

Abstract
Pituitary follicle-stimulating hormone (FSH) is an essential regulator of fertility in females and of quantitatively normal spermatogenesis in males. Pituitary-derived activins are thought to act as major stimulators of FSH synthesis by gonadotrope cells. In vitro, activins signal via SMAD3, SMAD4, and forkhead box L2 (FOXL2) to regulate transcription of the FSHβ subunit gene (Fshb). Consistent with this model, gonadotrope-specific Smad4 or Foxl2 knock-out mice have greatly reduced FSH and are subfertile. The role of SMAD3 in vivo is unresolved; however, residual FSH production in Smad4 conditional knock-out mice may derive from partial compensation by SMAD3 and its ability to bind DNA in the absence of SMAD4. To test this hypothesis and determine the role of SMAD3 in FSH biosynthesis, we generated mice lacking both the SMAD3 DNA binding domain and SMAD4 specifically in gonadotropes. Conditional knock-out females were hypogonadal, acyclic, and sterile and had thread-like uteri; their ovaries lacked antral follicles and corpora lutea. Knock-out males were fertile but had reduced testis weights and epididymal sperm counts. These phenotypes were consistent with those of Fshb knock-out mice. Indeed, pituitary Fshb mRNA levels were nearly undetectable in both male and female knock-outs. In contrast, gonadotropin-releasing hormone receptor mRNA levels were significantly elevated in knock-outs in both sexes. Interestingly, luteinizing hormone production was altered in a sex-specific fashion. Overall, our analyses demonstrate that SMAD3 is required for FSH synthesis in vivo.
Keywords: activin, follicle-stimulating hormone (FSH), gene knock-out, pituitary gland, SMAD transcription factor
Introduction
Pituitary follicle-stimulating hormone (FSH)2 is an essential regulator of gonadal function (1, 2). FSH acts on ovarian granulosa cells to regulate follicle development and on testicular Sertoli cells to regulate spermatogenesis (1, 3, 4). In both humans and rodents, mutations in genes regulating FSH synthesis or action cause primary amenorrhea because of the arrest of follicle development at the pre-antral or early antral stage (5–9). In contrast, the effects of these mutations in males are species-specific. For example, FSH-deficient mice are oligospermic but fertile (1), whereas FSH-deficient men are azoospermic and infertile (10–12).
FSH is a dimeric glycoprotein composed of the chorionic gonadotropin α subunit (CGA) non-covalently linked to the FSHβ subunit (FSHβ). The former is shared with other glycoprotein hormones; the latter is specific to FSH (13–16). Synthesis of the FSHβ subunit is rate-limiting in dimeric FSH production (17–19) and is regulated by several endocrine and autocrine/paracrine factors in the hypothalamic-pituitary-gonadal axis (20, 21). Among these factors, the pituitary-derived activins have been the most thoroughly investigated, both in vitro and in vivo. In recent years, particular focus has been placed on the mechanisms through which activins regulate transcription of the FSHβ subunit gene (Fshb) (16, 22–27).
As members of the TGFβ superfamily, activins signal through complexes of serine/threonine kinase receptors and SMAD signaling proteins (28, 29). According to current in vitro models, activins stimulate the phosphorylation and nuclear accumulation of the receptor-regulated SMAD protein SMAD3 in gonadotrope cells. In the nucleus, phospho-SMAD3 partners with the ubiquitous co-factor SMAD4 and the cell-specific forkhead box L2 (FOXL2) to promote transcription of the murine Fshb gene (30, 31). Specifically, SMAD4 and FOXL2 bind adjacent cis-elements in the proximal Fshb promoter (Fig. 1, top panel, SBE2/FBE2). The two proteins are then linked through their mutual association with the C-terminal Mad homology 2 (MH2) domain of SMAD3 (30, 32). The in vitro model further suggests that SMAD3 can regulate murine Fshb with SMAD4 at a canonical SMAD binding element (SBE1) located 144 bp upstream of SBE2/FBE2 (30) and with FOXL2 via a third response element (FBE1) ∼85 bp upstream of the SBE (33) (Fig. 1, top panel). Importantly, the actions of SMAD3 and SMAD4 at SBE1 appear to be independent of FOXL2, whereas the actions of SMAD3 and FOXL2 at FBE1 are independent of SMAD4 (30, 31). Thus, only at SBE2/FBE2 do all three proteins work together, but SMAD3 represents the common mediator of activin signaling at all three cis-elements (30, 31).
FIGURE 1.

Models of SMAD signaling to the Fshb promoter in murine gonadotrope cells of wild-type and Smad4 knock-out mice. Top panel, in wild-type mice, activins stimulate the formation of complexes of SMAD proteins and FOXL2, which act via at least three cis-elements in the proximal Fshb promoter. SMADs can bind DNA via SBEs, and FOXL2 binds via FBEs. FOXL2 binds the distal FBE1 site and recruits SMAD3 via protein-protein interaction. Complexes of SMAD3 and SMAD4 can bind SBE1, which contains binding sites for both proteins. More proximally, SMAD4 binds SBE2, FOXL2 binds FBE2, and the two proteins are linked through their mutual association with SMAD3. Bottom panel, in mice lacking SMAD4 in their gonadotropes (cKO), SMAD3/SMAD4 binding to SBE1 is lost. However, SMAD3/FOXL2 binding to FBE1 is intact. In the absence of SMAD4, SMAD3 can bind SBE2, enabling activins to stimulate Fshb production, although at reduced levels relative to wild type. MH1, DNA binding domain; MH2, protein-protein interaction domain.
Consistent with the in vitro model, conditional deletion of either Smad4 or Foxl2 from pituitary gonadotrope cells (hereafter, Smad4 cKO and Foxl2 cKO, respectively) produces profound FSH deficiency in mice in vivo (34, 35). Female cKOs are subfertile, producing smaller litters (both models) at reduced frequency (Foxl2 cKO) compared with wild-type littermates (34, 35). This contrasts with the more extreme phenotype of Fshb knock-out females, which are infertile (1). However, it is important to note that in both Smad4 and Foxl2 cKOs, females still produce some FSH, which may be sufficient to drive some ovarian follicle development (34, 35). This residual FSH production may reflect the fact that there are cis-elements in the Fshb promoter where FOXL2 and SMAD4 can act independently of one another (see above and Fig. 1). Consistent with this idea, ablation of Smad4 and Foxl2 together (hereafter, Smad4/Foxl2 cKO), which would affect protein complex binding to all three cis-elements, results in a more extreme FSH deficiency, as well as infertility in female mice (34).
In apparent contrast with the in vitro model, however, mice with a conditional deletion in the Smad3 gene in gonadotropes (hereafter Smad3 cKO) produce FSH at normal levels and are fertile (36). This was a surprising result because, a priori, these mice would be predicted to exhibit a phenotype comparable with that of Smad4/Foxl2 cKOs. The absence of a phenotype is not explained by compensation via the related receptor-regulated SMAD2, because mice with conditional deletions in both Smad2 and Smad3 in gonadotropes exhibit normal fertility and quantitatively normal FSH levels (36). Instead, the absence of a phenotype may be explained by the observation that the conditional deletion in the Smad3 gene does not remove all Smad3 expression (and hence function) in gonadotropes of these mice (36).
In the above described models, the Cre-lox system was used to remove functionally important exons in both the Smad2 and Smad3 genes in gonadotropes (36). Recombination of the Smad2 gene effectively abolished SMAD2 protein expression. In the case of Smad3, however, the floxed region (exons 2 and 3) encodes only one part of the SMAD3 protein: the N-terminal MH1 domain, which mediates SMAD3 binding to DNA (37). In gonadotropes of Smad3 cKO mice, the Smad3 transcript lacked exons 2 and 3, as predicted, but contained the remaining 7 exons (exons 1 and 4–9). Moreover, there was an increase in the abundance of this novel mRNA. Although translation is predicted to start in exon 1 and for the protein to be translated out of frame in exon 4, the translation start in exon 1 does not conform to a consensus Kozak sequence (38). In contrast, such a sequence exists in exon 4, allowing the C-terminal half of SMAD3 to be translated in frame. This includes the entirety of the MH2 domain, which is phosphorylated by activin type I receptors, and interacts with SMAD4 and FOXL2 (39, 40). In vitro, this truncated protein functions identically to full-length SMAD3, in the presence of SMAD4 (36). If this is also the case in vivo, it could explain the absence of an FSH or fertility phenotype in Smad3 cKO mice. This possibility is further supported by the in vitro model (Fig. 1), which indicates that SMAD3 does bind DNA at the FBE1 or SBE2/FBE2 cis-elements. Unfortunately, these data only indirectly suggest a role for SMAD3 in FSH synthesis in vivo.
To definitively rule in or out a role for SMAD3, we need to specifically and completely ablate SMAD3 protein function (including the MH2 domain) in gonadotropes of mice. To our knowledge, there are no other floxed Smad3 strains currently available that would enable this approach. Still, existing models may allow an assessment of SMAD3 function in FSH synthesis. Specifically, residual FSH production in Smad4 cKO mice may be explained by the ability of SMAD3 to partially compensate for the absence of SMAD4 (Fig. 1, bottom panel). In vitro, SMAD3 can bind to SBE2 in the Fshb promoter (30) (Fig. 1). Moreover, SMAD3-FOXL2 can stimulate transcription via SBE2/FBE2, although less potently than SMAD3-SMAD4-FOXL2 (30). If SMAD3 does in fact partially compensate for the absence of SMAD4 at this element and must bind DNA to do so, mice lacking SMAD4 and SMAD3 DNA binding activity should exhibit more extreme FSH deficiency and fertility phenotypes than mice lacking SMAD4 alone. Here, we tested this hypothesis using the Cre-lox system to simultaneously remove SMAD4 and the SMAD3 MH1 domain in gonadotrope cells.
Results
Truncated SMAD3 Stimulates Fshb Transcription in the Presence but Not the Absence of SMAD4 in Vitro
Mice lacking Smad3 exons 2 and 3 express a truncated form of SMAD3, which lacks the DNA binding MH1 domain (hereafter SMAD3-MH2) (36). We proposed that this protein functions equivalently to full-length SMAD3, but only in the presence of SMAD4, enabling transactivation of the Fshb gene. To further test this idea, we compared the activities of full-length SMAD3 and SMAD3-MH2 in a heterologous reporter assay. We co-transfected SW480.7 cells, a SMAD4-deficient human colon adenocarcinoma-derived cell line (41, 42), with a murine Fshb promoter-reporter and the indicated combinations of SMAD3 or SMAD3-MH2, SMAD4, FOXL2, and a constitutively active form of the canonical activin type I receptor (T206D or TD) (43). The latter was used as a surrogate for ligand. Co-transfection of SMAD3 and FOXL2 increased reporter activity, which was significantly stimulated by ALK4TD (Fig. 2). In contrast, the combination of SMAD3-MH2 and FOXL2 failed to turn on the reporter, whether or not ALK4TD was co-transfected. SMAD3 and SMAD3-MH2 were expressed at comparable levels (data not shown). Remarkably, when SMAD4 was introduced, SMAD3 and SMAD3-MH2 were comparable in their abilities to potently stimulate the Fshb reporter in the presence of FOXL2. Thus, as predicted, SMAD3-MH2 can activate the Fshb promoter, but only in the presence of SMAD4 (and FOXL2). These results reinforce the idea that SMAD3-MH2 should be unable to promote Fshb expression in the absence of SMAD4 in vivo.
FIGURE 2.
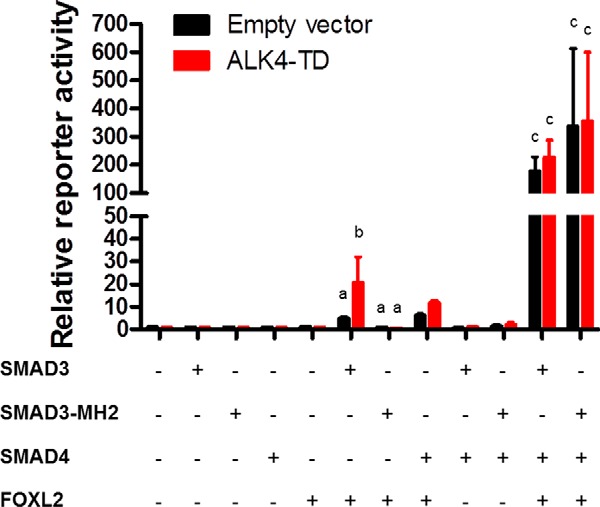
SW480.7 cells were transfected with the −1990/+1 murine Fshb-luc reporter and different combinations of FOXL2, SMAD3, SMAD3-MH2, and/or SMAD4. Cells were co-transfected with a constitutively active activin type I receptor (ALK4-TD, red bars) or empty expression plasmid (pcDNA3.0, black bars). Bars represent the means (±S.E.) of three independent experiments. Reporter activity was normalized to that of cells transfected with empty expression plasmids (first set of bars at left). Two-way analyses of variance were conducted to compare SMAD3/FOXL2 versus SMAD3-MH2/FOXL2 and SMAD3/SMAD4/FOXL2 versus SMAD3-MH2/SMAD4/FOXL2, followed by Bonferroni-corrected multiple comparison tests. Bars with different letters are significantly different from one another, whereas those without letters were not subjected to statistical analysis.
Smad3 and Smad4 Genes Were Efficiently and Specifically Recombined in Gonadotropes
To ablate the SMAD3-MH1 domain and SMAD4 in gonadotropes, we crossed mice harboring floxed alleles for Smad3 and Smad4 (Fig. 3A) with GRIC mice, which express Cre from the GnRH receptor locus. As anticipated, we observed recombination of the floxed alleles in pituitaries of both male and female cKO mice, as well as in the testes and epididymides of cKO males (Fig. 3B). Note that, in addition to gonadotropes, the GRIC allele is active in the male germ line (44). An analysis of gonadotropes (YFP+ cells) isolated from these animals revealed highly efficient depletion of Smad3 (Fig. 3C) and Smad4 (Fig. 3E) mRNAs, when examined using primers against the targeted exons. As reported previously (36), there was an overall up-regulation of the Smad3 mRNA levels in gonadotropes of cKO mice, although this transcript lacked the targeted exons (Fig. 3D). Note that Smad3 and Smad4 mRNA expression in non-gonadotrope cells of the pituitary (YFP−) was equivalent between genotypes (Fig. 3, C–E, white bars). The purity of YFP+ cells was 79.3% for the control and 78.3% for cKO mice, respectively. No YFP+ cells were observed in the YFP− population.
FIGURE 3.

Recombination of Smad3 and Smad4 alleles in mouse gonadotropes. A, schematic representation of the floxed Smad3 and Smad4 alleles pre- and postrecombination with Cre. Exons 2 and 3 of Smad3 and exon 8 of Smad4 were flanked with loxP sites (light gray triangles). Exons are shown as dark gray boxes. B, PCR detection of floxed and recombined Smad3 and Smad4 alleles from the indicated tissues of control and cKO mice. C–E, RT-qPCR analysis of mRNA levels of Smad3 and Smad4 in purified gonadotropes (YFP+) and non-gonadotropes (YFP−) from adult control (YFP/+;GRIC) and cKO (Smad3fx/fx;Smad4fx/fx;YFP/+;GRIC) mice. In C and E, primers were directed against the deleted region of the genes/transcripts. In D, primers were directed against non-targeted exons in Smad3.
cKO Mice Are FSH-deficient
Serum FSH levels were significantly depleted in adult cKO mice relative to littermate controls (Fig. 4, A and B). Similarly, intrapituitary FSH protein content was profoundly reduced in cKO animals as assessed by RIA (Fig. 4, C and D) or immunofluorescence (Fig. 5). Reductions in FSH protein derived from a robust decrease in pituitary Fshb mRNA expression in cKOs (Fig. 6, A and B).
FIGURE 4.

cKO males and females are FSH deficient. A, B, E, and F, FSH (A and B) and LH (E and F) levels in mouse serum were measured by multiplex ELISA. Individual data points are shown (n = 12 or 14 for control and cKO, respectively) as are the group means (horizontal lines). C and D, pituitary FSH content was assessed by RIA (n = 8 or 5 for control and cKO males; n = 11 or 7 for control and cKO females). All samples from cKO females were below the detection limit of the assay. G and H, pituitary LH content was measured by ELISA. The data were analyzed by Student's t test in each panel.
FIGURE 5.

FSH protein expression is greatly diminished in cKO pituitaries. Immunofluorescence staining for LHβ (green) and FSHβ (red) in pituitaries of adult control and cKO males (left panels) and female (right panels) mice. The nuclei are labeled with DAPI (blue). Scale bars, 100 μm.
FIGURE 6.

Fshb expression is abolished in pituitaries of cKO mice. Pituitaries were collected from 10-week-old control and cKO mice (n = 11 or 15 for control and cKO males. A, (n = 12) genotype for females; B, mRNA levels of gonadotropin subunits (Fshb, Lhb, and Cga), GnRH receptor (Gnrhr), Smad3, and Smad4 were measured by RT-qPCR. mRNA levels were normalized to the housekeeping gene Rpl19. Bars reflect group means (±S.E.). The data were analyzed by Student's t test. ***, p < 0.0001; **, p = 0.0004. n.s., not statistically significant.
Male cKO mice also showed diminished serum LH relative to controls (Fig. 4E). In contrast, serum LH was significantly elevated in cKO females (Fig. 4F). Consistent with the sex difference in LH secretion, pituitary Lhb and Cga mRNA levels were modestly decreased in cKO males but markedly increased in cKO females (Fig. 6, A and B). Interestingly, these RNA differences were not reflected at the level of intrapituitary LH protein content (Figs. 4, G and H, and 5). In both sexes, pituitary Gnrhr mRNA expression was up-regulated in cKOs relative to controls (Fig. 6, A and B). Expression of Smad3 or Smad4 did not differ between genotypes of either sex, except for Smad4, which was modestly decreased in cKO males (Fig. 6, A and B). Given the relatively small proportion of gonadotropes in the pituitary (∼5–10%) and the ubiquitous expression of SMADs, we did not expect to detect changes in Smad3 or Smad4 in the analysis of the whole gland, although, as described above, recombination in gonadotropes was highly efficient (Fig. 3).
Impaired Fertility in cKO Females and Males
8-week-old male and female control and cKO mice were paired with age-matched wild-type C57BL/6 mice for a period of 6 (males) or 4 (females) months. Littermate controls and cKOs were mated on the same day. cKO and control males sired equivalent numbers of litters at similar intervals over 6 months (Fig. 7A); however, cKOs fathered smaller litters (Fig. 7B). Note that two of the six cKO males only sired one litter and were excluded from the analysis. Control females were fertile and produced an average of 4.5 litters and 6.25 pups/litter over the course of 4 months. In contrast, cKO females were uniformly sterile; none produced a single pup or showed any overt evidence of pregnancy during the mating trial (Fig. 7C).
FIGURE 7.

Fertility is impaired in cKO mice. Control and cKO males (n = 6 or 4) and females (n = 4) were paired with wild-type C57BL6 mice for 6 or 4 months, respectively. A and B, average number of litters (A) and litter size (B) in control and cKO males. Different colors are used to indicate data from individual animals. C, average litter size in control and cKO females.
Spermatogenesis Is Impaired in cKO Males
Despite having equivalent body mass relative to littermate controls (supplemental Fig. S1A), cKO males exhibited a 50% reduction in testicular mass (Fig. 8, A and B). Cauda epididymal sperm count was similarly reduced by 50% in cKO males relative to controls (Fig. 8C). In contrast, there were no differences between genotypes in progressive sperm motility (Fig. 8D) or seminal vesicle weights (Fig. 8E). Similar results were seen in control and cKO males at the conclusion of the fertility trials (data not shown). Consistent with the normal seminal vesicle weights, but in apparent contrast with the reduced serum LH, cKO males had statistically normal serum testosterone levels (supplemental Fig. S2). Despite impairments in spermatogenesis, cKO males showed overtly normal testicular histology (Fig. 8F).
FIGURE 8.

cKO males are oligospermic. A, testes and seminal vesicles from adult control and cKO males. B, testicular weights of 10-week-old control and cKO males (n = 13/genotype). C, sperm counts from snap frozen epididymides of 10-week-old control and cKO males (n = 9/genotype). D, sperm motility assessment from fresh cauda epididymides in adult control and cKO males (n = 8 or 6 for control and cKO, respectively). E, seminal vesicle weights of 10-week-old control and cKO males (n = 13/genotype). F, H&E staining of testicular sections from representative control and cKO males. Scale bars, 100 μm.
cKO Females Are Acyclic and Hypogonadal
We next examined the underlying cause of infertility in cKO females. Adult cKO females exhibited normal body mass (supplemental Fig. S1B) but were profoundly hypogonadal with hypoplastic uteri (Fig. 9, A–C). The latter condition was emblematic of estrogen deficiency, although we did not measure levels of 17β-estradiol in these animals. In our experience, 17β-estradiol ELISAs lack sufficient sensitivity to accurately measure the hormone on cycle stages other than proestrus in the mouse. Although cKO and control littermates exhibited vaginal opening at roughly the same postnatal age (Fig. 9D), we were unable to reliably measure estrous cyclicity in cKO females (data not shown). Indeed, as indicated in ovarian tissue sections, cKO females showed an arrest in follicle development at the pre-antral or early antral stage and the complete absence of corpora lutea (Fig. 9E). cKO mice also showed impaired responsiveness (ovulation) to exogenous gonadotropins when treated as juveniles (postnatal days 24–28; Fig. 9F).
FIGURE 9.

cKO females do not ovulate. A, morphology of ovaries and uteri from adult control and cKO females. B and C, ovary and uterus weights from 10-week-old control and cKO females (n = 12/genotype). D, day of vaginal opening in control and cKO females (n = 6/genotype). E, H&E-stained ovaries from representative 10-week-old control and cKO females. Corpora lutea (CL) are labeled. Scale bars, 100 μm. F, COCs collected after exogenous gonadotropin stimulation in juvenile control and cKO females (n = 18 or 7 for control and cKO, respectively).
Basal and Activin A-stimulated Fshb Expression Are Reduced in cKO Pituitaries
To more directly assess the effects of the targeted mutations on activin signaling in gonadotropes, we cultured pituitary cells from male and female cKO and control mice. Basal Fshb mRNA expression in murine pituitary cultures depends on activins or related ligands (45). Here, basal Fshb expression was almost completely abolished in cKO cultures relative to controls (Fig. 10, A and B). Activin A induced significant increases in Fshb expression in gonadotropes of cultures from both control males and females (Fig. 10, A and B). In contrast, activin A modestly stimulated Fshb in cultures from male but not female cKOs (Fig. 10, A and B).
FIGURE 10.

Basal and activin A-stimulated Fshb expression were abolished in cKO pituitary cells. Primary pituitary cells were prepared from adult male (A) or female (B) control or cKO mice and treated with 1 nm activin A (red) or with vehicle (black). Fshb mRNA levels were measured by RT-qPCR. Bars represent the means (+ S.E.) of three independent experiments. Bars with different symbols (a, b, c, and d) differ significantly.
Discussion
SMAD3 Regulates FSH Synthesis in Vivo
The data presented here provide the first conclusive evidence that SMAD3 regulates FSH synthesis in vivo. Previously, we reported that Smad3 cKO mice are fertile with normal FSH synthesis (36). However, these animals express a truncated and functional form of SMAD3 (i.e. the C-terminal MH2 domain) that lacks DNA binding activity (i.e. the N-terminal MH1 domain) but can still cooperatively regulate Fshb promoter activity with SMAD4 and FOXL2 (Fig. 2). Although we will eventually require a model that removes all SMAD3 function in gonadotropes, here we were able to combine two existing mouse strains to probe the role of SMAD3 in FSH synthesis: the Smad3 mice described above (lacking the MH1 domain) and mice lacking Smad4 in gonadotropes (34).
The latter have reduced FSH and are subfertile (34). We argued that the residual FSH in these animals results from partial compensation by full-length SMAD3 and its ability to bind DNA via its MH1 domain (37) (Fig. 1, bottom panel). If this is true, then mice lacking the SMAD3 MH1 domain should be unable to compensate for the absence of SMAD4. Indeed, as we show here, Fshb mRNA and FSH protein are essentially undetectable in these mice. As a result, females are sterile; males are oligospermic and have small testes. These phenotypes are consistent with those of Fshb knock-outs (1). Our data therefore suggest that SMAD3 regulates FSH synthesis in vivo. They also suggest that the MH2 domain of SMAD3 is more critical for Fshb regulation than its MH1 domain and that SMAD complex binding to the Fshb promoter is mediated principally via SMAD4.
We contend that the SMAD3-MH1 domain plays a lesser role than its MH2 domain in Fshb regulation for at least two reasons. First, mice lacking the SMAD3-MH1 domain produce FSH at seemingly normal levels (36). A role for the SMAD3-MH1 domain in FSH synthesis is only observed when SMAD4 is absent (as demonstrated here). Second, the SMAD3-MH2 domain is phosphorylated by the type I receptor (in response to ligand binding and receptor complex activation) and mediates the protein interactions with both SMAD4 and FOXL2 (32, 39, 40, 46, 47). Based on these observations, one could argue that SMAD3 may not need to directly bind the Fshb promoter to regulate its transcription under normal conditions. Instead, SMAD complex binding activity is conferred principally by SMAD4 and FOXL2.
If this is true, the 8-bp SMAD binding element (SBE1 in Fig. 1) in the proximal promoter may not play a significant role in FSH synthesis in vivo, because SMAD3-SMAD4 complex binding to this site requires DNA binding activity of both proteins, at least in vitro (31). The role, if any, of this cis-element in vivo should be assessed, perhaps with the aid of newer gene editing technologies (e.g. CRISPR-Cas9). Interestingly, although this was the first activin-responsive cis-element described in the Fshb promoter (25), it has only been observed in rodents thus far (16). The Fshb promoters of other species, such as sheep and pigs, can be robustly stimulated by activins in the absence of this 8-bp SBE (24, 48, 49). Moreover, adding this element to the human promoter only modestly increases its activin and SMAD sensitivity (31). It is perhaps unsurprising then that it might not play an essential role in mice. Indeed, even point mutations in this element in in vitro promoter-reporter assays only partly attenuate activin responsiveness (17, 30, 31).
A Potential Role for SMAD2 in FSH Synthesis?
Our contention that the SMAD3-MH2 domain is essential for FSH synthesis is challenged by earlier findings. To date, four mouse strains with targeted mutations in Smad3 have been described. As we argued previously (36), we would predict that mice in two (50, 51), if not three (52), of these strains likely produce Smad3 transcripts that encode the MH2 domain, akin to what we describe in the present model. A fourth strain (hereafter Smad3Δex8), however, lacks exon 8 (of the 9 exon gene), and as a result, the terminal 89 amino acids of the MH2 domain of the protein (53). This includes the critical C-terminal serines that are phosphorylated by type I receptors. FSH has been measured in these mice, but not in great detail and with seemingly conflicting results. Female Smad3Δex8 mice appear to have increased circulating FSH, which may result from the loss of estrogen and/or inhibin feedback (i.e. these animals also have ovarian defects (54)). In contrast, pituitary Fshb mRNA levels are reduced by ∼30% in 8-week-old Smad3Δex8 males (55). Although the basis of the sex difference is not yet clear, the fact remains that these mice lack a functional SMAD3-MH2 domain and can still produce FSH protein or transcript at slightly reduced to elevated levels.
The prediction of our model is that a complete loss of SMAD3 (and in particular the MH2 domain) should abrogate FSH production, as we see in Smad4/Foxl2 cKO mice (34) and the Smad3/4 cKO animals described here. The Smad3Δex8 mice challenge this idea and raise the possibility that SMAD2 might compensate for the loss of functional SMAD3 in these animals. This is plausible for a few reasons. First, SMAD2 is abundantly expressed in gonadotropes and is phosphorylated in response to activin stimulation, at least in immortalized gonadotropes (22, 25). Second, SMAD2 interacts with both SMAD4 and FOXL2 (32, 39). The latter interaction appears to be weaker than the association between SMAD3 and FOXL2 but is still measureable. Moreover, the interaction might be enhanced in the absence of SMAD3. Recall that Smad3Δex8 mice would not express a functional SMAD3-MH2 domain, which is required for the FOXL2 interaction. Third, SMAD2 was previously discounted because it lacks DNA binding activity (36). However, as we discuss above, SMAD3 does not need to bind to DNA to regulate Fshb. Thus, if SMAD2 is indeed partially compensating for the loss of SMAD3 function in Smad3Δex8 mice, then crossing these animals with those lacking SMAD2 in gonadotropes (36) should lead to severe FSH deficiency.
We would still argue that, under normal circumstances, SMAD2 plays little to no role in Fshb regulation. Instead, SMAD3 is likely the primary transducer of type I receptor activation to the Fshb promoter. This concept is supported by the absence of an FSH phenotype in gonadotrope-specific Smad2 knock-out mice (36). Rather, the ability of SMAD2 to regulate Fshb may only be manifested when SMAD3 function is compromised, as in Smad3Δex8 mice. If this is true, it begs the question of why SMAD2 does not compensate in the Smad3/4 cKO mice described here. The simple answer is likely because it lacks DNA binding activity. That is, in Smad4 cKO mice, SMAD3 can partially compensate because it can bind DNA via its MH1 domain. In Smad3/4 cKO mice, the inability of SMAD2 to bind DNA and its weaker FOXL2 interaction would likely preclude compensation. There is a naturally occurring splice variant of SMAD2 that can bind DNA, but it is expressed at far lower levels than the full-length isoform (22, 25, 56, 57).
Shared and Unique Phenotypes of Smad3/4 versus Smad4/Foxl2 Knock-out Mice
The phenotypes of Smad3/4 cKO mice are highly similar but not identical to those of Smad4/Foxl2 cKO mice (34) (Table 1). In both models, FSH production is severely impaired, leading to oligospermia in males and sterility in females. Females exhibit a block in folliculogenesis at the pre-antral or early antral stage akin to what was described in Fshb knock-out mice (1). In both models, LH secretion and Lhb mRNA expression are increased in females. Cga mRNA expression and LH secretion are reduced in males of both models. Interestingly, however, Lhb mRNA levels are decreased in Smad3/4 cKO males but elevated in Smad4/Foxl2 cKO males. Lhb mRNA is not altered in males lacking Smad4 alone, suggesting that SMAD3 and FOXL2 play distinct roles in Lhb regulation. At present, there is no evidence that FOXL2 regulates Lhb expression (35). However, SMAD3 can potentiate the stimulatory effects of EGR1 (a mediator of GnRH action) on Lhb promoter activity (58) in vitro, and this effect is dependent upon SMAD binding elements (55). Therefore, the loss of SMAD3 binding to the Lhb promoter in Smad3/4 cKO males may contribute to the reduction in Lhb mRNA expression. Unfortunately, we did not measure Lhb mRNA in mice lacking SMAD3-MH1 (36), so it is unclear whether the down-regulation depends on coordinated loss of both SMAD3 and SMAD4 DNA binding activity. Moreover, it is not yet clear why Lhb levels are reduced in Smad3/4 cKO males but elevated in females, although this may reflect sex-specific endocrine effects.
TABLE 1.
Pituitary gene expression profiles in different conditional knockout models
The following abbreviations and symbols are used in the table: ND, not determined; ↑, increased in cKO relative to control (more arrows indicate larger effects); ↓, decreased in cKO relative to control; =, equivalent between cKO and control; +++, comparable litter sizes and frequency to controls; ?, males reproduce, but their fertility was not systematically characterized; ++, smaller litters than controls; +, smaller litters and at lower frequency than controls; −, sterile.
| Genotype |
Fshb |
Lhb |
Cga |
Gnrhr |
Fertility |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | Male | Female | Male | Female | Male | Female | |
| Fshb KO | ↓↓ | ↓↓ | = | ↑ | ND | ND | ND | ND | ? | − |
| Smad2/3 cKO | = | = | ND | ND | ND | ND | ND | ND | +++ | +++ |
| Foxl2 cKO | ↓ | ↓ | = | = | = | = | = | = | ++ | + |
| Smad4 cKO | ↓ | ↓ | = | = | ↓ | = | ↑ | ↑ | +++ | ++ |
| Smad4/Foxl2 cKO | ↓↓ | ↓↓ | ↑ | ↑ | ↓ | = | = | = | +++ | − |
| Smad3/Smad4 cKO | ↓↓ | ↓↓ | ↓ | ↑ | ↓ | ↑ | ↑ | ↑ | ++ | − |
Another major difference between Smad3/4 and Smad4/Foxl2 cKO models is in pituitary expression of Gnrhr, which is elevated in the former but unchanged in the latter. The Gnrhr is similarly up-regulated in pituitaries of mice lacking Smad4 alone (34) but is unaltered in mice lacking Foxl2 alone (35). These data suggest that the loss of Smad4 likely underlies the increase in Gnrhr in Smad3/4 cKO mice, although we cannot rule out the effects of the loss of SMAD3 DNA binding activity, because we did not measure Gnrhr in mice with the isolated Smad3 deletion (36). Interestingly, Gnrhr up-regulation is not observed in pituitaries cultured from Smad3/4 cKO males or females relative to controls (supplemental Fig. S3, E and F). This suggests that endocrine factors (e.g. GnRH) contribute to the increase in gene expression observed in vivo. As such, neither SMAD4 nor the SMAD3-MH2 domain appears to regulate Gnrhr expression directly.
In conclusion, the data presented here indicate that: 1) SMAD3 regulates FSH synthesis by gonadotrope cells in vivo, 2) FSH production in Smad4-deficient mice is partly rescued by full-length SMAD3, 3) SMAD4 DNA binding activity plays a more essential role in transcriptional regulation of Fshb than does SMAD3 DNA binding activity, 4) SMAD3/SMAD4 binding to the 8-bp SBE at −266/−259 of the murine Fshb promoter (SBE1) may be dispensable for transcription regulation of the gene, and 5) SMAD3 and SMAD4 play essential roles in Fshb expression in vivo. The gene deletions do not appear to markedly perturb gonadotrope cell development because expression of other gonadotrope-specific genes such as Gnrhr, Lhb, and Cga is largely intact. Because SMAD3 and SMAD4 are mediators of TGFβ superfamily signaling and have not previously been implicated in GnRH action, these data indicate that activins play a dominant role over GnRH in the regulation of FSH and/or that GnRH action is somehow dependent upon a functional activin signaling cascade.
Experimental Procedures
Materials
Pregnant mare's serum gonadotropin (or eCG, G4877), human chorionic gonadotropin (C1063), hyaluronidase (H3884), BSA (A4378), M199 medium with Hanks' salt (M7653), avertin (2,2,2-Tri bromoethanol, T48402), and paraformaldehyde (PFA, P6148) were from Sigma-Aldrich. EvaGreen (AC814737H) was from Applied Biological Materials Inc. (Richmond, Canada). Polyethylenimine (23966) was from Polysciences Inc. (Warrington, PA). RNasin (0000183771), Moloney murine leukemia virus reverse transcriptase (0000172807), DNase (0000156360), and random hexamer primers (0000184865) were from Promega Corporation (Madison, WI). TRIzol® reagent (15596026), FBS (10438026), Alexa Fluor 488 donkey anti-goat (A11055), Alexa Fluor 594 donkey anti-rabbit antibodies (A21207), ProLong® Gold Antifade reagent with DAPI (1266174), o-Phenylenediamine (002003) were from Life Technologies Inc. dNTPs (800-401-TL), HBSS (311-511-CL), and DMEM (319-005-CL) were from Wisent Inc. (St-Bruno, Canada). Rabbit anti-FSHβ antibody (AFP7798_1289P) was from the National Hormone & Peptide Program, NIDDK, National Institutes of Health (Bethesda, MD). Goat anti-Lutropin β (LHβ) antibody (sc-7824) used in immunofluorescence was from Santa Cruz Biotechnologies Inc. (Dallas, TX). Tissu-Tek® optimal cutting temperature (OCT, 4583) compound was from Sakura Finetek Europe B.V. (Alphen aan den Rijn, Netherlands). Recombinant activin A (338-AC-050) was from R&D Systems. Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA). Total RNA mini kit (FA32808-PS) was from Geneaid (New Taipei City, Taiwan). Adenoviruses expressing GFP (Ad-GFP) and Cre-IRES-GFP (Ad-Cre) were from Baylor College of Medicine Vector Development Laboratory (Houston, TX). Isoflurane (CP0406V2) was purchased from Fresenius Kabi (Homburg, Germany). Mouse LH reference (AFP5306A) and rabbit anti-LH (AFP240580Rb) used in the LH ELISA were provided by Dr. Parlow (National Hormone & Peptide Program). Monoclonal anti-bovine LH mAb (51887) used in the LH ELISA was provided by Dr. Roser (University of California-Los Angeles). Donkey anti-rabbit HRP (P0448) was purchased from Dako Canada ULC (Mississauga, Canada).
Constructs
The full-length and SMAD3-MH2 expression constructs were described previously (36). To circumvent expression differences between full-length and truncated SMAD3 constructs, we introduced a mutation in the start codon in exon 1 of the latter to bias translation initiation toward the AUG in exon 4. We used the QuikChange site-directed mutagenesis protocol and the following primer set on the MH2 expression vector (forward, CTT CGG TGC CAG CCT TGT CGT CCA TCC TG; reverse, CAG GAT GGA CGA CAA GGC TGG CAC CGA AG). The constitutively active ALK4 expression vector (T206D) and −1990/+1 mFshb-luc reporter were described previously (22, 43).
Cell Culture, Transfection, and Promoter-Reporter Assays
SW480.7 cells (human SMAD4-deficient colon carcinoma; gift from Dr. Fang Liu, Rutgers University, NJ) (41, 42) were cultured in DMEM with 10% FBS (v/v) at 37 °C and 5% CO2. For promoter-reporter experiments, the cells were seeded in 48-well plates at a density of 70,000 cells/well. Approximately 24 h after seeding, the cells were transfected with 225 ng/well of the −1990/+1 murine Fshb-luc reporter plasmid, along with 4.15 ng of FOXL2 and/or 25 ng of the indicated SMAD expression constructs using polyethylenimine in serum-free medium for 2 h. Where indicated, ALK4-TD was co-transfected at 10 ng/well. After transfection, serum-free medium was changed to complete medium. 48 h later, the cells were washed with PBS. The lysates were prepared (28, 59), and luciferase assays were performed as described (60) on an Orion II microplate luminometer (Berthold detection systems, Oak Ridge, TN). All conditions were performed in duplicate wells in three independent experiments.
Animals
The Smad3fx/fx, Smad4fx/fx, and GnrhrIRES-Cre/IRES-Cre (hereafter GRIC) mice were described previously (61–63). Smad3fx/fx were crossed with Smad4fx/fx animals to generate Smad3fx/+;Smad4fx/+ progeny, which were then crossed to produce Smad3fx/fx;Smad4fx/fx animals. GRIC females were crossed to Smad3fx/fx;Smad4fx/fx males. Their female GRIC/+;Smad3fx/+;Smad4fx/+ progeny were again crossed to Smad3fx/fx;Smad4fx/fx males to produce GRIC/+;Smad3fx/fx;Smad4fx/+ females. Conditional Smad3/4 double knock-outs (GRIC/+;Smad3fx/fx;Smad4fx/fx or cKO) and control (Smad3fx/fx;Smad4fx/fx) littermates were finally generated by crossing Smad3fx/fx;Smad4fx/fx males with GRIC/+;Smad3fx/fx;Smad4fx/+ females. To enable gonadotrope purification, Gt(ROSA)26Sortm1(EYFP)Cos/J males (hereafter ROSA26YFP/YFP) were crossed with GRIC/+;Smad3fx/fx;Smad4fx/+ females mice to generate GRIC/+;Smad3fx/+; Smad4fx/+;ROSA26YFP/+ mice (64). GRIC/+;Smad3fx/+;Smad4fx/+;ROSA26YFP/+ females were then crossed with Smad3fx/fx;Smad4fx/fx males to obtain experimental GRIC/+;Smad3fx/fx;Smad4fx/fx;ROSA26YFP/+ mice, in which gonadotropes deficient in SMAD3 and SMAD4 expressed enhanced YFP. Genotyping primers are listed in Table 2. All animal experiments were performed in accordance with institutional and federal guidelines and were approved by the McGill University and Goodman Cancer Centre Facility Animal Care Committee (protocol 5204).
TABLE 2.
Genotyping and qPCR primers
| 5′ → 3′ | |
|---|---|
| Genotyping | |
| Smad3 | |
| Forward | CTCCAGATCGTGGGCATACAGC |
| Reverse | GGTCACAGGGTCCTCTGTGCC |
| Recombined | TCGTCGATCGACCTCGAATAAC |
| Smad4 | |
| Forward | GGGCAGCGTAGCATATAAGA |
| Reverse | GACCCAAACGTCACCTTCAG |
| Recombined | AAGAGCCACAGGTCAAGCAG |
| GRIC | |
| Forward | GGACATGTTCAGGGATCGCCAGGC |
| Reverse | GCATAACCAGTGAAACAGCATTGCTG |
| ROSA26 eYFP | |
| Forward | AAAGTCGCTCTGAGTTGTTAT |
| WT reverse | GCGAAGAGTTTGTCCTCAACC |
| eYFP reverse | GGAGCGGGAGAAATGGATATG |
| qPCR | |
| Smad3 exons 3/4 | |
| Forward | CATTCCATTCCCGAGAACAC |
| Reverse | ATGCTGTGGTTCATCTGGTG |
| Smad3 exons 8/9 | |
| Forward | CATCCGTATGAGCTTCGTCA |
| Reverse | CATCTGGGTGAGGACCTTGT |
| Smad4 | |
| Forward | TCACAATGAGCTTGCATTCC |
| Reverse | CCATCCACAGTCACAACAGG |
| Fshb | |
| Forward | GTGCGGGCTACTGCTACACT |
| Reverse | CAGGCAATCTTACGGTCTCG |
| Rpl19 | |
| Forward | CGGGAATCCAAGAAGATTGA |
| Reverse | TTCAGCTTGTGGATGTGCTC |
| Gnrhr | |
| Forward | CACGGGTTTAGGAAAGCAAA |
| Reverse | TTCGCTACCTCCTTTGTCGT |
| Lhb | |
| Forward | AGCAGCCGGCAGTACTCGGA |
| Reverse | ACTGTGCCGGCCTGTCAACG |
| Cga | |
| Forward | TCCCTCAAAAAGTCCAQGAGC |
| Reverse | GAAGAGAATGAAGAATATGCAG |
FACS
Cell sorting was performed at the Cell Vision Core Facility for Flow Cytometry and Single Cell Analysis of the Life Science Complex at the Rosalind & Morris Goodman Cancer Research Centre. Pituitary cell suspensions were prepared from 14 GRIC/+;Smad3fx/fx;Smad4fx/fx;ROSA26YFP/+ male and female animals as described previously (35), and single cell suspensions were prepared in PBS. Fourteen GRIC/+;ROSA26YFP/+ male and female mice were used as controls. The cells were then passed through a 70-μm nozzle at 70 p.s.i. into a Becton Dickinson FACSAria (software version 8.0.1). The pituitary cells were then run and gated to sort YFP+ (gonadotropes) and YFP− (non-gonadotropes) cells from both control and experimental pituitaries. We obtained 1.7 × 104 YFP+ and 7.1 × 105 YFP− cells for the controls; 1.4 × 104 YFP+ and 6.8 × 105 YFP− cells for the experimental animals. Sorting purity was assessed by culturing 5 μl of both YFP+ and YFP− cells from control and cKO pituitaries in one well of a 96-well plate with M199 medium. 12 h later, the YFP+/− ratio was calculated in each group. Total RNA was extracted from YFP+ and YFP− cells using a total RNA mini kit according to the manufacturer's instructions. Pituitary selective gene expression was assessed by RT-qPCR.
Hormone Analyses
Blood was collected by cardiac puncture and allowed to clot at room temperature for 20 min. Serum was obtained following centrifugation at 3000 × g and stored at −20 °C until analysis. To measure pituitary FSH and LH content, pituitaries were isolated and homogenized in 300 μl of PBS, sonicated, and centrifuged for 10 min at 16,000 × g at 4 °C. Supernatant was removed and kept at −20 °C until assayed. All hormone assays were performed at the Ligand Assay and Analysis Core of the Center for Research in Reproduction at the University of Virginia (Charlottesville, VA), except for pituitary LH content (below). Serum FSH and LH levels were measured by multiplex ELISA. The reportable ranges were 2.40–300 and 0.24–30 ng/ml for FSH and LH, respectively. Intra-assay CVs were <5.0%. Testosterone was measured by ELISA. The reportable range was 10–1600 ng/ml, and the intra-assay CV was >20%. For pituitary FSH content, all samples were diluted 50 times and then measured by radioimmunoassay. The reportable range was 2.9–40 ng/ml. For pituitary LH content, all homogenized samples were diluted 106 times in 0.05% PBST supplemented with 0.2% BSA and measured with an in-house sandwich ELISA as described previously (65).
Immunofluorescence
10–12-week-old male and female animals were anesthetized with 200 mg/kg avertin by i.p. injection followed by an overdose of isoflurane through a nose cone. The animals were transcardially perfused with PBS followed by freshly made 4% PFA in PBS. Pituitaries were isolated and postfixed in 4% PFA for 3 h at room temperature. After a brief rinse with PBS, pituitaries were immersed in 30% sucrose overnight at 4 °C and then embedded in OCT on dry ice. Pituitaries were sectioned (5 μm) on a cryostat and mounted on prechilled Micro slides. The slides were then warmed up to room temperature, and sections were blocked in 1.5% donkey serum in PBS for 1 h. The sections were washed three times in 0.2% PBST and then incubated with rabbit anti-rat FSHβ (1:500) and goat anti-LHβ (1:500) overnight at room temperature. After three washes with PBST, the sections were incubated with Alexa Fluor 594-conjugated donkey anti-rabbit (1:600) and Alexa Fluor 488-conjugated donkey anti-goat (1:600) secondary antibodies for 1 h at room temperature. After another three washes with PBST, coverslips were mounted with ProLong Gold antifade reagent with DAPI and dried at 37 °C for 15 min in the dark. Images were acquired with an Axiocam 506 color camera using Zeiss blue edition (Zen 2 lite) software on a Zeiss Axio Imager M2 microscope.
RT-qPCR
Pituitaries from 10-week-old mice were extracted and immediately frozen in liquid nitrogen and stored at −80 °C. Control females were euthanized at 7 a.m. on the morning of estrus (as assessed by vaginal cytology). cKO females, which did not cycle, were euthanized the same day as control littermates. Samples were homogenized in 500 μl of TRIzol reagent, and the total RNA was extracted according to the manufacturer's protocol. RNA concentration was determined by NanoDrop. Two μg of RNA per sample were then reverse transcribed into cDNA using Moloney murine leukemia virus reverse transcriptase and random hexamer primers at a final volume of 40 μl. Two μl of cDNA were then used as template for qPCR analysis as described previously (22) on a Corbett Rotorgene 600 instrument (Corbett Life Science) using EvaGreen reagent and primers listed in Table 2. mRNA levels of target genes were determined using the 2−ΔΔCt method. Ribosomal protein L19 (Rpl19) was used as a control for normalization.
Puberty Onset, Estrous Cyclicity, and Fertility Assessment
To assess puberty onset, the females were monitored daily for vaginal opening starting on postnatal day 24 (66). Starting at 8 weeks of age, estrous cyclicity was assessed daily in the afternoon (∼3 p.m.) for 2–3 weeks by collecting vaginal cells with a cotton swab wet with sterile saline. The cells were smeared on a glass slide, stained with 0.1% methyl blue, and examined by light microscopy. Staging was assessed according to published guidelines (66). One cycle was defined as the sequential appearance of all estrous cycle stages, regardless of the number of days spent in each stage. To assess fertility, 8-week-old males (n = 6/genotype) and females (n = 4/genotype) were paired with age-matched, wild-type C57BL/6 mice (Charles River) for 6 or 4 months, respectively. Breeding cages were monitored daily, and the date of birth and number of newborn pups were recorded. The pups were removed from the breeding couple at postnatal day 15. At the end of the mating trials, control and cKO animals were dissected; blood samples were collected as described above. Pituitaries were isolated, snap frozen, and kept at −80 °C until processing for RNA extraction using TRIzol. One ovary or testis was collected, weighed, and fixed in 10% formalin (ovaries) or Bouin's buffer (testes).
Sperm Counting
Epididymal sperm heads were counted using a hemacytometer as described previously (35). Briefly, the left epididymis of the animal (n = 9/genotype) was immediately snap frozen in liquid nitrogen, weighed, and then homogenized (2 × 15 s, separated by 30 s) on ice in 2 ml 0.9% NaCl, 0.1% Thimerosal, and 0.5% Triton X-100 with a Polytron on power setting 4. The samples were then diluted 20 times in PBS. Ten μl of each sample were loaded and counted on each side of a hemacytometer.
Sperm Motility Analysis
Sperm motility of 10–12-week-old male control (n = 8) and cKO (n = 6) mice was analyzed using computer-assisted sperm analysis, as described previously (67). All materials and tools were prewarmed and kept at 37 °C before and during all steps. The left side cauda epididymis of each animal was clamped off and briefly rinsed in prewarmed Hanks' salt M199 medium supplemented with 0.5% (w/v) BSA and adjusted to pH 7.4. The tissue was cut 10 times with fine-point scissors, and sperm were allowed to disperse in fresh M199 medium with BSA for 5 min in a 35-mm Petri dish. The sperm suspension was then diluted four times before loading into a 2X-CEL 80-μm deep sperm analysis chamber. Subsequently, 20 μl of diluted sperm sample was analyzed using a Hamilton-Thorne IVOS automated semen analyzer (Hamilton-Thorne Biosciences, Beverly, MA). IVOS parameters were set according to the standards determined by The Jackson Laboratory. Two slides were analyzed per sample, and five views from each slide were analyzed.
Reproductive Organ Histology
Ovaries and testes were isolated from 10-week-old control and cKO females and males, respectively, weighed on a precision balance and fixed in 10% formalin or Bouin's buffer at room temperature. Tissues were then paraffin-embedded and cut into 5-μm sections. The sections were stained with H&E. The images were acquired with Leica DFC310 FX 1.4-megapixel digital color camera with a high quality transmitted light microscope (Leica DM1000 LED) using Leica Application Suite version 4.0.0 software.
Superovulation
Superovulation was performed in juvenile (postnatal days 23–28) females as described previously (35). Control and cKO females were injected i.p. with 5 IU of pregnant mare's serum gonadotropin at 5 pm. 48 h later, the mice were treated i.p. with 5 IU of human chorionic gonadotropin. After 14 h, mice were killed, and cumulus-oocyte complexes (COCs) were harvested in PBS from ampullae of the oviduct. COCs were enzymatically dissociated using 0.5 mg/ml hyaluronidase for 10 min at 37 °C. Oocyte number was then counted under an inverted microscope.
Primary Pituitary Cultures
Primary pituitary cultures were prepared as previously described (68). Briefly, pituitaries were isolated from adult control and cKO animals, single suspended cells were seeded at a density of 300,000 cells/well in 48-well plates. The cells were allowed to settle for 16 h in M199 culture medium supplement with 10% FBS. The media were then replaced with 2% FBS-containing media in the presence or absence of 1 nm activin A. Note that cultures from males and females were prepared and processed separately. Culture media were removed, the cells were harvested, and RNA was extracted using a total RNA mini kit following the manufacturer's instructions.
Statistical Analysis
All of the data were analyzed by unpaired Student's t tests except pituitary gene expression in the primary culture experiments, which was analyzed by multiple t tests. For the reporter assay, two-way analyses of variance were conducted, followed by Bonferroni-corrected multiple comparison tests. Statistical analyses were performed using GraphPad Prism 6 (GraphPad). Values of p < 0.05 were considered statistically significant.
Author Contributions
Y. L. and D. J. B. were responsible for the experimental design, data analyses, and manuscript preparation. Y. L. conducted the majority of the experiments. G. S. conducted the promoter-reporter assays and some qPCR experiments and edited the manuscript. U. B., C.-X. D., and J. G. generated the Cre and floxed Smad mouse strains and edited the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Drs. Bernard Robaire and Océane Albert for providing access to and training on the sperm computer-assisted sperm analysis system; Xiang Zhou for assistance with the cell sorting; Drs. Derik Steyn and Chirine Toufaily for help in establishing the in-house LH assay; Dr. Alfredo Ribeiro-da-Silva and Noosha Yousefpour for providing access to and training on the Zeiss Axio Imager M2 microscope.
This work was supported by Canadian Institutes of Health Research Operating Grant MOP-133394 (to D. J. B.), Natural Sciences and Engineering Research Council of Canada Discovery Grant 2015-05178 (to D. J. B.), and a Samuel Solomon Fellowship in Endocrinology from McGill University (to Y. L.). The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by Eunice Kennedy Shriver NICHD/National Institutes of Health (National Centers for Translational Research in Reproduction and Infertility) Grant P50-HD28934. The Cell Vision Core Facility for Flow Cytometry and Single Cell Analysis at the McGill Life Science Complex was supported by funding from the Canadian Foundation for Innovation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S3.
- FSH
- follicle-stimulating hormone
- CGA/Cga
- chorionic gonadotropin α
- cKO
- conditional knock-out
- COC
- cumulus-oocyte complex
- FOXL2/Foxl2
- forkhead box L2
- fx
- floxed
- Gnrhr
- gonadotropin-releasing hormone receptor
- GRIC
- GnRHR-IRES-Cre
- IRES
- internal ribosomal entry site
- LH
- luteinizing hormone
- MH
- Mad homology domain
- PFA
- paraformaldehyde
- Rpl19
- ribosomal protein L19
- SBE
- SMAD binding element
- qPCR
- quantitative PCR
- H&E
- hematoxylin and eosin
- FBE
- forkhead binding element.
References
- 1. Kumar T. R., Wang Y., Lu N., and Matzuk M. M. (1997) Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 15, 201–204 [DOI] [PubMed] [Google Scholar]
- 2. Matthews C. H., Borgato S., Beck-Peccoz P., Adams M., Tone Y., Gambino G., Casagrande S., Tedeschini G., Benedetti A., and Chatterjee V. K. (1993) Primary amenorrhoea and infertility due to a mutation in the β-subunit of follicle-stimulating hormone. Nat. Genet. 5, 83–86 [DOI] [PubMed] [Google Scholar]
- 3. Simoni M., Gromoll J., and Nieschlag E. (1997) The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr. Rev. 18, 739–773 [DOI] [PubMed] [Google Scholar]
- 4. Wreford N. G., Rajendra Kumar T., Matzuk M. M., and de Kretser D. M. (2001) Analysis of the testicular phenotype of the follicle-stimulating hormone β-subunit knockout and the activin type II receptor knock-out mice by stereological analysis. Endocrinology 142, 2916–2920 [DOI] [PubMed] [Google Scholar]
- 5. Abel M. H., Wootton A. N., Wilkins V., Huhtaniemi I., Knight P. G., and Charlton H. M. (2000) The effect of a null mutation in the follicle-stimulating hormone receptor gene on mouse reproduction. Endocrinology 141, 1795–1803 [DOI] [PubMed] [Google Scholar]
- 6. Dierich A., Sairam M. R., Monaco L., Fimia G. M., Gansmuller A., LeMeur M., and Sassone-Corsi P. (1998) Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc. Natl. Acad. Sci. U.S.A. 95, 13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huhtaniemi I. T., and Themmen A. P. (2005) Mutations in human gonadotropin and gonadotropin-receptor genes. Endocrine 26, 207–217 [DOI] [PubMed] [Google Scholar]
- 8. Themmen A. P. (2005) An update of the pathophysiology of human gonadotrophin subunit and receptor gene mutations and polymorphisms. Reproduction 130, 263–274 [DOI] [PubMed] [Google Scholar]
- 9. Trarbach E. B., Silveira L. G., and Latronico A. C. (2007) Genetic insights into human isolated gonadotropin deficiency. Pituitary 10, 381–391 [DOI] [PubMed] [Google Scholar]
- 10. Clark A. D., and Layman L. C. (2003) Analysis of the Cys82Arg mutation in follicle-stimulating hormone β (FSHβ) using a novel FSH expression vector. Fertil. Steril. 79, 379–385 [DOI] [PubMed] [Google Scholar]
- 11. Layman L. C., Porto A. L., Xie J., da Motta L. A., da Motta L. D., Weiser W., and Sluss P. M. (2002) FSHβ gene mutations in a female with partial breast development and a male sibling with normal puberty and azoospermia. J. Clin. Endocrinol. Metab. 87, 3702–3707 [DOI] [PubMed] [Google Scholar]
- 12. Lindstedt G., Nyström E., Matthews C., Ernest I., Janson P. O., and Chatterjee K. (1998) Follitropin (FSH) deficiency in an infertile male due to FSHβ gene mutation: a syndrome of normal puberty and virilization but underdeveloped testicles with azoospermia, low FSH but high lutropin and normal serum testosterone concentrations. Clin. Chem. Lab. Med. 36, 663–665 [DOI] [PubMed] [Google Scholar]
- 13. Bielinska M., Rzymkiewicz D., and Boime I. (1994) Human luteinizing hormone and chorionic gonadotropin are targeted to a regulated secretory pathway in GH3 cells. Mol. Endocrinol. 8, 919–928 [DOI] [PubMed] [Google Scholar]
- 14. Fiddes J. C., and Goodman H. M. (1981) The gene encoding the common α subunit of the four human glycoprotein hormones. J. Mol. Appl. Genet. 1, 3–18 [PubMed] [Google Scholar]
- 15. Rathnam P., and Saxena B. B. (1975) Primary amino acid sequence of follicle-stimulating hormone from human pituitary glands: I. α subunit. J. Biol. Chem. 250, 6735–6746 [PubMed] [Google Scholar]
- 16. Bernard D. J., Fortin J., Wang Y., and Lamba P. (2010) Mechanisms of FSH synthesis: what we know, what we don't, and why you should care. Fertil. Steril. 93, 2465–2485 [DOI] [PubMed] [Google Scholar]
- 17. McGillivray S. M., Thackray V. G., Coss D., and Mellon P. L. (2007) Activin and glucocorticoids synergistically activate follicle-stimulating hormone β-subunit gene expression in the immortalized LβT2 gonadotrope cell line. Endocrinology 148, 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaiser U. B., Jakubowiak A., Steinberger A., and Chin W. W. (1997) Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology 138, 1224–1231 [DOI] [PubMed] [Google Scholar]
- 19. Dalkin A. C., Burger L. L., Aylor K. W., Haisenleder D. J., Workman L. J., Cho S., and Marshall J. C. (2001) Regulation of gonadotropin subunit gene transcription by gonadotropin-releasing hormone: measurement of primary transcript ribonucleic acids by quantitative reverse transcription-polymerase chain reaction assays. Endocrinology 142, 139–146 [DOI] [PubMed] [Google Scholar]
- 20. Carroll R. S., Corrigan A. Z., Gharib S. D., Vale W., and Chin W. W. (1989) Inhibin, activin, and follistatin: regulation of follicle-stimulating hormone messenger ribonucleic acid levels. Mol. Endocrinol. 3, 1969–1976 [DOI] [PubMed] [Google Scholar]
- 21. Dalkin A. C., Haisenleder D. J., Gilrain J. T., Aylor K., Yasin M., and Marshall J. C. (1999) Gonadotropin-releasing hormone regulation of gonadotropin subunit gene expression in female rats: actions on follicle-stimulating hormone β messenger ribonucleic acid (mRNA) involve differential expression of pituitary activin (β-B) and follistatin mRNAs. Endocrinology 140, 903–908 [DOI] [PubMed] [Google Scholar]
- 22. Bernard D. J. (2004) Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β subunit in mouse gonadotrope cells. Mol. Endocrinol. 18, 606–623 [DOI] [PubMed] [Google Scholar]
- 23. Bernard D. J., and Tran S. (2013) Mechanisms of activin-stimulated FSH synthesis: the story of a pig and a FOX. Biol. Reprod. 88, 78. [DOI] [PubMed] [Google Scholar]
- 24. Lamba P., Fortin J., Tran S., Wang Y., and Bernard D. J. (2009) A novel role for the forkhead transcription factor FOXL2 in activin A-regulated follicle-stimulating hormone β subunit transcription. Mol. Endocrinol. 23, 1001–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suszko M. I., Balkin D. M., Chen Y., and Woodruff T. K. (2005) Smad3 mediates activin-induced transcription of follicle-stimulating hormone β-subunit gene. Mol. Endocrinol. 19, 1849–1858 [DOI] [PubMed] [Google Scholar]
- 26. Han S. O., and Miller W. L. (2009) Activin A induces ovine follicle stimulating hormone β using −169/−58 bp of its promoter and a simple TATA box. Reprod. Biol. Endocrinol. 7, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ghochani Y., Saini J. K., Mellon P. L., and Thackray V. G. (2012) FOXL2 is involved in the synergy between activin and progestins on the follicle-stimulating hormone β-subunit promoter. Endocrinology 153, 2023–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y., and Bernard D. J. (2012) Activin A induction of murine and ovine follicle-stimulating hormone β transcription is SMAD-dependent and TAK1 (MAP3K7)/p38 MAPK-independent in gonadotrope-like cells. Cell Signal. 24, 1632–1640 [DOI] [PubMed] [Google Scholar]
- 29. Pangas S. A., and Woodruff T. K. (2000) Activin signal transduction pathways. Trends Endocrinol. Metab. 11, 309–314 [DOI] [PubMed] [Google Scholar]
- 30. Tran S., Lamba P., Wang Y., and Bernard D. J. (2011) SMADs and FOXL2 synergistically regulate murine FSHβ transcription via a conserved proximal promoter element. Mol. Endocrinol. 25, 1170–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lamba P., Santos M. M., Philips D. P., and Bernard D. J. (2006) Acute regulation of murine follicle-stimulating hormone β subunit transcription by activin A. J. Mol. Endocrinol. 36, 201–220 [DOI] [PubMed] [Google Scholar]
- 32. Lamba P., Wang Y., Tran S., Ouspenskaia T., Libasci V., Hébert T. E., Miller G. J., and Bernard D. J. (2010) Activin A regulates porcine follicle-stimulating hormone β-subunit transcription via cooperative actions of SMADs and FOXL2. Endocrinology 151, 5456–5467 [DOI] [PubMed] [Google Scholar]
- 33. Corpuz P. S., Lindaman L. L., Mellon P. L., and Coss D. (2010) FoxL2 Is required for activin induction of the mouse and human follicle-stimulating hormone β-subunit genes. Mol. Endocrinol. 24, 1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fortin J., Boehm U., Deng C. X., Treier M., and Bernard D. J. (2014) Follicle-stimulating hormone synthesis and fertility depend on SMAD4 and FOXL2. FASEB J. 28, 3396–3410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tran S., Zhou X., Lafleur C., Calderon M. J., Ellsworth B. S., Kimmins S., Boehm U., Treier M., Boerboom D., and Bernard D. J. (2013) Impaired fertility and FSH synthesis in gonadotrope-specific Foxl2 knock-out mice. Mol. Endocrinol. 27, 407–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fortin J., Boehm U., Weinstein M. B., Graff J. M., and Bernard D. J. (2014) Follicle-stimulating hormone synthesis and fertility are intact in mice lacking SMAD3 DNA binding activity and SMAD2 in gonadotrope cells. FASEB J. 28, 1474–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shi Y., Wang Y. F., Jayaraman L., Yang H., Massagué J., and Pavletich N. P. (1998) Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell 94, 585–594 [DOI] [PubMed] [Google Scholar]
- 38. Kozak M. (1989) The scanning model for translation: an update. J. Cell Biol. 108, 229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chacko B. M., Qin B. Y., Tiwari A., Shi G., Lam S., Hayward L. J., De Caestecker M., and Lin K. (2004) Structural basis of heteromeric smad protein assembly in TGF-β signaling. Mol. Cell 15, 813–823 [DOI] [PubMed] [Google Scholar]
- 40. Blount A. L., Schmidt K., Justice N. J., Vale W. W., Fischer W. H., and Bilezikjian L. M. (2009) FoxL2 and Smad3 coordinately regulate follistatin gene transcription. J. Biol. Chem. 284, 7631–7645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calonge M. J., and Massagué J. (1999) Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-β antiproliferative responses in colon cancer cells. J. Biol. Chem. 274, 33637–33643 [DOI] [PubMed] [Google Scholar]
- 42. Goyette M. C., Cho K., Fasching C. L., Levy D. B., Kinzler K. W., Paraskeva C., Vogelstein B., and Stanbridge E. J. (1992) Progression of colorectal cancer is associated with multiple tumor suppressor gene defects but inhibition of tumorigenicity is accomplished by correction of any single defect via chromosome transfer. Mol. Cell. Biol. 12, 1387–1395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bernard D. J., Lee K. B., and Santos M. M. (2006) Activin B can signal through both ALK4 and ALK7 in gonadotrope cells. Reprod. Biol. Endocrinol. 4, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bull P., Morales P., Huyser C., Socías T., and Castellón E. A. (2000) Expression of GnRH receptor in mouse and rat testicular germ cells. Mol. Hum. Reprod. 6, 582–586 [DOI] [PubMed] [Google Scholar]
- 45. Rejon C. A., Ho C. C., Wang Y., Zhou X., Bernard D. J., and Hébert T. E. (2013) Cycloheximide inhibits follicle-stimulating hormone β subunit transcription by blocking de novo synthesis of the labile activin type II receptor in gonadotrope cells. Cell Signal. 25, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 46. Ellsworth B. S., Burns A. T., Escudero K. W., Duval D. L., Nelson S. E., and Clay C. M. (2003) The gonadotropin releasing hormone (GnRH) receptor activating sequence (GRAS) is a composite regulatory element that interacts with multiple classes of transcription factors including Smads, AP-1 and a forkhead DNA binding protein. Mol. Cell. Endocrinol. 206, 93–111 [DOI] [PubMed] [Google Scholar]
- 47. Liu X., Sun Y., Constantinescu S. N., Karam E., Weinberg R. A., and Lodish H. F. (1997) Transforming growth factor β-induced phosphorylation of Smad3 is required for growth inhibition and transcriptional induction in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10669–10674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. West B. E., Parker G. E., Savage J. J., Kiratipranon P., Toomey K. S., Beach L. R., Colvin S. C., Sloop K. W., and Rhodes S. J. (2004) Regulation of the follicle-stimulating hormone β gene by the LHX3 LIM-homeodomain transcription factor. Endocrinology 145, 4866–4879 [DOI] [PubMed] [Google Scholar]
- 49. Huang H. J., Sebastian J., Strahl B. D., Wu J. C., and Miller W. L. (2001) Transcriptional regulation of the ovine follicle-stimulating hormone-β gene by activin and gonadotropin-releasing hormone (GnRH): involvement of two proximal activator protein-1 sites for GnRH stimulation. Endocrinology 142, 2267–2274 [DOI] [PubMed] [Google Scholar]
- 50. Wang A., Ziyadeh F. N., Lee E. Y., Pyagay P. E., Sung S. H., Sheardown S. A., Laping N. J., and Chen S. (2007) Interference with TGF-β signaling by Smad3-knock-out in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am. J. Physiol. Renal Physiol. 293, F1657–F1665 [DOI] [PubMed] [Google Scholar]
- 51. Zhu Y., Richardson J. A., Parada L. F., and Graff J. M. (1998) Smad3 mutant mice develop metastatic colorectal cancer. Cell 94, 703–714 [DOI] [PubMed] [Google Scholar]
- 52. Datto M. B., Frederick J. P., Pan L., Borton A. J., Zhuang Y., and Wang X. F. (1999) Targeted disruption of Smad3 reveals an essential role in transforming growth factor β-mediated signal transduction. Mol. Cell. Biol. 19, 2495–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang X., Letterio J. J., Lechleider R. J., Chen L., Hayman R., Gu H., Roberts A. B., and Deng C. (1999) Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J. 18, 1280–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tomic D., Miller K. P., Kenny H. A., Woodruff T. K., Hoyer P., and Flaws J. A. (2004) Ovarian follicle development requires Smad3. Mol. Endocrinol. 18, 2224–2240 [DOI] [PubMed] [Google Scholar]
- 55. Coss D., Thackray V. G., Deng C. X., and Mellon P. L. (2005) Activin regulates luteinizing hormone β-subunit gene expression through Smad-binding and homeobox elements. Mol. Endocrinol. 19, 2610–2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dennler S., Huet S., and Gauthier J. M. (1999) A short amino-acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene 18, 1643–1648 [DOI] [PubMed] [Google Scholar]
- 57. Yagi K., Goto D., Hamamoto T., Takenoshita S., Kato M., and Miyazono K. (1999) Alternatively spliced variant of Smad2 lacking exon 3: comparison with wild-type Smad2 and Smad3. J. Biol. Chem. 274, 703–709 [DOI] [PubMed] [Google Scholar]
- 58. Fortin J., and Bernard D. J. (2010) SMAD3 and EGR1 physically and functionally interact in promoter-specific fashion. Cell Signal. 22, 936–943 [DOI] [PubMed] [Google Scholar]
- 59. Tran S., Wang Y., Lamba P., Zhou X., Boehm U., and Bernard D. J. (2013) The CpG island in the murine foxl2 proximal promoter is differentially methylated in primary and immortalized cells. PLoS One 8, e76642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang Y., Fortin J., Lamba P., Bonomi M., Persani L., Roberson M. S., and Bernard D. J. (2008) Activator protein-1 and smad proteins synergistically regulate human follicle-stimulating hormone β-promoter activity. Endocrinology 149, 5577–5591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li Q., Pangas S. A., Jorgez C. J., Graff J. M., Weinstein M., and Matzuk M. M. (2008) Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol. Cell. Biol. 28, 7001–7011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wen S., Schwarz J. R., Niculescu D., Dinu C., Bauer C. K., Hirdes W., and Boehm U. (2008) Functional characterization of genetically labeled gonadotropes. Endocrinology 149, 2701–2711 [DOI] [PubMed] [Google Scholar]
- 63. Yang X., Li C., Herrera P. L., and Deng C. X. (2002) Generation of Smad4/Dpc4 conditional knock-out mice. Genesis 32, 80–81 [DOI] [PubMed] [Google Scholar]
- 64. Srinivas S., Watanabe T., Lin C. S., William C. M., Tanabe Y., Jessell T. M., and Costantini F. (2001) Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Steyn F. J., Wan Y., Clarkson J., Veldhuis J. D., Herbison A. E., and Chen C. (2013) Development of a methodology for and assessment of pulsatile luteinizing hormone secretion in juvenile and adult male mice. Endocrinology 154, 4939–4945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Caligioni C. S. (2009) Assessing reproductive status/stages in mice. Curr. Protoc. Neurosci. Appendix 4, Appendix 4I [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oakes C. C., Kelly T. L., Robaire B., and Trasler J. M. (2007) Adverse effects of 5-aza-2′-deoxycytidine on spermatogenesis include reduced sperm function and selective inhibition of de novo DNA methylation. J. Pharmacol. Exp. Ther. 322, 1171–1180 [DOI] [PubMed] [Google Scholar]
- 68. Ho C. C., Zhou X., Mishina Y., and Bernard D. J. (2011) Mechanisms of bone morphogenetic protein 2 (BMP2) stimulated inhibitor of DNA binding 3 (Id3) transcription. Mol. Cell. Endocrinol. 332, 242–252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.