAGTCCTACTCGTTGCTGTTTAG-3′ and Rev 5′-CATTTGTGCAACAAATGTCTTTTC-3′ (1709 bp PCR product); KO allele (LacZ PCR): Fwd 5′-TGGTCGCTGGGGAATGAATC-3′ and Rev 5′-AACGGGGATACTGACGAAACG-3′ (318 bp PCR product).
FISH analysis
FISH analyses on the genetrapped ES cells were performed as described (Bracken et al, 2003). The probe specific for the BayGenomics insertion cassette vector pGT1lxf (http://www.baygenomics.ucsf.edu) was generated by PCR using genomic DNA of Suz12 genetrapped ES cells with the following primers: Fwd 5′-GCTGGCGTAATAGCGAAGAGG-3′ and Rev 5′-TTTTATGTTTCAGGTTCAGGGGG-3′. The PCR product was cloned into pCR®2.1 TOPO® vector (Invitrogen) and confirmed by sequencing. The plasmid was subsequently used to perform FISH analysis.
Embryo harvesting and histological analysis
Cloning and RNAi
The SUZ12-specific sequence (AAGCTGTTACCAAGCTCCGTG) was used to synthesize double-stranded siRNA oligos (MWG) or cloned into pRetroSuper (Brummelkamp et al, 2002). EZH2 and Eed open reading frames (ORFs) described by Bracken et al (2003) were cloned respectively into pFastBac1 and pFastBacHTb (Invitrogen). SUZ12 (NM_015355) and RbAp48 ORFs (NM_005610) were isolated from human primary TIG3 fibroblasts by PCR, cloned into pFastBac1 and the DNA sequences were verified by sequencing.
Tissue culture
U2OS, HeLa and TIG3-T (stably expressing TERT) cells were cultured in DMEM supplemented with 10% (v/v) FCS. Low-density assays of U2OS and TIG-3 cells were performed in 10 cm tissue culture dishes and stained with crystal violet.
Growth curves: TIG 3-T cells were infected with the indicated retroviral constructs, selected for 3 days in 500 μg/ml puromycin and plated in six-well dishes. Viable cells were counted by Trypan blue staining and lysed for WB analysis.
Transient RNAi: A total of 7 × 105 HeLa cells were plated in 10 cm dishes, transfected after 24 h with Oligofectamine® (Invitrogen) following the manufacturer's protocol and collected at the indicated time.
FACS analysis: TIG3-T was grown to 60–70% confluency, treated with 0.3 μM BrdU for 60 min (Sigma, catalog no. 59-14-3) and harvested. Cells (3 × 106) were treated and analyzed by FACS as described (Bracken et al, 2003).
Antibodies
The rabbit anti-EZH2 sera were generated against the N-terminal 286 aa of EZH2, which was fused to the maltose-binding protein (MBP). Mouse monoclonal antibodies were produced as described (Bracken et al, 2003). AC22 (anti-EZH2) was generated using aa 353–451 of EZH2 fused to MBP as an immunogen. AE25 (anti-EZH2) was generated using aa 1–286 of EZH2 fused to MBP as an immunogen. AI25-13 (anti-p53) was raised in p53−/− C57BL6 using mouse WT p53 as an immunogen. AA19 (anti-EED) and mouse monoclonal anti-di/trimethyl histone H3K27 have been described previously (Bracken et al, 2003; Okamoto et al, 2004). Other antibodies are commercially available: rabbit anti-Suz12 (Upstate), rabbit anti-histone H3 phospho-Ser10 (Upstate), mouse anti-β-Gal (Santa Cruz), rabbit anti-β-tubulin (Santa Cruz), mouse anti-BrdU (Beckson & Dickinson), mouse anti-H2AX (Upstate) and rabbit anti-monomethyl histone H3K27 (Upstate).
Biochemical and enzymatic analyses
Column separation: HeLa cells transfected with siRNA oligonucleotides were lysed in column buffer (25 mM HEPES pH 7.6, 1 mM EDTA, 10% glycerol, 300 mM NaCl) 48 h after transfection and separated on a Superose 6 PC 3.2/30 gel filtration column (Amersham). Fractions were analyzed by WB.
HMT assay: HeLa cells transfected with siRNA oligos were lysed in IP buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% Tween 20) 48 h after transfection. A 5 mg portion of the extracts was immunoprecipitated overnight with the indicated antibodies.
Sf9 cells (Invitrogen, catalog no. 25-0127) were infected with the indicated virus produced using Bac-to-Bac® HT Baculovirus Expression System following the manufacturer's procedure (Invitrogen). Cells were lysed in BPL2B buffer (20 mM Tris–HCl pH 8, 500 mM NaCl, 20% glycerol, 4 mM MgCl2, 3 mM 2-mercaptoethanol, 0.05% NP-40). Proteins were purified using Ni-NTA agarose (Invitrogen) and eluted in ERB (50 mM Tris–HCl pH 8.5, 150 mM NaCl, 5 mM MgCl2, 5 mM DTT, 300 mM imidazole).
Purified products, when indicated, were loaded on a Superdex 200 PC 3.2/30 gel filtration column (Amersham) and eluted in ERB in the absence of imidazole. Purified products and indicated fractions were incubated in 5 × HMT buffer for 60 min at 30°C (250 mM Tris–HCl pH 8.5, 25 mM MgCl2, 20 mM DTT, 15 μCi/ml S-adenosyl-L-[methyl-3H]methionine (Amersham, catalog no. TRK581), 0.1 μg/μl of histone mixture (Roche, catalog no. 0223565)). When indicated, the following substrates were used: histone recombinant octamers produced as described by Kuzmichev et al (2004); HeLa S3 oligonucleosomes produced as described by Fang et al (2004), oligonucleosomes with histone H1 as described by Kuzmichev et al (2004). The reaction products were denatured and separated on SDS–polyacrylamide gels. Proteins were transferred to PVDF membranes and histones were visualized by Coomassie staining. HMT activity was measured by film exposure of the membrane.
Supplementary Material
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
We thank BayGenomics for providing the Suz12 genetrap ES cells. We also thank Danny Reinberg for kindly providing the histone H3 K27 di- and trimethyl-specific antibody, Elena Prosperini and Daniele Piccini for assistance in generating the EZH2 and p53 antibodies, Sergio Bossi for technical help in producing baculovirus expressed proteins, and Claudio Ciferri and Andrea Musacchio for biochemical advice. We thank Claire Attwooll for critical reading of the manuscript and the Helin lab for discussions. DP is a fellow of Associazione Italiana per la Ricerca sul Cancro (AIRC) and ELD is a fellow of Fondazione Italiana per la Ricerca sul Cancro (FIRC). This work was supported by grants from AIRC, the Association for International Cancer Research, the Danish Medical Research Council and the Danish Ministry of Research.
References
- Birve A, Sengupta AK, Beuchle D, Larsson J, Kennison JA, Rasmuson-Lestander A, Muller J (2001) Su(z)12, a novel Drosophila Polycomb group gene that is conserved in vertebrates and plants. Development 128: 3371–3379 [DOI] [PubMed] [Google Scholar]
- Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K (2003) EZH2 is downstream of the pRB–E2F pathway, essential for proliferation and amplified in cancer. EMBO J 22: 5323–5335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brock HW, van Lohuizen M (2001) The Polycomb group—no longer an exclusive club? Curr Opin Genet Dev 11: 175–181 [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2: 243–247 [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298: 1039–1043 [DOI] [PubMed] [Google Scholar]
- Cao R, Zhang Y (2004) The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev 14: 155–164 [DOI] [PubMed] [Google Scholar]
- Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111: 185–196 [DOI] [PubMed] [Google Scholar]
- Erhardt S, Su IH, Schneider R, Barton S, Bannister AJ, Perez-Burgos L, Jenuwein T, Kouzarides T, Tarakhovsky A, Surani MA (2003) Consequences of the depletion of zygotic and embryonic enhancer of zeste 2 during preimplantation mouse development. Development 130: 4235–4248 [DOI] [PubMed] [Google Scholar]
- Fang J, Wang H, Zhang Y (2004) Purification of histone methyltransferases from HeLa cells. Methods Enzymol 377: 213–226 [DOI] [PubMed] [Google Scholar]
- Faust C, Lawson KA, Schork NJ, Thiel B, Magnuson T (1998) The Polycomb-group gene eed is required for normal morphogenetic movements during gastrulation in the mouse embryo. Development 125: 4495–4506 [DOI] [PubMed] [Google Scholar]
- Faust C, Schumacher A, Holdener B, Magnuson T (1995) The eed mutation disrupts anterior mesoderm production in mice. Development 121: 273–285 [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, van Lohuizen M (2002) Polycomb repression: from cellular memory to cellular proliferation and cancer. Biochim Biophys Acta 1602: 151–161 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Jones RS, Gelbart WM (1990) Genetic analysis of the enhancer of zeste locus and its role in gene regulation in Drosophila melanogaster. Genetics 126: 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM (2003) EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA 100: 11606–11611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koontz JI, Soreng AL, Nucci M, Kuo FC, Pauwels P, van Den Berghe H, Cin PD, Fletcher JA, Sklar J (2001) Frequent fusion of the JAZF1 and JJAZ1 genes in endometrial stromal tumors. Proc Natl Acad Sci USA 98: 6348–6353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A, Jenuwein T, Tempst P, Reinberg D (2004) Different ezh2-containing complexes target methylation of histone h1 or nucleosomal histone h3. Mol Cell 14: 183–193 [DOI] [PubMed] [Google Scholar]
- Kuzmichev A, Nishioka K, Erdjument-Bromage H, Tempst P, Reinberg D (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16: 2893–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K (2001) E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev 15: 267–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, Miller EL, O'Connor MB, Kingston RE, Simon JA (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111: 197–208 [DOI] [PubMed] [Google Scholar]
- O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T (2001) The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21: 4330–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, Otte AP, Allis CD, Reinberg D, Heard E (2004) Epigenetic dynamics of imprinted X inactivation during early mouse development. Science 303: 644–649 [DOI] [PubMed] [Google Scholar]
- Pasini D, Bracken AP, Helin K (2004) Polycomb group proteins in cell cycle progression and cancer. Cell Cycle 3: 396–400 [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Rastelli L, Chan CS, Pirrotta V (1993) Related chromosome binding sites for zeste, suppressors of zeste and Polycomb group proteins in Drosophila and their dependence on Enhancer of zeste function. EMBO J 12: 1513–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J, Chiang A, Bender W (1992) Ten different Polycomb group genes are required for spatial control of the abdA and AbdB homeotic products. Development 114: 493–505 [DOI] [PubMed] [Google Scholar]
- van Lohuizen M (1998) Functional analysis of mouse Polycomb group genes. Cell Mol Life Sci 54: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA, Chinnaiyan AM (2002) The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419: 624–629 [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ (2001) Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol Cell Biol 21: 6820–6832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sonoda M, Inokuchi J, Shirasawa S, Sasazuki T (2004) Polycomb group suppressor of zeste 12 links heterochromatin protein 1alpha and enhancer of zeste 2. J Biol Chem 279: 401–406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2