

Abstract
Nivolumab is a fully human monoclonal antibody that inhibits programmed death‐1 activation. The clinical pharmacology profile of nivolumab was analyzed by a population pharmacokinetics model that assessed covariate effects on nivolumab concentrations in 1,895 patients who received 0.3–10.0 mg/kg nivolumab in 11 clinical trials. Nivolumab pharmacokinetics is linear with a time‐varying clearance. A full covariate model was developed to assess covariate effects on pharmacokinetic parameters. Nivolumab clearance and volume of distribution increase with body weight. The final model included the effects of baseline performance status (PS), baseline body weight, and baseline estimated glomerular filtration rate (eGFR), sex, and race on clearance, and effects of baseline body weight and sex on volume of distribution in the central compartment. Sex, PS, baseline eGFR, age, race, baseline lactate dehydrogenase, mild hepatic impairment, tumor type, tumor burden, and programmed death ligand‐1 expression had a significant but not clinically relevant (<20%) effect on nivolumab clearance.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Nivolumab is the first anti‐programmed death‐1 antibody that demonstrated improved survival in multiple tumor types.
WHAT QUESTIONS DID THIS STUDY ADDRESS?
☑ The analysis characterized pharmacokinetics (PK) and effects of covariates on PK of this novel antibody to better define dose adjustment and use in the various segments of the population.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
☑ This study is the first peer‐reviewed report of nivolumab clinical PK and includes development, evaluation, and application of a robust population PK model to support clinical pharmacology sections in prescriber information. The analysis shows that nivolumab PK is similar among patients across different tumor types and also shows that hepatic and renal status have no effect on nivolumab PK and exposure.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
☑ This analysis assessed the clinical relevance of demographic and pathophysiological covariates affecting PK of nivolumab. The model also explored the PK of nivolumab across tumor types and was used to determine individual exposures in patients to support exposure–response analyses for target populations. This analysis serves as an example for characterizing time‐varying clearance for monoclonal antibodies.
One of the mechanisms by which tumors evade immune surveillance is via modulation of inhibitory checkpoint pathways regulating immune responses. The programmed death‐1 (PD‐1) membrane receptor is a key component of one such pathway, and is a negative regulatory molecule expressed by activated T and B lymphocytes.1 Binding of PD‐1 to its ligands, programmed death ligand‐1 (PD‐L1) and −2 (PD‐L2), results in the downregulation of lymphocyte activation. Anti‐ PD‐1 monoclonal antibodies that inhibit interaction between PD‐1 and its ligands prevent the downregulation of lymphocyte activation and reactivate exhausted effector T cells, thus promoting immune responses and antigen‐specific T‐cell responses.1, 2, 3, 4 Animal tumor models and in vitro studies employing a variety of human tumor types have demonstrated that blockade of the PD‐1 receptor potentiates antitumor immune response.5, 6 This suggests that antitumor immunotherapy via PD‐1 blockade is not limited, in principle, to any single tumor type but may augment the immune response to a number of histologically distinct tumors.7 In addition, expression of PD‐1 has been shown to be a negative prognostic factor in patients with malignant melanoma.8
Nivolumab (Opdivo, Bristol‐Myers Squibb, Princeton, NJ, and Ono Pharmaceutical, Trenton, NJ) is a fully human immunoglobulin G4 (IgG4) monoclonal antibody that selectively binds to PD‐1 and prevents interactions between PD‐1 and PD‐L1 or PD‐L2 on tumors, thus preventing T‐cell exhaustion and reactivation of exhausted effector T cells.5, 9 The clinical activity of nivolumab was initially evaluated in malignant melanoma and squamous non‐small cell lung cancer (NSCLC), and the remarkable response rates, prolonged survival, and better safety profile were the basis of regulatory approval.10, 11, 12 Nivolumab is approved for the treatment of unresectable or metastatic melanoma for patients with first‐line and disease progression following anti‐cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4) treatment with ipilimumab and with a BRAF inhibitor (if positive for the BRAF V600 mutation); for the treatment of patients with metastatic squamous NSCLC with progression on or after platinum‐based chemotherapy, and for the treatment of patients with advanced renal cell carcinoma (RCC), among other tumor types.11, 13 Nivolumab in combination with the CTLA‐4 checkpoint inhibitor ipilimumab is approved for the treatment of unresectable or metastatic melanoma.
Pharmacokinetics (PK), clinical activity, and safety of nivolumab have been assessed in phase I, phase II, and phase III studies in adult patients with NSCLC, melanoma, and RCC, together with additional tumor types.7, 10, 11, 14, 15, 16 The population PK (PPK) model supporting the clinical development of nivolumab for these indications17 was revised to reflect the finding of time‐varying nivolumab clearance (CL).18 Development, evaluation, and application of the nivolumab PPK model with time‐varying CL are presented in this article, including the assessment of the potential effect of intrinsic and extrinsic factors on nivolumab PK and exposure.
METHODS
Data
The nivolumab PPK model was developed using data from 1,895 patients for whom nivolumab concentrations were available in three phase I studies (MDX1106‐01 (ClinicalTrials.gov identifier: NCT00441337), ONO‐4538‐01 (NCT00836888), and MDX1106‐03 (NCT00730639)), three phase II studies (CA209010 (NCT01354431), CA209063 (NCT01721759), and ONO‐4538‐02 (JapicCTI‐111681)), and five phase III studies (CA209017 (NCT01642004), CA209037 (NCT01721746), CA209025 (NCT01668784), CA209057 (NCT01673867), CA209066 (NCT01721772)) of nivolumab. Table 2 provides a summary of the studies and specified PK sampling intervals.
Table 2.
Summary of studies included in PPK analyses
| Study | Patients included in PPK analysis, N | Dosing regimen | Number of PK samples |
|---|---|---|---|
| MDX1106‐01 – Phase I, open‐label, multicenter, dose‐escalation study for selected refractory or relapsed MEL, NSCLC, RCC, CRC, mCRPC | 39 | 0.3, 1.0, 3.0, or 10.0 mg/kg IV infusion administered over 60 minutes | 800 |
| ONO‐4538‐01 – Phase I single‐dose study in patients with progressive or recurrent solid tumors | 17 | 1.0, 3.0, 10.0, and 20.0 mg/kg, 1‐hour IV infusion Regimen: 3‐week for first dose, followed by Q2W | 268 |
| MDX1106‐03 – Phase I, open‐label, multicenter, multidose, dose‐escalation study in patients with selected advanced or recurrent MEL, NSCLC, RCC, CRC, mCRPC | 304 | 0.1, 0.3, 1.0, 3.0, or 10.0 mg/kg IV infusion depending upon tumor type, dosed over 60 minutes Q2W for up to twelve 8‐week cycles | 3,218 |
| CA209010 – A randomized, blinded, phase II dose‐ranging study in patients with progressive advanced/metastatic clear‐cell RCC who had received prior anti‐angiogenic therapy | 167 | 0.3, 2.0, and 10.0 mg/kg, 1‐hour IV infusion Regimen: Q3W | 1,509 |
| CA209017–‐ An open‐label, randomized phase III trial of BMS‐936558 (nivolumab) versus docetaxel in previously treated advanced or metastatic squamous cell non‐small cell lung cancer (NSCLC) | 125 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 454 |
| CA209025 – A randomized, open‐label, phase III study of nivolumab (BMS‐936558) vs everolimus in patients with advanced or metastatic clear‐cell renal cell carcinoma who have received prior anti‐angiogenic therapy | 403 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 2,375 |
| CA209037 – A randomized, open‐label phase III study of nivolumab vs. investigator's choice in advanced melanoma patients progressing post‐anti‐CTLA‐4 therapy | 232 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 773 |
| CA209057 – An open‐label, randomized phase III trial of BMS‐936558 (nivolumab) versus docetaxel in previously treated advanced or metastatic non‐squamous cell non‐small cell lung cancer (NSCLC) | 280 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 1,060 |
| CA209063 – A single‐arm phase II study in patients with advanced or metastatic squamous NSCLC cancer who had received ≥2 prior systemic regimens | 115 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 430 |
| ONO‐4538‐02 – A multicenter, open‐label uncontrolled study in patients with unresectable, advanced (stage III or IV) or recurrent malignant melanoma | 35 | 2.0 mg/kg, 1‐hour IV infusion Regimen: Q3W | 740 |
| CA209066 – A phase III, randomized, double‐blind study of BMS‐936558 (nivolumab) vs dacarbazine in patients with previously untreated, unresectable or metastatic melanoma | 178 | 3.0 mg/kg, 1‐hour IV infusion Regimen: Q2W | 665 |
CRC, colorectal cancer; CTLA‐4, cytotoxic T lymphocyte‐associated antigen 4; IV, intravenous; mCRPC, metastatic castration‐resistant prostate cancer; MEL, melanoma; NSCLC, non‐small cell lung cancer; PK, pharmacokinetic; PPK, population pharmacokinetic; RCC, renal cell carcinoma; Q2W, every 2 weeks; Q3W, every 3 weeks.
PPK model development
The PPK model was developed in three stages, consisting of the base, full, and final models. First, a base model was developed to describe the PK of nivolumab without consideration of covariate effects. Second, a full covariate model was developed by incorporating the effect of all prespecified covariate parameter relationships. In the third stage, the final PPK model was developed by retaining covariates that improved a goodness‐of‐fit statistic (BIC).19
The first step of PPK analysis was base model development, which consisted of the development of a structural model, interindividual variability (IIV) model, and a residual error model. Structural model development included the assessment of temporal changes in nivolumab CL, which consisted of the selection of the functional form of the temporal effect in comparison to the model with constant CL. In addition to the model with constant CL, two alternative functional forms, which were used to describe time‐dependent CL (hyperbolic‐Emax and sigmoid‐Emax (described below)), were tested relative to the base model with constant CL:
| (1) |
| (2) |
The parameter of patient i is given by the following expression:
| (3) |
where represents the population (typical value) estimate of the maximal change in CL; and ∼N(0, ) is a normally distributed random variable, with mean 0, and variance , which represents the interindividual variability in . The parameter represents the time at which the change in CL is 50% of , and represents the sigmoidicity of the relationship with time.
The full model was developed to obtain unbiased estimates of the magnitude of covariate effects on model parameters by simultaneously incorporating all prespecified covariate–parameter relationships of interest into the model.20, 21 The analysis included the effect of the following baseline covariates on the PK of nivolumab: body weight (BW), age, sex, race, renal function, hepatic function status, performance status (PS), lactate dehydrogenase (LDH), tumor type, PD‐L1 expression status, and tumor burden. The value of PS is identical to the Eastern Cooperative Oncology Group (ECOG) for patients in studies in which performance status was assessed according to ECOG criteria (all studies except CA209025). The performance status of patients in study CA209025 was assessed by Karnofsky performance status (KPS) criteria, so the PS values of patients in this study were derived by mapping the KPS values to the ECOG scale.22 Baseline hepatic impairment status was specified on the basis of criteria established by the National Cancer Institute Organ Dysfunction Working Group23 and baseline estimated glomerular filtration rate (eGFR) was estimated using the CKD‐EPI equation.24 Covariates that had an effect of >20% on model parameters were considered potentially clinically important and were assessed further in the model application. Table 3 describes the baseline demographic characteristics and laboratory and disease status variables that comprised the covariates included in the model development.
Table 3.
Summary of baseline demographic and laboratory covariates
| Covariate | PPK analyses index dataset (N = 1895) |
|---|---|
| Continuous | |
| Age, years, mean (SD) | 61.12 (11.12) |
| Body weight, kg, mean (SD) | 79.09 (19.28) |
| eGFR, mL/min/1.73 m2, mean (SD) | 78.49 (21.63) |
| Lactate dehydrogenase, U/L, mean (SD) | 350.85 (397.57) |
| Liver dysfunction groups, n (%) | |
| Missing | 18 (0.95) |
| Group A: normal | 1688 (89.08) |
| Group B: mild | 186 (9.82) |
| Group C: moderate | 2 (0.11) |
| Group D: severe | 1 (0.05) |
| Categorical | |
| Sex, n (%) | |
| Male | 1264 (66.7) |
| Female | 631 (33.3) |
| Race, n (%) | |
| White | 1685 (88.92) |
| African American/Black | 53 (2.8) |
| Asian | 122 (6.44) |
| Other | 33 (1.74) |
| Baseline performance status, n (%) | |
| 0 | 734 (38.73) |
| 1 | 1109 (58.52) |
| 2 | 52 (2.74) |
| Tumor type, n (%) | |
| MEL | 565 (29.82) |
| NSCLC | 659 (34.78) |
| RCC | 605 (31.93) |
| Other | 66 (3.48) |
eGFR, estimated glomerular filtration rate; MEL, melanoma; NSCLC, non‐small cell lung cancer; PPK, population pharmacokinetic; RCC, renal cell carcinoma; SD, standard deviation.
The functional relationships between continuous covariates and structural model parameters were modeled using the following equation:
| (4) |
where and are fixed‐effect parameters, and is the reference value of the covariate.
The relationship between the typical value of a parameter and a binary time‐invariant covariate (R) was characterized with the following equation:
| (5) |
where and are fixed‐effects parameters and is the indicator variable. Inferences regarding the effect of covariates were based on the full model parameter estimates.
The final model was developed from the full model by backward elimination of covariates one at a time, such that the model with the lowest BIC was selected. The PPK model parameters were estimated using the first‐order conditional estimation with interaction method implemented in NONMEM (v. 7.3, ICON Development Solutions, Hanover, MD). Diagnostic plots were prepared using R (v. 3.0.2)25 or S‐PLUS software (v. 8.0, TIBCO Software, Palo Alto, CA).
The effect of immunogenicity on nivolumab CL was assessed as a time‐varying covariate, and the CL of ith patient on the jth occasion at which ADA data were available was modeled as follows:
| (6) |
where is the typical value of CL; is an indicator variable for the ADA status of patient i at occasion j (ADA positive: 1, negative: 0), is the estimated effect of ADA on CL; where is a normally distributed random variable with zero mean and variance , which represents the IIV in CL. Three different ADA assays were employed over the course of the nivolumab development program, with progressive improvements in the drug tolerance of the assay.26 The effect of ADA for each assay developed and used in different studies was estimated separately, and the estimated parameter for ADA for each assay represents the fractional change within‐patient CL due to the presence of ADA. Samples with missing ADA were assessed separately where the term in the above equation was replaced by , which represents a nuisance parameter. This approach avoided having to exclude PK records for which ADA values were not available, or having to impute ADA values for such records.
PPK model evaluation
Model evaluation was performed by using standard goodness‐of‐fit plots including conditional weighted residuals with interaction (CWRESI) vs. actual time after first dose, CWRESI vs. actual time after previous dose, CWRESI vs. predicted (typical) serum concentration, observed vs. predicted population average and individual concentrations, and visual predictive check (VPC) to provide an evaluation of model assumptions and population parameter estimates.27 VPC was performed with 500 simulated datasets that were obtained by using parameter values from the final model.
The VPC provides a graphical assessment of the agreement between the time course of model predictions and observations at the recommended dose of 3.0 mg/kg Q2W as well as at 10.0 mg/kg Q2W. The check was performed by plotting the 5th, 50th, and 95th percentiles of observed plasma concentration–time data with their corresponding 90% prediction intervals by dose level.
PPK model application
The final PPK model was applied to estimate individual measures of nivolumab exposure (time‐averaged nivolumab concentrations at steady state obtained with nominal nivolumab dosing regimen (Cavgss)), and for ad hoc analyses to estimate covariate–parameter relationships of interest that could not be assessed in the full model. Cavgss was calculated by dividing the area under the steady‐state concentration–time curve (AUCss) with the nominal dosing interval given every 21 days (Q3W) and Q2W. The AUCss of each patient was determined for the purpose of computing Cavgss by dividing the nivolumab dose with the maximum a posteriori Bayesian estimate of CL.
Data for PD‐L1 expression were not available for all patients. The effect of PD‐L1 on PK was only assessed using graphical analysis for patients with available verified PD‐L1 assay results. Albumin (ALB) was not available for studies CA209037 and CA209066, and LDH was not available in CA209010 and CA209025; therefore, the effect of LDH and ALB on PK was assessed in a sensitivity analysis.
RESULTS
PPK model development
The PK of nivolumab was determined to be linear, such that CL is independent of dose within the dose range of 0.1–20.0 mg/kg (Figure 1 a). Analysis of dose proportionality during base‐model development indicated that models describing the elimination of nivolumab by a nonlinear model incorporating a Michaelis–Menten elimination term representing target mediated drug disposition did not improve the goodness‐of‐fit compared to a linear model.
Figure 1.
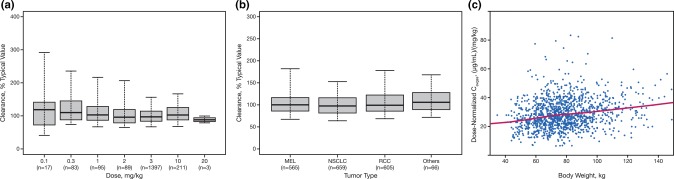
(a) PPK‐based estimates of individual nivolumab clearance vs. dose based on final PPK model. The boxplots represent median (bold line), 25th, and 75th percentiles of clearance distribution. The whiskers represent 5th and 95th percentiles of the distribution. PPK, population pharmacokinetic. (b) Distribution of nivolumab clearance estimates across tumor types using final PPK model. The boxplots represent median (bold line), 25th, and 75th percentiles of the distribution. The whiskers represent 5th and 95th percentiles of the distribution. (c) Nivolumab dose‐normalized Cavgss vs. body weight for body weight‐based, Q2W dose regimens. Normalized exposure is Cavgss. Solid line represents locally weighted smooth line and is used to visualize relationships between dose‐normalized Cavgss and body weight. Cavgss, average concentration at steady state; CRC, colorectal cancer; CRPC, castration‐resistant prostate cancer; MEL, melanoma; NSCLC, non‐small cell lung cancer; PPK, population pharmacokinetic; Q2W, every 2 weeks; RCC, renal cell carcinoma.
The base model is a two‐compartment model with zero‐order i.v. infusion and first‐order elimination, parameterized in terms of CL, volume of central compartment (VC), intercompartmental CL (Q), and volume of peripheral compartment (VP). The CL of nivolumab was determined to be time‐varying, as the BIC values of the constant CL model was higher than the BIC values of the models with time‐varying CL. The BIC value for the sigmoid‐Emax model was markedly lower than that of the model with constant CL (by 261 points) and hyperbolic (Emax) effect on CL (by 35 points); therefore, time‐varying CL with sigmoid‐Emax function form was selected for the subsequent model development. The IIV parameters of the base model were specified by a lognormal model and nonzero covariance between the IIV on CL, VC, and Emax (maximal change in CL from baseline), as specified in the Methods section. The residual error was best described by a proportional error model, in comparison with additive and combined residual error models.
Covariate effects were assessed by a full‐model approach, in which all prespecified covariate‐parameter relationships were simultaneously estimated. Results of the full model indicate that the magnitude of the effect of covariates on CL was within ±20% of the reference value for all covariates except BW, and of the remaining covariates, the 95% confidence interval (CI) was within ±20% for all covariates except baseline PS, baseline eGFR, and sex (Figure 2 a). PS appears to be an important covariate for CL, showing a >20% increase of CL in patients with PS >0. Sex appears to have a similar effect on CL and VC, and the magnitude of the effect was outside the boundary of ±20%. Male patients included in this analysis had a higher CL and VC than females; however, these effects are unlikely to be of clinical relevance, as exposures are similar between male and female patients. Other covariates that were within the ±20% boundaries were race, tumor type, hepatic function, and age, suggesting these covariates are also not of clinical relevance.
Figure 2.
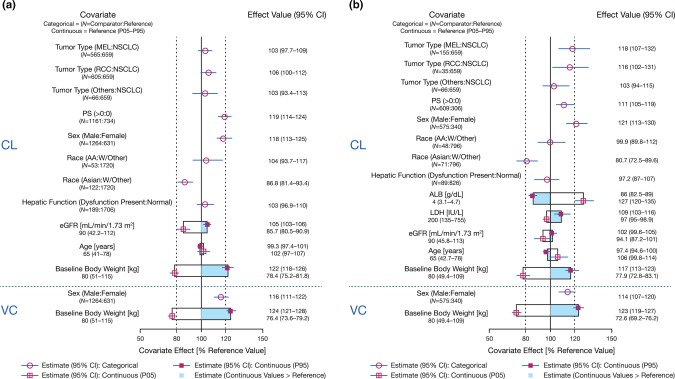
(a,b) Covariate effects on PK model parameters from full population PK model. Categorical covariate effects (95% CI) are represented by open symbols (horizontal lines). Continuous covariate effects (95% CI) at the 5th/95th percentiles of the covariate are represented by the end of horizontal boxes (horizontal lines). Open/shaded area of boxes represents the range of covariate effects from the median to the 5th/95th percentile of the covariate. The reference patient is a 65‐year‐old Caucasian/other female weighing 80 kg with a baseline performance status of 0, LDH of 200 IU/L, baseline ALB of 4 g/dL, estimated GFR of 90 mL/min/1.73 m2, and normal hepatic function. Parameter estimate in reference patient is considered 100% (vertical solid line), and dashed vertical lines are at 80% and 120% of this value. AA, African American; ALB, albumin; CI, confidence interval; CL, clearance; eGFR, estimated glomerular filtration rate; LDH, lactate dehydrogenase; MEL, melanoma; NSCLC, non‐small cell lung cancer; PS, baseline performance status; RCC, renal cell carcinoma; PK, pharmacokinetics; VC, volume of central compartment; W, white.
Tumor type has less than a 20% effect on CL. This finding is similar with the results from a previous report, in which the dose‐normalized nivolumab concentrations at steady state (Cavgss) were similar between patients with NSCLC and melanoma.17 The CL (% of typical value, baseline CL) was similar across tumor types (Figure 1 b), indicating that PK is independent of tumor types. The median CL (baseline CL) estimates in patients with NSCLC, melanoma, and RCC were 10.5, 10.8, and 11.5 mL/h, respectively.
The final model was developed by backward elimination of the covariates in the full PPK model, based on BIC. The final PPK model contained baseline BW, eGFR, sex, race, and PS on CL and baseline BW and sex on VC; the parameter estimates from the final PPK model are provided in Table 1. The IIV on CL and VC of the final model were reduced by 30% and 21%, respectively, compared with the base model. The extent of shrinkage for IIV on CL, VC, and VP, and residual variability in the final and full model, were within 30% except for the VP, which was 41%, indicating the parameter estimates are reliable.
Table 1.
PPK final model parameter estimates
| Parametera [units] | Estimateb | 95% confidence intervalc |
|---|---|---|
| Structural model parameters | ||
| CL REF [mL/h] | 9.4 | 8.7 – 10 |
| VC REF [L] | 3.63 | 3.5 – 3.75 |
| Q REF [mL/h] | 32.1 | 25.9 – 37.4 |
| VP REF [L] | 2.78 | 2.58 – 3.04 |
| CLBW | 0.566 | 0.479 – 0.658 |
| CL eGFR | 0.186 | 0.126 – 0.25 |
| CL SEX | 0.165 | 0.119 – 0.216 |
| CL BPS | 0.172 | 0.132 – 0.212 |
| CL RAAS | −0.125 | −0.189 – −0.057 |
| CL EMAX | −0.295 | −0.395 – −0.216 |
| CL T50 | 1.41 × 103 | 1.21 × 103 – 1.85 × 103 |
| CL HILL | 3.15 | 1.65 – 7.52 |
| VCBW | 0.597 | 0.514 – 0.681 |
| VC sex | 0.152 | 0.11 – 0.2 |
| Interindividual variability model parameters | ||
| ω2 CL | 0.123 (0.35) | 0.106 – 0.143 |
| ω2 VC | 0.123 (0.351) | 0.0929 – 0.156 |
| ω2VP [‐] | 0.258 (0.508) | 0.197 – 0.315 |
| ω2 EMAX | 0.0719 (0.268) | 0.0488 – 0.119 |
| ω CL :ωVC | 0.0432 (0.352) | 0.0344 – 0.0547 |
| Residual error model parameters | ||
| Proportional error [‐] | 0.215 | 0.203 – 0.229 |
BW, body weight; BPS, baseline performance status; CL, clearance; eGFR, estimated glomerular filtration rate; Q, inter‐compartmental clearance; RAAS, Race (Asian); VC, volume of central compartment; VP, volume of peripheral compartment.
ETA shrinkage: ETA_CL: 14.2, ETA_VC: 13.4, ETA_VP: 41.4, ETA_EMAX: 48.5, and EPS shrinkage (%): 15.5. CLREF and VCREF are typical values of CL and VC at the reference values. Covariate effect was estimated relative to a white female reference weighing 80 kg with an eGFR of 90 mL/min/1.73 m2, and BPS of 0.
Estimate values in parentheses are standard deviation for estimated variances and correlation for estimated covariances.
Confidence interval values are taken from bootstrap calculations (1,918 successful out of a total of 2,000).
The final model was as follows:
| (7) |
| (8) |
| (9) |
| (10) |
where and are the typical values of baseline CL and VC at the reference values of BW, PS, sex, race (Asian), and eGFR, respectively, and , , , , , , and are model parameters. represents the estimate of the maximal change in CL. The parameter represents the time at which the change in is 50% of , and represents the sigmoidicity of the relationship with time. represents baseline CL of patient i; represents the CL of patient i at a given time t, and represents the steady‐state CL of patient i. The reference values of covariates are shown in Figure 2 a and were chosen close to median values in the PPK dataset.
In the final model, nivolumab CL decreases over time, with a mean maximal reduction from baseline values of ∼24.5%. Following intravenous (i.v.) administration, nivolumab undergoes a biphasic elimination consisting of a rapid distribution phase with a geometric mean (coefficient of variation (CV) %) terminal half‐life t½(α) of 32.5 hours (24.8%) and a slow elimination phase with a geometric mean (CV%) t½(β) of 25 days (77.5%) at steady state.
PPK model evaluation
The PPK model was evaluated using standard diagnostic plots. The diagnostic plots of the final PPK model show that the two‐compartment model with zero‐order infusion appropriately characterizes nivolumab PK (Supplemental Figures S1–S3).
The visual predictive check (VPC) plots showed that the observed concentration–time course of nivolumab at the 5th, 50th, and 95th percentiles fell within their corresponding 90% prediction intervals, indicating that the model adequately characterized the observed data. Figure 3 shows a representative VPC of concentration vs. actual time following the previous dose at 3.0 mg/kg and 10.0 mg/kg every 2 weeks (Q2W).
Figure 3.
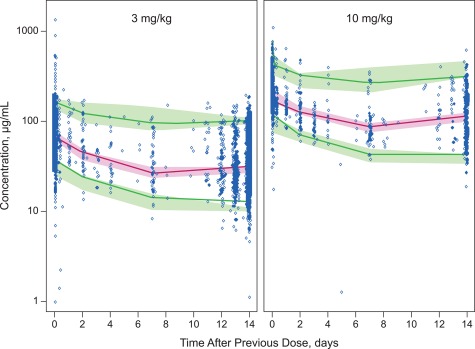
Model validation representative visual predictive check from final population pharmacokinetic model: nivolumab concentration vs. actual time after dose. Representative visual predictive check of concentration (log scale) vs. actual time after previous dose for 3.0 mg/kg and 10.0 mg/kg Q2W doses. Dots are observed data and the solid lines represent the 5th, 50th, and 95th percentiles of observed data, respectively. The shaded areas represent the simulation‐based 90% confidence intervals for the 5th, 50th, and 95th percentiles of the predicted data. Q2W, every 2 weeks.
PPK model application
BW‐normalized dosing produced approximately uniform exposures for Cavgss in patients with BW in the range of 34 − 168 kg receiving Q2W and every‐3‐week (Q3W) dosing regimens, as shown in Figure 1 c. The effects of PS are shown in the online supplement (Supplemental Figure S4). The lack of effect of parameters, including renal function status, hepatic function status, and PD‐L1 expression on nivolumab CL are also shown in the online supplement (Supplemental Figures S5–S7).
A sensitivity analysis was done to evaluate the effect of baseline ALB and baseline LDH, as these values were not collected for some studies. The sensitivity analysis showed that ALB has a significant effect on CL (>20%) and that nivolumab CL was greater in patients with lower ALB levels (Figure 2 b). Nivolumab exposures (dose‐normalized Cavgss) in patients with lower than normal ALB were markedly lower than in patients with normal ALB levels. The effect of LDH was also found to be significant, with nivolumab CL higher in patients with higher LDH levels; however, the magnitude of the effect was within 20%.
The effect of immunogenicity (antidrug antibodies (ADAs)) on nivolumab CL was assessed as a time‐varying covariate using the final model. ADA has minimal effect on nivolumab CL (<20%), and the estimated effect for ADA on CL was 114% (95% CI: 106–122%) relative to occasions when ADAs were not present. Given the modest impact of ADAs on CL, for a fraction of the treatment period over which ADA is positive, and the small number of patients with a low titer of ADA‐positive samples, the effect of immunogenicity on the PK of nivolumab was not considered to be clinically relevant in this analysis.
DISCUSSION
Nivolumab concentration–time data were well described by a linear, two‐compartment, zero‐order i.v. infusion model with first‐order elimination, and time‐varying CL. The change in CL with time was evaluated by comparing the goodness‐of‐fit of models incorporating hyperbolic and sigmoid Emax temporal changes in CL with that of a model with constant CL. The BIC values of both hyperbolic and sigmoid Emax models were markedly lower than that of the model with constant CL, indicating that the models with time‐varying CL provided a better description of the data. The magnitude of the decrease in BIC was greatest with the sigmoid‐Emax model, indicating that the temporal change in CL was best described by this model; nivolumab CL decreases over time with ∼25% maximal reduction from baseline values. The final PPK model was evaluated using VPC, which showed that the linear two‐compartment model with zero‐order infusion adequately characterized the data.
The previous PPK model did not include time‐varying CL, and the assessment of stationarity in nivolumab CL was based on diagnostic plots and incorporation of interoccasion variability, which appeared to indicate that nivolumab CL did not change with time. However, a reassessment of nivolumab PK motivated by a report of time‐varying PK of anti‐PD‐1 agents18 and feedback from the US Food and Drug Administration reviewers (see Acknowledgments) confirmed that incorporation of time‐varying CL resulted in a statistically significant improvement in the goodness‐of‐fit, as measured by BIC. Therefore, the base model was redeveloped using this empirical‐based approach to include time‐varying CL, and covariate effects were reassessed on the PK parameters of this base model. These covariate effects were found to be consistent with that of the previous stationary model.17
Although there is no clear mechanistic understanding on the time‐varying CL of nivolumab, we hypothesize that the decrease in nivolumab CL over the course of treatment may be associated with improvement in disease status, and the corresponding decrease in the rate of cancer‐related cachexia. Cachexia is a complex metabolic syndrome associated with underlying disease, characterized by loss of muscle mass due to a hypermetabolic state.28, 29 One indicator of the level of cachexia and the hypermetabolic state is low serum ALB,28, 30 which has been shown to be due to higher turnover in cachexic patients (and not due to a decrease in synthesis rate).31 The strong association between nivolumab CL and ALB (Figure 2 b) suggests that elimination or turnover of these proteins is higher in cachexic patients due to their hypermetabolic state. The higher rate of nivolumab CL in putatively cachexic patients is consistent with reports of higher rates of whole‐body protein turnover in cancer patients.31 A decrease in nivolumab CL may be indicative of decreased cachexia, and improvement in disease state.32, 33
Other than time‐varying CL, nivolumab exhibits similar PK properties to those typically associated with IgG therapeutics.34, 35 However, unlike some IgG monoclonal antibodies that have been approved for therapeutic use such as efalizumab, nivolumab PK was found to be linear. Following i.v. administration, nivolumab undergoes a biphasic elimination consisting of a rapid distribution phase and a slow elimination, with a derived steady‐state terminal t1/2 of 25 days, which is consistent with those seen for typical IgG therapeutics.35, 36, 37
BW, PS, sex, and eGFR were found to be covariates that accounted for a portion of the IIV of nivolumab CL in the final PPK model, while only sex and BW accounted for a portion of the IIV on VC. The IIV for nivolumab on CL and VC was reduced by 30% and 21%, respectively, when these covariates were incorporated into the PPK model. Consistent with the mechanism of elimination for monoclonal antibodies, both CL and VC are higher in patients with higher BW.36 Although the PPK analysis showed that both CL and VC increase with BW, nivolumab steady‐state exposure (Cavgss, estimated with 400 dosing, allowing all patients to reach steady state) was comparable across the range of BW (34.1 − 168.2 kg) of the patients included in this PPK analysis dataset. The slight increase in exposure for patients with high BW is due to a less than proportional increase of CL as BW increases. During model development, it was found that PS and sex had a significant effect on nivolumab CL. The full PPK model showed that CL in patients with PS >0 appeared to be 19% higher compared to patients with PS of 0. CL in male patients appeared to be 18% higher relative to that of female patients, and CL in patients with Asian origin appeared to be 15% lower compared to white patients; however, the magnitude of PS, sex, and race effect was less than 20%, indicating that this effect was not clinically relevant.
No clinically important differences in the CL and exposure of nivolumab were found between patients with renal impairment and those with normal renal function. The full model showed that eGFR was unlikely to be a clinically relevant covariate, as the estimated change in CL in patients with renal impairment was within 20% (although the 95% CI was slightly lower at the 5th percentile of eGFR). However, during development of the final model eGFR and race (Asian) were found to be a statistically significant covariate based on the BIC used to select the final model. Therefore, eGFR was included as a covariate on CL in the final model. The potential impact of renal impairment on nivolumab exposure was also tested to determine whether there was a relationship between renal function and nivolumab CL. The range of eGFR in the dataset covered the normal to moderate and had very few patients with severely impaired renal function based on the categorization of eGFR using the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) equation. There was no relationship between renal function status and nivolumab CL and exposure, indicating that renal function has no impact on nivolumab PK. Furthermore, the absence of a relationship between eGFR and nivolumab CL is entirely consistent with renal physiology, as the large size of nivolumab (144 kDa) is expected to prevent it from being filtered through the glomeruli of the kidney and eliminated via the urine.
The relatively modest covariate effects of about 20% in magnitude are not expected to be clinically significant. First, the safety and tolerability of nivolumab have been established up to a dose level of 10 mg/kg Q2W, which is more than 3‐fold higher than the recommended 3 mg/kg Q2W dose across indications. Second, the exposure–response analysis of efficacy in multiple tumor types has shown that the exposure–response relationship is relatively flat for exposures produced by a 3 mg/kg Q2W dosing regimen. Thus, these findings taken together indicate that no dose adjustment is warranted in patients with renal impairment. LDH data were not available in study CA209010 and CA209025, and serum ALB data were not available in study CA209037 and CA209066; therefore, their effects on nivolumab CL were assessed in a sensitivity analysis. It was found that ALB level had a significant effect on CL (>20%), and nivolumab CL was greater in patients with lower ALB levels (Figure 2 b). Lower than normal ALB (<3.4 g/dL) could be a sign of liver or kidney disease, increased catabolic activity, or low neonatal Fc receptor expression level or activity. Nivolumab exposures (dose‐normalized Cavgss) in patients with lower than normal ALB were ∼45% lower than in patients with normal ALB levels. Therefore, ALB was considered a potentially clinically relevant covariate for nivolumab PK. Furthermore, there appeared to be some correlation between PS and ALB, as ALB levels were found to be lower in patients with higher PS (correlation coefficient −0.27). It is likely that the potential effect of ALB on CL was represented to some extent by the effect of PS.
Other covariates including age, LDH, mild hepatic impairment, and tumor type did not have clinically relevant (<20%) effects on nivolumab CL and were not included in the final PPK model. Further, a graphical assessment of baseline tumor burden, baseline tumor volume, and PD‐L1 expression on nivolumab PK parameters showed that these factors have no impact on nivolumab PK.
In conclusion, this report presents the PPK model of nivolumab PK data in patients with solid tumors and demonstrates a similar level of nivolumab exposure in both patients with melanoma and NSCLC, with nivolumab PK being linear with time‐varying CL. Nivolumab CL and VC increased with increasing BW, and BW‐normalized dosing produced approximately uniform nivolumab exposures over the studied range of BW. The lack of effect of mild hepatic and renal impairment on nivolumab PK suggests that dose adjustments will not be required in these patient subpopulations.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Acknowledgments
This study was supported by Bristol‐Myers Squibb. Rosemary Reinke on behalf of inScience Communications, Springer Healthcare (Philadelphia, PA), provided medical writing and editorial support funded by Bristol‐Myers Squibb. We thank the following FDA scientists who initially identified the time‐varying PK property of nivolumab: Chao Liu, Hongshan Li, Jingyu Yu, and Yaning Wang. The nivolumab PPK model was revised based on this new finding.
Conflict of Interest
Y.F. is employed as a scientific director in Clinical Pharmacology and Pharmacometrics at Bristol‐Myers Squibb. G.B., X.W., S.A., M.G., and A.R. are all employees of Bristol‐Myers Squibb.
Author Contributions
G. Bajaj and Y. Feng wrote the article. G. Bajaj and Y. Feng performed the analysis. G. Bajaj, Y. Feng, M. Gupta, S. Agrawal, X. Wang, and A. Roy contributed to the design of the analysis and reviewed the analysis.
References
- 1. Sharpe, A.H. , Wherry, E.J. , Ahmed, R. & Freeman, G.J. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 8, 239–245 (2007). [DOI] [PubMed] [Google Scholar]
- 2. Momtaz, P. & Postow, M.A. Immunologic checkpoints in cancer therapy: focus on the programmed death‐1 (PD‐1) receptor pathway. Pharmgenomics Pers Med. 7, 357–365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ott, P.A. , Hodi, F.S. & Robert, C. CTLA‐4 and PD‐1/PD‐L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 19, 5300–5309 (2013). [DOI] [PubMed] [Google Scholar]
- 4. Jurado, J.O. et al Programmed death (PD)−1:PD‐ligand 1/PD‐ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J Immunol. 181, 116–125 (2008). [DOI] [PubMed] [Google Scholar]
- 5. Wang, C. et al In vitro characterization of the anti‐PD‐1 antibody nivolumab, BMS‐936558, and in vivo toxicology in non‐human primates. Cancer Immunol Res. 2, 846–856 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Iwai, Y. , Ishida, M. , Tanaka, Y. , Okazaki, T. , Honjo, T. & Minato, N. Involvement of PD‐L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD‐L1 blockade. Proc Natl Acad Sci U S A. 99, 12293–12297 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Topalian, S.L. et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med. 366, 2443–2454 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hino, R. et al Tumor cell expression of programmed cell death‐1 ligand 1 is a prognostic factor for malignant melanoma. Cancer. 116, 1757–1766 (2010). [DOI] [PubMed] [Google Scholar]
- 9. Shih, K. , Arkenau, H.T. & Infante, J.R. Clinical impact of checkpoint inhibitors as novel cancer therapies. Drugs. 74, 1993–2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Topalian, S.L. et al Survival, durable tumor remission, and long‐term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 32, 1020–1030 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robert, C. et al Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 372, 320–330 (2015). [DOI] [PubMed] [Google Scholar]
- 12. Rizvi, N.A. et al Activity and safety of nivolumab, an anti‐PD‐1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non‐small‐cell lung cancer (CheckMate 063): a phase 2, single‐arm trial. Lancet Oncol. 16, 257–265 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Opdivo® [prescribing information]. May, 2016. Princeton, NJ: Bristol‐Myers Squibb. [Google Scholar]
- 14. Wolchok, J.D. et al Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 369, 122–133 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ansell, S.M. et al PD‐1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 372, 311–319 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brahmer, J.R. et al Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med. 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng, Y. , Bajaj, G. , Wang, X. , Agrawal, S. , Gupta, M. & Roy, A. Model‐based analysis of nivolumab pharmacokinetics to support clinical pharmacology profiling in patients with solid tumors [abstract]. Clin Pharmacol Ther. 97, PI–071 (2015). [Google Scholar]
- 18. Wang, Y. Special considerations for modeling exposure‐response relationships for biologics. Presented at the 117th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, San Diego, CA, March 8, 2016.
- 19. Schwarz, G. Estimating the dimension of a model. Ann Stat. 6, 461–464 (1978). [Google Scholar]
- 20. Harrell, F.E., Jr. Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis. New York: Springer; 2001. [Google Scholar]
- 21. Gastonguay, M. Full covariate models as an alternative to methods relying on statistical significance for inferences about covariate effects: a review of methodology and 42 case studies [abstract], Annual Meeting of the Population Approach Group in Europe. Athens, Greece, 2011, Abstract 2229.
- 22. Ma, C. et al Interconversion of three measures of performance status: an empirical analysis. Eur J Cancer. 46, 3175–3183 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Ramalingam, S.S. et al Phase I study of vorinostat in patients with advanced solid tumors and hepatic dysfunction: a National Cancer Institute Organ Dysfunction Working Group study. J Clin Oncol. 28, 4507–4512 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stevens, L.A. et al Evaluation of the Chronic Kidney Disease Epidemiology Collaboration equation for estimating the glomerular filtration rate in multiple ethnicities. Kidney Int. 79, 555–562 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. R Core Team . R: A language and environment for statistical computing. Vienna, Austria, R Foundation for Statistical Computing (http://www.R-project.org/) 2016.
- 26. Agrawal, S. et al Evaluation of immunogenicity of nivolumab monotherapy and its clinical relevance in patients with metastatic solid tumors. J Clin Pharmacol.; e‐pub ahead of print 2016. [DOI] [PubMed] [Google Scholar]
- 27. Bergstrand, M. , Hooker, A.C. , Wallin, J.E. & Karlsson, M.O. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 13, 143–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans, W.J. et al Cachexia: a new definition. Clin Nutr. 27, 793–799 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Fearon, K.C. Cancer cachexia: developing multimodal therapy for a multidimensional problem. Eur J Cancer. 44, 1124–1132 (2008). [DOI] [PubMed] [Google Scholar]
- 30. McMillan, D.C. The systemic inflammation‐based Glasgow Prognostic Score: a decade of experience in patients with cancer. Cancer Treat Rev. 39, 534–540 (2013). [DOI] [PubMed] [Google Scholar]
- 31. Fearon, K.C. et al Influence of whole body protein turnover rate on resting energy expenditure in patients with cancer. Cancer Res. 48, 2590–2595 (1988). [PubMed] [Google Scholar]
- 32. Meibohm, B. : Dose response: the confluence of disease, endpoints, pharmacology, modality and their impact. Presented at the 117th Annual Meeting of the American Society for Clinical Pharmacology and Therapeutics, San Diego, CA, March 8–12, 2016.
- 33. Miyamoto, Y. , Hanna, D.L. , Zhang, W. , Baba, H. & Lenz, H.J. Molecular pathways: cachexia signaling — a targeted approach to cancer treatment. Clin Cancer Res. 22, 3999–4004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feng, Y. , Masson, E. , Dai, D. , Parker, S.M. , Berman, D. & Roy, A. Model‐based clinical pharmacology profiling of ipilimumab in patients with advanced melanoma. Br J Clin Pharmacol. 78, 106–117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Joshi, A. et al An overview of the pharmacokinetics and pharmacodynamics of efalizumab: a monoclonal antibody approved for use in psoriasis. J Clin Pharmacol. 46, 10–20 (2006). [DOI] [PubMed] [Google Scholar]
- 36. Lobo, E.D. , Hansen, R.J. & Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 93, 2645–2668 (2004). [DOI] [PubMed] [Google Scholar]
- 37. Mager, D.E. & Jusko, W.J. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn. 28, 507–532 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information