

Abstract
Radiation therapy is one of the major tools of cancer treatment, and is widely used for a variety of malignant tumours. Radiotherapy causes DNA damage directly by ionization or indirectly via the generation of reactive oxygen species (ROS), thereby destroying cancer cells. However, ionizing radiation (IR) paradoxically promotes metastasis and invasion of cancer cells by inducing the epithelial-mesenchymal transition (EMT). Metastasis is a major obstacle to successful cancer therapy, and is closely linked to the rates of morbidity and mortality of many cancers. ROS have been shown to play important roles in mediating the biological effects of IR. ROS have been implicated in IR-induced EMT, via activation of several EMT transcription factors—including Snail, HIF-1, ZEB1, and STAT3—that are activated by signalling pathways, including those of TGF-β, Wnt, Hedgehog, Notch, G-CSF, EGFR/PI3K/Akt, and MAPK. Cancer cells that undergo EMT have been shown to acquire stemness and undergo metabolic changes, although these points are debated. IR is known to induce cancer stem cell (CSC) properties, including dedifferentiation and self-renewal, and to promote oncogenic metabolism by activating these EMT-inducing pathways. Much accumulated evidence has shown that metabolic alterations in cancer cells are closely associated with the EMT and CSC phenotypes; specifically, the IR-induced oncogenic metabolism seems to be required for acquisition of the EMT and CSC phenotypes. IR can also elicit various changes in the tumour microenvironment (TME) that may affect invasion and metastasis. EMT, CSC, and oncogenic metabolism are involved in radioresistance; targeting them may improve the efficacy of radiotherapy, preventing tumour recurrence and metastasis. This study focuses on the molecular mechanisms of IR-induced EMT, CSCs, oncogenic metabolism, and alterations in the TME. We discuss how IR-induced EMT/CSC/oncogenic metabolism may promote resistance to radiotherapy; we also review efforts to develop therapeutic approaches to eliminate these IR-induced adverse effects.
Keywords: Radiotherapy, Epithelial-mesenchymal transition, Metastasis, Cancer stem cells, Oncogenic metabolism, Tumour microenvironment, Reactive oxygen species, Radioresistance, Snail
Background
Ionizing radiation (IR) is an effective and common therapeutic tool for cancer treatment. More than half of cancer patients are treated with IR at some point during their treatment, either alone or in combination with surgery and/or chemotherapy [1–6]. In radiotherapy, fractionated treatment regimens have been established. The standard fractionation schedule is the delivery of 1.8–2.0 Gy per day, five days per week. This reduces side effects, and allows damaged normal cells to recover before additional doses are given [4, 5]. Fractionated radiotherapy increases damage to the tumour; it may reoxygenate the tumour cells and re-distribute their cell cycles into more sensitive phases. It also minimises repopulation of the tumour during therapy [2, 4, 7].
Nuclear DNA is the primary target of IR; it causes DNA damage (genotoxic stress) by direct DNA ionization. IR also indirectly induces DNA damage by stimulating reactive oxygen species (ROS) production [8–15]. The therapeutic effects of IR are traditionally associated with the DNA double-strand breaks (DSBs) that are the most lethal form of damage to tumour cells. Much evidence has shown that p53 is activated in response to IR-induced DNA damage [8–11]. p53 is a multifunctional transcription factor and it acts principally as a tumour suppressor. It increases the expression of several genes to induce cell cycle arrest (p21, 14-3-3σ), apoptosis (PUMA, NOXA, BAX), autophagy (phosphatase and tensin homolog [PTEN], TSC1, DRAM), or senescence (p21), depending on the cell type and the severity of damage [9, 10]. These are important therapeutic effects of IR.
ROS have been shown to play an important role in mediating the biological effects of IR [12–19]. IR can increase ROS production both by inducing extracellular water radiolysis and by causing intracellular metabolic changes or damage to mitochondria. IR induces delayed (24 h onward), persistent (for days) increases in mitochondrial ROS production, while ROS generated from water have very short life spans (10-9 s) [15, 17]. IR also induces a reversible mitochondrial permeability transition that stimulates ROS production [16]. IR-induced mitochondrial ROS production is associated with partial deactivation of mitochondrial respiratory complexes I and III of the electron transport chain [18, 19]. In turn, excess ROS can disrupt intracellular oxidation/reduction systems and cause oxidative damage to biomolecules, including DNA [12–15]. Activation of the mitochondrial permeability transition also increases levels of reactive nitrogen species (RNS), such as nitric oxide (NO) [16].
Although IR is used as a standard treatment for a variety of malignant tumours, IR paradoxically also promotes tumour recurrence and metastasis [20–28]. The epithelial-mesenchymal transition (EMT) has been shown to endow cancer cells with migratory and invasive properties, enabling the initiation of metastasis [29–31]. IR is known to induce EMT in vitro [20–26]. EMT may be closely linked to cancer stem cells (CSCs) and the metabolic reprogramming of cancer cells, although there is disagreement in the field on these points.
IR is known to induce stemness and metabolic alterations in cancer cells; IR can also cause various changes in the tumour microenvironment (TME) that may promote tumour invasion and metastasis. Oncogenic metabolism has been shown to play important roles in the acquisition of EMT and CSC phenotypes; thus, IR seems to induce EMT and CSC phenotypes by regulating cellular metabolism. EMT, stemness, and oncogenic metabolism are known to be associated with resistance to radiotherapy and chemotherapy. Therefore, understanding the molecular mechanisms of IR-induced EMT/CSC/oncogenic metabolism and changes in the TME is required to improve the efficacy of radiotherapy. Here, we review recent advances in the understanding of the molecular mechanisms of IR-induced EMT, CSC, oncogenic metabolism, and changes in TME, and we discuss a relationship between EMT/CSC/oncogenic metabolism and radioresistance.
Induction of EMT, invasion, and metastasis by IR
EMT, invasion, and metastasis
Cancer cells can acquire multiple biological capabilities during their multistage development. Hanahan and Weinberg proposed ten hallmarks of cancer that alter cell physiology to enhance malignant growth: 1) sustained proliferation, 2) evasion of growth suppression, 3) cell death resistance, 4) replicative immortality, 5) evasion of immune destruction, 6) tumour-promoting inflammation, 7) activation of invasion and metastasis, 8) induction of angiogenesis, 9) genome instability, and 10) alteration of metabolism [32, 33]. Recently, it has also been suggested that cancer is characterised by a breakdown of multicellular cooperation by instances of cellular “cheating” that disrupt all of the following: proliferation inhibition, regulation of cell death, division of labour, resource transport, and maintenance of the extracellular environment. Furthermore, it has also been suggested that deregulation of differentiation is another important aspect of tumourigenesis [34] (Fig. 1).
Fig. 1.
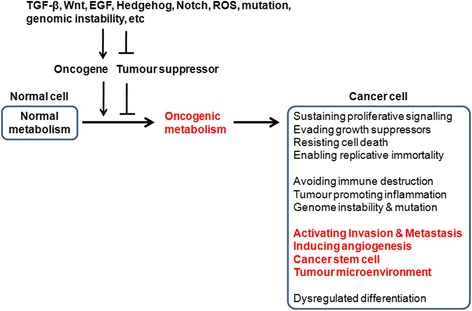
Epithelial-mesenchymal transition (EMT), metastasis, cancer stem cells (CSCs), and oncogenic metabolism. Cancer cells can acquire multiple capabilities, including sustained proliferation, evasion of growth suppression, cell death resistance, replicative immortality, evasion of immune destruction, tumour-promoting inflammation, activation of invasion and metastasis, induction of angiogenesis, genome instability, and alteration of metabolism. Deregulation of differentiation, acquisition of stem cell phenotypes, and their tumour microenvironment are also important aspects of tumourigenesis. Several signal pathways (such as those of TGF-β, Wnt, EGF, Hedgehog, Notch, and ROS) and mutation/genomic instability are closely associated with tumourigenesis and tumour progression. These signals could activate oncogenes and inactivate tumour suppressors. Activation of oncogenes, or loss of tumour suppressors, can drive tumour progression, particularly via metabolic reprogramming. Metabolic reprogramming may be required for malignant transformation and tumour development, including invasion and metastasis, CSC phenotype, and TME
Among the known characteristics of cancer, metastasis is the major obstacle to therapeutic access [29, 35, 36]. EMT is closely linked to the induction of metastasis. EMT is a developmental process that plays critical roles in embryogenesis, wound healing, and organ fibrosis [29–31]. EMT confers mesenchymal properties on epithelial cells; it is characterised by the loss of epithelial morphology and markers (including E-cadherin, desmoplakin, Muc-1, cytokeratin-18, occludins, claudins, and ZO-1), and by the acquisition of mesenchymal markers (including N-cadherin, vimentin, fibronectin, vitronectin, α-smooth muscle actin [α-SMA], and FSP1). Thus, cancer cells undergoing EMT acquire invasive and metastatic properties [29–31].
EMT programs are regulated by a network of signalling pathways that involve components such as growth factors (transforming growth factor-β [TGF-β], epidermal growth factor [EGF]) and their associated signalling proteins (Wnt, Notch, Hedgehog, nuclear-factor kappa B [NF-κB], extracellular signal-regulated kinase [ERK], and phosphatidylinositol 3-kinase [PI3K]/Akt) in response to stresses involved in tumourigenesis, including hypoxia, oncogenic or metabolic stress, inflammation, and physical constraints [30, 31, 37–39].
These signals activate EMT-inducing transcription factors, including Snail/Slug, ZEB1/δEF1, ZEB2/SIP1, Twist1/2, and E12/E47 [40–42]. EMT-inducing transcription factors regulate the expression of proteins involved in cell polarity, cell-cell contact, cytoskeletal structural maintenance, and extracellular matrix (ECM) degradation, and they suppress key epithelial genes. Loss of E-cadherin is considered a hallmark of EMT; these EMT-inducing transcription factors bind to E-box elements in the E-cadherin gene promoter to repress its transcription. Of particular note, Snail is an early marker of EMT that is involved in the initial cell-migratory phenotype, and it occasionally induces other factors [40–42].
In addition to having pro-metastatic roles, these EMT-inducing transcription factors are also implicated in tumour initiation and early tumour development. Their oncogenic potential has proven to be associated with the capacity to inhibit tumour-suppressive 'fail-safe' programs (senescence and apoptosis), and to induce stemness properties and metabolic alterations. The Twist protein is known to inhibit senescence and apoptosis. Although the roles of Snail and ZEB in senescence are debated, these proteins have been shown to confer resistance to cell death. Snail, ZEB, and Twist also induce malignant transformation, as well as the acquisition of stemness properties [40, 43]. Emerging evidence also shows that Snail can promote metabolic alterations [42, 43]. The roles of these proteins in the CSC phenotype, metabolic alteration, and resistance to therapy will be addressed in more detail below.
Induction of EMT, invasion, and metastasis by IR
IR has been shown to induce EMT to enhance the motility and invasiveness of several cancer cells, including those of breast, lung, and liver cancer, and glioma cells [20–27]. Clinical and preclinical evidence suggests that IR may increase metastasis in both the primary tumour site and in normal tissues under some circumstances [20, 23, 27]. Even sublethal doses of IR have been shown to enhance the migratory and invasive behaviours of glioma cells [21, 22].
ROS are known to play an important role in IR-induced EMT [44, 45]. ROS act as second messengers in intracellular signalling that induce tumourigenicity and sustain tumour progression. ROS have been closely associated with tumourigenesis and tumour progression. ROS can act as signalling molecules that regulate cell proliferation and death [46–52]. Mitochondrial ROS production is known to be activated by hypoxia, oncogenes, loss of tumour suppressors, or mitochondrial mutations to increase tumourigenicity [50, 51]. High levels of ROS trigger cell death by causing irreversible damage to cellular components such as proteins, nucleic acids, and lipids, whereas low levels of ROS have been shown to promote tumour progression—including tumour growth, invasion, and metastasis [46–52]. It has been noted that cancer cells also express high levels of antioxidant proteins to inhibit ROS-induced cytotoxicity [47–49, 51]. Therefore, ROS levels are crucial for radiotherapy outcomes. ROS promote EMT to allow cancer cells to avoid hostile environments [46–49, 52].
IR can induce ROS production directly and indirectly, by extracellular water radiolysis and by intracellular metabolic alterations or mitochondrial dysfunction [15, 17]. Treatment with the N-acetylcysteine (NAC), a general ROS scavenger, prevents IR-induced EMT, adhesive affinity, and invasion of breast cancer cells, suggesting an important role for ROS in IR-induced EMT [44, 45].
Snail has been shown to play a crucial role in IR-induced EMT, migration, and invasion [53–56]. ROS are also involved in IR-induced Snail expression. IR-induced ROS activate ERK1/2, which inactivates glycogen synthase kinase 3β (GSK3β), an endogenous inhibitor of Snail, thereby upregulating Snail [53]. Sustained elevation of Snail expression is required for IR-induced ERK activation and GSK3β inhibition, suggesting that ERK/GSK3β/Snail might form a positive feedback loop [54]. Several signalling pathways have also been implicated in IR-induced Snail expression, including TGF-β, Wnt, Hedgehog, Notch, granulocyte-colony stimulating factor (G-CSF), EGFR/PI3K/Akt, mitogen-activated protein kinase (MAPK), and p21-activated kinase 1 (PAK1), as discussed below. IR activates the p38 MAPK pathway, which contributes to the induction of Snail expression to promote EMT and invasion [56]. PAK1 is also activated by IR, after which it directly binds to Snail, which increases the transcriptional repression activity of Snail, thereby repressing E-cadherin expression [55].
Snail is known to be regulated by distal-less homeobox-2 (Dlx-2) [57, 58]. Dlx-2 is a homeobox transcription factor and is involved in embryonic and tumour development [59–63]. We previously showed that Dlx-2 acts as an upstream regulator of Snail [57, 58]. In addition, IR has been shown to upregulate Dlx-2 by activating Smad2/3 signalling that induces EMT in A549 and MDA-MB-231 cell lines [64]. We also found that Dlx-2 is implicated in IR-induced EMT by activating Snail; Dlx-2 expression was increased by IR-induced ROS. Dlx-2 shRNA suppressed the IR-induced EMT phenotype, and was accompanied by downregulation of Snail (data not shown; see the abstract of MSIP reports (No. 2012M2B2A9A02029802; http://www.ndsl.kr/ndsl/search/detail/report/reportSearchResultDetail.do?cn=TRKO201300032641 and No. 2013M2B2A9A03050902; http://www.ndsl.kr/ndsl/search/detail/report/reportSearchResultDetail.do?cn=TRKO201600009259). These results suggest that IR induces EMT via ROS-dependent activation of Dlx-2 and Snail.
In addition, ultraviolet (UV) radiation, a form of non-IR, which is considered the main cause of skin cancer, is also known to enhance cell migration by increasing ROS levels, similar to IR. UV radiation-induced ROS activates NF-κB signalling that promotes cell migration [65]. NF-κB is known to increase Snail stabilisation by preventing the ubiquitination and degradation of Snail, which promotes cell migration and invasion [66, 67]. Snail also plays an important role in UV radiation-induced EMT. UV radiation induces Snail expression by activating the EGFR, ERK, and p38 MAPK cascades [68–70]. MAPK signalling activates AP-1 transcription factor to directly increase Snail expression in keratinocytes [69].
ZEB1 is also implicated in IR-induced EMT [71, 72]. IR-induced GSK3β inactivation has been shown to contribute to the induction of ZEB1 expression [72]. IR also promotes Akt phosphorylation to elevate ZEB1 expression, which promotes EMT. Indeed, following radiotherapy, high levels of ZEB1 and phosphorylated Akt (S473) are correlated with recurrence and distance metastasis in patients with nasopharyngeal carcinoma [71].
In addition, hypoxia-inducible factor-1 (HIF-1) is involved in IR-induced EMT [73–82]. HIF-1 is a heterodimer composed of an oxygen-sensitive α subunit and a constitutively expressed β subunit. Under normoxia, HIF-1α is rapidly degraded, whereas hypoxia induces stabilisation and accumulation of HIF-1α [73–76]. Several mechanisms are known to induce HIF-1 activation by increasing the translation of HIF-1α mRNA or inhibiting HIF-1α degradation; levels of HIF-1α mRNA are enhanced by activation of the PI3K/Akt/mammalian target of rapamycin (mTOR) pathway and by the binding of YB-1, an RNA and DNA binding protein. HIF-1α protein degradation has been prevented by ROS and NO. Inactivation of von Hippel-Lindau tumour suppressor protein (pVHL, an E3 ubiquitin ligase targeting HIF-1α) and activation of WSB1 (an E3 ligase targeting pVHL) and ubiquitin C-terminal hydrolase-L1 (UCHL1, a HIF-1 deubiquitinating enzyme) are also known to induce HIF-1α stabilisation and activation [73–77].
IR is known to increase stabilisation and nuclear accumulation of HIF-1α, since hypoxia is a major condition for HIF-1 activation [73, 75]. IR induces vascular damage that causes hypoxia. In addition, ROS is implicated in IR-induced HIF-1 activation; IR causes the reoxygenation of hypoxic cancer cells to increase ROS production, which leads to the stabilisation and nuclear accumulation of HIF-1 [77, 78]. IR-induced reoxygenation also enhances the translation of HIF-1-regulated transcripts [77]. In addition, IR increases glucose availability under reoxygenated conditions that promote HIF-1α translation by activating the Akt/mTOR pathway [78]. Furthermore, IR upregulates Nijmegen breakage syndrome protein 1 (NBS1), which directly interacts with HIF-1α and stabilises it [80]. The stabilised HIF-1α then translocates to the nucleus, dimerizes with HIF-1β, and increases gene expression— including the expression of essential EMT regulators such as Snail—to induce EMT, migration, and invasion [73, 83].
A number of signalling pathways, including those of TGF-β, Wnt, Hedgehog, Notch, G-CSF, EGFR/PI3K/Akt, CXCL12/CXCR4, PAI-1, and MAPK, have been implicated in IR-induced EMT [45, 84–117] (Fig. 2). TGF-β signalling has been shown to play a crucial role in IR-induced EMT [84–94]. Among three isoforms of TGF-β (TGF-β1, TGF-β2, and TGF-β3), IR is known to specifically induce TGF-β1 [84, 85]. AP-1 transcription factor is involved in IR-induced TGF-β1 expression [84]. After it is synthesised, TGF-β is secreted as an inactive homodimer that binds to latent TGF-β binding protein (LTBP), forming a latent complex. The latent TGF-β complexes can be activated by extracellular stimuli (ROS and acidic conditions) or by the proteolytic activity of proteases (matrix metalloproteinase [MMP]-2 and MMP-9) [87, 88].
Fig. 2.
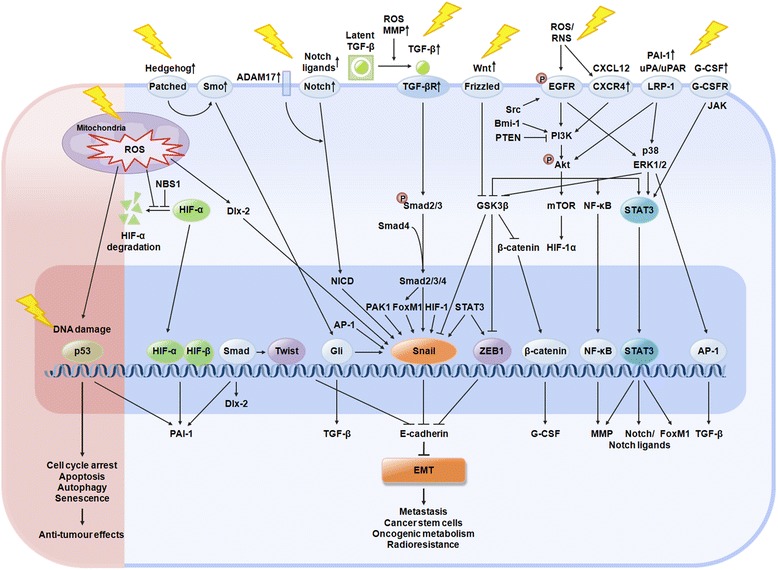
Signalling pathways of IR-induced EMT, metastasis, CSCs, and oncogenic metabolism. Ionizing radiation (IR) causes DNA damage directly, by ionization, or indirectly, by the production of reactive oxygen species (ROS) in tumours. In response to DNA damage, p53 is activated and it exerts the therapeutic effects of IR: induction of cell cycle arrest, apoptosis, autophagy, or senescence. However, IR is also known to enhance the metastatic potential of cancer cells by inducing EMT. IR-induced EMT is mediated by transcription factors (including Snail, HIF-1, ZEB1, Twist, and STAT3) that are activated by signalling pathways (including those of TGF-β, Wnt, Hedgehog, Notch, G-CSF, EGFR/PI3K/Akt, CXCL12/CXCR4, PAI-1, and MAPK). ROS are implicated in IR-induced EMT via the activation of these transcription factors and signalling pathways. Cancer cells that undergo EMT also acquire stemness and oncogenic metabolisms. In addition, EMT, CSCs, and oncogenic metabolism are known to contribute to the radioresistance of cancer cells
IR-induced ROS is known to promote the extracellular proteolytic cleavage of latent complexes so that the biologically activated TGF-β can bind to its receptors [86]. TGF-β binds with the TGF-β type II receptor (TβRII); this receptor-ligand complex recruits and phosphorylates a TGF-β type I receptor, ALK5. ALK5 then phosphorylates the proteins Smad2 and 3, which bind to Smad4 and translocate to the nucleus, where they transactivate target genes [87, 88]. In addition to activation of the synthesis and secretion of TGF-β1, IR promotes intracellular TGF-β signalling, as evidenced by the phosphorylation of Smad 2/3C and the upregulation of its target genes (TβRII and ALK5), thereby leading to hyperactivation of TGF-β signalling [93]. Furthermore, IR elevates FoxM1, which acts downstream of TGF-β1/Smad signalling. IR-induced FoxM1 directly binds to the Snail promoter and increases Snail expression to mediate TGF-β-induced EMT [92].
Wnt/β-catenin signalling is also implicated in IR-induced EMT [95–98]. IR has been shown to increases Wnt signalling by increasing Wnt ligand expression [96]. Generally, Wnt binds to its receptor Frizzled and to its co-receptor, lipoprotein receptor-related protein (LRP) 5/6 that suppresses GSK3β-mediated phosphorylation of β-catenin [39]. IR is known to enhance β-catenin stabilisation [95, 97]. Subsequently, the stabilised β-catenin is translocated to the nucleus and binds to T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors to activate target gene expression [39]. IR also induces the nuclear translocation and accumulation of β-catenin, and increases β-catenin/TCF transcriptional activities [95, 98].
In addition, Wnt signalling increases Snail protein stability in the nucleus by activating an Axin2 pathway, thereby inducing EMT. Axin2 acts as a chaperone for nuclear export of GSK3β, the dominant kinase responsible for Snail protein turnover and activity [118, 119]. Recently, we showed that the Dlx-2/Snail cascade is implicated in TGF-β- and Wnt3a-induced EMT [57]. IR-induced Wnt/β-catenin signalling elevates Snail to promote EMT, migration, and invasiveness of progeny from irradiated colorectal cancer cells [95, 97].
Notch signalling is known to be involved in IR-induced EMT [99, 100]. Notch signalling is activated by interaction between transmembrane Notch receptors (Notch 1–4) and ligands (Jagged-1, Jagged-2, Delta-like 1, Delta-like 3, Delta-like 4) on contacting cells. IR activates the IL-6/JAK/signal transducer and activator of transcription 3 (STAT3) pathway to upregulate Notch-2, Jagged1, and Delta-like 4, and induces EMT [100]. IR also increases Notch-1 expression [99]. Notch-1 is known to induce EMT by upregulating Snail. Treatment with two Notch-1-regulating radiosensitizers, rhamnetin and cirsiliol, induces the miR-34a-mediated downregulation of Notch-1, preventing IR-induced EMT [99].
IR has also been shown to activate Hedgehog (Hh) signalling to induce EMT [101]. IR increases expression of the Hh ligand (Indian Hh, Sonic Hh), the Hh receptor (Smoothened), and the Hh-target gene (Gli2), with enhanced expression of the EMT-stimulating factor (TGF-β) and mesenchymal markers (N-cadherin, α-SMA). Blocking Hh activity suppresses the IR-induced expression of EMT-stimulating genes, suggesting a potential role for Hh signalling in IR-induced EMT [101].
Furthermore, EGFR activation is known to be associated with IR-induced EMT, cell migration, and invasion by activating two downstream pathways: PI3K/Akt and Raf/MEK/ERK [45, 102–108]. Ligand binding to EGFR generally induces receptor dimerization, activation of its kinase domain, and consequent autophosphorylation [102, 103]. IR promotes EGFR heterodimerization with ErbB2 in a ligand-independent manner [104].
ROS and RNS are also implicated in IR-induced EGFR activation [45, 105]. IR-induced ROS are known to promote phosphorylation of EGFR or ErbB2 Y877 [45, 105]. IR-induced RNS also induce autophosphorylation on EGFR Y1173. Following IR, NO is generated within minutes, which is necessary for the rapid activation of EGFR [105]. UV-induced ROS are also implicated in IR-induced EGFR activation. NAC prevents UV-mediated EGFR phosphorylation at Y992 and Snail expression [70]. These studies suggest important roles for ROS and RNS in IR-induced activation of the EGFR pathway that may upregulate Snail to induce EMT and invasion. In addition, IR has been shown to induce Src activation [45, 106]. Src is a non-receptor tyrosine kinase that acts both upstream and downstream of EGFR and ErbB2. IR-induced Src activation promotes phosphorylation of EGFR and ErbB2 [45, 106]. Furthermore, IR-induced EGFR and IGFR-1 activation are known to promote the PI3K-dependent Rho signalling pathway, which enhances the invasive potential of glioblastoma cells [107].
IR has been shown to induce Akt activation through several signalling pathways (EGFR, C-X-C chemokine receptor type 4 [CXCR4]/C-X-C motif chemokine 12 [CXCL12], plasminogen activator inhibitor 1 [PAI-1]) and upstream regulators (Bmi1, PTEN) that promote EMT and invasion [81, 104, 109–111]. IR-mediated activation of EGFR leads to Akt activation through phosphorylation at two key regulatory residues, T308 and S473 [104]. ROS is also involved in IR-mediated Akt activation to enhance invasiveness. IR-induced ROS upregulates CXCR4, which interacts with its ligand, CXCL12, and activates the PI3K/Akt and ERK1/2 pathways [109].
PAI-1 signalling is also implicated in IR-induced Akt activation that increases Snail levels to induce EMT [81]. IR increases the expression and secretion of PAI-1 by upregulating HIF-1α, p53, and phospho-Smad3. PAI-1 secreted from radioresistant NSCLC cells induces EMT and the radioresistance of nearby cells in a paracrine manner; extracellular PAI-1 associates with the urokinase-type plasminogen activator (uPA)/uPAR complex and then binds to its receptor, low density LRP-1, which subsequently activates Akt and ERK1/2 to upregulate Snail, thereby inducing EMT and cell survival in radiosensitive cells [81]. IR also increases the expression of Bmi-1, which acts as an upstream regulator of the PI3K/Akt pathway. Bmi-1 is known as a key gene involved in EMT and the self-renewal of cancer cells [110]. In addition, IR downregulates PTEN to activate the PI3K/Akt pathway, which then inactivates GSK3β to increase Snail expression and induce EMT [111].
The IR-induced PI3K/Akt pathway also stabilises β-catenin, which directly binds to the promoter region of G-CSF. Subsequently, G-CSF is secreted and binds to G-CSFR to activate the JAK/STAT3 pathway [112]. STAT3 activation is also mediated by EGFR-Akt, as well as by the EGFR-p38/ERK pathway, in response to IR [113]. STAT3 is known to be involved in IR-induced EMT and invasion by upregulating the molecules governing EMT (N-cadherin, vimentin, uPA), invasion (MMP-2, MMP-9), and angiogenesis (vascular endothelial growth factor [VEGF], iNOS) [113–115]. In addition, IR-induced STAT3 also increases FoxM1 expression and it interacts and co-localizes with FoxM1 in the nucleus [117]. IR-induced FoxM1 directly binds to Snail promoter to induce Snail expression, thereby showing the involvement of STAT3/FoxM1 complex in EMT [92]. In addition, in radioresistant cervical cancer cells, IR induces K-Ras activation that promotes the c-Raf/p38 pathway to increase cell migration and metastatic potential [116].
Induction of CSCs by IR
CSCs
CSCs possess a capacity for self-renewal, and they can persistently proliferate to initiate tumours upon serial transplantation, thus enabling them to maintain the whole tumour. Under certain microenvironment, CSCs exhibit plasticity; mutations in normal stem cells, progenitor cells, and/or differentiated cells can give rise to CSCs, and these newly generated CSCs produce daughter CSCs as well as differentiated bulk cancer cells [120–124]. Notably, some CSCs can spontaneously arise from normal and neoplastic nonstem cells, suggesting a bidirectional interconversion between the stem and non-stem cell state. Thus, different types of CSC coexist and contribute to tumour heterogeneity [120–123, 125]. Conventional cancer treatments kill most cancer cells, but CSCs survive due to their resistance to therapy, eventually leading to tumour relapse and metastasis [126–131].
For the identification of CSCs, three types of markers are utilised: cell surface molecules, transcription factors, and signalling pathway molecules [132–140]. CSCs express distinct and specific surface markers; commonly used ones are CD24, CD34, CD38, CD44, CD90, CD133, and ALDH. These markers enable CSCs to be distinguished from other tumour cells and from normal stem cells [132–140]. For example, breast CSCs express CD44+CD24-, while pancreatic or ovarian CSCs express CD44+CD24+EpCAM+ [135–137].
Transcription factors, including Oct4, Sox2, Nanog, c-Myc, and Klf4, and signalling pathways, including those of TGF-β, Wnt, Hedgehog, Notch, platelet-derived growth factor receptor (PDGFR), and JAK/STAT, are known to play crucial roles in maintaining the self-renewal abilities and pluripotency of stem cells [132–134]. These transcription factors and signalling pathways are also frequently used as CSC markers. In addition, several microRNAs (miRNAs), including let-7, miR-22, miR-34a, miR-128, the miR-200 family, and miR-451, are known to regulate the self-renewal, differentiation, and tumourigenicity of CSCs [141–143].
The CSC state can be regulated by cell-autonomous forces (genetic, epigenetic, and metabolic regulation) and by external forces (niche factors and the immune system) [120–123]. Non-CSCs can be reprogrammed to become CSCs by epigenetic and genetic changes that are involved in phenotypic heterogenicity among cancer cells [141–145]. Epigenetic changes, including DNA methylation, histone modifications, and miRNAs, play important roles in the acquisition of CSC properties.
In particular, miRNAs have been shown to play important roles in stemness and tumour metastasis; they modulate the expression of many target genes that regulate tumour cell EMT, motility, invasion, intravasation, resistance to anoikis, extravasation, and metastatic colonisation, as well as cell stemness, dormancy, metabolic reprogramming, and the TME. Through these means, miRNA can positively or negatively regulate tumour progression and tumour metastasis [141–143, 146–149]. In addition, long noncoding RNAs (lncRNAs) have been associated with numerous functions in cells [147, 150–154]. LncRNAs are known to positively or negatively affect the expression of nearby genes, control protein activity or localisation, and serve as organisational frameworks of subcellular structures. Many lncRNAs are also processed to yield small RNAs or to modulate other RNAs to be processed [154]. In particular, MALAT1, HOTAIR, and H19 lncRNAs are known to control stemness, cell migration and invasion, EMT, and metastasis by epigenetic regulation, alternative splicing, chromatin modification, and translational control [147, 150–153].
EMT and CSCs
EMT has been shown to play important roles in the acquisition of stemness in cancer cells [155–160]. EMT-inducing transcription factors, such as Snail, ZEB1, and Twist1, are known to confer CSC properties [161–165]. In addition to its role in EMT, Snail is known to induce the CSC phenotype in colorectal carcinoma cells, where it enhances stemness properties—including self-renewal, tumourigenicity, and resistance to radiotherapy/chemotherapy—with an increased metastatic potential [161–163].
ZEB1 is implicated in maintaining stemness and EMT properties in pancreatic and colorectal cancer cells [164]. ZEB1 represses the expression of stemness-inhibiting miRNAs, including miR-183, miR-200c, and miR-203, thereby upregulating the stem-cell factors Sox2 and Klf4. Knockdown of ZEB1 prevents not only EMT, invasion, and metastasis, but also the stemness phenotype [164]. In addition, Twist1 is known to link EMT to stem-like features. Twist1 directly increases Bmi-1 expression, and acts cooperatively with Bmi-1 to induce EMT and stemness properties [165].
Signalling pathways involved in EMT, including those of TGF-β, Wnt, and Notch, have been shown to play important roles in inducing the CSC phenotype [166–168]. TGF-β1 not only increases EMT markers (Slug, Twist1, β-catenin, N-cadherin), but also upregulates CSC markers (Oct4, Sox2, Nanog, Klf4) in breast and lung cancer cells [166, 167].
Wnt/β-catenin signalling also plays critical roles in increasing the stemness properties of liver CSCs by activating Notch1 [168]. Blocking Wnt/β-catenin and/or Notch decreases the expression of transcription factors involving EMT (such as Snail) and stemness (such as Sox2 and Nanog). These changes result in reduced metastatic potential in vivo, and they inhibit CSC properties, including self-renewal and tumourigenicity. This suggests a role for EMT in the acquisition of CSC phenotypes [168].
However, in heterogeneous solid tumours, some CSC subpopulations arise independently of EMT [169, 170]. This suggests that CSC populations may be heterogeneous, and may contain a significant proportion of epithelial stem cells in which stemness is entirely uncoupled from EMT. These epithelial stem cells may cooperatively interact with non-CSCs, thereby potentiating the metastatic behaviours of the combined tumour cell populations [171–174]. Therefore, other mechanisms are likely involved in the induction of CSC in an EMT transcription factors-independent manner.
Induction of the CSC phenotype by IR
IR has been shown to induce the CSC phenotype in many cancers, including breast, lung, and prostate cancers, as well as melanoma [175–181]. Genotoxic stress due to IR or chemotherapy promotes a CSC-like phenotype by increasing ROS production [179]. IR has been shown to induce reprogramming of differentiated cancer cells into CSCs [181]. In prostate cancer patients, radiotherapy increases the CD44+ cell population that exhibit CSC properties [175]. IR also induces the re-expression of stem cell regulators, such as Sox2, Oct4, Nanog, and Klf4, to promote stemness in cancer cells [176, 181].
EMT has been implicated in the acquisition of the IR-induced CSC phenotype [178, 179]. After IR, surviving cells exhibit a complex phenotype combining the properties of EMT and CSC with high expression levels of Snail, CD24, CD44, and PDGFR-β in NSCLC cells [178]. In addition, the subset of CD24+ ovarian cancer cells or CD133+ colorectal cancer cells that possess CSC properties exhibit the EMT phenotype—including higher levels of expression of Snail, Twist, and vimentin, and lower levels of expression of E-cadherin [159, 160].
EMT-inducing transcription factors and signalling pathways, including Snail, STAT3, Notch signalling, the PI3K/Akt pathway, and the MAPK cascade, have been shown to play important roles in IR-induced CSC properties [180–184]. STAT3 has been shown to be involved in the IR-induced increase of CSCs [180], and is known to activate Snail to induce the CSC phenotype. STAT3 directly binds to the Snail promoter and increases Snail transcription, which induces the EMT and CSC phenotypes, in cisplatin-selected resistant cells [163]. Inhibition of the DNA-binding activity of STAT3 prevents IR-induced increases of the CSC population, and sensitises cells to radiotherapy [180].
Notch signalling is also implicated in the IR-induced de novo generation of CSCs [181, 184]. Inhibition of Notch signalling partially prevents the IR-induced re-expression of Oct4, Sox2, Nanog, and Klf4 [181]. Notch signalling also plays important roles in the IR-induced metastatic potential of CSCs. IR upregulates disintegrin and metalloproteinase-17 (ADAM17) to activate Notch signalling, which increases the migration and invasiveness of CSCs [182].
The PI3K/Akt pathway and the MAPK cascade are involved in the IR-induced CSC and EMT phenotypes. IR promotes Src activity to trigger the PI3K/AKT and p38 MAPK pathways that induce both CSC status and EMT [183]. Therefore, EMT transcription factors and signalling pathways may enable CSCs to acquire the ability to invade, migrate, and disseminate.
Induction of oncogenic metabolism by IR
Oncogenic metabolism
Most cancer cells produce their energy predominantly by high rate of glycolysis rather than by oxidative phosphorylation, even in the presence of oxygen: a phenomenon that has been termed the Warburg effect, aerobic glycolysis, or the glycolytic switch [185–194]. Other oncogenic metabolic pathways, including glutamine metabolism, the pentose phosphate pathway (PPP), and synthesis of fatty acids and cholesterol, are also enhanced in many cancers. These alterations are known to contribute to cell survival and sustain the increased demands of cell proliferation by providing biosynthetic precursors for nucleic acids, lipids, and proteins [186–196].
The activation of oncogenes and the loss of tumour suppressors have been shown to drive tumour progression; in particular, they seem to drive metabolic reprogramming. Several transcription factors, including HIF-1α, p53, and c-Myc, are known to contribute to oncogenic metabolism [186–194]. Emerging evidence suggests that metabolic reprogramming is one of the hallmarks of cancer, and may be required to convert a normal cell into a malignant cell [186–194].
Although the Warburg effect has been considered a metabolic signature of tumour cells, increasing evidence indicates that tumour cells exhibit high mitochondrial metabolism as well as aerobic glycolysis. These contradictory findings have even been reported as occurring within the same tumour [197–208]. In addition, CSCs exhibit unique metabolic features in a tumour type-dependent manner. CSCs can be highly glycolytic-dependent or oxidative phosphorylation (OXPHOS)-dependent. In any case, mitochondrial function is crucial for maintaining CSC functionality [209–212]. To explain such contradiction, reverse Warburg effects and metabolic symbiosis have been proposed [197–208, 212].
According to this model, cancer cells depend on mitochondrial metabolism and increase mitochondrial production of ROS that cause pseudo-hypoxia. Tumour tissue is a heterogeneous population of cells consisting of cancer cells and surrounding stromal cells, with various genetic and epigenetic backgrounds. These ROS reduce caveolin-1 expression in cancer-associated fibroblasts (CAFs), which are the main component of tumour stroma. Loss of caveolin-1 in CAFs leads to further increases in ROS production, which stabilise HIF-1α (and by extension, this increases levels of the HIF-1 heterodimer). HIF-1 then enhances glycolysis in CAFs. Furthermore, tumour cell-derived ROS also induce autophagy in CAFs. Autophagy is a lysosomal self-degradation process that removes damaged mitochondria through mitophagy. Thus, CAFs have defective mitochondria that lead to the cells exhibiting the Warburg effect; the cells take up glucose, and then secrete lactate to 'feed' adjacent cancer cells [197–207].
In tumour tissue, epithelial cancer cells and CAFs express different subtypes of the lactate transporter, monocarboxylate transporter (MCT). This heterogeneity of MCT expression induces metabolic symbiosis between epithelial cancer cells and CAFs. Metabolic symbiosis is required for adaptation to changes in the nutrient microenvironment that is caused by cancer treatment. Epithelial cancer cells express MCT1, while CAFs express MCT4. MCT4-positive, hypoxic CAFs secrete lactate by aerobic glycolysis, and MCT1-expressing epithelial cancer cells then uptake and use that lactate as a substrate for the tricarboxylic acid (TCA) cycle [197–201].
However, the reverse Warburg effect may not be pervasive in all cancers. MCT4-expressing tumour cells or the mesenchymal phenotype do not lead to the reverse Warburg phenomenon. Rather, hierarchical metabolic heterogeneity may be observed in cancer cells; MCT4-positive cancer cells depend on glycolysis and then efflux lactate, while MCT1-positive cells uptake lactate and rely on OXPHOS. Therefore, metabolic heterogeneity induces a lactate shuttle between hypoxic/glycolytic cells and oxidative/aerobic tumour cells. This kind of lactate shuttle has also been observed between neurons and astrocytes in normal brain tissue [198, 200].
This interaction between cancer cells and stromal cells can contribute to tumour progression—including tumour EMT, invasion, growth, and angiogenesis. Cancer cells interact with stromal cells and use their environment to sustain tumour growth. In addition, cells in the tissues surrounding the tumour, such as CAFs and adipocytes, create a nutrient-rich microenvironment that feeds the cancer cells; cancer cells then secrete waste products (e.g., CO2, H+, ammonia, polyamines) that further promote EMT, invasion, and angiogenesis [198, 200, 208].
MCT1-positive cancer cells are also involved in the stem-like phenotypes observed within heterogeneous tumour populations. While bulk tumour cells exhibit a glycolytic phenotype, with increased conversion of glucose to lactate (and enhanced lactate efflux through MCT4), CSC subsets depend on oxidative phosphorylation; most of the glucose entering the cells is converted to pyruvate to fuel the TCA cycle and the electron transport chain (ETC), thereby increasing mitochondrial ROS production [198, 209, 212]. In these cells, the major fraction of glucose is directed into the pentose phosphate pathway, to produce redox power through the generation of NADPH and ROS scavengers [212]. Therefore, this activated mitochondrial metabolism provides enough energy for CSC self-renewal, invasion, and metastasis.
EMT/CSC regulators involved in oncogenic metabolism
Several transcription factors, including HIF-1α, p53, and c-Myc, are known to contribute to oncogenic metabolism. Many regulatory molecules involved in EMT and CSCs, including Snail, Dlx-2, HIF-1, STAT3, TGF-β, Wnt, and Akt, are implicated in the metabolic reprogramming of cancer cells. The induction of EMT is involved in the acquisition of CSC properties, as well as in reduced mitochondrial metabolism and induction of the glycolytic switch [57, 58, 213–222].
Snail has been shown to induce mitochondrial repression and glucose metabolism by downregulating cytochrome C oxidase (COX) subunits or fructose-1,6-bisphosphatase 1 (FBP1). Snail has also been shown to induce the EMT phenotype [57, 58, 213–215].
HIF-1 induces the expression of glycolytic enzymes, including the glucose transporter GLUT, hexokinase, lactate dehydrogenase (LDH), and MCT, resulting in the glycolytic switch. In addition, HIF-1 represses the expression of pyruvate dehydrogenase kinase (PDK), which inhibits pyruvate dehydrogenase (PDH), thereby inhibiting mitochondrial activity [216, 217].
STAT3 has been implicated in EMT-induced metabolic changes as well [218]. Stable EMT cells are generated through mammosphere culture in epithelial breast cancer cells. These EMT-derived cancer cells exhibit elevated activation of STAT3 and enhanced aerobic glycolysis, with upregulation of certain enzymes and transporters related to glycolysis (such as MCT2); these cells also show downregulation of gluconeogenesis and some anabolic side-pathways. Inhibition of STAT3 suppresses certain EMT-related metabolic alterations in the expression of MCT2 and ZEB1, suggesting a role for STAT3 in EMT-induced metabolic changes [218].
Emerging evidence suggests that TGF-β and Wnt play important roles in the metabolic alteration of cancer cells [57, 58, 214, 219–221]. TGF-β and Wnt are known to induce mitochondrial repression and the glycolytic switch by activating Dlx-2 and Snail [57, 58]. TGF-β/Wnt-induced mitochondrial repression is mediated by inhibition of mitochondrial complex IV (COX) [57, 214]. Wnt also directly targets PDK1, thereby inhibiting mitochondrial respiration and promoting the glycolytic switch [219, 221].
Akt is also implicated in the glycolytic switch and in promoting cancer cell invasiveness [222]. Overexpression of Akt impairs mitochondrial function, promotes glycolytic metabolism with upregulation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and converts radial growth (i.e., noninvasive) melanoma into vertical growth (i.e., invasive) melanoma [222].
Oncogenic metabolism plays a critical role(s) in EMT and CSC phenotypes
Accumulating evidence suggests that metabolic alteration is one of the hallmarks of cancer, and may contribute to malignant transformation and tumour development—including the induction of EMT, invasion, metastasis, and stemness [58, 211–213, 223–233] (Fig. 1). Metabolic reprogramming of cells toward aerobic glycolysis has been shown to support the invasive phenotype of malignant melanoma [224]. A glycolytic mechanism is also known to modulate the angiogenic switch for metastatic growth [225].
Several glycolytic enzymes, including pyruvate kinase M2 (PKM2), LDH, and pyruvate carboxylase (PC), are implicated in the induction of the EMT and CSC phenotypes [234–237]. PKM2 is a less active isoform of pyruvate kinase and is primarily expressed in embryonic and cancer cells. This decreased activity of PKM2 is known to promote an overall shift in metabolism to aerobic glycolysis. EMT-inducing stimuli cause nuclear translocation of PKM2, which promotes EMT; nuclear PKM2 directly interacts with TGF-β-induced factor homeobox 2 (TGIF2), a transcriptional repressor of TGF-β signalling, and recruits histone deacetylase 3 to the E-cadherin promoter to suppress E-cadherin transcription [234].
LDH catalyses the bidirectional conversion of lactate to pyruvate [237]. LDHA is one of the predominant isoforms of LDH; it is also known to be implicated in the Warburg effect, as well as in cell invasion and migration. High levels of LDHA are positively correlated with the expression of EMT and CSC markers in invasive bladder cell lines and in muscle-invasive bladder cancer specimens, suggesting a critical role for LDHA in the activation of EMT and CSC [237].
In addition, PC is implicated in cell migration and invasion [236]. PC is a key enzyme of anaplerosis that converts pyruvate to oxaloacetate, which replenishes the TCA cycle. Knockdown of PC inhibits proliferation, migration, and invasion behaviours in invasive breast cancer cells; conversely, the overexpression of PC promotes proliferation, migration, and invasion abilities in noninvasive breast cancer cells [236].
Furthermore, the misregulation of lipogenic metabolism is involved in the regulation of EMT [238, 239]. Fatty acid synthase (FASN) is a key lipogenic enzyme that catalyses de novo synthesis of fatty acids. FASN signalling is known to modulate subcellular structural components that determine the epithelial or mesenchymal state of a cell. Transient knockdown of FASN suppresses structural hallmarks of EMT in stem-like cells. Loss of FASN signalling also reverses a tumour phenotype to a normal-like tissue phenotype, and efficiently suppresses the tumourigenicity of metastatic breast cancer cells in vivo [238]. Mechastically, FASN increases TGF-β levels and TGF-β, in turn, elevates FASN expression. These results suggest that a FASN-TGF-β-FASN positive loop contributes to high EMT/metastatic potential in cisplatin resistant cancer cells [239].
Interestingly, the respiratory enzymes citrate synthase (CS) and succinate dehydrogenase subunit B (SDHB), and the gluconeogenesis regulatory enzyme FBP, are known to negatively regulate the EMT and CSC phenotypes [215, 240, 241]. Loss of CS has been shown to induce EMT and the glycolytic switch. CS is a mitochondrial respiratory enzyme that catalyses the first step of the TCA cycle. CS knockdown cells exhibit EMT, mitochondrial repression, and the glycolytic switch, with concomitant upregulation of Snail and Twist, and downregulation of p53 and its target genes (TIGAR and SCO2). p53 is known to prevent glycolysis and promote mitochondrial respiration by increasing the expression of TIGAR and SCO2. p53 reactivation inhibits CS-knockdown-induced EMT, suggesting a role for p53 in these metabolic alterations and in malignant transformation [240].
SDHB is also implicated in EMT, glucose and glutamine metabolism, and mitochondrial dysfunction. SDH is a mitochondrial metabolic enzyme complex that participates in both the TCA cycle and the electron transport chain; it converts succinate into fumarate in the TCA cycle and catalyses the transfer of electrons to the ubiquinone pool in the electron transport chain. SDH mutations have frequently been observed in many cancers. Knockdown of SDHB leads to alterations of the epigenome; this promotes EMT, induces altered glucose and glutamine utilisation, and induces mitochondrial dysfunction [241].
In addition, loss of FBP has been associated with the EMT-driven CSC phenotype. FBP catalyses the conversion of fructose 1,6-bisphosphate to fructose-6-phosphate. Snail induces epigenetic silencing of FBP1; this enhances glycolysis, suppresses oxygen consumption and ROS production, and promotes the EMT and CSC phenotypes [215].
We also showed that glutamine metabolism plays an important role in the induction of EMT [58]. Glutaminase 1 (GLS1) converts glutamine to glutamate. The inhibition of glutamine metabolism (via GLS1 knockdown, glutamine deprivation, or glutamine metabolism inhibitors) suppressed Dlx-2-, TGF-β-, Wnt-, and Snail-induced EMT and the glycolytic switch. In addition, GLS1 knockdown also suppressed tumour growth and metastasis in vivo. Dlx-2 knockdown and glutamine metabolism inhibition decreased Snail mRNA levels through the p53-dependent upregulation of Snail-targeting microRNAs (miR-23b, miR-29b, miR-30, miR-34, miR-125b, miR-148a, miR-153, miR-200, and miR-203). These results indicate that the Dlx-2/GLS1/glutamine metabolic axis is a crucial regulator of TGF-β/Wnt-induced, Snail-dependent EMT, metastasis, and the glycolytic switch [58].
Oncogenic metabolism, including glutamine metabolism, is known to endow cancer cells with growth advantages by providing biosynthetic precursors [187–196]. Given that GLS1 knockdown suppressed tumour growth and metastasis in vivo, it is possible that knockdown of any component enzyme in oncogenic metabolism results in a pronounced suppression of metastasis. Like GLS1, other enzymes in oncogenic metabolism may also regulate p53-dependent modulation of Snail-targeting microRNAs to mediate Snail-induced EMT. Therefore, we propose that all oncogenic metabolic pathways are interconnected so that inhibition of any component enzyme within the overall oncogenic metabolism may suppress EMT. Further studies are needed to determine which enzyme inhibition is the most effective in producing EMT inhibition.
IR induces oncogenic metabolism
IR has been shown to induce metabolic changes in cancer cells [242–247]. IR enhances glycolysis by upregulating GAPDH (a glycolysis enzyme), and it increases lactate production by activating LDHA, which converts pyruvate to lactate. IR also elevates MCT1 expression that exports lactate into the extracellular environment, leading to acidification of the tumour microenvironment. These changes are associated with IR-induced invasion of the non-irradiated, surrounding breast cancer tissues and normal endothelial cells [243].
IR increases intracellular glucose, glucose 6-phosphate, fructose, and products of pyruvate (lactate and alanine), suggesting a role for IR in the upregulation of cytosolic aerobic glycolysis; this was also revealed in the metabolomic profile of hepatoma cells [246]. Lactate can activate latent TGF-β through a pH-dependent mechanism so that LDHA inhibition prevents radiation-induced activation of TGF-β [247]. In addition, lactate stimulates cell migration and enhances secretion of hyaluronan from CAF that promote tumour metastasis [235]. In addition to glycolysis, IR has been shown to affect other components of oncogenic metabolism. For example, radioresistant head and neck squamous cells exhibit profound alterations in their metabolism; they demonstrate increased glucose uptake, enhanced PPP signalling, and increased fatty acid biosynthesis, while also showing decreased mitochondrial oxidative phosphorylation [245].
ROS are known to play important roles in the IR-induced glycolytic switch [242]. IR-induced ROS generation increases tumour glucose uptake in vivo. An antioxidant SOD mimic prevents IR-induced glucose uptake, forestalls the glycolytic switch, and inhibits invasiveness [242]. IR-induced ROS generation is known to increase the activity of transcription factors and inducers that are involved in the EMT and CSC phenotypes, such as Snail, Dlx-2, HIF-1, and TGF-β. These factors have been shown to regulate the enzymes involved in glycolysis and mitochondrial oxidative phosphorylation, which may be involved in the IR-induced glycolytic switch.
Snail has been shown to induce the glycolytic switch with EMT phenotypes [57, 58, 213–215]. Because Snail is known to be induced by IR [53–56], we investigated whether Snail affected the IR-induced glycolytic switch (data not shown). We found that IR increases glucose consumption and lactate production, and decreases O2 consumption; this indicates that IR induces mitochondrial repression and the glycolytic switch in MCF-7 cells. Conversely, Snail shRNA prevented IR-induced mitochondrial repression and glycolytic switch, indicating that IR induces these phenomena via Snail.
Dlx-2 shRNA also decreased the IR-induced glycolytic switch and mitochondrial repression, and resulted in the downregulation of Snail. Thus, the Dlx-2/Snail axis seems to be implicated in the IR-induced glycolytic switch. Using cDNA microarray technology, we also found that Dlx-2 elevates a key enzyme in glutamine metabolism, GLS1, and that the Dlx-2/GLS1/Gln metabolic axis plays important roles in TGF-β/Wnt/Snail-dependent EMT and in the glycolytic switch [58]. These results suggest that Dlx-2 may be implicated in IR-induced alterations of other oncogenic metabolic pathways. In addition, we found that GLS1 knockdown inhibits IR-induced EMT (data not shown).
HIF-1 is also implicated in IR-induced metabolic alterations [244]. IR increases HIF-1α expression that inhibits PDH and the tricarboxylic acid cycle, and triggers a metabolic switch to increase lactate production [244].
As described above, metabolic changes have been implicated as being closely involved in the acquisition of the EMT and CSC phenotypes [58, 211–213, 223–233]. IR may indirectly activate several signalling pathways through ROS production, and may induce the activation of oncogenes or the inactivation of tumour suppressors, which then leads to metabolic alterations, EMT, and stemness phenotypes. Therefore, IR appears to induce the EMT and CSC phenotypes by promoting oncogenic metabolism.
Similarly, UV radiation is known to induce the Warburg effect to promote melanoma invasion. UV radiation increases glucose consumption and lactate production, which is partly mediated by ROS. Lactic acid then enhances the invasive potential of melanoma cells. UV radiation also upregulates Transketolase (an enzyme of the PPP) and activates Akt, both of which are involved in metabolic changes [248].
Changes in TME by IR
Crosstalk between cancer cells and their microenvironment is critical for invasive growth and metastasis. The TME is composed of ECM and multiple cell types, including fibroblasts, vascular endothelial cells, immune cells, pericytes, and adipocytes. Cancer cells secrete multiple factors, such as growth factors, cytokines, and chemokines, that regulate the phenotype and function of tumour-resident cells and that influence the composition and organisation of the ECM, thereby regulating such qualities as tumour stiffness [36, 249–253]. IR can elicit various changes in the TME. These changes contribute to creating a favourable microenvironment for tumour metastasis and for the self-renewal and maintenance of cancer stem cells [87, 249–253].
TME
-
Cancer-associated fibroblasts (CAFs)
Fibroblasts are derived from mesenchyme; they form the structural framework in tissues, and typically prevent tumour formation. Unlike normal fibroblasts, CAFs do the following: promote tumour survival, growth, invasion, and metastasis; enhance the stiffness of the ECM; contribute to angiogenesis; and induce inflammation by releasing several growth factors and cytokines (TGF-β, VEGF, hepatocyte growth factor [HGF], PDGF, and stromal cell-derived factor 1 [SDF1]), as well as MMP [249, 254–256]. Recent studies have demonstrated that CAFs also exert tumour-suppressive effects through direct suppression of cancer cells and via regulation of immune cell behaviour. Although some debate exists on this subject, CAFs are predominantly assigned a tumour-promoting function [254].
-
2.
Vascular endothelial cells
The tumour vascular network is dynamic and is associated with tumour growth. A growing tumour requires a constant supply of oxygen, nutrients, and blood-borne mitogens, and requires an effective way to remove toxic metabolites. Thus, tumours recruit the host tissue’s blood vessel network to perform four mechanisms: angiogenesis (formation of new vessels), vasculogenesis (de novo formation of blood vessels from endothelial precursor cells), co-option, and modification of existing vessels within tissues. These mechanisms are required for continuous tumour growth and metastatic potential [36, 249, 252].
-
3.
Immune cells
The immune infiltrate can be composed of a variety of different cell types. These cell populations can have both pro- and anti-tumour functions, and can vary in their activation status and their localisation within the tumour. Innate (macrophages, dendritic cells, myeloid-derived suppressor cells (MDSCs), natural killer cells, etc.) and adaptive (T and B cells) immune system components play major roles in the regulation of tumour growth [257, 258]. Although immune cells have commonly been accepted to exert anti-tumour responses, mechanisms of immune suppression can prevent this process. These immune suppression networks include the immunosuppressive cells such as tumour-associated macrophages (TAM), MDSCs, and regulatory T cells, and the immunosuppressive cytokines, TGF-β and interleukin-10 (IL-10) [259]. Cancer cells interact with the immune system, and can either reduce its intrinsic immunogenicity or induce tolerance [249, 260, 261].
This tumour-host immune relationship is referred to as ‘cancer immunoediting’, which is described by three phases: 1) elimination, 2) equilibrium, and 3) escape. In the elimination phase, highly immunogenic transformed cells are immediately recognised and destroyed by both the innate and adaptive immune systems. In the equilibrium phase, some tumours elude the initial host defences and coexist with the adaptive immune system. In this phase, tumours try to grow but they are inhibited by the immune system. The third phase, tumour escape, is mediated by antigen loss, immunosuppressive cells (TAM, MDSCs, and regulatory T cells), and immunosuppressive cytokines (TGF-β and IL-10). Various types of immunotherapy try to shift the tumour from the escape phase and equilibrium phase to the elimination phase [36, 261]. Heterogeneity in the tumour immune system is associated with various factors, including CAF-secreted factors, vasculature permeability, and the tumour cells themselves [249].
Changes in TME by IR
IR can elicit various changes in the TME, such as CAF activity-mediated ECM remodelling and fibrosis, cycling hypoxia, and an inflammatory response [87, 249–253] (Fig. 3). IR activates CAFs to promote the release of growth factors and ECM modulators, including TGF-β and MMP. TGF-β is a major CAF-secreted factor [87, 255, 256]. TGF-β directly influences tumour cells and CAFs, promotes tumour immune escape, and activates HIF-1 signalling [87, 252, 255]. MMPs degrade ECM that facilitates angiogenesis, tumour cell invasion, and metastasis [262]. IR also promotes MMP-2/9 activation in cancer cells to promote EMT, invasion, and metastasis [54, 106, 263–266]. IR enhances MMP-2 transcription and protein secretion by activating the EGFR/p38/Akt and EGFR/PI3K/Akt signalling pathways, which enhance the invasion of glioma cells [106]. IR-induced Snail increases MMP-2 expression to promote EMT [54]. IR also increases MMP-9 expression by activating the PI3K/Akt/NF-κB pathway, which enhances hepatocellular carcinoma cell invasion [263]. IR-induced MMP-2/MMP-9 expression not only degrades ECM proteins, but also cleaves latent TGF-β1 to activate the TME [266].
Fig. 3.
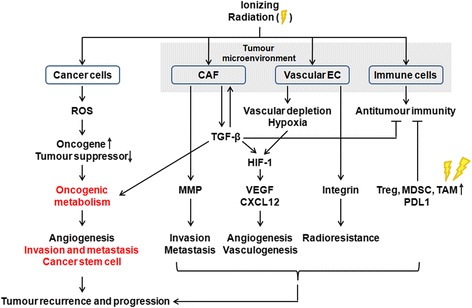
IR-induced side effects on cancer cells and the tumour microenvironment (TME). Radiotherapy has the paradoxical side-effect of increasing tumour aggressiveness. IR promotes ROS production in cancer cells, which may induce the activation of oncogenes and the inactivation of tumour suppressors, which further promote oncogenic metabolism. Metabolic alterations are involved in tumour progression, and include growth, invasion, metastasis, and the acquisition of the CSC phenotype, thereby contributing to tumour recurrence and distant metastasis. Given that IR induces EMT and CSC properties in cancer cells, it is possible that IR-induced oncogenic metabolism is required for the acquisition of the EMT and CSC phenotypes. IR can also elicit various changes in the TME, such as: 1) the emergence of cancer-associated fibroblasts (CAFs), activity-mediated extracellular matrix (ECM) remodelling, and fibrosis, 2) cycling hypoxia, and 3) an inflammatory response. IR activates cancer-associated fibroblasts (CAFs) to promote the release of growth factors, including transforming growth factor-β (TGF-β), and extracellular matrix (ECM) modulators, including matrix metalloproteinase (MMP). TGF-β directly affects tumour cells and CAFs, enhances tumour immune escape, and activates hypoxia-inducible factor-1 (HIF-1) signalling. MMPs degrade the ECM, facilitating tumour invasion and metastasis. IR can also cause damage to the vascular endothelial cells (EC), leading to hypoxia that further promotes HIF-1 signalling. HIF-1 increases the expression of vascular endothelial growth factor (VEGF) and chemokine (C-X-C motif) ligand 12 (CXCL12), both of which induce angiogenesis and vasculogenesis. IR also upregulates integrins on ECs that enhance survival and confer radioresistance. Although IR activates an antitumour immune response, this signalling is frequently suppressed by tumour escape mechanisms (such as programmed cell death protein 1 ligand 1 [PDL1] signalling) and by suppressive immune cells (regulatory T cells [Treg], myeloid-derived suppressor cells [MDSC], and tumour-associated macrophages [TAM]), which are relatively less radiosensitive than other lymphocyte subsets. These IR-mediated changes in the TME may constitute additional adverse effects of IR on the patient by promoting angiogenesis, invasion, metastasis, and radioresistance
IR can also damage endothelial cells, resulting in hypoxia that further promotes HIF-1 signalling. HIF-1 induces angiogenesis and vasculogenesis through the upregulation of VEGF and CXCL12 [75, 267–271]. VEGF is known to be induced by various upstream activators, such as environmental cues, growth factors, cytokines, hormones, and oncogenes. IR increases VEGF expression by upregulating HIF-1α and NF-κB in prostate cancer [269, 270]. As mentioned above, IR also induces the reoxygenation of hypoxic cancer cells to activate HIF-1 signalling. IR-induced reoxygenation also enhances the translation and secretion of HIF-1-regulated genes and VEGF, thereby increasing endothelial cell radioresistance [77]. Inhibition of HIF-1α/VEGF-A signalling enhances radiosensitivity [271]. Notably, the hypoxic regions of tumours can function as a refuge for CSCs, and increase their survival during chemotherapy. In addition, stem cell-like properties could be induced by paracrine signalling from endothelial cells, thereby increasing chemoresistance [249]. Furthermore, IR also upregulates integrins on endothelial cells, which enhances their survival and confers radioresistance [249, 252].
Endothelial cell damage also leads to initiation of inflammatory signalling and increased attraction of innate immune cells [75, 267, 268]. Although IR stimulates an immune response by inducing damage-associated molecular pattern (DAMP) and NKG2D signalling in cancer cells, this signalling is frequently suppressed by regulatory T cells, which leads to immune tolerance. Other tumour escape mechanisms, such as programmed cell death protein 1 ligand 1 (PDL1) signalling and MDSC/TAM-derived IL-10 immunosuppression, also remain intact. In addition, after radiotherapy, the number of these locally immunosuppressive cells (TAM, MDSCs, and regulatory T cells) is relatively high owing to their lower radiosensitivity compared to other lymphocyte subsets [252, 260, 261].
These IR-mediated changes in the TME may be additional adverse effects of IR by promoting radioresistance, tumour recurrence, and metastasis. The roles of the TME in determining radiotherapy outcomes have been reviewed elsewhere, and are not discussed in detail here.
The roles of EMT, CSC, and oncogenic metabolism in radioresistance
More than half of cancer patients receive radiotherapy, with varying success. The dose of IR delivered to the tumour is limited by the risk of damage to the surrounding normal tissues. Therefore, radiation therapy aims to minimise the toxicity to normal tissues in the first approach, while maximising doses to cancer cells in the second approach. Three main biological factors of tumours can influence treatment outcome: 1) the intrinsic radioresistance of the cancer cells, 2) the repopulation capacity of surviving cancer cells during the intervals between treatments, and 3) the degree of hypoxia in the tissue environment [4, 272].
Radioresistance has been shown to arise from the activation of several different pathways, including survival pathways (PI3K/Akt, ERK), DNA DSB repair pathways (homologous recombination and non-homologous end-joining [NHEJ]), glycolysis, and autophagy. Radioresistance has also been shown to arise from the induction of cell cycle redistribution, and the inactivation of the apoptosis pathway, that follows exposure to radiation [272–275]. EMT, CSCs, and oncogenic metabolism play important roles in the development of cancer radioresistance by activating these pathways. Understanding these mechanisms is important to be able to develop new strategies to enhance cancer radiotherapy.
The roles of EMT signalling pathways in radioresistance
EMT has been shown to confer resistance to radiation and chemotherapy in many cancers [273–277]. After IR, surviving cells exhibit an EMT phenotype with upregulation of EMT markers, including Snail, Slug, ZEB1, Twist1, vimentin, and N-cadherin, in lung adenocarcinoma cells [277]. Cells undergoing EMT also exhibit increased radioresistance by acquiring stem-like properties, preventing apoptosis, enhancing survival pathways, and activating signalling pathways involved in cell cycle progression and DNA damage repair [273–275].
EMT-promoting transcription factors, including Snail, Slug, ZEB1, and ZEB2, are known to be associated with radioresistance [43, 278–286]. Snail is known to play important roles in radioresistance by inhibiting p53-mediated apoptosis, activating survival pathways, and inducing stem cell properties [278, 279]. IR induces apoptosis by upregulating the p53 target gene PTEN, a negative regulator of the PI3K/Akt survival pathway. Snail protein is stabilised by IR and subsequently binds to the PTEN promoter that inhibit p53 binding to the PTEN promoter. Thus, Snail prevents IR-mediated PTEN upregulation and activates the Akt pathway, thereby increasing radioresistance [278].
Slug is also known to be involved in radioresistance by inhibiting p53-mediated apoptosis and activating stem cell properties [279–282]. Slug knockout mice exhibited increased radiosensitivity [280, 281]. IR upregulates Slug by activating p53; Slug then directly represses p53-target gene PUMA transcription, thereby preventing IR-induced apoptosis [281]. Slug also induces CSC activity and radioresistance [279, 282]. Long non-coding RNA MALAT1 regulates Slug expression by reciprocally repressing miR-1, which contributes to CSC activity and radioresistance [282]. IR-induced Snail and Slug also promote EMT and stem cell properties, and they suppress p53-mediated apoptosis [279]. All these events help cancer cells to escape to newer and less adverse niches, generate the critical tumour mass necessary to form macrometastases, and survive under stress conditions in the primary tumour [279].
In addition, ZEB1 and ZEB2 have been associated with radioresistance [283–286]. ZEB1 is known to confer radioresistance by activating DNA damage repair pathways [283]. IR-induced DNA damage increases ATM activation that stabilises ZEB1. ZEB1, in turn, directly binds to USP7 deubiquitinase to stabilise CHK1, thereby activating the recombination-dependent DNA repair response. ZEB1 inhibition enhances radiosensitivity, but has no effect on EMT [283]. Consistent with this observation, ZEB2 also protects cancer cells from IR-induced apoptosis by inhibiting ATM/ATR activation in an EMT-independent manner [285]. These observations suggest EMT-independent roles for these transcription factors in radioresistance, but contradictory evidence also exists: ZEB1-induced EMT is involved in the radioresistance of nasopharyngeal carcinoma cells [284]. Therefore, further studies are needed to precisely determine the contribution of EMT and EMT-inducing transcription factors in responses to cancer therapy.
The signalling pathways involved in EMT, including those of TGF-β, Wnt, Notch, Hedgehog, and EGFR, are also known to be involved in radioresistance [90, 287–302]. TGF-β has been shown to play critical roles in radioresistance by inducing CSC properties and by activating DNA repair pathways [287–291]. TGF-β is known to promote IR-induced self-renewal pathways, including Notch1, and to induce effective DNA damage responses that lead to the radioresistance of glioblastoma-initiating cells [288]. TGF-β activates the NHEJ DNA repair pathway upon IR, by upregulating LIG4 (a DNA ligase in DNA DSB repair), thereby protecting cells from IR [290].
It is generally agreed that TGF-β switches from a tumour suppressor (in an early stage of tumourigenesis) to a tumour promoter (in a later stage of tumourigenesis) [37, 38]. Thus, the role of the TGF-β pathway in radiotherapy is still a matter of debate. However, inhibition of TGF-β signalling has been shown to increase radiosensitivity in vitro and enhance IR-induced tumour growth delay in vivo [287]. Inhibition of TGF-β also prevents IR-induced metastases in tumour-bearing mice [90]. In addition, increased circulating TGF-β levels during radiotherapy have been strongly correlated with poor prognoses for patients with non-small cell lung cancer [291].
Wnt/β-catenin signalling has been shown to confer radioresistance by enhancing stemness, by activating survival pathways, and by activating DNA damage repair pathways [292–296]. High Wnt signalling activity is associated with increased stemness and radioresistance in colorectal cancer cells and intestinal stem cells [296]. IR selectively increases β-catenin expression and nuclear localisation in progenitor cells, but not in nonprogenitor cells. β-catenin then enhances cell survival, partly by upregulating survivin, an apoptosis inhibitor [292, 293]. β-catenin also promotes the self-renewal of progenitor cells [293]. These behaviours may lead to increases in the IR-induced enrichment of progenitor cells, and may further enhance their radioresistance [292]. In addition, β-catenin activates the NHEJ DNA repair pathway by directly promoting LIG4 transcription, thereby increasing radioresistance [296]. Supporting this observation, nuclear β-catenin expression has been highly correlated with poor outcomes after radiotherapy in patients with cervical squamous cell carcinoma [294].
In addition, Notch signalling is associated with radioresistance by preventing apoptosis and enhancing survival pathways. Notch signalling confers radioresistance to glioma cells by activating the PI3K/Akt pathway and increasing expression of Mcl-1, an anti-apoptotic Bcl-2 family protein [297]. Akt activation is also mediated by EGFR signalling, and also increases radioresistance [298]. The PI3K/Akt/mTOR pathway promotes the EMT and CSC phenotypes via elevated levels of Snail, thereby increasing radioresistance [299]. Increased Akt Ser (473) phosphorylation and mTORC1 protein expression are also associated with enhanced EMT and radioresistance [301].
Furthermore, Hedgehog signalling is involved in radioresistance [302]. GLI1 proteins are the transcription factors of the Hedgehog effector. IR triggers the mTOR/S6K1 pathway that increases expression and nuclear translocation of GLI1, accompanied by increased expression of Snail. These events and components mediate radioresistance and IR-induced tumour repopulation in vivo [302].
The roles of CSC signalling pathways in radioresistance
Several lines of evidence support the assertion that CSCs are implicated in radioresistance [126–129, 303–306]. Clinical studies showed that the expression of CSC markers, including CD44, CD133, and ALDH1, is correlated with a poor prognosis after radiotherapy in patients with lung and larynx cancer [303, 304]. The radioresistance of CSCs is associated with both the intrinsic properties of CSCs (increased DNA repair capability, cell cycle status, upregulated ROS scavengers, inhibited apoptosis, induced autophagy, induced survival pathways) and the adaptive responses of CSCs that are caused by IR and by microenvironmental changes (e.g., changes in endothelial cells, ECM, cytokine levels, NO levels, oxygen levels) [198, 305, 307–315]. As mentioned above, CSCs can exhibit additional metabolic reprogramming in response to cancer treatment, and this can lead to adaptive and acquired resistance [198]. IR can also modify the TME, and these factors affect the IR response of CSCs [305].
In particular, CSCs exhibit several biological features that are responsible for resistance to conventional anti-tumour therapies. CSCs commonly express high levels of genes involved in DNA damage response (ATM, SMC1, CHK1, CHK2, p53) and in DNA DSB repair pathways, including homologous recombination genes (BRCA1, Exo1, Rad51, Rad52) and genes involved in NHEJ (XLF), that contribute to radioresistance [307–311]. In addition, overexpression of stem cell factors, such as ALDH, increases the clonogenic capacity of CSCs and decreases their growth rates, thereby also conferring radioresistance [312].
ROS scavengers are also highly expressed in the CSCs in some tumours, and these protect them from ROS-induced damage [313, 314]. Pharmacological depletion of ROS scavengers decreases the colony-forming ability of CSCs and enhances their radiosensitivity, indicating that ROS levels are involved in CSC radioresistance [313].
In addition, regulation of the apoptosis and survival pathways is involved in CSC radioresistance. CD133+ liver CSCs exhibit elevated levels of anti-apoptotic Bcl-2, and show activation of the PI3K and ERK pathways, compared with CD133- cells [314]. Furthermore, autophagy is implicated in CSC radioresistance. IR induces a larger degree of autophagy in CD133+ CSCs, with upregulation of the autophagy-related proteins LC3, ATG5, and ATG12, as compared with CD133- cells. Inhibition of autophagy enhances the radiosensitivity of CD133+ CSCs, suggesting a role for autophagy in radioresistance [315].
The roles of oncogenic metabolism signalling pathways in radioresistance
Metabolic alteration leads to adaptive and acquired resistance to cancer treatment. Accumulating evidence suggests that alterations in cancer cell metabolism are associated with radioresistance [245, 316–329]. Radioresistant cells have been shown to exhibit the Warburg effect, with increased glucose uptake and decreased mitochondrial oxidative phosphorylation to support their growth [245]. Consistent with this observation, mitochondrial respiration-deficient ρ(0) cells are more resistant to radiation than ρ(+) cells [317].
High glucose levels are also known to prevent IR-induced cell death and to promote EMT by increasing levels of the DANGER protein (also known as ITPRIP or ‘inositol 1,4,5-trisphosphate receptor [IP3R] interacting protein’), resulting in radioresistance [322]. DANGER is known to bind directly to death-associated protein kinase (DAPK) and disrupts the catalytic activity of DAPK, which mediates anoikis (anchorage-dependent apoptosis). IR increases DAPK activity, which enhances p53 transcriptional activity, which leads to anoikis. High glucose levels upregulate DANGER and inhibit DAPK activity, which prevents anoikis and promotes EMT, thereby increasing radioresistance. Much clinical evidence has supported the assertion that high glucose uptake in a tumour translates to a poor prognosis for the patient [322]. Thus, inhibition of the glycolytic switch could be a promising therapeutic strategy for treating many cancers, by enhancing their radiosensitivity [320, 323–328].
In addition, glutamine metabolism has been shown to play critical role in radioresistance. Glutamate is a precursor for glutathione synthesis, which regulates redox homeostasis and thereby contributes to cellular defense systems. Thus, inhibition of GLS markedly enhances the radiosensitivity of cancer cells, suggesting an important role of glutamine metabolism in radioresistance [329]. Because the same metabolic pathways are required for both proliferating normal cells and proliferating cancer cells, understanding the molecular mechanisms of cancer metabolism opens a new therapeutic window to the development of better and more successful cancer treatments, by enabling the targeting of oncogenic metabolic pathways.
Conclusions
Many types of therapy are used to treat cancer, including surgery, chemotherapy, and ionizing radiation (IR) therapy. IR is a major therapeutic tool for treating a variety of malignant tumours. However, IR paradoxically also enhances the migration and invasiveness of cancer cells by inducing EMT. IR induces stromal, vascular, and immunological changes in the TME that present additional adverse effects for the cancer patient by promoting tumour recurrence and metastasis. These side effects are also commonly observed after chemotherapy.
Cancer cells that undergo EMT not only exhibit enhanced metastatic ability, but also acquire stemness and metabolic alterations. EMT, CSCs, oncogenic metabolism, and the TME have all been shown to play important roles in determining cancer treatment outcomes. It is now understood that metabolic changes are associated with malignant transformation, tumour invasion, and metastasis. Oncogenic metabolism has been shown to drive the EMT and CSC phenotypes; these changes may cause resistance to radiotherapy and promote tumour recurrence. Supporting this view, dysregulated metabolism is known to have played important roles in the evolution of cell motility. Cells with higher metabolic rates evolve to have increased motility in premalignant neoplasms, and this may enable cells to preadapt for subsequent invasion and metastasis [330].
Thus, targeting CSCs, EMT, and oncogenic metabolic pathways may reduce primary tumour recurrence, prevent invasion, and prevent distant metastasis. For example, inhibition of TGF-β signalling with a selective inhibitor of ALK5 seems to enhance radiosensitivity by preventing EMT, disrupting self-renewal capabilities, blocking the DNA damage response, and increasing apoptosis [331–333]. Blocking Akt with an inhibitor, such as GSK690693, may also prove useful in suppressing IR-induced EMT and increasing radiosensitivity [71]. A dual PI3K/mTOR inhibitor, BEZ235, is also known to enhance the radiosensitivity of prostate cancer cells with reduced EMT/CSC phenotypes [299].
IR can increase ROS production, which can loop back and mediate most of the biological effects of IR itself [12–19]. ROS have been closely associated with tumorigenesis and tumour progression. High levels of ROS trigger cell death by causing irreversible damage to cellular components such as proteins, nucleic acids, and lipids, whereas low levels of ROS have been shown to promote tumour progression via growth, invasion, and metastasis [46–52]. Thus, this review suggests that IR-induced ROS may play important roles in the induction of EMT, CSCs, and oncogenic metabolic pathways as undesired side effects. Notably, cancer cells express high levels of antioxidant proteins to detoxify themselves against ROS [47, 49, 51]. Therefore, use of radiotherapy must include considerations of the unique redox status of the target tumour.
p53 is one of the most important tumour suppressors. It is well known that p53 is activated in response to IR-induced DNA damage; p53 induces growth arrest, apoptosis, or senescence-like, irreversible growth-arrest in cancer cells, and these actions constitute the therapeutic effects of IR [8–11]. A recent study showed that elephants are cancer-resistant, potentially because of their multiple copies (40 alleles) of TP53, compared with the smaller number of copies (2 alleles) in humans. Thus, in response to IR-induced DNA damage, elephant cells exhibit higher rates of apoptotic death than human cells, suggesting a role for tumour suppressor p53 in cancer resistance [334]. In addition, while wild-type p53 is a tumour suppressor, the mutant form of p53 has been shown to represent not just a loss-of-function phenotype of the protein, but also a gain-of-function phenotype in terms of pro-oncogenic activities [335]. Interestingly, a recent study showed an opposite result, in which IR-induced p53 played an important role in the development of lymphomas. After IR, p53 promoted bone marrow cell death; this created a favourable environment for the expansion of tumour-initiating cells in the thymus, by decreasing cell competition from the bone marrow. Through this mechanism, p53 promoted the IR-induced development of lymphoma [336]. Thus, radiotherapy needs to be carefully considered regarding its effects on p53.
This review concludes that IR can induce EMT, CSCs, and oncogenic metabolism in many cancer cells, as a side-effect; several other studies also raise the possibility that IR causes unwanted side effects. Therefore, a better understanding of the mechanisms involved in IR-induced EMT, CSCs, and oncogenic metabolism may help improve the effectiveness of radiotherapy.
Furthermore, after chemotherapy, surviving cells have been shown to display EMT and CSC phenotypes, oncogenic metabolism, and additional metabolic reprogramming. Similar roles for the EMT and CSC phenotypes, and for oncogenic metabolism, have been demonstrated in cancer cell chemoresistance. Chemotherapy is known to induce the EMT and CSC phenotypes [163, 337–342]. EMT leads cancer cells to become quiescent circulating tumour cells (CTCs) that enter the bloodstream. These CTCs are transformed into CSCs that display both the EMT phenotype and chemoresistance. Thus, surviving CSCs repopulate the tumour and cause a relapse [337]. For example, cisplatin-resistant cancer cells are known to display enhanced EMT features and CSC properties, via the activation of the Akt/β-catenin/Snail signalling pathway [341]. Chemotherapy is also known to induce metabolic alterations [343–347]. For example, taxol-resistant breast cancer cells exhibit higher LDHA expression and activity than do taxol-sensitive cells. Inhibition of LDHA can resensitise these resistant cells to taxol, suggesting a role for metabolic alteration in chemoresistance [345]. Furthermore, chemotherapy can induce the reverse Warburg effect [348–351]; chemotherapy drives stromal fibroblasts to become CAFs that subsequently exhibit the glycolytic switch, activating the HIF-1, STAT3, TGF-β, JNK/AP1, and NF-κB pathways. These CAFs, in turn, set up synergistic relationships with adjacent epithelial cancer cells to acquire stemness [350]. Therefore, chemotherapy also causes undesired side effects in cancer cells by inducing EMT, CSCs, and oncogenic metabolic pathways, in a manner similar to IR. In the long term, any therapeutic strategy that affects EMT/CSC/oncogenic metabolic behaviour will require patient-personalized considerations of how to best utilize radiotherapy and chemotherapy.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant, funded by the Korean government (MSIP) (Grant Nos. 2011-0011084, 2012M2B2A9A02029802, 2013M2B2A9A03050902, 2015M2B2A9028108, 2012R1A1A2044246, and 2015R1A2A2A01004468), and by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare, Republic of Korea (1320040).
Availability of data and materials
Not applicable.
Authors’ contributions
SYL, EKJ, and MKJ contributed equally to this work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- ALK5
TGF-β type I receptor kinase
- CAFs
Cancer-associated fibroblasts
- COX
Cytochrome C oxidase
- CS
Citrate synthase
- CSC
Cancer stem cell
- CTCs
Circulating tumour cells
- CXCL12
C-X-C motif chemokine ligand 12
- CXCR4
C-X-C chemokine receptor type 4
- DAMP
Damage-associated molecular pattern
- DAPK
Death-associated protein kinase
- Dlx-2
Distal-less homeobox-2
- DSBs
DNA double-strand breaks
- ECM
Extracellular matrix
- EGF
Epidermal growth factor
- EMT
Epithelial-mesenchymal transition
- ERK
Extracellular signal-regulated kinase
- FASN
Fatty acid synthase
- FBP1
Fructose-1,6-bisphosphatase 1
- GAPDH
Glyceraldehyde-3-phosphate dehydrogenase
- G-CSF
Granulocyte-colony stimulating factor
- GLS1
Glutaminase 1
- GSK3β
Glycogen synthase kinase3β
- HIF-1
Hypoxia-inducible factor-1
- IR
Ionizing radiation
- LDH
Lactate dehydrogenase
- LncRNAs
Long noncoding RNAs
- LRP
Lipoprotein receptor-related protein
- MAPK
Mitogen-activated protein kinase
- MCT
Monocarboxylate transporter
- MDSCs
Myeloid-derived suppressor cells
- MiRNAs
MicroRNAs
- MMP
Matrix metalloproteinase
- MTOR
Mammalian target of rapamycin
- NAC
N-acetylcysteine
- NF-κB
Nuclear factor-kappa B
- NHEJ
Non-homologous end-joining
- NO
Nitric oxide
- OXPHOS
Oxidative phosphorylation
- PAI-1
Plasminogen activator inhibitor-1
- PAK1
p21-activated kinase 1
- PC
Pyruvate carboxylase
- PDGFR
Platelet-derived growth factor receptors
- PDH
Pyruvate dehydrogenase
- PDK
Pyruvate dehydrogenase kinase
- PI3K
Phosphatidylinositol 3-kinase
- PKM2
Pyruvate kinase M2
- PPP
Pentose phosphate pathway
- PTEN
Phosphatase and tensin homolog
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SDHB
Succinate dehydrogenase subunit B
- STAT3
Signal transducer and activator of transcription 3
- TCA
Tricarboxylic acid
- TGF-β
Transforming growth factor-β
- TME
Tumour microenvironment
- UPA
Urokinase-type plasminogen activator
- UV
Ultraviolet
- VEGF
Vascular endothelial growth factor
- α-SMA
α-smooth muscle actin
References
- 1.Bartelink H, Horiot JC, Poortmans P, Struikmans H, Van den Bogaert W, Barillot I, et al. Recurrence rates after treatment of breast cancer with standard radiotherapy with or without additional radiation. N Engl J Med. 2001;345(19):1378–87. doi: 10.1056/NEJMoa010874. [DOI] [PubMed] [Google Scholar]
- 2.Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nat Rev Cancer. 2004;4(9):737–47. doi: 10.1038/nrc1451. [DOI] [PubMed] [Google Scholar]
- 3.Delaney G, Jacob S, Featherstone C, Barton M. The role of radiotherapy in cancer treatment: estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer. 2005;104(6):1129–37. doi: 10.1002/cncr.21324. [DOI] [PubMed] [Google Scholar]
- 4.Huber SM, Butz L, Stegen B, Klumpp D, Braun N, Ruth P, et al. Ionizing radiation, ion transports, and radioresistance of cancer cells. Front Physiol. 2013;4:212. doi: 10.3389/fphys.2013.00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hubenak JR, Zhang Q, Branch CD, Kronowitz SJ. Mechanisms of injury to normal tissue after radiotherapy: a review. Plast Reconstr Surg. 2014;133(1):49e–56e. doi: 10.1097/01.prs.0000440818.23647.0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernier J, Poortmans PM. Surgery and radiation therapy of triple-negative breast cancers: From biology to clinics. Breast. 2016;28:148–55. doi: 10.1016/j.breast.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 7.Good JS, Harrington KJ. The hallmarks of cancer and the radiation oncologist: updating the 5Rs of radiobiology. Clin Oncol (R Coll Radiol) 2013;25(10):569–77. doi: 10.1016/j.clon.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 8.Santivasi WL, Xia F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal. 2014;21(2):251–9. doi: 10.1089/ars.2013.5668. [DOI] [PubMed] [Google Scholar]
- 9.Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32(33):3789–97. doi: 10.1038/onc.2012.556. [DOI] [PubMed] [Google Scholar]
- 10.Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3(2):117–29. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- 11.Fei P, El-Deiry WS. P53 and radiation responses. Oncogene. 2003;22(37):5774–83. doi: 10.1038/sj.onc.1206677. [DOI] [PubMed] [Google Scholar]
- 12.Feinendegen LE. Reactive oxygen species in cell responses to toxic agents. Hum Exp Toxicol. 2002;21(2):85–90. doi: 10.1191/0960327102ht216oa. [DOI] [PubMed] [Google Scholar]
- 13.Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23(3-4):311–22. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 14.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6(9):702–13. doi: 10.1038/nrc1950. [DOI] [PubMed] [Google Scholar]
- 15.Kam WW, Banati RB. Effects of ionizing radiation on mitochondria. Free Radic Biol Med. 2013;65:607–19. doi: 10.1016/j.freeradbiomed.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 16.Leach JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001;61(10):3894–901. [PubMed] [Google Scholar]
- 17.Tulard A, Hoffschir F, de Boisferon FH, Luccioni C, Bravard A. Persistent oxidative stress after ionizing radiation is involved in inherited radiosensitivity. Free Radic Biol Med. 2003;35(1):68–77. doi: 10.1016/S0891-5849(03)00243-0. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T, Goto S, Kawakatsu M, Urata Y, Li TS. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free Radic Res. 2012;46(2):147–53. doi: 10.3109/10715762.2011.645207. [DOI] [PubMed] [Google Scholar]
- 19.Barjaktarovic Z, Schmaltz D, Shyla A, Azimzadeh O, Schulz S, Haagen J, et al. Radiation-induced signaling results in mitochondrial impairment in mouse heart at 4 weeks after exposure to X-rays. PLoS One. 2011;6(12):e27811. doi: 10.1371/journal.pone.0027811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Essen CF. Radiation enhancement of metastasis: a review. Clin Exp Metastasis. 1991;9(2):77–104. doi: 10.1007/BF01756381. [DOI] [PubMed] [Google Scholar]
- 21.Moncharmont C, Levy A, Guy JB, Falk AT, Guilbert M, Trone JC, et al. Radiation-enhanced cell migration/invasion process: a review. Crit Rev Oncol Hematol. 2014;92(2):133–42. doi: 10.1016/j.critrevonc.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 22.Wild-Bode C, Weller M, Rimner A, Dichgans J, Wick W. Sublethal irradiation promotes migration and invasiveness of glioma cells: implications for radiotherapy of human glioblastoma. Cancer Res. 2001;61(6):2744–50. [PubMed] [Google Scholar]
- 23.Madani I, De Neve W, Mareel M. Does ionizing radiation stimulate cancer invasion and metastasis? Bull Cancer. 2008;95(3):292–300. doi: 10.1684/bdc.2008.0598. [DOI] [PubMed] [Google Scholar]
- 24.De Bacco F, Luraghi P, Medico E, Reato G, Girolami F, Perera T, et al. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. J Natl Cancer Inst. 2011;103(8):645–61. doi: 10.1093/jnci/djr093. [DOI] [PubMed] [Google Scholar]
- 25.Kawamoto A, Yokoe T, Tanaka K, Saigusa S, Toiyama Y, Yasuda H, et al. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol Rep. 2012;27(1):51–7. doi: 10.3892/or.2011.1485. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Li X, Zhang N, Yang Q, Moran MS. Low doses ionizing radiation enhances the invasiveness of breast cancer cells by inducing epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2011;412(1):188–92. doi: 10.1016/j.bbrc.2011.07.074. [DOI] [PubMed] [Google Scholar]
- 27.Park JK, Jang SJ, Kang SW, Park S, Hwang SG, Kim WJ, et al. Establishment of animal model for the analysis of cancer cell metastasis during radiotherapy. Radiat Oncol. 2012;7:153. doi: 10.1186/1748-717X-7-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chargari C, Goodman KA, Diallo I, Guy JB, Rancoule C, Cosset JM, et al. Risk of second cancers in the era of modern radiation therapy: does the risk/benefit analysis overcome theoretical models? Cancer Metastasis Rev. 2016;35(2):277–88. doi: 10.1007/s10555-016-9616-2. [DOI] [PubMed] [Google Scholar]
- 29.Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–206. doi: 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13(2):97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 31.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 33.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 34.Aktipis CA, Boddy AM, Jansen G, Hibner U, Hochberg ME, Maley CC, et al. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond B Biol Sci. 2015;370(1673). doi:10.1098/rstb.2014.0219 [DOI] [PMC free article] [PubMed]
- 35.Weber GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Lett. 2013;328(2):207–11. doi: 10.1016/j.canlet.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 36.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 38.Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13(11):788–99. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013;13(1):11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- 40.Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16(6):488–94. doi: 10.1038/ncb2976. [DOI] [PubMed] [Google Scholar]
- 41.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Shi J, Chai K, Ying X, Zhou BP. The Role of Snail in EMT and Tumorigenesis. Curr Cancer Drug Targets. 2013;13(9):963–72. doi: 10.2174/15680096113136660102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ansieau S, Collin G, Hill L. EMT or EMT-Promoting Transcription Factors, Where to Focus the Light? Front Oncol. 2014;4:353. doi: 10.3389/fonc.2014.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng H, Lee SH, Wu S. Effects of N-acetyl-L-cysteine on adhesive strength between breast cancer cell and extracellular matrix proteins after ionizing radiation. Life Sci. 2013;93(21):798–803. doi: 10.1016/j.lfs.2013.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kambach DM, Sodi VL, Lelkes PI, Azizkhan-Clifford J, Reginato MJ. ErbB2, FoxM1 and 14-3-3zeta prime breast cancer cells for invasion in response to ionizing radiation. Oncogene. 2014;33(5):589–98. doi: 10.1038/onc.2012.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res. 1994;54(7 Suppl):1890s–4s. [PubMed] [Google Scholar]
- 47.Pani G, Giannoni E, Galeotti T, Chiarugi P. Redox-based escape mechanism from death: the cancer lesson. Antioxid Redox Signal. 2009;11(11):2791–806. doi: 10.1089/ars.2009.2739. [DOI] [PubMed] [Google Scholar]
- 48.Cannito S, Novo E, di Bonzo LV, Busletta C, Colombatto S, Parola M. Epithelial-mesenchymal transition: from molecular mechanisms, redox regulation to implications in human health and disease. Antioxid Redox Signal. 2010;12(12):1383–430. doi: 10.1089/ars.2009.2737. [DOI] [PubMed] [Google Scholar]
- 49.Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell’s escape from oxidative stress. Cancer Metastasis Rev. 2010;29(2):351–78. doi: 10.1007/s10555-010-9225-4. [DOI] [PubMed] [Google Scholar]
- 50.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35(9):505–13. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mori K, Shibanuma M, Nose K. Invasive potential induced under long-term oxidative stress in mammary epithelial cells. Cancer Res. 2004;64(20):7464–72. doi: 10.1158/0008-5472.CAN-04-1725. [DOI] [PubMed] [Google Scholar]
- 53.Nagarajan D, Melo T, Deng Z, Almeida C, Zhao W. ERK/GSK3beta/Snail signaling mediates radiation-induced alveolar epithelial-to-mesenchymal transition. Free Radic Biol Med. 2012;52(6):983–92. doi: 10.1016/j.freeradbiomed.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mahabir R, Tanino M, Elmansuri A, Wang L, Kimura T, Itoh T, et al. Sustained elevation of Snail promotes glial-mesenchymal transition after irradiation in malignant glioma. Neuro Oncol. 2014;16(5):671–85. doi: 10.1093/neuonc/not239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim E, Youn H, Kwon T, Son B, Kang J, Yang HJ, et al. PAK1 tyrosine phosphorylation is required to induce epithelial-mesenchymal transition and radioresistance in lung cancer cells. Cancer Res. 2014;74(19):5520–31. doi: 10.1158/0008-5472.CAN-14-0735. [DOI] [PubMed] [Google Scholar]
- 56.Pan Y, Zhou C, Yuan D, Zhang J, Shao C. Radiation exposure promotes hepatocarcinoma cell invasion through epithelial mesenchymal transition mediated by H2S/CSE pathway. Radiat Res. 2016;185(1):96–105. doi: 10.1667/RR14177.1. [DOI] [PubMed] [Google Scholar]
- 57.Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Yoo MA, et al. Dlx-2 is implicated in TGF-beta- and Wnt-induced epithelial-mesenchymal, glycolytic switch, and mitochondrial repression by Snail activation. Int J Oncol. 2015;46(4):1768–80. doi: 10.3892/ijo.2015.2874. [DOI] [PubMed] [Google Scholar]
- 58.Lee SY, Jeon HM, Ju MK, Jeong EK, Kim CH, Park HG, et al. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget. 2016;7(7):7925–39. doi: 10.18632/oncotarget.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Merlo GR, Zerega B, Paleari L, Trombino S, Mantero S, Levi G. Multiple functions of Dlx genes. Int J Dev Biol. 2000;44(6):619–26. [PubMed] [Google Scholar]
- 60.Panganiban G, Rubenstein JL. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002;129(19):4371–86. doi: 10.1242/dev.129.19.4371. [DOI] [PubMed] [Google Scholar]
- 61.Yilmaz M, Maass D, Tiwari N, Waldmeier L, Schmidt P, Lehembre F, et al. Transcription factor Dlx2 protects from TGFbeta-induced cell-cycle arrest and apoptosis. EMBO J. 2011;30(21):4489–99. doi: 10.1038/emboj.2011.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tang P, Huang H, Chang J, Zhao GF, Lu ML, Wang Y. Increased expression of DLX2 correlates with advanced stage of gastric adenocarcinoma. World J Gastroenterol. 2013;19(17):2697–703. doi: 10.3748/wjg.v19.i17.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yan ZH, Bao ZS, Yan W, Liu YW, Zhang CB, Wang HJ, et al. Upregulation of DLX2 confers a poor prognosis in glioblastoma patients by inducing a proliferative phenotype. Curr Mol Med. 2013;13(3):438–45. [PubMed] [Google Scholar]
- 64.Choi YJ, Baek GY, Park HR, Jo SK, Jung U. Smad2/3-Regulated Expression of DLX2 Is Associated with Radiation-Induced Epithelial-Mesenchymal Transition and Radioresistance of A549 and MDA-MB-231 Human Cancer Cell Lines. PLoS One. 2016;11(1):e0147343. doi: 10.1371/journal.pone.0147343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yao J, Liu Y, Wang X, Shen Y, Yuan S, Wan Y, et al. UVB radiation induces human lens epithelial cell migration via NADPH oxidase-mediated generation of reactive oxygen species and up-regulation of matrix metalloproteinases. Int J Mol Med. 2009;24(2):153–9. doi: 10.3892/ijmm_00000218. [DOI] [PubMed] [Google Scholar]
- 66.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15(5):416–28. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102(4):639–44. doi: 10.1038/sj.bjc.6605530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hudson LG, Choi C, Newkirk KM, Parkhani J, Cooper KL, Lu P, et al. Ultraviolet radiation stimulates expression of Snail family transcription factors in keratinocytes. Mol Carcinog. 2007;46(4):257–68. doi: 10.1002/mc.20257. [DOI] [PubMed] [Google Scholar]
- 69.Li Y, Liu Y, Xu Y, Voorhees JJ, Fisher GJ. UV irradiation induces Snail expression by AP-1 dependent mechanism in human skin keratinocytes. J Dermatol Sci. 2010;60(2):105–13. doi: 10.1016/j.jdermsci.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 70.Shirley SH, Hammiller B, Hansen LA, Crysup B, Hudson LG, Kusewitt DF. Role of the epidermal growth factor receptor in ultraviolet radiation induction of Snail family transcription factors. J Dermatol Sci. 2014;76(2):149–51. doi: 10.1016/j.jdermsci.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 71.Chen W, Wu S, Zhang G, Wang W, Shi Y. Effect of AKT inhibition on epithelial-mesenchymal transition and ZEB1-potentiated radiotherapy in nasopharyngeal carcinoma. Oncol Lett. 2013;6(5):1234–40. doi: 10.3892/ol.2013.1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu W, Huang YJ, Liu C, Yang YY, Liu H, Cui JG, et al. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab Invest. 2014;94(4):362–70. doi: 10.1038/labinvest.2013.153. [DOI] [PubMed] [Google Scholar]
- 73.Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175–80. doi: 10.1126/science.aaf4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. 2012;33(4):207–14. doi: 10.1016/j.tips.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harada H. How can we overcome tumor hypoxia in radiation therapy? J Radiat Res. 2011;52(5):545–56. doi: 10.1269/jrr.11056. [DOI] [PubMed] [Google Scholar]
- 76.Ghattass K, Assah R, El-Sabban M, Gali-Muhtasib H. Targeting hypoxia for sensitization of tumors to radio- and chemotherapy. Curr Cancer Drug Targets. 2013;13(6):670–85. doi: 10.2174/15680096113139990004. [DOI] [PubMed] [Google Scholar]
- 77.Moeller BJ, Cao Y, Li CY, Dewhirst MW. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer Cell. 2004;5(5):429–41. doi: 10.1016/S1535-6108(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 78.Harada H, Itasaka S, Kizaka-Kondoh S, Shibuya K, Morinibu A, Shinomiya K, et al. The Akt/mTOR pathway assures the synthesis of HIF-1alpha protein in a glucose- and reoxygenation-dependent manner in irradiated tumors. J Biol Chem. 2009;284(8):5332–42. doi: 10.1074/jbc.M806653200. [DOI] [PubMed] [Google Scholar]
- 79.Harada H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J Radiat Res. 2016;57(Suppl 1):i99–i105. doi: 10.1093/jrr/rrw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuo YC, Wu HT, Hung JJ, Chou TY, Teng SC, Wu KJ. Nijmegen breakage syndrome protein 1 (NBS1) modulates hypoxia inducible factor-1alpha (HIF-1alpha) stability and promotes in vitro migration and invasion under ionizing radiation. Int J Biochem Cell Biol. 2015;64:229–38. doi: 10.1016/j.biocel.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 81.Kang J, Kim W, Kwon T, Youn H, Kim JS, Youn B. Plasminogen activator inhibitor-1 enhances radioresistance and aggressiveness of non-small cell lung cancer cells. Oncotarget. 2016. doi:10.18632/oncotarget.8208. [DOI] [PMC free article] [PubMed]
- 82.Harada H, Inoue M, Itasaka S, Hirota K, Morinibu A, Shinomiya K, et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat Commun. 2012;3:783. doi: 10.1038/ncomms1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Luo D, Wang J, Li J, Post M. Mouse snail is a target gene for HIF. Mol Cancer Res. 2011;9(2):234–45. doi: 10.1158/1541-7786.MCR-10-0214. [DOI] [PubMed] [Google Scholar]
- 84.Martin M, Vozenin MC, Gault N, Crechet F, Pfarr CM, Lefaix JL. Coactivation of AP-1 activity and TGF-beta1 gene expression in the stress response of normal skin cells to ionizing radiation. Oncogene. 1997;15(8):981–9. doi: 10.1038/sj.onc.1201433. [DOI] [PubMed] [Google Scholar]
- 85.O'Malley Y, Zhao W, Barcellos-Hoff MH, Robbins ME. Radiation-induced alterations in rat mesangial cell Tgfb1 and Tgfb3 gene expression are not associated with altered secretion of active Tgfb isoforms. Radiat Res. 1999;152(6):622–8. doi: 10.2307/3580257. [DOI] [PubMed] [Google Scholar]
- 86.Jobling MF, Mott JD, Finnegan MT, Jurukovski V, Erickson AC, Walian PJ, et al. Isoform-specific activation of latent transforming growth factor beta (LTGF-beta) by reactive oxygen species. Radiat Res. 2006;166(6):839–48. doi: 10.1667/RR0695.1. [DOI] [PubMed] [Google Scholar]
- 87.Dancea HC, Shareef MM, Ahmed MM. Role of Radiation-induced TGF-beta Signaling in Cancer Therapy. Mol Cell Pharmacol. 2009;1(1):44–56. doi: 10.4255/mcpharmacol.09.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du S, Barcellos-Hoff MH. Tumors as organs: biologically augmenting radiation therapy by inhibiting transforming growth factor beta activity in carcinomas. Semin Radiat Oncol. 2013;23(4):242–51. doi: 10.1016/j.semradonc.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Andarawewa KL, Erickson AC, Chou WS, Costes SV, Gascard P, Mott JD, et al. Ionizing radiation predisposes nonmalignant human mammary epithelial cells to undergo transforming growth factor beta induced epithelial to mesenchymal transition. Cancer Res. 2007;67(18):8662–70. doi: 10.1158/0008-5472.CAN-07-1294. [DOI] [PubMed] [Google Scholar]
- 90.Biswas S, Guix M, Rinehart C, Dugger TC, Chytil A, Moses HL, et al. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J Clin Invest. 2007;117(5):1305–13. doi: 10.1172/JCI30740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhou YC, Liu JY, Li J, Zhang J, Xu YQ, Zhang HW, et al. Ionizing radiation promotes migration and invasion of cancer cells through transforming growth factor-beta-mediated epithelial-mesenchymal transition. Int J Radiat Oncol Biol Phys. 2011;81(5):1530–7. doi: 10.1016/j.ijrobp.2011.06.1956. [DOI] [PubMed] [Google Scholar]
- 92.Balli D, Ustiyan V, Zhang Y, Wang IC, Masino AJ, Ren X, et al. Foxm1 transcription factor is required for lung fibrosis and epithelial-to-mesenchymal transition. EMBO J. 2013;32(2):231–44. doi: 10.1038/emboj.2012.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carl C, Flindt A, Hartmann J, Dahlke M, Rades D, Dunst J, et al. Ionizing radiation induces a motile phenotype in human carcinoma cells in vitro through hyperactivation of the TGF-beta signaling pathway. Cell Mol Life Sci. 2016;73(2):427–43. doi: 10.1007/s00018-015-2003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chen Y, Liu W, Wang P, Hou H, Liu N, Gong L, et al. Halofuginone inhibits radiotherapy-induced epithelial-mesenchymal transition in lung cancer. Oncotarget. 2016. doi:10.18632/oncotarget.11217. [DOI] [PMC free article] [PubMed]
- 95.de Marcondes PG, Morgado-Diaz JA. The Role of EphA4 Signaling in Radiation-Induced EMT-Like Phenotype in Colorectal Cancer Cells. J Cell Biochem. 2016 doi: 10.1002/jcb.25738. [DOI] [PubMed] [Google Scholar]
- 96.Lento W, Ito T, Zhao C, Harris JR, Huang W, Jiang C, et al. Loss of beta-catenin triggers oxidative stress and impairs hematopoietic regeneration. Genes Dev. 2014;28(9):995–1004. doi: 10.1101/gad.231944.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bastos LG, de Marcondes PG, De-Freitas-Junior JC, Leve F, Mencalha AL, De Souza WF, et al. Progeny from irradiated colorectal cancer cells acquire an EMT-like phenotype and activate Wnt/beta-catenin pathway. J Cell Biochem. 2014;115(12):2175–87. doi: 10.1002/jcb.24896. [DOI] [PubMed] [Google Scholar]
- 98.Dong Z, Zhou L, Han N, Zhang M, Lyu X. Wnt/beta-catenin pathway involvement in ionizing radiation-induced invasion of U87 glioblastoma cells. Strahlenther Onkol. 2015;191(8):672–80. doi: 10.1007/s00066-015-0858-7. [DOI] [PubMed] [Google Scholar]
- 99.Kang J, Kim E, Kim W, Seong KM, Youn H, Kim JW, et al. Rhamnetin and cirsiliol induce radiosensitization and inhibition of epithelial-mesenchymal transition (EMT) by miR-34a-mediated suppression of Notch-1 expression in non-small cell lung cancer cell lines. J Biol Chem. 2013;288(38):27343–57. doi: 10.1074/jbc.M113.490482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kim RK, Kaushik N, Suh Y, Yoo KC, Cui YH, Kim MJ, et al. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget. 2016. doi:10.18632/oncotarget.10802. [DOI] [PMC free article] [PubMed]
- 101.Wang S, Lee Y, Kim J, Hyun J, Lee K, Kim Y, et al. Potential role of Hedgehog pathway in liver response to radiation. PLoS One. 2013;8(9):e74141. doi: 10.1371/journal.pone.0074141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nijkamp MM, Span PN, Bussink J, Kaanders JH. Interaction of EGFR with the tumour microenvironment: implications for radiation treatment. Radiother Oncol. 2013;108(1):17–23. doi: 10.1016/j.radonc.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 103.Cuneo KC, Nyati MK, Ray D, Lawrence TS. EGFR targeted therapies and radiation: Optimizing efficacy by appropriate drug scheduling and patient selection. Pharmacol Ther. 2015;154:67–77. doi: 10.1016/j.pharmthera.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toulany M, Minjgee M, Kehlbach R, Chen J, Baumann M, Rodemann HP. ErbB2 expression through heterodimerization with erbB1 is necessary for ionizing radiation- but not EGF-induced activation of Akt survival pathway. Radiother Oncol. 2010;97(2):338–45. doi: 10.1016/j.radonc.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 105.Lee HC, An S, Lee H, Woo SH, Jin HO, Seo SK, et al. Activation of epidermal growth factor receptor and its downstream signaling pathway by nitric oxide in response to ionizing radiation. Mol Cancer Res. 2008;6(6):996–1002. doi: 10.1158/1541-7786.MCR-08-0113. [DOI] [PubMed] [Google Scholar]
- 106.Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66(17):8511–9. doi: 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- 107.Zhai GG, Malhotra R, Delaney M, Latham D, Nestler U, Zhang M, et al. Radiation enhances the invasive potential of primary glioblastoma cells via activation of the Rho signaling pathway. J Neurooncol. 2006;76(3):227–37. doi: 10.1007/s11060-005-6499-4. [DOI] [PubMed] [Google Scholar]
- 108.Pickhard AC, Margraf J, Knopf A, Stark T, Piontek G, Beck C, et al. Inhibition of radiation induced migration of human head and neck squamous cell carcinoma cells by blocking of EGF receptor pathways. BMC Cancer. 2011;11:388. doi: 10.1186/1471-2407-11-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gu Q, He Y, Ji J, Yao Y, Shen W, Luo J, et al. Hypoxia-inducible factor 1alpha (HIF-1alpha) and reactive oxygen species (ROS) mediates radiation-induced invasiveness through the SDF-1alpha/CXCR4 pathway in non-small cell lung carcinoma cells. Oncotarget. 2015;6(13):10893–907. doi: 10.18632/oncotarget.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Yuan W, Yuan Y, Zhang T, Wu S. Role of Bmi-1 in regulation of ionizing irradiation-induced epithelial-mesenchymal transition and migration of breast cancer cells. PLoS One. 2015;10(3):e0118799. doi: 10.1371/journal.pone.0118799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.He E, Pan F, Li G, Li J. Fractionated Ionizing Radiation Promotes Epithelial-Mesenchymal Transition in Human Esophageal Cancer Cells through PTEN Deficiency-Mediated Akt Activation. PLoS One. 2015;10(5):e0126149. doi: 10.1371/journal.pone.0126149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cui YH, Suh Y, Lee HJ, Yoo KC, Uddin N, Jeong YJ, et al. Radiation promotes invasiveness of non-small-cell lung cancer cells through granulocyte-colony-stimulating factor. Oncogene. 2015;34(42):5372–82. doi: 10.1038/onc.2014.466. [DOI] [PubMed] [Google Scholar]
- 113.Cho JH, Hong WG, Jung YJ, Lee J, Lee E, Hwang SG, et al. Gamma-Ionizing radiation-induced activation of the EGFR-p38/ERK-STAT3/CREB-1-EMT pathway promotes the migration/invasion of non-small cell lung cancer cells and is inhibited by podophyllotoxin acetate. Tumour Biol. 2016;37(6):7315–25. doi: 10.1007/s13277-015-4548-y. [DOI] [PubMed] [Google Scholar]
- 114.Ho JN, Kang GY, Lee SS, Kim J, Bae IH, Hwang SG, et al. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010;101(6):1417–23. doi: 10.1111/j.1349-7006.2010.01552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nambiar DK, Rajamani P, Singh RP. Silibinin attenuates ionizing radiation-induced pro-angiogenic response and EMT in prostate cancer cells. Biochem Biophys Res Commun. 2015;456(1):262–8. doi: 10.1016/j.bbrc.2014.11.069. [DOI] [PubMed] [Google Scholar]
- 116.Su WH, Chuang PC, Huang EY, Yang KD. Radiation-induced increase in cell migration and metastatic potential of cervical cancer cells operates via the K-Ras pathway. Am J Pathol. 2012;180(2):862–71. doi: 10.1016/j.ajpath.2011.10.018. [DOI] [PubMed] [Google Scholar]
- 117.Maachani UB, Shankavaram U, Kramp T, Tofilon PJ, Camphausen K, Tandle AT. FOXM1 and STAT3 interaction confers radioresistance in glioblastoma cells. Oncotarget. 2016. doi:10.18632/oncotarget.12670. [DOI] [PMC free article] [PubMed]
- 118.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, et al. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8(12):1398–406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 119.Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6(10):931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 120.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 121.Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009;15(9):1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 122.Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–17. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang T, Shigdar S, Gantier MP, Hou Y, Wang L, Li Y, et al. Cancer stem cell targeted therapy: progress amid controversies. Oncotarget. 2015;6(42):44191–206. doi: 10.18632/oncotarget.6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015;7(1):27–36. doi: 10.4252/wjsc.v7.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108(19):7950–5. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lyakhovich A, Lleonart ME. Bypassing Mechanisms of Mitochondria-Mediated Cancer Stem Cells Resistance to Chemo- and Radiotherapy. Oxid Med Cell Longev. 2016;2016:1716341. doi: 10.1155/2016/1716341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chang L, Graham P, Hao J, Ni J, Deng J, Bucci J, et al. Cancer stem cells and signaling pathways in radioresistance. Oncotarget. 2016;7(10):11002–17. doi: 10.18632/oncotarget.6760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cojoc M, Mabert K, Muders MH, Dubrovska A. A role for cancer stem cells in therapy resistance: cellular and molecular mechanisms. Semin Cancer Biol. 2015;31:16–27. doi: 10.1016/j.semcancer.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 129.Ogawa K, Yoshioka Y, Isohashi F, Seo Y, Yoshida K, Yamazaki H. Radiotherapy targeting cancer stem cells: current views and future perspectives. Anticancer Res. 2013;33(3):747–54. [PubMed] [Google Scholar]
- 130.Kurth I, Hein L, Mabert K, Peitzsch C, Koi L, Cojoc M, et al. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget. 2015;6(33):34494–509. doi: 10.18632/oncotarget.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2016. doi:10.1016/j.addr.2016.02.002 [DOI] [PubMed]
- 132.Hadjimichael C, Chanoumidou K, Papadopoulou N, Arampatzi P, Papamatheakis J, Kretsovali A. Common stemness regulators of embryonic and cancer stem cells. World J Stem Cells. 2015;7(9):1150–84. doi: 10.4252/wjsc.v7.i9.1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jaggupilli A, Elkord E. Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol. 2012;2012:708036. doi: 10.1155/2012/708036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells - what challenges do they pose? Nat Rev Drug Discov. 2014;13(7):497–512. doi: 10.1038/nrd4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 137.Gao MQ, Choi YP, Kang S, Youn JH, Cho NH. CD24+ cells from hierarchically organized ovarian cancer are enriched in cancer stem cells. Oncogene. 2010;29(18):2672–80. doi: 10.1038/onc.2010.35. [DOI] [PubMed] [Google Scholar]
- 138.Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106(38):16281–6. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, et al. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009;7(3):330–8. doi: 10.1158/1541-7786.MCR-08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Luo J, Wang P, Wang R, Wang J, Liu M, Xiong S, et al. The Notch pathway promotes the cancer stem cell characteristics of CD90+ cells in hepatocellular carcinoma. Oncotarget. 2016;7(8):9525–37. doi: 10.18632/oncotarget.6672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Takahashi RU, Miyazaki H, Ochiya T. The role of microRNAs in the regulation of cancer stem cells. Front Genet. 2014;4:295. doi: 10.3389/fgene.2013.00295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51. doi: 10.1016/j.biocel.2012.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sun X, Jiao X, Pestell TG, Fan C, Qin S, Mirabelli E, et al. MicroRNAs and cancer stem cells: the sword and the shield. Oncogene. 2014;33(42):4967–77. doi: 10.1038/onc.2013.492. [DOI] [PubMed] [Google Scholar]
- 144.Munoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6(6):620–36. doi: 10.1016/j.molonc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Quintana E, Shackleton M, Foster HR, Fullen DR, Sabel MS, Johnson TM, et al. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell. 2010;18(5):510–23. doi: 10.1016/j.ccr.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zhou L, Liu F, Wang X, Ouyang G. The roles of microRNAs in the regulation of tumor metastasis. Cell Biosci. 2015;5:32. doi: 10.1186/s13578-015-0028-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lin CW, Lin PY, Yang PC. Noncoding RNAs in Tumor Epithelial-to-Mesenchymal Transition. Stem Cells Int. 2016;2016:2732705. doi: 10.1155/2016/2732705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Arora A, Singh S, Bhatt AN, Pandey S, Sandhir R, Dwarakanath BS. Interplay Between Metabolism and Oncogenic Process: Role of microRNAs. Transl Oncogenomics. 2015;7:11–27. doi: 10.4137/TOG.S29652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xia H, Hui KM. MicroRNAs involved in regulating epithelial-mesenchymal transition and cancer stem cells as molecular targets for cancer therapeutics. Cancer Gene Ther. 2012;19(11):723–30. doi: 10.1038/cgt.2012.58. [DOI] [PubMed] [Google Scholar]
- 150.Ghosh Z, Mallick B. Cancer Stem Cells and Regulatory RNAs Crosstalk: Fostering Possibilities for Cancer Therapies. Bioscience. 2014;64(12):1138–49. doi: 10.1093/biosci/biu149. [DOI] [Google Scholar]
- 151.Zhu Y, Luo M, Brooks M, Clouthier SG, Wicha MS. Biological and clinical significance of cancer stem cell plasticity. Clin Transl Med. 2014;3(1):32. doi: 10.1186/s40169-014-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dhamija S, Diederichs S. From junk to master regulators of invasion: lncRNA functions in migration, EMT and metastasis. Int J Cancer. 2016;139(2):269–80. doi: 10.1002/ijc.30039. [DOI] [PubMed] [Google Scholar]
- 153.Xu Q, Deng F, Qin Y, Zhao Z, Wu Z, Xing Z, et al. Long non-coding RNA regulation of epithelial-mesenchymal transition in cancer metastasis. Cell Death Dis. 2016;7(6):e2254. doi: 10.1038/cddis.2016.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Genes Dev. 2009;23(13):1494–504. doi: 10.1101/gad.1800909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 157.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012;22(5-6):396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wang SS, Jiang J, Liang XH, Tang YL. Links between cancer stem cells and epithelial-mesenchymal transition. Onco Targets Ther. 2015;8:2973–80. doi: 10.2147/OTT.S91863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kang KS, Choi YP, Gao MQ, Kang S, Kim BG, Lee JH, et al. CD24(+) ovary cancer cells exhibit an invasive mesenchymal phenotype. Biochem Biophys Res Commun. 2013;432(2):333–8. doi: 10.1016/j.bbrc.2013.01.102. [DOI] [PubMed] [Google Scholar]
- 160.Zhi Y, Mou Z, Chen J, He Y, Dong H, Fu X, et al. B7H1 Expression and Epithelial-To-Mesenchymal Transition Phenotypes on Colorectal Cancer Stem-Like Cells. PLoS One. 2015;10(8):e0135528. doi: 10.1371/journal.pone.0135528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hwang WL, Yang MH, Tsai ML, Lan HY, Su SH, Chang SC, et al. SNAIL regulates interleukin-8 expression, stem cell-like activity, and tumorigenicity of human colorectal carcinoma cells. Gastroenterology. 2011;141(1):279–91. doi: 10.1053/j.gastro.2011.04.008. [DOI] [PubMed] [Google Scholar]
- 162.Kyjacova L, Hubackova S, Krejcikova K, Strauss R, Hanzlikova H, Dzijak R, et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Differ. 2015;22(6):898–911. doi: 10.1038/cdd.2014.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu WH, Chen MT, Wang ML, Lee YY, Chiou GY, Chien CS, et al. Cisplatin-selected resistance is associated with increased motility and stem-like properties via activation of STAT3/Snail axis in atypical teratoid/rhabdoid tumor cells. Oncotarget. 2015;6(3):1750–68. doi: 10.18632/oncotarget.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 165.Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY, Yang WH, et al. Bmi1 is essential in Twist1-induced epithelial-mesenchymal transition. Nat Cell Biol. 2010;12(10):982–92. doi: 10.1038/ncb2099. [DOI] [PubMed] [Google Scholar]
- 166.Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, et al. Epithelial to mesenchymal transition by TGFbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One. 2011;6(6):e21548. doi: 10.1371/journal.pone.0021548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Park SJ, Kim JG, Kim ND, Yang K, Shim JW, Heo K. Estradiol, TGF-beta1 and hypoxia promote breast cancer stemness and EMT-mediated breast cancer migration. Oncol Lett. 2016;11(3):1895–902. doi: 10.3892/ol.2016.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang R, Sun Q, Wang P, Liu M, Xiong S, Luo J, et al. Notch and Wnt/beta-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget. 2016;7((5):5754–68. doi: 10.18632/oncotarget.6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Papagerakis S, Pannone G, Zheng L, About I, Taqi N, Nguyen NP, et al. Oral epithelial stem cells - implications in normal development and cancer metastasis. Exp Cell Res. 2014;325(2):111–29. doi: 10.1016/j.yexcr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Aguilar E, Marin de Mas I, Zodda E, Marin S, Morrish F, Selivanov V, et al. Metabolic Reprogramming and Dependencies Associated with Epithelial Cancer Stem Cells Independent of the Epithelial-Mesenchymal Transition Program. Stem Cells. 2016;34(5):1163–76. doi: 10.1002/stem.2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Celia-Terrassa T, Meca-Cortes O, Mateo F, Martinez de Paz A, Rubio N, Arnal-Estape A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122(5):1849–68. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cleary AS, Leonard TL, Gestl SA, Gunther EJ. Tumour cell heterogeneity maintained by cooperating subclones in Wnt-driven mammary cancers. Nature. 2014;508(7494):113–7. doi: 10.1038/nature13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Tsuji T, Ibaragi S, Shima K, Hu MG, Katsurano M, Sasaki A, et al. Epithelial-mesenchymal transition induced by growth suppressor p12CDK2-AP1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008;68(24):10377–86. doi: 10.1158/0008-5472.CAN-08-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chapman A, Fernandez del Ama L, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 2014;8(3):688–95. doi: 10.1016/j.celrep.2014.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Cho YM, Kim YS, Kang MJ, Farrar WL, Hurt EM. Long-term recovery of irradiated prostate cancer increases cancer stem cells. Prostate. 2012;72(16):1746–56. doi: 10.1002/pros.22527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lee Y, Kim KH, Kim DG, Cho HJ, Kim Y, Rheey J, et al. FoxM1 Promotes Stemness and Radio-Resistance of Glioblastoma by Regulating the Master Stem Cell Regulator Sox2. PLoS One. 2015;10(10):e0137703. doi: 10.1371/journal.pone.0137703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ghisolfi L, Keates AC, Hu X, Lee DK, Li CJ. Ionizing radiation induces stemness in cancer cells. PLoS One. 2012;7(8):e43628. doi: 10.1371/journal.pone.0043628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gomez-Casal R, Bhattacharya C, Ganesh N, Bailey L, Basse P, Gibson M, et al. Non-small cell lung cancer cells survived ionizing radiation treatment display cancer stem cell and epithelial-mesenchymal transition phenotypes. Mol Cancer. 2013;12(1):94. doi: 10.1186/1476-4598-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446. doi: 10.1038/cddis.2012.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wu X, Tang W, Marquez RT, Li K, Highfill CA, He F, et al. Overcoming chemo/radio-resistance of pancreatic cancer by inhibiting STAT3 signaling. Oncotarget. 2016;7(10):11708–23. doi: 10.18632/oncotarget.7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lagadec C, Vlashi E, Della Donna L, Dekmezian C, Pajonk F. Radiation-induced reprogramming of breast cancer cells. Stem Cells. 2012;30(5):833–44. doi: 10.1002/stem.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hong SW, Hur W, Choi JE, Kim JH, Hwang D, Yoon SK. Role of ADAM17 in invasion and migration of CD133-expressing liver cancer stem cells after irradiation. Oncotarget. 2016. doi:10.18632/oncotarget.8112. [DOI] [PMC free article] [PubMed]
- 183.Kim RK, Cui YH, Yoo KC, Kim IG, Lee M, Choi YH, et al. Radiation promotes malignant phenotypes through SRC in breast cancer cells. Cancer Sci. 2015;106(1):78–85. doi: 10.1111/cas.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98(24):1777–85. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 185.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124(3215):269–70. [PubMed] [Google Scholar]
- 186.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–7. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 187.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 189.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 190.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 191.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26(9):877–90. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schulze A, Harris AL. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature. 2012;491(7424):364–73. doi: 10.1038/nature11706. [DOI] [PubMed] [Google Scholar]
- 193.Finley LW, Zhang J, Ye J, Ward PS, Thompson CB. SnapShot: cancer metabolism pathways. Cell Metab. 2013;17(3):466–e2. doi: 10.1016/j.cmet.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 194.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277(34):30409–12. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- 196.Jitrapakdee S, Vidal-Puig A, Wallace JC. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell Mol Life Sci. 2006;63(7-8):843–54. doi: 10.1007/s00018-005-5410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lee M, Yoon JH. Metabolic interplay between glycolysis and mitochondrial oxidation: The reverse Warburg effect and its therapeutic implication. World J Biol Chem. 2015;6(3):148–61. doi: 10.4331/wjbc.v6.i3.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111. doi: 10.1186/s13046-015-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. 2014;41(2):195–216. doi: 10.1053/j.seminoncol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 200.Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol. 2014;25:47–60. doi: 10.1016/j.semcancer.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 201.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 202.Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer associated fibroblasts. Cell Cycle. 2010;9(10):1960–71. doi: 10.4161/cc.9.10.11601. [DOI] [PubMed] [Google Scholar]
- 203.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the "reverse Warburg effect": a transcriptional informatics analysis with validation. Cell Cycle. 2010;9(11):2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 204.Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate "fuel" tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle. 2010;9(17):3506–14. doi: 10.4161/cc.9.17.12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9(17):3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174(6):2035–43. doi: 10.2353/ajpath.2009.080924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174(6):2023–34. doi: 10.2353/ajpath.2009.080873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Icard P, Kafara P, Steyaert JM, Schwartz L, Lincet H. The metabolic cooperation between cells in solid cancer tumors. Biochim Biophys Acta. 2014;1846(1):216–25. doi: 10.1016/j.bbcan.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 209.Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114(12):1305–12. doi: 10.1038/bjc.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ciavardelli D, Rossi C, Barcaroli D, Volpe S, Consalvo A, Zucchelli M, et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014;5:e1336. doi: 10.1038/cddis.2014.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514(7524):628–32. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pasto A, Bellio C, Pilotto G, Ciminale V, Silic-Benussi M, Guzzo G, et al. Cancer stem cells from epithelial ovarian cancer patients privilege oxidative phosphorylation, and resist glucose deprivation. Oncotarget. 2014;5(12):4305–19. doi: 10.18632/oncotarget.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. 2015;356(2 Pt A):156–64. doi: 10.1016/j.canlet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, et al. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012;72(14):3607–17. doi: 10.1158/0008-5472.CAN-12-0006. [DOI] [PubMed] [Google Scholar]
- 215.Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell. 2013;23(3):316–31. doi: 10.1016/j.ccr.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–13. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 217.Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18(20):5585–94. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- 218.Kondaveeti Y, Guttilla Reed IK, White BA. Epithelial-mesenchymal transition induces similar metabolic alterations in two independent breast cancer cell lines. Cancer Lett. 2015;364(1):44–58. doi: 10.1016/j.canlet.2015.04.025. [DOI] [PubMed] [Google Scholar]
- 219.Thompson CB. Wnt meets Warburg: another piece in the puzzle? EMBO J. 2014;33(13):1420–2. doi: 10.15252/embj.201488785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sherwood V. WNT signaling: an emerging mediator of cancer cell metabolism? Mol Cell Biol. 2015;35(1):2–10. doi: 10.1128/MCB.00992-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, et al. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014;33(13):1454–73. doi: 10.15252/embj.201488598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Govindarajan B, Sligh JE, Vincent BJ, Li M, Canter JA, Nickoloff BJ, et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J Clin Invest. 2007;117(3):719–29. doi: 10.1172/JCI30102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Multiple biological activities of lactic acid in cancer: influences on tumor growth, angiogenesis and metastasis. Curr Pharm Des. 2012;18(10):1319–30. doi: 10.2174/138161212799504902. [DOI] [PubMed] [Google Scholar]
- 224.Bettum IJ, Gorad SS, Barkovskaya A, Pettersen S, Moestue SA, Vasiliauskaite K, et al. Metabolic reprogramming supports the invasive phenotype in malignant melanoma. Cancer Lett. 2015;366(1):71–83. doi: 10.1016/j.canlet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 225.Wang J, Wang J, Dai J, Jung Y, Wei CL, Wang Y, et al. A glycolytic mechanism regulating an angiogenic switch in prostate cancer. Cancer Res. 2007;67(1):149–59. doi: 10.1158/0008-5472.CAN-06-2971. [DOI] [PubMed] [Google Scholar]
- 226.Zhou H, Zhang B, Zheng J, Yu M, Zhou T, Zhao K, et al. The inhibition of migration and invasion of cancer cells by graphene via the impairment of mitochondrial respiration. Biomaterials. 2014;35(5):1597–607. doi: 10.1016/j.biomaterials.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 227.Ren Y, Hao P, Dutta B, Cheow ES, Sim KH, Gan CS, et al. Hypoxia modulates A431 cellular pathways association to tumor radioresistance and enhanced migration revealed by comprehensive proteomic and functional studies. Mol Cell Proteomics. 2013;12(2):485–98. doi: 10.1074/mcp.M112.018325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast Cancer Res. 2016;18(1):55. doi: 10.1186/s13058-016-0712-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wu Z, Wei D, Gao W, Xu Y, Hu Z, Ma Z, et al. TPO-Induced Metabolic Reprogramming Drives Liver Metastasis of Colorectal Cancer CD110+ Tumor-Initiating Cells. Cell Stem Cell. 2015;17(1):47–59. doi: 10.1016/j.stem.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 230.Li D, Fu Z, Chen R, Zhao X, Zhou Y, Zeng B, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. 2015;6(31):31151–63. doi: 10.18632/oncotarget.5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ginestier C, Monville F, Wicinski J, Cabaud O, Cervera N, Josselin E, et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells. 2012;30(7):1327–37. doi: 10.1002/stem.1122. [DOI] [PubMed] [Google Scholar]
- 232.Li X, Zhong Y, Lu J, Axcrona K, Eide L, Syljuasen RG, et al. MtDNA depleted PC3 cells exhibit Warburg effect and cancer stem cell features. Oncotarget. 2016;7(26):40297–313. doi: 10.18632/oncotarget.9610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zou ZW, Ma C, Medoro L, Chen L, Wang B, Gupta R, et al. LncRNA ANRIL is up-regulated in nasopharyngeal carcinoma and promotes the cancer progression via increasing proliferation, reprograming cell glucose metabolism and inducing side-population stem-like cancer cells. Oncotarget. 2016. doi:10.18632/oncotarget.11437. [DOI] [PMC free article] [PubMed]
- 234.Hamabe A, Konno M, Tanuma N, Shima H, Tsunekuni K, Kawamoto K, et al. Role of pyruvate kinase M2 in transcriptional regulation leading to epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2014;111(43):15526–31. doi: 10.1073/pnas.1407717111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. 2011;71(22):6921–5. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 236.Phannasil P, Thuwajit C, Warnnissorn M, Wallace JC, MacDonald MJ, Jitrapakdee S. Pyruvate Carboxylase Is Up-Regulated in Breast Cancer and Essential to Support Growth and Invasion of MDA-MB-231 Cells. PLoS One. 2015;10(6):e0129848. doi: 10.1371/journal.pone.0129848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Jiang F, Ma S, Xue Y, Hou J, Zhang Y. LDH-A promotes malignant progression via activation of epithelial-to-mesenchymal transition and conferring stemness in muscle-invasive bladder cancer. Biochem Biophys Res Commun. 2016;469(4):985–92. doi: 10.1016/j.bbrc.2015.12.078. [DOI] [PubMed] [Google Scholar]
- 238.Gonzalez-Guerrico AM, Espinoza I, Schroeder B, Park CH, Kvp CM, Khurana A, et al. Suppression of endogenous lipogenesis induces reversion of the malignant phenotype and normalized differentiation in breast cancer. Oncotarget. 2016. doi:10.18632/oncotarget.9463. [DOI] [PMC free article] [PubMed]
- 239.Yang L, Zhang F, Wang X, Tsai Y, Chuang KH, Keng PC, et al. A FASN-TGF-beta1-FASN regulatory loop contributes to high EMT/metastatic potential of cisplatin-resistant non-small cell lung cancer. Oncotarget. 2016:55543-54. doi:10.18632/oncotarget.10837 [DOI] [PMC free article] [PubMed]
- 240.Lin CC, Cheng TL, Tsai WH, Tsai HJ, Hu KH, Chang HC, et al. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci Rep. 2012;2:785. doi: 10.1038/srep00785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Aspuria PJ, Lunt SY, Varemo L, Vergnes L, Gozo M, Beach JA, et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014;2:21. doi: 10.1186/2049-3002-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zhong J, Rajaram N, Brizel DM, Frees AE, Ramanujam N, Batinic-Haberle I, et al. Radiation induces aerobic glycolysis through reactive oxygen species. Radiother Oncol. 2013;106(3):390–6. doi: 10.1016/j.radonc.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Liao EC, Hsu YT, Chuah QY, Lee YJ, Hu JY, Huang TC, et al. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014;5:e1255. doi: 10.1038/cddis.2014.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Dittmann K, Mayer C, Paasch A, Huber S, Fehrenbacher B, Schaller M, et al. Nuclear EGFR renders cells radio-resistant by binding mRNA species and triggering a metabolic switch to increase lactate production. Radiother Oncol. 2015;116(3):431–7. doi: 10.1016/j.radonc.2015.08.016. [DOI] [PubMed] [Google Scholar]
- 245.Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW, et al. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res. 2015;183(3):291–304. doi: 10.1667/RR13828.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Wang M, Keogh A, Treves S, Idle JR, Beyoglu D. The metabolomic profile of gamma-irradiated human hepatoma and muscle cells reveals metabolic changes consistent with the Warburg effect. PeerJ. 2016;4:e1624. doi: 10.7717/peerj.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Judge JL, Owens KM, Pollock SJ, Woeller CF, Thatcher TH, Williams JP, et al. Ionizing radiation induces myofibroblast differentiation via lactate dehydrogenase. Am J Physiol Lung Cell Mol Physiol. 2015;309(8):L879–87. doi: 10.1152/ajplung.00153.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kamenisch Y, Baban TS, Schuller W, von Thaler AK, Sinnberg T, Metzler G, et al. UVA-irradiation induces melanoma invasion via enhanced Warburg effect. J Invest Dermatol. 2016. doi:10.1016/j.jid.2016.02.815 [DOI] [PubMed]
- 249.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501(7467):346–54. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 250.Barcellos-Hoff MH, Park C, Wright EG. Radiation and the microenvironment - tumorigenesis and therapy. Nat Rev Cancer. 2005;5(11):867–75. doi: 10.1038/nrc1735. [DOI] [PubMed] [Google Scholar]
- 251.Kuonen F, Secondini C, Ruegg C. Molecular pathways: emerging pathways mediating growth, invasion, and metastasis of tumors progressing in an irradiated microenvironment. Clin Cancer Res. 2012;18(19):5196–202. doi: 10.1158/1078-0432.CCR-11-1758. [DOI] [PubMed] [Google Scholar]
- 252.Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15(7):409–25. doi: 10.1038/nrc3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Leroi N, Lallemand F, Coucke P, Noel A, Martinive P. Impacts of Ionizing Radiation on the Different Compartments of the Tumor Microenvironment. Front Pharmacol. 2016;7:78. doi: 10.3389/fphar.2016.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, et al. Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther. 2015;147:22–31. doi: 10.1016/j.pharmthera.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 256.Yarnold J, Brotons MC. Pathogenetic mechanisms in radiation fibrosis. Radiother Oncol. 2010;97(1):149–61. doi: 10.1016/j.radonc.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 257.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pistoia V, Morandi F, Bianchi G, Pezzolo A, Prigione I, Raffaghello L. Immunosuppressive microenvironment in neuroblastoma. Front Oncol. 2013;3:167. doi: 10.3389/fonc.2013.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Butt AQ, Mills KH. Immunosuppressive networks and checkpoints controlling antitumor immunity and their blockade in the development of cancer immunotherapeutics and vaccines. Oncogene. 2014;33(38):4623–31. doi: 10.1038/onc.2013.432. [DOI] [PubMed] [Google Scholar]
- 260.Schaue D, McBride WH. Links between innate immunity and normal tissue radiobiology. Radiat Res. 2010;173(4):406–17. doi: 10.1667/RR1931.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kalbasi A, June CH, Haas N, Vapiwala N. Radiation and immunotherapy: a synergistic combination. J Clin Invest. 2013;123(7):2756–63. doi: 10.1172/JCI69219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 263.Cheng JC, Chou CH, Kuo ML, Hsieh CY. Radiation-enhanced hepatocellular carcinoma cell invasion with MMP-9 expression through PI3K/Akt/NF-kappaB signal transduction pathway. Oncogene. 2006;25(53):7009–18. doi: 10.1038/sj.onc.1209706. [DOI] [PubMed] [Google Scholar]
- 264.Asuthkar S, Nalla AK, Gondi CS, Dinh DH, Gujrati M, Mohanam S, et al. Gadd45a sensitizes medulloblastoma cells to irradiation and suppresses MMP-9-mediated EMT. Neuro Oncol. 2011;13(10):1059–73. doi: 10.1093/neuonc/nor109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Chou CH, Teng CM, Tzen KY, Chang YC, Chen JH, Cheng JC. MMP-9 from sublethally irradiated tumor promotes Lewis lung carcinoma cell invasiveness and pulmonary metastasis. Oncogene. 2012;31(4):458–68. doi: 10.1038/onc.2011.240. [DOI] [PubMed] [Google Scholar]
- 266.Yue H, Hu K, Liu W, Jiang J, Chen Y, Wang R. Role of matrix metalloproteinases in radiation-induced lung injury in alveolar epithelial cells of Bama minipigs. Exp Ther Med. 2015;10(4):1437–44. doi: 10.3892/etm.2015.2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Wang J, Boerma M, Fu Q, Hauer-Jensen M. Significance of endothelial dysfunction in the pathogenesis of early and delayed radiation enteropathy. World J Gastroenterol. 2007;13(22):3047–55. doi: 10.3748/wjg.v13.i22.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120(3):694–705. doi: 10.1172/JCI40283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Kunz M, Ibrahim SM. Molecular responses to hypoxia in tumor cells. Mol Cancer. 2003;2:23. doi: 10.1186/1476-4598-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Shi M, Guo XT, Shu MG, Chen FL, Li LW. Cell-permeable hypoxia-inducible factor-1 (HIF-1) antagonists function as tumor radiosensitizers. Med Hypotheses. 2007;69(1):33–5. doi: 10.1016/j.mehy.2006.10.062. [DOI] [PubMed] [Google Scholar]
- 271.Miyasaka A, Oda K, Ikeda Y, Sone K, Fukuda T, Inaba K, et al. PI3K/mTOR pathway inhibition overcomes radioresistance via suppression of the HIF1-alpha/VEGF pathway in endometrial cancer. Gynecol Oncol. 2015;138(1):174–80. doi: 10.1016/j.ygyno.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 272.Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11(4):239–53. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- 273.Dave B, Mittal V, Tan NM, Chang JC. Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res. 2012;14(1):202. doi: 10.1186/bcr2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Marie-Egyptienne DT, Lohse I, Hill RP. Cancer stem cells, the epithelial to mesenchymal transition (EMT) and radioresistance: potential role of hypoxia. Cancer Lett. 2013;341(1):63–72. doi: 10.1016/j.canlet.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 275.Chang L, Graham PH, Hao J, Bucci J, Cozzi PJ, Kearsley JH, et al. Emerging roles of radioresistance in prostate cancer metastasis and radiation therapy. Cancer Metastasis Rev. 2014;33(2-3):469–96. doi: 10.1007/s10555-014-9493-5. [DOI] [PubMed] [Google Scholar]
- 276.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Gomez-Casal R, Epperly MW, Wang H, Proia DA, Greenberger JS, Levina V. Radioresistant human lung adenocarcinoma cells that survived multiple fractions of ionizing radiation are sensitive to HSP90 inhibition. Oncotarget. 2015;6(42):44306–22. doi: 10.18632/oncotarget.6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Escriva M, Peiro S, Herranz N, Villagrasa P, Dave N, Montserrat-Sentis B, et al. Repression of PTEN phosphatase by Snail1 transcriptional factor during gamma radiation-induced apoptosis. Mol Cell Biol. 2008;28(5):1528–40. doi: 10.1128/MCB.02061-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY, et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27(9):2059–68. doi: 10.1002/stem.154. [DOI] [PubMed] [Google Scholar]
- 280.Inoue A, Seidel MG, Wu W, Kamizono S, Ferrando AA, Bronson RT, et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell. 2002;2(4):279–88. doi: 10.1016/S1535-6108(02)00155-1. [DOI] [PubMed] [Google Scholar]
- 281.Wu WS, Heinrichs S, Xu D, Garrison SP, Zambetti GP, Adams JM, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123(4):641–53. doi: 10.1016/j.cell.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 282.Jin C, Yan B, Lu Q, Lin Y, Ma L. The role of MALAT1/miR-1/slug axis on radioresistance in nasopharyngeal carcinoma. Tumour Biol. 2016;37(3):4025–33. doi: 10.1007/s13277-015-4227-z. [DOI] [PubMed] [Google Scholar]
- 283.Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16(9):864–75. doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Lu Y, Li T, Wei G, Liu L, Chen Q, Xu L, et al. The long non-coding RNA NEAT1 regulates epithelial to mesenchymal transition and radioresistance in through miR-204/ZEB1 axis in nasopharyngeal carcinoma. Tumour Biol. 2016. doi:10.1007/s13277-015-4773-4 [DOI] [PubMed]
- 285.Sayan AE, Griffiths TR, Pal R, Browne GJ, Ruddick A, Yagci T, et al. SIP1 protein protects cells from DNA damage-induced apoptosis and has independent prognostic value in bladder cancer. Proc Natl Acad Sci U S A. 2009;106(35):14884–9. doi: 10.1073/pnas.0902042106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Zhang P, Wang L, Rodriguez-Aguayo C, Yuan Y, Debeb BG, Chen D, et al. miR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat Commun. 2014;5:5671. doi: 10.1038/ncomms6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bouquet F, Pal A, Pilones KA, Demaria S, Hann B, Akhurst RJ, et al. TGFbeta1 inhibition increases the radiosensitivity of breast cancer cells in vitro and promotes tumor control by radiation in vivo. Clin Cancer Res. 2011;17(21):6754–65. doi: 10.1158/1078-0432.CCR-11-0544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Hardee ME, Marciscano AE, Medina-Ramirez CM, Zagzag D, Narayana A, Lonning SM, et al. Resistance of glioblastoma-initiating cells to radiation mediated by the tumor microenvironment can be abolished by inhibiting transforming growth factor-beta. Cancer Res. 2012;72(16):4119–29. doi: 10.1158/0008-5472.CAN-12-0546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Zakharchenko O, Cojoc M, Dubrovska A, Souchelnytskyi S. A role of TGFss1 dependent 14-3-3sigma phosphorylation at Ser69 and Ser74 in the regulation of gene transcription, stemness and radioresistance. PLoS One. 2013;8(5):e65163. doi: 10.1371/journal.pone.0065163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Kim MR, Lee J, An YS, Jin YB, Park IC, Chung E, et al. TGFbeta1 protects cells from gamma-IR by enhancing the activity of the NHEJ repair pathway. Mol Cancer Res. 2015;13(2):319–29. doi: 10.1158/1541-7786.MCR-14-0098-T. [DOI] [PubMed] [Google Scholar]
- 291.Zhao L, Ji W, Zhang L, Ou G, Feng Q, Zhou Z, et al. Changes of circulating transforming growth factor-beta1 level during radiation therapy are correlated with the prognosis of locally advanced non-small cell lung cancer. J Thorac Oncol. 2010;5(4):521–5. doi: 10.1097/JTO.0b013e3181cbf761. [DOI] [PubMed] [Google Scholar]
- 292.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104(2):618–23. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, et al. Wnt/beta-catenin mediates radiation resistance of Sca1+ progenitors in an immortalized mammary gland cell line. J Cell Sci. 2007;120(Pt 3):468–77. doi: 10.1242/jcs.03348. [DOI] [PubMed] [Google Scholar]
- 294.Zhang Y, Liu B, Zhao Q, Hou T, Huang X. Nuclear localizaiton of beta-catenin is associated with poor survival and chemo-/radioresistance in human cervical squamous cell cancer. Int J Clin Exp Pathol. 2014;7(7):3908–17. [PMC free article] [PubMed] [Google Scholar]
- 295.Cojoc M, Peitzsch C, Kurth I, Trautmann F, Kunz-Schughart LA, Telegeev GD, et al. Aldehyde Dehydrogenase Is Regulated by beta-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015;75(7):1482–94. doi: 10.1158/0008-5472.CAN-14-1924. [DOI] [PubMed] [Google Scholar]
- 296.Jun S, Jung YS, Suh HN, Wang W, Kim MJ, Oh YS, et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat Commun. 2016;7:10994. doi: 10.1038/ncomms10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28(1):17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Toulany M, Dittmann K, Kruger M, Baumann M, Rodemann HP. Radioresistance of K-Ras mutated human tumor cells is mediated through EGFR-dependent activation of PI3K-AKT pathway. Radiother Oncol. 2005;76(2):143–50. doi: 10.1016/j.radonc.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 299.Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013;4:e875. doi: 10.1038/cddis.2013.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Chang L, Graham PH, Ni J, Hao J, Bucci J, Cozzi PJ, et al. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit Rev Oncol Hematol. 2015;96(3):507–17. doi: 10.1016/j.critrevonc.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 301.Jiang X, Wang J, Zhang K, Tang S, Ren C, Chen Y. The role of CD29-ILK-Akt signaling-mediated epithelial-mesenchymal transition of liver epithelial cells and chemoresistance and radioresistance in hepatocellular carcinoma cells. Med Oncol. 2015;32(5):141. doi: 10.1007/s12032-015-0595-x. [DOI] [PubMed] [Google Scholar]
- 302.Gan GN, Eagles J, Keysar SB, Wang G, Glogowska MJ, Altunbas C, et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014;74(23):7024–36. doi: 10.1158/0008-5472.CAN-14-1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.de Jong MC, Pramana J, van der Wal JE, Lacko M, Peutz-Kootstra CJ, de Jong JM, et al. CD44 expression predicts local recurrence after radiotherapy in larynx cancer. Clin Cancer Res. 2010;16(21):5329–38. doi: 10.1158/1078-0432.CCR-10-0799. [DOI] [PubMed] [Google Scholar]
- 304.Shien K, Toyooka S, Ichimura K, Soh J, Furukawa M, Maki Y, et al. Prognostic impact of cancer stem cell-related markers in non-small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer. 2012;77(1):162–7. doi: 10.1016/j.lungcan.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 305.Mannino M, Chalmers AJ. Radioresistance of glioma stem cells: intrinsic characteristic or property of the 'microenvironment-stem cell unit'? Mol Oncol. 2011;5(4):374–86. doi: 10.1016/j.molonc.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Ni J, Cozzi PJ, Hao JL, Beretov J, Chang L, Duan W, et al. CD44 variant 6 is associated with prostate cancer metastasis and chemo-/radioresistance. Prostate. 2014;74(6):602–17. doi: 10.1002/pros.22775. [DOI] [PubMed] [Google Scholar]
- 307.Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB, et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013;73(3):1219–31. doi: 10.1158/0008-5472.CAN-12-1408. [DOI] [PubMed] [Google Scholar]
- 308.Desai A, Webb B, Gerson SL. CD133+ cells contribute to radioresistance via altered regulation of DNA repair genes in human lung cancer cells. Radiother Oncol. 2014;110(3):538–45. doi: 10.1016/j.radonc.2013.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Gemenetzidis E, Gammon L, Biddle A, Emich H, Mackenzie IC. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget. 2015;6(41):43964–77. doi: 10.18632/oncotarget.6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Chen Y, Zhang F, Tsai Y, Yang X, Yang L, Duan S, et al. IL-6 signaling promotes DNA repair and prevents apoptosis in CD133+ stem-like cells of lung cancer after radiation. Radiat Oncol. 2015;10:227. doi: 10.1186/s13014-015-0534-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 311.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 312.Voulgaridou GP, Kiziridou M, Mantso T, Chlichlia K, Galanis A, Koukourakis MI, et al. Aldehyde dehydrogenase 3A1 promotes multi-modality resistance and alters gene expression profile in human breast adenocarcinoma MCF-7 cells. Int J Biochem Cell Biol. 2016. doi:10.1016/j.biocel.2016.06.004 [DOI] [PubMed]
- 313.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Piao LS, Hur W, Kim TK, Hong SW, Kim SW, Choi JE, et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012;315(2):129–37. doi: 10.1016/j.canlet.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 315.Lomonaco SL, Finniss S, Xiang C, Decarvalho A, Umansky F, Kalkanis SN, et al. The induction of autophagy by gamma-radiation contributes to the radioresistance of glioma stem cells. Int J Cancer. 2009;125(3):717–22. doi: 10.1002/ijc.24402. [DOI] [PubMed] [Google Scholar]
- 316.Marchiq I, Pouyssegur J. Hypoxia, cancer metabolism and the therapeutic benefit of targeting lactate/H(+) symporters. J Mol Med (Berl) 2016;94(2):155–71. doi: 10.1007/s00109-015-1307-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Xu H, He W, Jiang HG, Zhao H, Peng XH, Wei YH, et al. Prognostic value of mitochondrial DNA content and G10398A polymorphism in non-small cell lung cancer. Oncol Rep. 2013;30(6):3006–12. doi: 10.3892/or.2013.2783. [DOI] [PubMed] [Google Scholar]
- 318.Tian WN, Braunstein LD, Apse K, Pang J, Rose M, Tian X, et al. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am J Physiol. 1999;276(5 Pt 1):C1121–31. doi: 10.1152/ajpcell.1999.276.5.C1121. [DOI] [PubMed] [Google Scholar]
- 319.Boros LG, Torday JS, Lim S, Bassilian S, Cascante M, Lee WN. Transforming growth factor beta2 promotes glucose carbon incorporation into nucleic acid ribose through the nonoxidative pentose cycle in lung epithelial carcinoma cells. Cancer Res. 2000;60(5):1183–5. [PubMed] [Google Scholar]
- 320.Sattler UG, Meyer SS, Quennet V, Hoerner C, Knoerzer H, Fabian C, et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol. 2010;94(1):102–9. doi: 10.1016/j.radonc.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 321.Bhatt AN, Chauhan A, Khanna S, Rai Y, Singh S, Soni R, et al. Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer. 2015;15:335. doi: 10.1186/s12885-015-1368-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Kwon T, Youn H, Son B, Kim D, Seong KM, Park S, et al. DANGER is involved in high glucose-induced radioresistance through inhibiting DAPK-mediated anoikis in non-small cell lung cancer. Oncotarget. 2016;7(6):7193–206. doi: 10.18632/oncotarget.6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Zhai X, Yang Y, Wan J, Zhu R, Wu Y. Inhibition of LDH-A by oxamate induces G2/M arrest, apoptosis and increases radiosensitivity in nasopharyngeal carcinoma cells. Oncol Rep. 2013;30(6):2983–91. doi: 10.3892/or.2013.2735. [DOI] [PubMed] [Google Scholar]
- 324.Meng MB, Wang HH, Guo WH, Wu ZQ, Zeng XL, Zaorsky NG, et al. Targeting pyruvate kinase M2 contributes to radiosensitivity of non-small cell lung cancer cells in vitro and in vivo. Cancer Lett. 2015;356(2 Pt B):985–93. doi: 10.1016/j.canlet.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 325.Bola BM, Chadwick AL, Michopoulos F, Blount KG, Telfer BA, Williams KJ, et al. Inhibition of monocarboxylate transporter-1 (MCT1) by AZD3965 enhances radiosensitivity by reducing lactate transport. Mol Cancer Ther. 2014;13(12):2805–16. doi: 10.1158/1535-7163.MCT-13-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Zhao F, Ming J, Zhou Y, Fan L. Inhibition of Glut1 by WZB117 sensitizes radioresistant breast cancer cells to irradiation. Cancer Chemother Pharmacol. 2016;77(5):963–72. doi: 10.1007/s00280-016-3007-9. [DOI] [PubMed] [Google Scholar]
- 327.Allen KT, Chin-Sinex H, DeLuca T, Pomerening JR, Sherer J, Watkins JB, 3rd, et al. Dichloroacetate alters Warburg metabolism, inhibits cell growth, and increases the X-ray sensitivity of human A549 and H1299 NSC lung cancer cells. Free Radic Biol Med. 2015;89:263–73. doi: 10.1016/j.freeradbiomed.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 328.Goetze K, Fabian CG, Siebers A, Binz L, Faber D, Indraccolo S, et al. Manipulation of tumor metabolism for therapeutic approaches: ovarian cancer-derived cell lines as a model system. Cell Oncol (Dordr) 2015;38(5):377–85. doi: 10.1007/s13402-015-0237-5. [DOI] [PubMed] [Google Scholar]
- 329.Sappington DR, Siegel ER, Hiatt G, Desai A, Penney RB, Jamshidi-Parsian A, et al. Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim Biophys Acta. 2016;1860(4):836–43. doi: 10.1016/j.bbagen.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Aktipis CA, Maley CC, Pepper JW. Dispersal evolution in neoplasms: the role of disregulated metabolism in the evolution of cell motility. Cancer Prev Res (Phila) 2012;5(2):266–75. doi: 10.1158/1940-6207.CAPR-11-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Zhang M, Lahn M, Huber PE. Translating the combination of TGFbeta blockade and radiotherapy into clinical development in glioblastoma. Oncoimmunology. 2012;1(6):943–5. doi: 10.4161/onci.19789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Calone I, Souchelnytskyi S. Inhibition of TGFbeta signaling and its implications in anticancer treatments. Exp Oncol. 2012;34(1):9–16. [PubMed] [Google Scholar]
- 333.Zhang M, Herion TW, Timke C, Han N, Hauser K, Weber KJ, et al. Trimodal glioblastoma treatment consisting of concurrent radiotherapy, temozolomide, and the novel TGF-beta receptor I kinase inhibitor LY2109761. Neoplasia. 2011;13(6):537–49. doi: 10.1593/neo.11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Abegglen LM, Caulin AF, Chan A, Lee K, Robinson R, Campbell MS, et al. Potential Mechanisms for Cancer Resistance in Elephants and Comparative Cellular Response to DNA Damage in Humans. JAMA. 2015;314(17):1850–60. doi: 10.1001/jama.2015.13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26(12):1268–86. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Lee CL, Castle KD, Moding EJ, Blum JM, Williams N, Luo L, et al. Acute DNA damage activates the tumour suppressor p53 to promote radiation-induced lymphoma. Nat Commun. 2015;6:8477. doi: 10.1038/ncomms9477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Mitra A, Mishra L, Li S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget. 2015;6(13):10697–711. doi: 10.18632/oncotarget.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13(4-5):109–18. doi: 10.1016/j.drup.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Blagosklonny MV. Oncogenic resistance to growth-limiting conditions. Nat Rev Cancer. 2002;2(3):221–5. doi: 10.1038/nrc743. [DOI] [PubMed] [Google Scholar]
- 340.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, et al. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006;12(14 Pt 1):4147–53. doi: 10.1158/1078-0432.CCR-06-0038. [DOI] [PubMed] [Google Scholar]
- 341.Wang H, Zhang G, Zhang H, Zhang F, Zhou B, Ning F, et al. Acquisition of epithelial-mesenchymal transition phenotype and cancer stem cell-like properties in cisplatin-resistant lung cancer cells through AKT/beta-catenin/Snail signaling pathway. Eur J Pharmacol. 2014;723:156–66. doi: 10.1016/j.ejphar.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 342.Latifi A, Abubaker K, Castrechini N, Ward AC, Liongue C, Dobill F, et al. Cisplatin treatment of primary and metastatic epithelial ovarian carcinomas generates residual cells with mesenchymal stem cell-like profile. J Cell Biochem. 2011;112(10):2850–64. doi: 10.1002/jcb.23199. [DOI] [PubMed] [Google Scholar]
- 343.Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532. doi: 10.1038/cddis.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Hirschey MD, DeBerardinis RJ, Diehl AM, Drew JE, Frezza C, Green MF, et al. Dysregulated metabolism contributes to oncogenesis. Semin Cancer Biol. 2015;35(Suppl):S129–50. doi: 10.1016/j.semcancer.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer. 2010;9:33. doi: 10.1186/1476-4598-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Peng X, Gong F, Chen Y, Jiang Y, Liu J, Yu M, et al. Autophagy promotes paclitaxel resistance of cervical cancer cells: involvement of Warburg effect activated hypoxia-induced factor 1-alpha-mediated signaling. Cell Death Dis. 2014;5:e1367. doi: 10.1038/cddis.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Hua G, Liu Y, Li X, Xu P, Luo Y. Targeting glucose metabolism in chondrosarcoma cells enhances the sensitivity to doxorubicin through the inhibition of lactate dehydrogenase-A. Oncol Rep. 2014;31(6):2727–34. doi: 10.3892/or.2014.3156. [DOI] [PubMed] [Google Scholar]
- 348.Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30(9):1002–19. doi: 10.1101/gad.279737.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Allan JM, Travis LB. Mechanisms of therapy-related carcinogenesis. Nat Rev Cancer. 2005;5(12):943–55. doi: 10.1038/nrc1749. [DOI] [PubMed] [Google Scholar]
- 350.Peiris-Pages M, Sotgia F, Lisanti MP. Chemotherapy induces the cancer-associated fibroblast phenotype, activating paracrine Hedgehog-GLI signalling in breast cancer cells. Oncotarget. 2015;6(13):10728–45. doi: 10.18632/oncotarget.3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Denise C, Paoli P, Calvani M, Taddei ML, Giannoni E, Kopetz S, et al. 5-fluorouracil resistant colon cancer cells are addicted to OXPHOS to survive and enhance stem-like traits. Oncotarget. 2015;6(39):41706–21. doi: 10.18632/oncotarget.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.