

Abstract
Pseudomonas aeruginosa has two genes encoding ferredoxin NADP(+) reductases, denoted fprA and fprB. We show here that P. aeruginosa fprA is an essential gene. However, the ΔfprA mutant could only be successfully constructed in PAO1 strains containing an extra copy of fprA on a mini-Tn7 vector integrated into the chromosome or carrying it on a temperature-sensitive plasmid. The strain containing an extra copy of the ferredoxin gene (fdx1) could suppress the essentiality of FprA. Other ferredoxin genes could not suppress the requirement for FprA, suggesting that Fdx1 mediates the essentiality of FprA. The expression of fprA was highly induced in response to treatments with a superoxide generator, paraquat, or sodium hypochlorite (NaOCl). The induction of fprA by these treatments depended on FinR, a LysR-family transcription regulator. In vivo and in vitro analysis suggested that oxidized FinR acted as a transcriptional activator of fprA expression by binding to its regulatory box, located 20 bases upstream of the fprA -35 promoter motif. This location of the FinR box also placed it between the -35 and -10 motifs of the finR promoter, where the reduced regulator functions as a repressor. Under uninduced conditions, binding of FinR repressed its own transcription but had no effect on fprA expression. Exposure to paraquat or NaOCl converted FinR to a transcriptional activator, leading to the expression of both fprA and finR. The ΔfinR mutant showed an increased paraquat sensitivity phenotype and attenuated virulence in the Drosophila melanogaster host model. These phenotypes could be complemented by high expression of fprA, indicating that the observed phenotypes of the ΔfinR mutant arose from the inability to up-regulate fprA expression. In addition, increased expression of fprB was unable to rescue essentiality of fprA or the superoxide-sensitive phenotype of the ΔfinR mutant, suggesting distinct mechanisms of the FprA and FprB enzymes.
Introduction
Pseudomonas aeruginosa is one of the most common opportunistic bacterial pathogens that cause deadly infections in patients with impaired immune systems or in critical condition. Nosocomial infections caused by P. aeruginosa are increasing worldwide [1, 2]. The ability of a pathogen to overwhelmingly invade the host is often associated with its ability to rapidly adapt and evade or overcome host immune systems. During pathogen-host interactions, several transcriptional regulators are differentially expressed to fine-tune gene expression networks required for adaptive responses to host-generated stresses [3]. Reactive oxygen species (ROS) are key components of host innate immune responses generated within the phagolysosomes of phagocytes to attack invading microbes. Additionally, normal aerobic metabolism produces ROS as by-products [4]. Consequently, bacteria have evolved mechanisms to protect themselves from oxidative stress. An array of either antioxidant enzymes, such as catalases, superoxide dismutases, and thiol peroxidases or antioxidant molecules, such as glutathione and vitamins are involved in removal of ROS. In addition, there are extensive repaired and rebuilding systems for oxidatively damaged molecules, such as iron-sulfur cluster biosynthesis (Isc), DNA repair (the Mut systems) and proteins repair (methionine sulfoxide reductases, Msr). These systems are necessary for bacterial survivals under stressful conditions [5, 6]. Various transcriptional regulators are involved in coordinating the complex processes of sensing and responding to stresses. The LysR-type transcriptional regulators (LTTRs) represents the most abundant type of transcriptional regulator with an N-terminal DNA-binding helix-turn-helix motif and a C-terminal co-inducer-binding domain as conserved structures. LTTRs exhibit a negative autoregulation and regulate a diverse set of genes, including those involved in virulence, metabolism, quorum sensing and motility [7–15]. A major regulator in hydrogen peroxide (H2O2) defense is OxyR, a LysR-type transcriptional regulator, while the transcription factor SoxR triggers a global stress response against superoxide anions as well as redox cycling drugs [16–19]. Many proteobacterial genomes contain another LysR-type oxidative stress sensing transcriptional regulator, FinR, which is located next to fprA (ferredoxin NADP+ reductase, Fpr), an enzyme catalyzing the reversible electron-transferring reaction between NADPH and one-electron carriers such as ferredoxin or flavodoxin. The enzymes are important in maintain NADP(+)/NADPH ratio. Fpr also catalyzes the irreversible electron transfer in diaphorase reaction which drives the oxidation of NADPH in a wide variety of adventitious electron acceptors [20]. In bactria, Fpr has been shown to control the ratio of NADP(+)/NADPH. Fpr participates in many cellular processes, including iron acquisition, iron-sulfur cluster biogenesis and oxidative stress defense [21, 22]. FinR is required for the induction of fprA expression upon exposure to superoxide anion stress generated by paraquat [21, 23–25]. However, Escherichia coli fpr is a member of the SoxRS regulon, and inactivation of fpr increases sensitivity to paraquat [26, 27].
Pseudomonas putida KT2440 contains at least two genes encoding Fpr, namely fprA and fprB [23, 28]. The fprA mutant confers high sensitivity to oxidative and osmotic stresses, while the fprB mutant is susceptible only to high osmotic conditions [23, 28]. Like P. putida, P. aeruginosa PAO1 possesses both fprA and fprB. Two types of Fprs have their preferred electron transport and redox partners. FprA achieves higher catalytic efficiency when flavodoxin is its redox partner. FprB is important in defenses against multiple stresses including metal, oxidative, and osmotic stresses and is required for the full function of iron-sulfur cluster ([Fe-S])-containing enzymes via its redox partner, Fdx2, involving in the ISC [Fe-S] biogenesis system [29]. For example, in an iron storage complex, the [Fe-S] cluster of bacterioferritin-associated ferredoxin (Bdf) transfers electrons to the heme in bacterioferritin (BfrB) and promotes the release of Fe2+ from BfrB by mediating electrons from FprA to BfrB [30]. Moreover, roles for FprA in sulfate assimilation and siderophore biosynthesis in pseudomonads have been characterized [31]. The expression of fprB could be induced by exposure to oxidative stress in an [Fe-S] biogenesis regulator IscR-dependent manner [29]. The physiological function of P. aeruginosa FprA remains mysterious due to unsuccessful construction of the fprA mutant [31, 32]. This observation raised the possibility that the activity of FprA is essential for bacterial viability. In this communication, P. aeruginosa fprA was shown to be essential and was determined to be regulated by FinR.
Results and discussion
fprA is an essential gene in P. aeruginosa
P. aeruginosa PAO1 has two annotated fpr genes, fprA (PA3397) and fprB [33]. An open reading frame located next to fprA in the opposite orientation was annotated as a putative LysR-type transcriptional regulator, FinR (PA3398). P. aeruginosa FinR shares 80.8% and 80.5% amino acid sequence identity with FinR from P. putida and Azotobacter vinelandii, respectively (S1 Fig). Several attempts to construct the fprA mutant in pseudomonads have been met with mixed results. No mutants were obtained in P. aeruginosa, but a mutant was constructed in P. putida [31, 32]. These observations suggest the essentiality of fprA in PAO1. We made several unsuccessful attempts to construct either insertion inactivation or deletion fprA mutants. Hence, the notion of the essentiality of fprA was tested. A new PAO1 parental strain was constructed that had an extra copy of fprA cloned into a mini-Tn7 vector [34], and the recombinant Tn7T-fprA transposed into the PAO1 chromosome attTn7 site, giving PAO1::Tn7T-fprA. The antibiotic marker of the mini-Tn7 vector was removed by the Flp-FRT system [35]. fprA gene deletion by allelic exchange was made by electroporating pUCΔfprA::Gmr (Table 1) into PAO1::Tn7T-fprA and selecting for gentamicin resistance (Gmr). Several Gmr and carbenicillin susceptible (Cbs) colonies were screened by PCR and found to have deleted the functional copy of the chromosomal fprA gene. The ΔfprA mutant (ΔfprA::Tn7T-fprA) was successfully constructed. In the control strain, which contains only the mini-Tn7 vector (PAO1::Tn7T), no Gmr transformants or fprA mutants were recovered (Table 1). The results support the notion that fprA is an essential gene in PAO1.
Table 1. Efficiency of fprA deletion in P. aeruginosa strains carrying an extra copy of various genes.
| P. aeruginosa strains | Efficiency of fprA deletiona |
|---|---|
| PAO1::Tn7T | ND |
| PAO1::Tn7T-finR | ND |
| PAO1::Tn7T-fprA | 1.6 × 102 |
| PAO1::Tn7T-fprB | ND |
| PAO1::Tn7T-fdx1 | 2.1 × 101 |
| PAO1::Tn7T-fdx2 | ND |
| PAO1::Tn7T-rnfB | ND |
| PAO1::Tn7T-fdxA | ND |
a Indicated strains of PAO1 were transformed with 1 μg pΔfprA::Gmr plasmid using electroporation. The transformants with fprA deletion were selected by the Gmr and Cbs phenotypes. The efficiency of fprA deletion is defined as the number of ΔfprA mutant obtained per 1 μg pUCΔfprA::Gmr plasmid. The data shown are means from triple independent experiments. ND, not detectable.
An independent approach was conducted to assure the essentiality of fprA in P. aeruginosa using a plasmid vector with a temperature-sensitive origin of replication that has been recently developed in P. aeruginosa [36]. We firstly constructed a temperature-sensitive replication plasmid by cloning the temperature-sensitive replicon mSFts1 from pSS255 [36] into the broad-host-range vector pBBR1MCS-4 [37], yielding pTS. The plasmid can be maintained at 30°C but not at the non-permissive temperature of 45°C. The full-length fprA was cloned into the plasmid pTS, generating pTS-fprA. Transformants harboring pTS-fprA were grown and maintained at 30°C. Growing bacterial cultures at the non-permissive temperature of 45°C resulted in the loss of pTS-fprA. pUCΔfprA::Gmr was introduced into PAO1 harboring pTS-fprA, and Gmr transformants were selected and screened for double crossing over and marker exchange events, giving ΔfprA::Gmr/pTS-fprA. The ΔfprA::Gmr/pTS-fprA mutant strain had normal growth at 30°C. This mutant strain could not grow on either an agar plate or in LB broth medium when the incubation temperature was shifted to the non-permissive temperature of 45°C for pTS-fprA, indicating the essentiality of fprA (Fig 1). The results confirmed that fprA is an essential gene that is required for the growth of PAO1. Although P. aeruginosa FprA shares the greatest amino acid sequence identity with FrpA from P. putida and A. vinelandii (S1 Fig), there is no evidence suggesting that it is essential in these two bacteria [31, 38].
Fig 1. fprA is an essential gene in P. aeruginosa.

The viability of exponential-phase cultures of P. aeruginosa PAO1 and ΔfprA mutant strains harboring an extra copy of fprA or fdx1 was determined using viable cell count on LB agar plates incubated at either 30°C or 45°C. The viability is expressed as a percentage of the CFU of the tested strain over the CFU of the PAO1::Tn7T or PAO1/pTS control.
Since PAO1 has both fprA and fprB, we tested the potential cross-functional complementation between fprB and fprA. Similar approaches that were successfully used to construct the ΔfprA mutant were applied to test cross-complementation between fprA and fprB. A PAO1::Tn7T-fprB strain carrying an extra copy of fprB was used for ΔfprA mutant construction using pUCΔfprA::Gmr. After several attempts, no fprA mutant was obtained. This indicated the essential function of fprA for bacterial growth and showed that expression of fprB could not complement fprA. This suggests that FprA and FprB have different biochemical and physiological functions. Fpr have essential functions in maintenance of the NAD(P)/NAD(P)H ratio via their reactions with ferredoxins and flavodoxins. In the fprA mutant, alterations in the ratio of reduced/ oxidized ferredoxins could contribute to the mutant lethality under tested conditions.
The requirement for fprA could be complemented by fdx1 expression
Fpr catalyzes reversible electron transfer between NADPH and electron carriers such as ferredoxins (Fdx), thereby maintaining a balance between NADPH and reduced Fdx pools. Since Fpr is important in maintaining reduced Fdx, we determined whether the expression of fdx genes could suppress the essentiality of fprA. PAO1 has several genes encoding Fdx of different families, e.g., fdx1 (PA0362), encoding two[4Fe-4S]-containing bacterial ferredoxin; fdx2 (PA3809) (a member of the isc operon that is involved in iron-sulfur cluster biogenesis), encoding a [2Fe-2S]-containing ferredoxin; rnfB (PA3490), encoding a ferredoxin-like protein; and fdxA (PA3621), encoding a [4Fe-4S] cluster-containing ferredoxin [33],[6, 39–42]. We tested whether the essentiality of the fprA gene was due to its Fdx1 redox partners. Using a similar strategy as used for the construction of the fprA mutant, P. aeruginosa PAO1 strains were constructed with an extra copy of fdx1 (PAO1::Tn7-fdx1), fdx2 (PAO1::Tn7-fdx2), rnfB (PAO1::Tn7-rnfB) or fdxA (PAO1::Tn7-fdxA) and used to test whether ΔfprA mutants could be constructed with a suicide plasmid pUCΔfprA::Gmr. The fprA mutant construction was accomplished only in the parental strains PAO1::Tn7-fdx1 and PAO1::Tn7T-fprA (Table 1). In other parental strains tested, no fprA mutant could be recovered. The functional complementation of fprA by fdx1 was confirmed by the fact that the ΔfprA mutant harboring pTS-fdx1 could grow at 30°C and at the non-permissive 45°C (Fig 1). This finding indicated that expression of fdx1 can suppress the essential function of fprA and permit the growth of the ΔfprA mutants. It is likely that Fdx1 functions as a redox partner of FprA. We speculate that deletion of fprA severely affects the redox status of Fdx1 by shifting the ratio between reduced and oxidized forms. Increased expression of fdx1, either from Tn7T-fdx1 or pTS-fdx1 in the mutant was sufficient to compensate for the loss of FprA function by restoring the ratio of reduced/oxidized ferredoxins to a viable levels for P. aeruginosa. Fdx1 has been shown to be essential for the viability of PAO1 [40]. The physiological role of Fdx1 in P. aeruginosa remains unclear.
fprA and finR expression increased in response to exposure to paraquat and NaOCl
The expression patterns of fprA under stressful growth conditions were evaluated using real-time RT-PCR. The expression profiles of PAO1 fprA challenged with superoxide anion-generating agents (plumbagin, menadione, and paraquat), H2O2, organic hydroperoxides (cumene hydroperoxide, and t-butyl hydroperoxide), the iron-chelating agent 2,2’-dipyridyl, high salt (NaCl and KCl), and a bleaching agent (NaOCl) were determined. The results in Fig 2A illustrate that fprA expression was considerably induced by exposure to paraquat (7.6 ± 2.6-fold) and NaOCl (11.3 ± 2.8-fold), but not by the other oxidants, 2,2’-dipyridyl, or high salt conditions. The expression profiles of finR in response to stresses were also determined by real-time RT-PCR. The expression of finR could be induced only by exposure to paraquat (6.9 ± 1.9-fold) or NaOCl (9.9 ±1.4-fold) treatments (Fig 2B). Other oxidants and stresses did not significantly (2-fold or less) induce finR expression. This induction pattern is similar to the stress response pattern of fprA. A previous report indicated that paraquat induction of fprA in Pseudomonas spp. is significantly affected by the addition of various sources of sulfur [31]. Nonetheless, how intracellular sulfur affects the induction of gene expression by superoxide generator is being investigated.
Fig 2. Expression analysis finR and fprA in response to various stresses.

The expression levels of finR (A) and fprA (B) were determined using real-time RT-PCR. Exponential-phase cultures of P. aeruginosa PAO1 were subjected to various stress conditions, including 1 mM H2O2, superoxide anion-generating agents (0.5 mM plumbagin [PB], 0.5 mM menadione [MD] and 0.5 paraquat [PQ]), organic hydroperoxides (1 mM cumene hydroperoxide [CHP] and 1 mM t-butyl hydroperoxide [tBH]), 1 mM 2,2’-dipyridyl (DIPY), high salts (0.5 M NaCl and 0.5 M KCl), or 0.04% NaOCl for 15 minutes prior to RNA preparation for real-time RT-PCR analysis. Relative expression was analyzed using the 16S rRNA gene as the normalizing gene and was expressed as the fold expression relative to the level of uninduced (UN) PAO1. Data shown are means ± SD of three independent experiments.
FinR regulates the expression of fprA and itself
To assess whether FinR mediates induction of fprA expression upon exposure to oxidative stress, fprA expression levels were determined in the ΔfinR mutant (ΔfinR::Tn7T) and the complemented mutant (ΔfinR::Tn7T-finR) using real-time RT-PCR. The results showed that paraquat- and NaOCl-induced expression of fprA was abolished in the ΔfinR mutant and that this could be restored in the complemented mutant strain (Fig 3A). The levels of fprA expression in the ΔfinR mutant in all tested conditions were comparable to those of the uninduced wild-type PAO1 (Fig 3A). Moreover, the fprA level in a complemented strain (ΔfinR::Tn7T-finR) was comparable to wild type and a ΔfinR mutant strain. Thus, oxidized FinR likely functions as a transcriptional activator on the fprA promoter in the presence of the inducers paraquat and NaOCl. However, reduced FinR neither represses nor activates fprA expression.
Fig 3. Expression analysis of fprA and finR in P. aeruginosa strains.
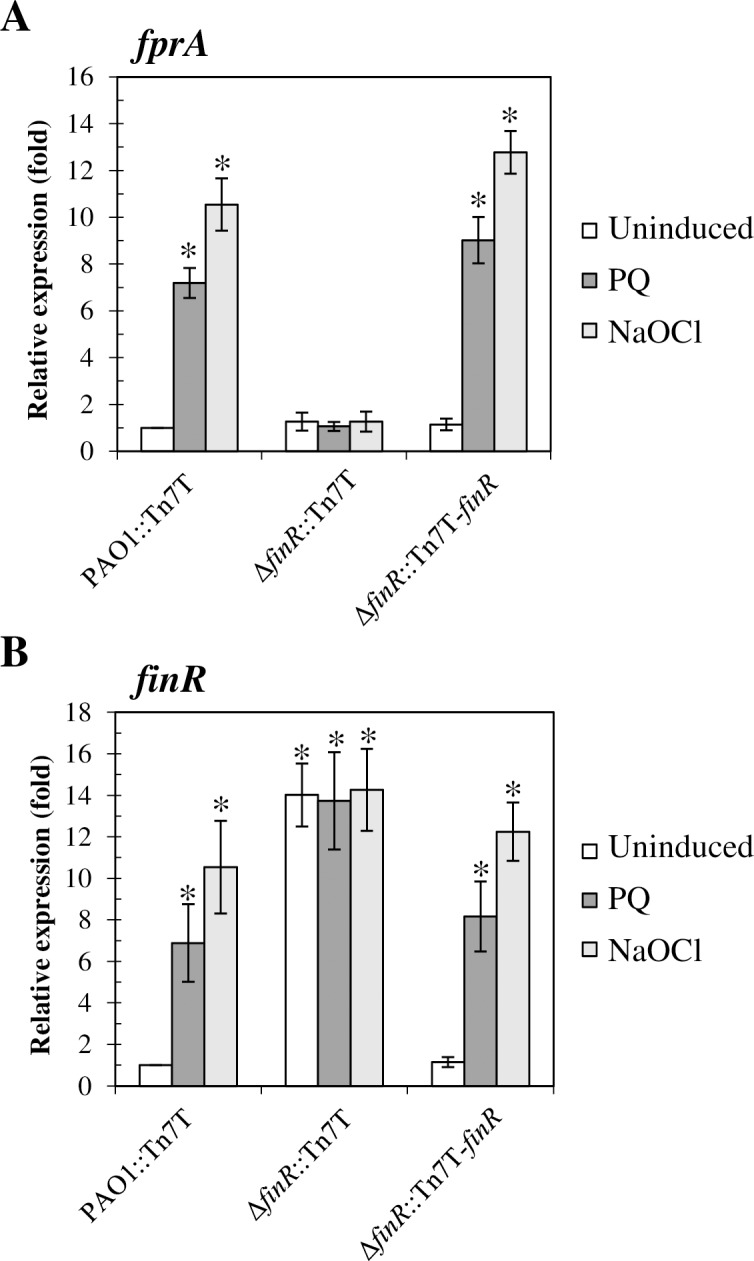
Expression levels of fprA (A) and finR (B) in PAO1 wild type (PAO1::Tn7T), ΔfinR mutant (ΔfinR::Tn7T) and the complemented mutant (ΔfinR::Tn7T-finR) grown under uninduced, 0.5 mM paraquat (PQ), or 0.04% NaOCl (NaOCl) induced conditions. Relative expression was analyzed using the 16S rRNA gene as the normalizing gene and is expressed as fold expression relative to the level of uninduced PAO1. Data shown are means ± SD of three independent experiments. The asterisks indicate statistically significant differences (p < 0.01) compared with the uninduced condition.
The expression levels of finR in response to oxidants were also evaluated in the ΔfinR mutant and the complemented mutant using real-time RT-PCR with primers located immediately downstream of the transcriptional start site (+1) of finR and next to the deletion site (BT3334 and EBI62). The expression levels of finR in the ΔfinR mutant were constitutively high (~14-fold over wild-type PAO1 levels) in both uninduced and oxidant-induced samples (Fig 3B). The constitutively high expression levels in the finR mutant strongly suggest that reduced FinR functions as a transcriptional repressor of its own promoter. Paraquat- and NaOCl-induced finR expression could be restored in the complemented ΔfinR mutant strain (ΔfinR::Tn7T-finR) (Fig 3B). This suggests that reduced FinR functions as a repressor of finR expression while oxidized FinR either activates expression or derepresses finR expression. The results indicate that finR is negatively auto-regulated, which is similar to other transcriptional regulators in the LysR-family [25, 43–45].
FinR binds directly to finR-fprA promoter region
fprA is located next to a divergently transcribed gene, finR, with a 273-bp intergenic region. To characterize the fprA and finR promoters, the putative +1 sites were determined using 5’ RACE. The +1 site of fprA was mapped to an A residue located 54 bp upstream of its translational start codon ATG (Fig 4A). Two sequences (TTTTGC and TAAAAT, separated by 18 bp) that resemble the E. coli δ70–35 and -10 promoter motifs were identified. Using a similar technique, the +1 site of finR was mapped to a G residue situated 125 bp upstream of the finR start codon (ATG) and 97 bp upstream of the putative fprA +1 (Fig 4A). The -35 and -10 promoter motifs were identified as TGCTTA and GATAAC and were separated by 18 bp. The fprA and finR promoter motifs did not overlap with each other (Fig 4A).
Fig 4. Characterization and binding of purified FinR to the finR-fprA promoter.

(A) Nucleotide sequence showing the finR-fprA promoter structure. +1 indicates the transcriptional start site, and the bold sequences are the putative -35 and -10 promoter motifs. CAT and ATG are the translational start codons of FinR and FprA, respectively. The box shaded gray represents the proposed FinR binding site. (B), (C), and (D) Electrophoretic mobility shift assay using purified FinR. A 32P –labeled DNA fragment (B), mutagenized MU1 and MU2 fragments (C), or the promoter fragments (EBI61-62), with (EBI 61–70) and without (EBI 62–69) proposed FinR binding site (D) spanning the finR-fprA promoter was incubated with increasing amounts of FinR. BSA represents an unrelated protein (2.5 μg BSA). CP and UD signify the cold probe (100 ng unlabeled promoter fragment) and unrelated DNA (1 μg of pUC18 plasmid), respectively, that were added to the binding reaction mixture containing 100 nM FinR. F and B indicate free and bound probes, respectively.
The ability of purified FinR to bind to the fprA-finR promoter was investigated using electrophoretic mobility shift assays (EMSA). A 6His-tagged FinR protein was purified using an E. coli system (25]. A [P32]-labeled DNA probe (398 bp) spanning the fprA-finR promoters was used in the binding experiments. Purified FinR could bind to the fprA-finR promoter sequence at nanomolar concentrations (Fig 4B). The binding specificity of FinR was illustrated by the ability of excess unlabeled fprA-finR promoter fragment (CP) but not excess of unrelated DNA (pUC18 plasmid, UP) to compete with the binding of FinR to labeled promoter fragments. Addition of an excess amount of unrelated protein (2.5 μg bovine serum albumin [BSA]) did not affect binding of purified FinR to the fprA-finR promoter (Fig 4B). Thus, FinR functions as a transcriptional regulator of fprA and finR itself through a direct binding to the fprA-finR promoter region.
To our knowledge, no consensus sequence for FinR binding box on target gene promoters has been identified. FinR is a member of LysR family of transcription regulators, which often use palindromic DNA sequences as a binding box that the regulator in LysR family binds to modulate expression of the target gene [46]. We identified DNA sequences with two overlaps and almost perfect dyadic symmetry, 5’TATCCATATTCTGGATAAGCATTATCCAGA3’, consisting of the first palindrome 5’TATCCATATTCTGGATA3’ and the second palindrome 5’TCTGGATAAGCATTATCCAGA3’ located between positions -22 and -51 of the finR promoter and -46 and -83 of the fprA promoter (Fig 4A). The involvement of these two dyadic symmetries in the binding of FinR was evaluated. Site-directed mutagenesis was performed to mutate the putative binding site for FinR from 5’TATCCATATTCTGGATAAGCATTATCCAGA3’ to 5’GCGAACTATTCTGGATAAGCATTATCCA3’ (referred to as MU1) and to 5’TATCCATATTCTGGATAAGCATGCGAACGA3’ (referred to as MU2) using pPfprA as a DNA template. The mutations essentially changed the first palindrome sequence in MU1 and the second palindrome sequence in MU2. [P32]-labeled fprA-finR promoter containing MU1 or MU2 sequences was used in the EMSA experiments. The results in Fig 4C showed that purified FinR bound to the promoter containing MU2 in a similar manner as the native promoter. However, purified FinR at concentration of 200 nM was unable to bind the mutagenized MU1 promoter (Fig 4C). This suggests that the sequence TATCCA of the first palindromic sequence 5’TATCCATATTCTGGATA3’ is important for in vitro binding of P. aeruginosa FinR. To confirm the putative binding site of FinR, the EMSA experiments were performed using the promoter fragment with and without proposed FinR binding site. The results in Fig 4D showed that the purified FinR could bind to the promoter fragment with the proposed FinR-binding site. No FinR binding could be detected when the DNA fragment without the binding site was used (Fig 4D). This supports the site-directed mutagenesis results that in vitro FinR binds specifically to the palindromic sequence 5’TATCCATATTCTGGATA3’.
ΔfinR mutant shows an increased paraquat sensitivity phenotype that could be suppressed by increasing fprA expression
Next, the physiological function of finR was assessed using the ΔfinR mutant. Since FinR is involved in sensing various oxidant resistance levels, the ΔfinR mutant resistance to oxidants was determined using a plate sensitivity assay. The ΔfinR mutant exhibited similar levels of resistance to various oxidants, including H2O2, cumene hydroperoxide, and NaOCl, as the wild-type PAO1 (data not shown). Nonetheless, Fig 5A shows that the ΔfinR mutant (ΔfinR::Tn7T) was much more sensitive (104-fold) to paraquat (150 μM) than its parental strain PAO1 (PAO1::Tn7T), and this hypersensitive phenotype of the mutant was fully restored by the expression of a single copy of functional finR in a mini-Tn7 vector (ΔfinR::Tn7T-finR). These results indicate a crucial role of finR for survival under paraquat stress and are consistent with the previously reported resistance of a finR mutant of P. putida [25].
Fig 5. Determination of paraquat resistance levels in P. aeruginosa strains.
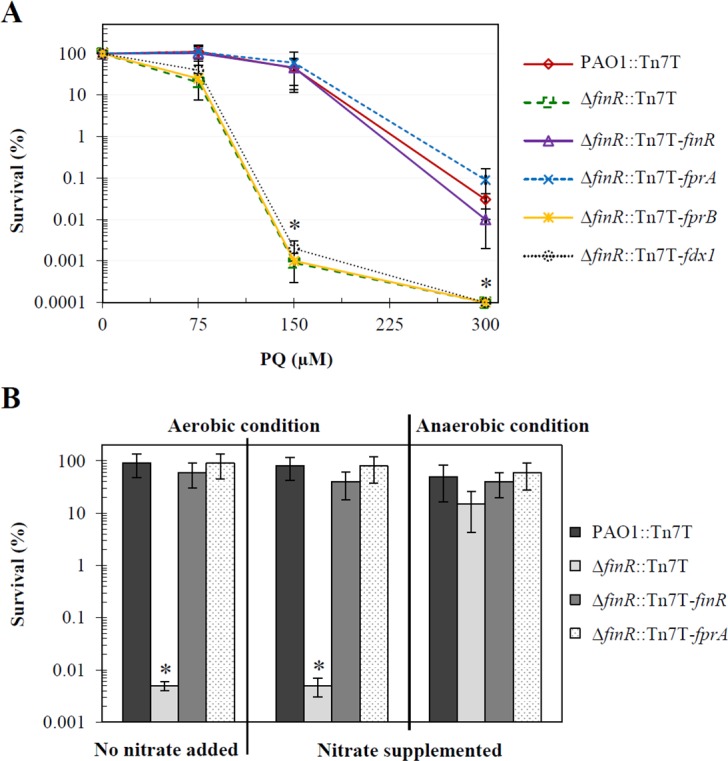
(A) Paraquat resistance levels in PAO1 containing the mnin-Tn7 vector control (PAO1::Tn7T, red) and ΔfinR mutants containing Tn7T (dotted green), Tn7T-finR (purple), Tn7T-fprA (dotted blue), Tn7T-fprB (yellow), or Tn7T-fdx1 (dotted black) were determined using plate sensitivity assays. (B) Paraquat (150 μM) resistance levels of P. aeruginosa strains were determined using LB with and without 1% (w/v) KNO3 supplementation and incubated under aerobic and anaerobic atmospheres. The survival is expressed as a percentage of the CFU on LB plates containing paraquat over the CFU on plates without paraquat. Data shown are means ± SD from three independent experiments.
Paraquat is a redox cycling drug that has been recognized as a superoxide anion-generating agent in the presence of oxygen by disrupting normal electron flow in aerobic respiration [17]. The drug itself can undergo intracellular transformations and is toxic to cells [19]. The question was raised as to whether hypersensitivity of the finR mutant to paraquat was due to reduced ability to detoxify superoxide anions generated from the drug or direct toxicity of the drug. An approach previously described in E. coli was used to test the likely mechanism responsible for paraquat sensitivity in the ΔfinR mutant; this approach used anaerobic cultivation to distinguish between the direct toxicity of the drug and the generation of superoxide anions, which requires oxygen [19]. P. aeruginosa did not grow under anaerobic conditions unless nitrate was added to the culture medium [47]. Plate sensitivity assays were performed to re-examine the paraquat sensitivity levels using LB medium supplemented with potassium nitrate (KNO3, 1% w/v) under anaerobic conditions. All P. aeruginosa grew anaerobically (data not shown). The results of the paraquat sensitivity assay under aerobic and anaerobic conditions are shown in Fig 5B. The ΔfinR mutant (ΔfinR::Tn7T) was much more sensitive (104-fold) to paraquat (150 μM) under aerobic growth than the parental PAO1 (PAO1::Tn7T), whereas no significant change was observed when the plates were incubated under anaerobic conditions. Thus, the killing effects of paraquat are oxygen-dependent and likely occur by generating superoxide anions. Hence, the observed increased sensitivity to paraquat in the ΔfinR mutant most likely is a result of superoxide killing. PAO1 produces two superoxide dismutase (Sod) isozymes, namely SodA (manganese-containing Sod) and SodB (iron-containing Sod); mutations of either sodA or sodB enhance sensitivity to superoxide anions generated from paraquat [48]. We tested whether the paraquat-sensitive phenotype of the finR mutant was due to lower level of Sod activity; total Sod activity was measured in the finR mutant cultivated aerobically. The results showed non-significant differences in the levels of total Sod activity in the finR mutant relative to wild-type PAO1 (data not shown). Therefore, alterations in levels of paraquat resistance of the finR mutant are independent of total Sod enzyme activity.
We have shown that fdx1 could suppress the ΔfprA essentiality phenotype. Hence, we tested whether expression of fprB or the ferredoxin-encoding genes fdx1, fdx2, fdxA and rnfB could complement the paraquat hypersensitivity phenotype of the ΔfinR mutant, and the results showed that expression of these genes could not complement the finR mutant phenotype (data not shown). Here, we have established that FinR positively regulates fprA expression, and therefore, we speculate that the paraquat hypersensitive phenotype of the ΔfinR mutant could arise from loss of the ability to activate fprA expression upon exposure to paraquat. Expression of fprA under the control of the lac promoter in a mini-Tn7 vector was transposed into the ΔfinR mutant, generating ΔfinR::Tn7T-fprA. The paraquat resistance levels of this strain were evaluated. The results in Fig 5A illustrate that increased expression of fprA completely restored the paraquat sensitivity of the ΔfinR mutant to the levels that were attained by the ΔfinR::Tn7T-finR and a wild-type control (PAO1::Tn7T). Since basal levels of fprA expression in the ΔfinR mutant and the parental strain are similar (Fig 3A), the results suggest that the paraquat-hypersensitive phenotype of the ΔfinR mutant could be due to the inability of the mutant to up-regulate the expression of fprA in response to stressful conditions. This suggests that the levels of FprA are critically important, especially under certain stress conditions (i.e., paraquat and NaOCl).
ΔfinR mutant shows attenuated virulence in a Drosophila host model
FinR positively regulates the expression of an essential gene, fprA, in response to oxidative stress; therefore, the contribution of finR to the bacterial pathogenicity of P. aeruginosa was evaluated using the fruit fly (Drosophila melanogaster) as a pathogen-host model as previously described [5, 49]. As shown in Fig 6, feeding the flies with PAO1 cultures resulted in 26.1 ± 3.9% fly survival compared with 99.4 ± 1.0% fly survival when LB medium was fed to the flies as a negative control. Feeding the flies with ΔfinR mutant cultures resulted in a 2-fold increase in fly survival (54.4 ± 9.2%) compared with feeding with PAO1. Thus, deletion of finR attenuated the virulence of P. aeruginosa PAO1 in the tested model (p < 0.01). The attenuated virulence phenotype of the ΔfinR mutant could be restored in the complemented mutant strain (ΔfinR::Tn7T-finR), which expressed a functional copy of finR (27.8 ± 2.6% fly survival). Additionally, expressing fprA could complement the attenuated virulence phenotype of the finR mutant (ΔfinR::Tn7T-fprA), as shown by 28.9 ± 4.2% fly survival, while expressing fprB (ΔfinR::Tn7T-fprB) could not (49.4 ±5.1% fly survival) (Fig 6). The phenotype of attenuated virulence was consistent with that of paraquat sensitivity levels, in which expression of fprA restored the ΔfinR mutant phenotype (Fig 5A). The facts that increased expression of fprA could restore the ΔfinR mutant phenotype and that basal expression of fprA (Fig 4A) in the ΔfinR mutant was comparable to that of the PAO1 wild type suggested that loss of adaptive expression of fprA, which is modulated by FinR, is responsible for the virulence attenuation as well as the paraquat hypersensitivity of the ΔfinR mutant. We also present here that the paraquat-sensitive phenotype of the ΔfinR mutant involved superoxide anion-mediated toxicity (Fig 5B). In several plant and animal pathogenic bacteria, defects in superoxide anion detoxification systems, such as knockout of superoxide dismutase genes, render the mutant strains attenuated for virulence in the model hosts [50–52]. Superoxide anions are one of the key components of innate immunity generated by host cells to eradicate invading microbes. In human hosts, superoxide anions are produced within the phagolysosomes of phagocytic cells to kill the engulfed pathogens [53]. Thus, defects in protection against superoxide toxicity of the bacteria would reduce the ability to survive within the host. Hence, the attenuated phenotype could result from the reduced ability of the ΔfinR mutant to cope with exposure to superoxides during host interactions.
Fig 6. Virulence of P. aeruginosa strains.

The virulence of PAO1 containing the Tn7T vector control (PAO1::Tn7T) and ΔfinR mutants containing Tn7T, Tn7T-finR, Tn7T-fprA, Tn7T-fprB, or Tn7T-fdx1 were determined using the Drosophila melanogaster feeding method. The percent fly survival was scored after 18 hours of incubation. Data presented are means ± SD of three independent experiments. The asterisk indicates statistically significant difference (p < 0.01) compared with PAO1::Tn7T. LB represents feeding the flies with LB medium.
Alternatively, in pseudomonads, FprA plays a role in sulfur metabolism and cysteine biosynthesis, which are important components of [Fe-S] cluster biogenesis [31, 54]. [Fe-S] clusters, which are key cofactors of proteins that are implicated in diverse cellular processes, including respiration and central metabolism, are prone to oxidative damage when cells are exposed to reactive oxygen species (ROS) such as superoxide anions and H2O2 [54, 55]. Therefore, impaired [Fe-S] cluster biogenesis during exposure to oxidative stress due to lack of FinR-mediated increased expression of fprA would lead to lowered ability of the bacteria to survive oxidative stress generated by the host’s innate immune system. Similar mechanisms could also account for the observed paraquat hypersensitive phenotype. Mutants defective in [Fe-S] cluster biogenesis or repair, for example, deletion of the IscR coding gene, which regulates [Fe-S] cluster biogenesis, show attenuated virulence in host models, including such mutants of P. aeruginosa [56–59].
Since fprA is essential in PAO1, a direct analysis of the mutant phenotypes is difficult. Analysis of the ΔfinR mutant provides insight into the importance of fprA. ΔfinR mutant phenotypes (paraquat sensitivity and attenuated virulence) most likely occur as a result of the inability of fprA to be up-regulated during stressful conditions. This suggests that the level of FprA is crucial in P. aeruginosa. An fdx1 encoding putative reaction partner of FprA is also an essential gene in PAO1 [40]. Thus, the link between FprA and Fdx1 is important to PAO1 physiology.
Materials and methods
Bacterial strains, plasmids and growth conditions
Both E. coli and P. aeruginosa (PAO1, ATCC15692) strains were aerobically cultivated in Luria-Bertani (LB) broth (BD Difco, USA) at 37°C with shaking at 180 rpm unless otherwise stated. To produce synchronous growth, an overnight culture was inoculated into fresh LB medium to give an optical density at 600 nm (OD600) of 0.1. Exponential phase cells (OD600 of about 0.6, after 3 h of growth) were used in all experiments. All plasmids used in this study are listed in Table 2.
Table 2. List of plasmids used in this study.
| Plasmid | Relevant characteristic(s) | Source or Reference |
|---|---|---|
| pBBR1MCS-4 | Broad-host-range expression vector, Apr | [37] |
| pSS255 | Expression vector with a temperature sensitive replicon (mSFts1), Apr | [36] |
| pTS | pBBR1MCS-4 carrying mSFts1, Apr | This study |
| pTS-fprA | pTS carrying fprA | This study |
| pTS-fprB | pTS carrying fprB | This study |
| pTS-fdx1 | pTS carrying fdx1 | This study |
| pTS-fdx2 | pTS carrying fdx2 | This study |
| pTS-rnfB | pTS carrying rnfB | This study |
| pTS-fdxA | pTS carrying fdxA | This study |
| pUCΔfinR::Gmr | pUC18 containing Gmr inserted into deleted finR, Gmr | This study |
| pUCΔfprA::Gmr | pUC18 containing Gmr inserted into deleted fprA, Gmr | This study |
| pCM351 | vector containing the loxP-Gmr-loxP region, Gmr | [61] |
| pCM157 | vector containing the Cre-encoding gene, Tetr | [61] |
| pUC18-mini-Tn7T::Gm-LAC | mini-Tn7 vector with Ptac expression cassette, Gmr | [34] |
| pTNS2 | Helper plasmid for Tn7 insertion, Apr | [34] |
| pTn-finR | pUC18-mini-TN7T::Gm-LAC containing finR | This study |
| pTn-fprA | pUC18-mini-TN7T::Gm-LAC containing fprA | This study |
| pTn-fprB | pUC18-mini-TN7T::Gm-LAC containing fprB | This study |
| pTn-fdx1 | pUC18-mini-TN7T::Gm-LAC containing fdx1 | This study |
| pTn-fdx2 | pUC18-mini-TN7T::Gm-LAC containing fdx2 | This study |
| pTn-rnfB | pUC18-mini-TN7T::Gm-LAC containing rnfB | This study |
| pTn-fdxA | pUC18-mini-TN7T::Gm-LAC containing fdxA | This study |
| pPfprA | pUC18 carrying fprA promoter | This study |
| pPfprAMU1 | pUC18 carrying mutagenized fprA promoter MU1 | This study |
| pPfprAMU2 | pUC18 carrying mutagenized fprA promoter MU2 | This study |
| pQE30Xa | Vector for expressing N-terminal 6His tagged protein in E. coli, Apr, Cmr | Qiagen (Germany) |
| pQE30Xa-finR | pQE30XA carrying full-length finR | This study |
Gmr, gentamicin resistance; Apr, ampicillin resistance; Tetr, tetracycline resistance; Cmr chloramphenicol resistance.
Molecular techniques
General molecular techniques including DNA and RNA preparations, DNA cloning, PCR amplification, Southern analyses and bacterial transformation were performed according to standard protocols [60]. The oligonucleotide primers used in this study are listed in Table 3.
Table 3. List of primers used in this study.
| Name | Sequence 5’→3’ | Purpose |
| BT2781 | GCCCGCACAAGCGGTGGAG | Forward primer for 16S rRNA |
| BT2782 | ACGTCATCCCCACCTTCCT | Reverse primer for 16S rRNA |
| BT3332 | ACGTGCACAACACCGCCC | Forward primer for full-length finR |
| BT3333 | CAGGCGGATGTTCAGCGG | Reverse primer for full-length finR |
| BT3334 | TAGACGAGGAAGCCTGGATG | Forward primer for finR fragment |
| BT3335 | TGTCCCTGGCCAACTGAG | Reverse primer for finR fragment |
| BT3336 | GGAGTTCTTCAGCATCAAGG | Forward primer for full-length fprA |
| BT3337 | GAAGTACTCGTGTTCCGGCA | Reverse primer for full-length fprA |
| BT3456 | GTCTGCTGCTGTTGGTGTG | Forward primer for fprA expression |
| BT3457 | GGCAGGGGCTTTCTTCG | Reverse primer for fprA expression |
| BT4443 | GTGGCTGTCCGTCGCGGTTG | Forward primer for full-length fdx1 |
| BT4444 | CAGGCGCCGGCGGGGATCAG | Reverse primer for full-length fdx1 |
| BT4479 | CCTTGATGCTGAAGAACTCC | Sp2 primer for fprA |
| BT4780 | GCAAAATGAATTGTCGTTCGCATGCTTAT | Forward primer for mutated finR promoter MU2 |
| BT4781 | CTGGATAAGCATGCGAACGACAATTC | Reverse primer for mutated finR promoter MU2 |
| BT4782 | CTTATCCAGAATAGTTCGCTGGGATAA | Forward primer for mutated finR promoter MU1 |
| BT4783 | CGTGTTATCCCAGCGAACTATTCTGG | Reverse primer for mutated finR promoter MU1 |
| BT3499 | GTGCTTTGCGGGACACTAGG | Forward primer for full-length fprB |
| BT3500 | GCTATCCGCCGCTACTGC | Reverse primer for full-length fprB |
| BT5019 | CCTGGGCGGTGTTGTGCA | Sp1 primer for finR |
| BT5201 | GAGGAGAGAACTAGAAAATG | Forward primer for full-length fdxA |
| BT5309 | CTTGGCGTATCAGCGCTC | Reverse primer for full-length fdxA |
| EBI01 | CATGGGCTTCAGCGGGTTGG | Forward primer for full-length rnfB |
| EBI02 | GTGCAGGGCGCTCATGCC | Reverse primer for full-length rnfB |
| EBI53 | GGGAATTCGAAGTACTCGTGTTCCGGCA | Forward primer for upstream fragment of finR |
| EBI54 | GGCCATGGGAACAGCTTGCAGTCGAACTG | Reverse primer for upstream fragment of finR |
| EBI57 | CCGAATTCTCCAGCTCGTAGTGGGCGAC | Forward primer for upstream fragment of fprA |
| EBI58 | GGCCATGGTAGTTCGGGCTGGCAATGCTG | Reverse primer for upstream fragment of fprA |
| EBI61 | AGCTGAACAGGGTGTCGT | Forward primer for finR promoter |
| EBI62 | AGACGCTCTCCTGCTGGG | Reverse primer for finR promoter |
| EBI69 | GGGATAACACGATATCGGTCGG | Forward primer for finR promoter |
| EBI70 | CGATATCGTGTTATCCCATATCC | Reverse primer for finR promoter |
| EBI73 | CCATCGATCGATCAAGCGTGCCGTGGAG | Forward primer for downstream fragment of finR |
| EBI74 | CCGGAGCTCTGCTGCTGGGGATCGTCCTG | Reverse primer for downstream fragment of finR |
| EBI75 | CCATCGATGGCAAGCTGTTCGAGGACATC | Forward primer for downstream fragment of fprA |
| EBI76 | CCGGAGCTCCCTCAGCCAGGGTCACCTGAGC | Reverse primer for downstream fragment of fprA |
| EBI269 | GAACTGTCGAGGAATAAGCGAAGATGCC | Forward primer for full-length fdx2 |
| EBI270 | ATTGCACGCTCCTCTACTAC | Reverse primer for full-length fdx2 |
| EBI292 | GCGCCTGCAGTCAGGGAATCAGCGGCA | Reverse primer for FinR protein expression |
| EBI322 | ATGAAATTCACCCTCCGC | Forward primer for FinR protein expression |
Construction of P. aeruginosa ΔfinR mutant
The finR deletion mutant was constructed using homologous recombination with an unmarked Cre-loxP antibiotic marker system as previously described [61]. The primer pairs, EBI73-EBI74 and EBI53-EBI54, were designed to amplify a finR fragment containing the C-terminus and N-terminus, respectively, of the finR coding region, plus additional flanking regions from the PAO1 genomic DNA. The 1030-bp PCR fragment of the C-terminus was digested with ClaI and SacI and cloned into pUC18Gmr (pUC18 containing loxP-flanked Gmr, which was constructed by inserting SacI-EcoRI fragments containing loxP-flanked Gmr from pCM351 [61] into pUC18 cut with the same enzymes) at the ClaI and SacI sites, yielding pUCfinRC::Gmr. The 926-bp PCR fragment of the N-terminus was digested with EcoRI and NcoI and cloned into pUCfinRC::Gmr at the EcoRI and NcoI sites, yielding pUCΔfinR::Gmr. The constructed plasmid resulted in the deletion of 526 bp of the coding region of finR. pUCΔfinR::Gmr was transferred into PAO1, and the putative ΔfinR mutants that arose from a double crossover event were selected for the Gmr and Cbs phenotypes. An unmarked ΔfinR mutant was created using the Cre-loxP system to excise the Gmr gene as previously described [61], and deletion of finR was confirmed by Southern blot analysis.
Construction of the P. aeruginosa ΔfprA mutant
The fprA deletion mutant was constructed using homologous recombination with an unmarked Cre-loxP antibiotic marker system using the same protocol as the construction of the ΔfinR mutant but using primer pairs, EBI75-EBI76 and EBI57-EBI58, which were designed to amplify the fprA fragment containing the fprA coding region plus additional flanking regions. The restriction enzyme sites and plasmids were same as those used in the construction of the ΔfinR mutant. The obtained plasmid, pUCΔfprA::Gmr, was used to transform PAO1 wild type and strains containing either an expression plasmid or a temperature-sensitive expression plasmid. PAO1 strains containing an extra copy of various genes (fdx, fdxA, fdx2, rnfB, finR, fprA, fprB) was used to test the essentiality of the fprA gene and were constructed by transposition of a mini-Tn7 vector containing a target gene into the PAO1 chromosome and the subsequent removal of the Gmr antibiotic resistance marker gene of mini-Tn7 using the Flp-FRT recombinase as previously described [35].
Construction of plasmid and mini-Tn7 harboring gene coding regions and promoters
To construct pTS, a temperature-sensitive replicon cassette-containing plasmid, a broad-host-range plasmid pBBR1MCS-4 [37] was inserted with a BamHI fragment containing the temperature-sensitive (TS) regulon isolated from vector pTS225 [36] at the BamHI site. pTS-fprA was constructed by amplifying the full-length fprA from the PAO1 genomic DNA with primers BT3456-BT3457. The 866-bp PCR products were cloned into the pTS cut with SmaI. A similar protocol was used to construct pTS-fdx1, pTS-fdx2, pTS-fdxA, pTS-rnfB and pTS-fprB for trans expression of fdx1 (PA0362), fdx2 (PA3809), fdxA (PA3621), rnfB (PA3490) and fprB, respectively. The specific primer pairs for PCR amplification of full-length fdx1, fdx2, fdxA, rnfB, and fprB genes were BT4443-BT4444, EBI269-EBI270, BT5201-BT5309, EBI01-EBI02 and BT3499-BT3500, respectively.
Single-copy complementation was performed using a mini-Tn7 system [34]. The full-length PCR fragments of various genes were PCR amplified with specific primer pairs as described above (and BT3334-BT3335 for finR) and cloned into pUC18-mini-Tn7T-Gm-LAC [34], generating pTn-fprA, pTn-fdx1, pTn-fdx2, pTn-fdxA, pTn-rnfB, pTn-fprB and pTn-finR. The mini-Tn based recombinant plasmid were then transposed into either PAO1 or mutant strains, generating the complemented strains ΔfinR::Tn7T-finR and ΔfinR::Tn7T-fprA. Confirmation of transposition was carried out as previously described [34].
To construct the plasmids containing the fprA promoter region, a putative fprA promoter fragment was amplified from the PAO1 genomic DNA with primers EBI61 and EBI62. The 398-bp PCR product was ligated into EcoRV-digested pUC18 and was named pPfprA. PCR-based site-directed mutagenesis at the putative FinR-binding site was performed as previously described [6] using primers BT4782-BT4783 and BT4780-BT4781, and these vectors are referred to as pPfprA-MU1 and pPfprA-MU2, respectively.
5’ rapid amplification of cDNA ends (RACE)
5’ RACE was performed using a 5’/3’ RACE kit (Roche, Germany) as previously described [62]. Essentially, DNase I-treated total RNA was reverse transcribed using specific primers BT3311 and BT3337 as SP1 primers for finR and fprA, respectively. The first-strand DNA (cDNA) was purified, and poly(A) was added to the 5’-terminus of the cDNA using terminal transferase. Next, poly(A)-tailed cDNA was PCR-amplified using the specific SP2 primer BT4438 for finR and BT4479 for fprA and an anchored oligo(dT) primer. The purified PCR product was cloned into the pGemT vector, and the +1 site was identified from the DNA sequences.
Real-time RT-PCR
Reverse transcription was performed as described for end-point RT-PCR [63]. Real-time RT-PCR was conducted using 10 ng of cDNA as template, a specific primer pair and a KAPA SYBR® FAST qPCR kit (Kapa Biosystems, USA). The reaction was run on an Applied Biosystems StepOnePlus thermal cycler under the following conditions: denaturation at 95°C for 20 s, annealing at 60°C for 30 s, and extension at 60°C for 30 s, for 40 cycles. The specific primer pairs used for finR and fprA were BT3334-EBI69 and BT3336-BT3337, respectively. The primer pair for the 16S rRNA gene was BT2781-BT2782, which was used as the normalizing gene. Relative expression analysis was calculated using StepOne software version 2.1 and is presented as expression fold-change relative to the level of PAO1 wild type grown under uninduced conditions. Experiments were repeated independently three times, and the data shown are the means with standard deviations (SD).
Expression and purification of P. aeruginosa FinR
The 6His-tagged FinR from P. aeruginosa was purified using the pQE-30Xa expression system (Qiagen, Germany). The full-length finR gene was amplified from PAO1 genomic DNA with primers EBI322 and EBI292. A 937-bp PCR product was digested with PstI before ligation into pQE-30Xa digested with StuI (blunt ended) and PstI to generate pQE30Xa-finR for high-level expression of finR containing an N-terminal 6His-tag. An E. coli DH5α strain harboring pQE30Xa-finR was grown in LB medium containing 100 μg/ml ampicillin at 37°C to an OD600 of 1.0 before being induced with 100 μM IPTG for 60 min. Purification of 6His-tagged FinR was carried out using a nickel-nitrilotriacetic acid (Ni-NTA) agarose column as previously described [63]. The purity of the FinR protein was more than 90%, as judged by a major band corresponding to the 36.9-kDa protein observed on SDS-PAGE.
Gel mobility shift assay
Gel mobility shift assays were performed using a labeled probe containing either native or mutagenized fprA-promoters amplified from pPfprA, pPfprA-MU1, or pPfprA-MU2 as a template and using 32P-labeled BT4691 and BT4692 primers. The promoter fragments (EBI61and EBI62) with and without proposed FinR binding site were amplified from genomic PAO1 using primers, EBI61-EBI70 and EBI62-EBI69, respectively. Binding reactions were conducted using 3 fmol of labeled probe in 25 μl of reaction buffer containing 20 mM Tris-HCl (pH 8.0), 50 mM KCl, 4 mM MgCl2, 0.5 mM EDTA, 0.02 mg ml-1 bovine serum albumin (BSA), 5 mM dithiothreitol (DTT), 10% (v/v) glycerol, and 200 ng of poly(dI-dC). Various amounts of purified FinR were added, and the reaction mixture was incubated at 25°C for 20 min. Protein-DNA complexes were separated by electrophoresis on a 5% non-denaturing polyacrylamide gel in 0.5× Tris-borate-EDTA buffer at 4°C and were visualized by exposure to Hyperfilm (GE Healthcare).
Plate sensitivity assay
A plate sensitivity assay was performed to determine the oxidant resistance level as previously described [63]. Briefly, exponential phase cells were adjusted to OD600 of 0.1 before making 10-fold serial dilutions. 10 μl of each dilution was then spotted onto LB agar plate containing appropriate concentrations of testing reagents. The plates were incubated overnight at 37°C before the colony forming units (CFU) were scored. Percent survival was defined as the CFU on plates containing oxidant divided by the CFU on plates without oxidant and multiply by 100.
Drosophila virulence tests
The virulence of P. aeruginosa was investigated using the Drosophila melanogaster feeding assay as previously described [5]. Briefly, exponential phase cultures of each P. aeruginosa strains were adjusted to an OD600 of 0.5 before 800 μL of the bacterial cells were overlaid to completely cover the surface of the preservative-free corn flour Drosophila medium at the bottom of a glass fly culture vial. Approximately one-week-old adult flies were starved for 3 h prior to the feeding assay. Twenty flies were added to each vial, and each strain of P. aeruginosa was tested for at least three replications. Then, all of the tested flies were incubated at 25°C for 18 h before the number of the viable flies was observed. The experiments were performed in a double-blind fashion and were analyzed from nine experiments using three different batches of flies.
Statistical analysis
The significance of differences between strains, cultured conditions, or changes of expression level was statistically determined using Student’s t-test. P < 0.05 is considered significant difference and indicated as an asterisk.
Supporting information
The deduced amino acid sequence of P. aeruginosa (A) FprA ferredoxin NADP(+) reductase A and (B) FinR transcriptional regulator was aligned with those in Pseudomonas putida and Azotobacter vinelandii by using CLASTAL Omega alignment. The asterisk, colon, and period symbols indicate identical residues, conserved substitutions, and semi-conserved substitutions, respectively.
(PDF)
Acknowledgments
The authors gratefully acknowledge Dr. Sang-Jin Suh for providing temperature-sensitive plasmid pSS255. Parts of this work are from S.B. dissertation submitted for a Ph.D. degree from Mahidol University.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by grants from the Chulabhorn Research Institute (http://www.cri.or.th/en/index.php), the Faculty of Science (http://science.mahidol.ac.th/), and the Mahidol University (https://www.mahidol.ac.th). AR was supported by the grant from the joint funding of Office of the Higher Education Commission and Thailand Research Fund (MRG5980047) with SM as his mentor. SB was supported by the Royal Thai Government Scholarship under the Development and Promotion of Science and Technology Talents Project (DPST), Thailand (www.dpstcenter.org). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Oliver A, Mulet X, Lopez-Causape C, Juan C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat. 2015; 21–22: 41–59. 10.1016/j.drup.2015.08.002 [DOI] [PubMed] [Google Scholar]
- 2.Suwantarat N, Carroll KC. Epidemiology and molecular characterization of multidrug-resistant Gram-negative bacteria in Southeast Asia. Antimicrob Resist Infect Control. 2016; 5: 15 10.1186/s13756-016-0115-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imlay JA. Transcription Factors That Defend Bacteria Against Reactive Oxygen Species. Annu Rev Microbiol. 2015; 69: 93–108. 10.1146/annurev-micro-091014-104322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farr SB, Kogoma T. Oxidative stress responses in Escherichia coli and Salmonella typhimurium. Microbiol Rev. 1991; 55(4): 561–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romsang A, Atichartpongkul S, Trinachartvanit W, Vattanaviboon P, Mongkolsuk S. Gene expression and physiological role of Pseudomonas aeruginosa methionine sulfoxide reductases during oxidative stress. J Bacteriol. 2013; 195(15): 3299–308. 10.1128/JB.00167-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romsang A, Duang-Nkern J, Leesukon P, Saninjuk K, Vattanaviboon P, Mongkolsuk S. The iron-sulphur cluster biosynthesis regulator IscR contributes to iron homeostasis and resistance to oxidants in Pseudomonas aeruginosa. PLoS One. 2014; 9(1): e86763 10.1371/journal.pone.0086763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindquist S, Lindberg F, Normark S. Binding of the Citrobacter freundii AmpR regulator to a single DNA site provides both autoregulation and activation of the inducible ampC beta-lactamase gene. J Bacteriol. 1989; 171(7): 3746–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovacikova G, Skorupski K. A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J Bacteriol. 1999; 181(14): 4250–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deghmane AE, Petit S, Topilko A, Pereira Y, Giorgini D, Larribe M, et al. Intimate adhesion of Neisseria meningitidis to human epithelial cells is under the control of the crgA gene, a novel LysR-type transcriptional regulator. EMBO J. 2000; 19(5): 1068–78. 10.1093/emboj/19.5.1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc Natl Acad Sci U S A. 2001; 98(25): 14613–8. 10.1073/pnas.251465298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Kim JG, Kang Y, Jang JY, Jog GJ, Lim JY, et al. Quorum sensing and the LysR-type transcriptional activator ToxR regulate toxoflavin biosynthesis and transport in Burkholderia glumae. Mol Microbiol. 2004; 54(4): 921–34. 10.1111/j.1365-2958.2004.04338.x [DOI] [PubMed] [Google Scholar]
- 12.Russell DA, Byrne GA, O'Connell EP, Boland CA, Meijer WG. The LysR-type transcriptional regulator VirR is required for expression of the virulence gene vapA of Rhodococcus equi ATCC33701. J Bacteriol. 2004; 186(17): 5576–84. 10.1128/JB.186.17.5576-5584.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byrne GA, Russell DA, Chen X, Meijer WG. Transcriptional regulation of the virR operon of the intracellular pathogen Rhodococcus equi. J Bacteriol. 2007; 189(14): 5082–9. 10.1128/JB.00431-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Z, Takeuchi M, Sato T. The LysR-type transcriptional regulator YofA controls cell division through the regulation of expression of ftsW in Bacillus subtilis. J Bacteriol. 2007; 189(15): 5642–51. 10.1128/JB.00467-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sperandio B, Gautier C, McGovern S, Ehrlich DS, Renault P, Martin-Verstraete I, et al. Control of methionine synthesis and uptake by MetR and homocysteine in Streptococcus mutans. J Bacteriol. 2007; 189(19): 7032–44. 10.1128/JB.00703-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid Redox Signal. 2011; 14(6): 1049–63. 10.1089/ars.2010.3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dietrich LE, Kiley PJ. A shared mechanism of SoxR activation by redox-cycling compounds. Mol Microbiol. 2011; 79(5): 1119–22. 10.1111/j.1365-2958.2011.07552.x [DOI] [PubMed] [Google Scholar]
- 18.Dubbs JM, Mongkolsuk S. Peroxide-sensing transcriptional regulators in bacteria. J Bacteriol. 2012; 194(20): 5495–503. 10.1128/JB.00304-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu M, Imlay JA. The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol Microbiol. 2011; 79(5): 1136–50. 10.1111/j.1365-2958.2010.07520.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yeom J, Park W. Biochemical characterization of ferredoxin-NADP(+) reductase interaction with flavodoxin in Pseudomonas putida. BMB Rep. 2012; 45(8): 476–81. 10.5483/BMBRep.2012.45.8.071 [DOI] [PubMed] [Google Scholar]
- 21.Yeom J, Jeon CO, Madsen EL, Park W. In vitro and in vivo interactions of ferredoxin-NADP+ reductases in Pseudomonas putida. J Biochem. 2009; 145(4): 481–91. 10.1093/jb/mvn185 [DOI] [PubMed] [Google Scholar]
- 22.Romsang A, Leesukon P, Duangnkern J, Vattanaviboon P, Mongkolsuk S. Mutation of the gene encoding monothiol glutaredoxin (GrxD) in Pseudomonas aeruginosa increases its susceptibility to polymyxins. Int J Antimicrob Agents. 2015; 45(3): 314–8. 10.1016/j.ijantimicag.2014.10.024 [DOI] [PubMed] [Google Scholar]
- 23.Lee Y, Pena-Llopis S, Kang YS, Shin HD, Demple B, Madsen EL, et al. Expression analysis of the fpr (ferredoxin-NADP+ reductase) gene in Pseudomonas putida KT2440. Biochem Biophys Res Commun. 2006; 339(4): 1246–54. 10.1016/j.bbrc.2005.11.135 [DOI] [PubMed] [Google Scholar]
- 24.Yeom J, Jeon CO, Madsen EL, Park W. Ferredoxin-NADP+ reductase from Pseudomonas putida functions as a ferric reductase. J Bacteriol. 2009; 191(5): 1472–9. 10.1128/JB.01473-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yeom S, Yeom J, Park W. Molecular characterization of FinR, a novel redox-sensing transcriptional regulator in Pseudomonas putida KT2440. Microbiology. 2010; 156(Pt 5): 1487–96. 10.1099/mic.0.034181-0 [DOI] [PubMed] [Google Scholar]
- 26.Park W, Pena-Llopis S, Lee Y, Demple B. Regulation of superoxide stress in Pseudomonas putida KT2440 is different from the SoxR paradigm in Escherichia coli. Biochem Biophys Res Commun. 2006; 341(1): 51–6. 10.1016/j.bbrc.2005.12.142 [DOI] [PubMed] [Google Scholar]
- 27.Takeda K, Sato J, Goto K, Fujita T, Watanabe T, Abo M, et al. Escherichia coli ferredoxin-NADP+ reductase and oxygen-insensitive nitroreductase are capable of functioning as ferric reductase and of driving the Fenton reaction. Biometals. 2010; 23(4): 727–37. 10.1007/s10534-010-9339-8 [DOI] [PubMed] [Google Scholar]
- 28.Lee Y, Yeom J, Kang YS, Kim J, Sung JS, Jeon CO, et al. Molecular characterization of fprB (ferredoxin-NADP+ reductase) in Pseudomonas putida KT2440. J Microbiol Biotechnol. 2007; 17(9): 1504–12. [PubMed] [Google Scholar]
- 29.Romsang A, Duang-Nkern J, Wirathorn W, Vattanaviboon P, Mongkolsuk S. Pseudomonas aeruginosa IscR-Regulated Ferredoxin NADP(+) Reductase Gene (fprB) Functions in Iron-Sulfur Cluster Biogenesis and Multiple Stress Response. PLoS One. 2015; 10(7): e0134374 10.1371/journal.pone.0134374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weeratunga SK, Lovell S, Yao H, Battaile KP, Fischer CJ, Gee CE, et al. Structural studies of bacterioferritin B from Pseudomonas aeruginosa suggest a gating mechanism for iron uptake via the ferroxidase center. Biochemistry. 2010; 49(6): 1160–75. 10.1021/bi9015204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis TA, Glassing A, Harper J, Franklin MJ. Role for ferredoxin:NAD(P)H oxidoreductase (FprA) in sulfate assimilation and siderophore biosynthesis in Pseudomonads. J Bacteriol. 2013; 195(17): 3876–87. 10.1128/JB.00528-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee SA, Gallagher LA, Thongdee M, Staudinger BJ, Lippman S, Singh PK, et al. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2015; 112(16): 5189–94. 10.1073/pnas.1422186112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000; 406(6799): 959–64. 10.1038/35023079 [DOI] [PubMed] [Google Scholar]
- 34.Choi KH, Schweizer HP. mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat Protoc. 2006; 1(1): 153–61. 10.1038/nprot.2006.24 [DOI] [PubMed] [Google Scholar]
- 35.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998; 212(1): 77–86. [DOI] [PubMed] [Google Scholar]
- 36.Silo-Suh LA, Elmore B, Ohman DE, Suh SJ. Isolation, characterization, and utilization of a temperature-sensitive allele of a Pseudomonas replicon. J Microbiol Metho. 2009; 78(3): 319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM 2nd, et al. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995; 166(1): 175–6. [DOI] [PubMed] [Google Scholar]
- 38.Isas JM, Yannone SM, Burgess BK. Azotobacter vinelandii NADPH:ferredoxin reductase cloning, sequencing, and overexpression. J Biol Chem. 1995; 270(36): 21258–63. [DOI] [PubMed] [Google Scholar]
- 39.Biegel E, Muller V. Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc Natl Acad Sci U S A. 2010; 107(42): 18138–42. 10.1073/pnas.1010318107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elsen S, Efthymiou G, Peteinatos P, Diallinas G, Kyritsis P, Moulis JM. A bacteria-specific 2[4Fe-4S] ferredoxin is essential in Pseudomonas aeruginosa. BMC Microbiol. 2010; 10: 271 10.1186/1471-2180-10-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vollack KU, Xie J, Hartig E, Romling U, Zumft WG. Localization of denitrification genes on the chromosomal map of Pseudomonas aeruginosa. Microbiology. 1998; 144 (Pt 2): 441–8. [DOI] [PubMed] [Google Scholar]
- 42.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. 2016; 44(D1): D646–53. 10.1093/nar/gkv1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schell MA. Molecular biology of the LysR family of transcriptional regulators. Annu Rev Microbiol. 1993; 47: 597–626. 10.1146/annurev.mi.47.100193.003121 [DOI] [PubMed] [Google Scholar]
- 44.Storz G, Tartaglia LA. OxyR: a regulator of antioxidant genes. J Nutr. 1992; 122(3 Suppl): 627–30. [DOI] [PubMed] [Google Scholar]
- 45.Braz VS, da Silva Neto JF, Italiani VC, Marques MV. CztR, a LysR-type transcriptional regulator involved in zinc homeostasis and oxidative stress defense in Caulobacter crescentus. J Bacteriol. 2010; 192(20): 5480–8. 10.1128/JB.00496-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008; 154(Pt 12): 3609–23. 10.1099/mic.0.2008/022772-0 [DOI] [PubMed] [Google Scholar]
- 47.Yoon MY, Lee KM, Park Y, Yoon SS. Contribution of cell elongation to the biofilm formation of Pseudomonas aeruginosa during anaerobic respiration. PLoS One. 2011; 6(1): e16105 10.1371/journal.pone.0016105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hassett DJ, Schweizer HP, Ohman DE. Pseudomonas aeruginosa sodA and sodB mutants defective in manganese- and iron-cofactored superoxide dismutase activity demonstrate the importance of the iron-cofactored form in aerobic metabolism. J Bacteriol. 1995; 177(22): 6330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Apidianakis Y, Rahme LG. Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. Nat Protoc. 2009; 4(9): 1285–94. 10.1038/nprot.2009.124 [DOI] [PubMed] [Google Scholar]
- 50.Bakshi CS, Malik M, Regan K, Melendez JA, Metzger DW, Pavlov VM, et al. Superoxide dismutase B gene (sodB)-deficient mutants of Francisella tularensis demonstrate hypersensitivity to oxidative stress and attenuated virulence. J Bacteriol. 2006; 188(17): 6443–8. 10.1128/JB.00266-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang IH, Kim JS, Lee JK. The virulence of Vibrio vulnificus is affected by the cellular level of superoxide dismutase activity. J Microbiol Biotechnol. 2007; 17(8): 1399–402. [PubMed] [Google Scholar]
- 52.Saenkham P, Eiamphungporn W, Farrand SK, Vattanaviboon P, Mongkolsuk S. Multiple superoxide dismutases in Agrobacterium tumefaciens: functional analysis, gene regulation, and influence on tumorigenesis. J Bacteriol. 2007; 189(24): 8807–17. 10.1128/JB.00960-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Forman HJ, Torres M. Redox signaling in macrophages. Mol Aspects Med. 2001; 22(4–5): 189–216. [DOI] [PubMed] [Google Scholar]
- 54.Roche B, Aussel L, Ezraty B, Mandin P, Py B, Barras F. Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim Biophys Acta. 2013; 1827(3): 455–69. 10.1016/j.bbabio.2012.12.010 [DOI] [PubMed] [Google Scholar]
- 55.Romsang A, Dubbs JM, Mongkolsuk S. The iron-sulfur cluster biosynthesis regulator IscR contributes to iron homeostasis and resistance to oxidants in Pseudomonas aeruginosa In: de Bruijn FJ, editor. Stress and Environmental Control of Gene Expression in Bacteria. 2 USA: John Wiley & Sons; 2016. pp. 1090–102. [Google Scholar]
- 56.Fuangthong M, Jittawuttipoka T, Wisitkamol R, Romsang A, Duang-nkern J, Vattanaviboon P, et al. IscR plays a role in oxidative stress resistance and pathogenicity of a plant pathogen, Xanthomonas campestris. Microbiol Res. 2015; 170: 139–46. 10.1016/j.micres.2014.08.004 [DOI] [PubMed] [Google Scholar]
- 57.Kim SH, Lee BY, Lau GW, Cho YH. IscR modulates catalase A (KatA) activity, peroxide resistance and full virulence of Pseudomonas aeruginosa PA14. J Microbiol Biotechnol. 2009; 19(12): 1520–6. [DOI] [PubMed] [Google Scholar]
- 58.Runyen-Janecky L, Daugherty A, Lloyd B, Wellington C, Eskandarian H, Sagransky M. Role and regulation of iron-sulfur cluster biosynthesis genes in Shigella flexneri virulence. Infect Immun. 2008; 76(3): 1083–92. 10.1128/IAI.01211-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Velayudhan J, Karlinsey JE, Frawley ER, Becker LA, Nartea M, Fang FC. Distinct roles of the Salmonella enterica serovar Typhimurium CyaY and YggX proteins in the biosynthesis and repair of iron-sulfur clusters. Infect Immun. 2014; 82(4): 1390–401. 10.1128/IAI.01022-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sambrook J, Green MR. Molecular cloning, a laboratory manual. 4th ed. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- 61.Marx CJ, Lidstrom ME. Broad-host-range cre-lox system for antibiotic marker recycling in gram-negative bacteria. Biotechniques. 2002; 33(5): 1062–7. [DOI] [PubMed] [Google Scholar]
- 62.Srijaruskul K, Charoenlap N, Namchaiw P, Chattrakarn S, Giengkam S, Mongkolsuk S, et al. Regulation by SoxR of mfsA, which encodes a major facilitator protein involved in paraquat resistance in Stenotrophomonas maltophilia. PLoS One. 2015; 10(4): e0123699 10.1371/journal.pone.0123699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Somprasong N, Jittawuttipoka T, Duang-Nkern J, Romsang A, Chaiyen P, Schweizer HP, et al. Pseudomonas aeruginosa thiol peroxidase protects against hydrogen peroxide toxicity and displays atypical patterns of gene regulation. J Bacteriol. 2012; 194(15): 3904–12. 10.1128/JB.00347-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The deduced amino acid sequence of P. aeruginosa (A) FprA ferredoxin NADP(+) reductase A and (B) FinR transcriptional regulator was aligned with those in Pseudomonas putida and Azotobacter vinelandii by using CLASTAL Omega alignment. The asterisk, colon, and period symbols indicate identical residues, conserved substitutions, and semi-conserved substitutions, respectively.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.