

SUMMARY
The embryonic basal ganglia generates multiple projection neurons and interneuron subtypes from distinct progenitor domains. Combinatorial interactions of transcription factors and chromatin are thought to regulate gene expression. In the medial ganglionic eminence, the NKX2-1 transcription factor controls regional identity and, with LHX6, is necessary to specify pallidal projection neurons and forebrain interneurons. Here, we dissected the molecular functions of NKX2-1 by defining its chromosomal binding, regulation of gene expression, and epigenetic state. NKX2-1 binding at distal regulatory elements led to a repressed epigenetic state and transcriptional repression in the ventricular zone. Conversely, NKX2-1 is required to establish a permissive chromatin state and transcriptional activation in the sub-ventricular and mantle zones. Moreover, combinatorial binding of NKX2-1 and LHX6 promotes transcriptionally permissive chromatin and activates genes expressed in cortical migrating interneurons. Our integrated approach provides a foundation for elucidating transcriptional networks guiding the development of the MGE and its descendants.
INTRODUCTION
Control of cell fate during development is mediated by a hierarchical network of transcription factors (TFs). Whole-genome approaches provide unbiased approaches toward identifying chromosomal loci where TFs bind to regulatory elements (REs; e.g., promoters and enhancers), epigenetic modifications associated with gene activity or repression, and changes in transcription after TF knockout (KO) (Nord et al., 2015). Here, we applied these methods to investigate transcriptional circuitry that orchestrates development of the medial ganglionic eminence (MGE), an evolutionary conserved region of the forebrain that generates GABAergic and cholinergic projection neurons and interneurons populating the forebrain (Marín and Rubenstein, 2003). The genetically encoded wiring of the brain is of great significance, as perturbations to transcription pathways during brain development is implicated in disorders such as autism and intellectual disability (De Rubeis et al., 2014).
Mouse genetic experiments elucidated the functions of many TFs in MGE development (Campbell, 2003). Briefly, MGE progenitors in the ventricular zone (VZ) require the homeodomain (HD) protein NKX2-1 for regional specification and to repress alternative identities. NKX2-1 also promotes GABAergic and cholinergic cell fate via induction of Lhx6 and Lhx8 Lim-homeobox TFs (Sussel et al., 1999); LHX6 is required for maturation of cortical interneurons (CINs), in part through promoting Arx and Cxcr7 expression (Vogt et al., 2014). LHX8 drives cholinergic neuron fate (Fragkouli et al., 2009; Zhao et al., 2003). Together, LHX6 and LHX8 are required for globus pallidus (GP) differentiation and sonic hedgehog (sHH)-mediated generation of CINs (Flandin et al., 2011). These studies have outlined a map of TF pathways guiding brain development; however, little is known about how these functions are accomplished at the genomic level.
While NKX2-1 and LHX6 confer a core pathway for MGE neuronal identity, systematic whole-genome analysis of their MGE function is lacking (i.e., direct and indirect targets and genomic binding properties). NKX2-1 is reported to directly activate Lhx6, and LHX6 directly activates Arx and Cxcr7 (Du et al., 2008; Vogt et al., 2014). A genome-wide regulatory function of NKX2-1 and LHX6 could reveal how these TFs interact to drive MGE development. Using a combination of genetic models, neuroanatomical studies, and genomics, we characterized NKX2-1 and LHX6 functions in the mouse MGE at a stage when it actively generates forebrain interneurons. Via microarray and in situ experiments using a conditional Nkx2-1 null mutant, we examined differential RNA expression, identifying many novel target genes that may orchestrate MGE development. Next, we used TF chromatin immunoprecipitation followed by DNA sequencing (chromatin immunoprecipitation sequencing [ChIP-seq]) to identify REs bound by NKX2-1 and LHX6. We further compared histone ChIP-seq results from the MGE of wild-type (WT) and Nkx2-1 conditional mutants to identify epigenetic modifications associated with repression and/or activation. Herein, we present a model where NKX2-1 binds to distal REs to repress alternative fates in progenitor cells within the VZ of the MGE. In addition, NKX2-1 is required for the expression of LHX6 and associated activation of lineage- and region-specific genes in the subventricular zone (SVZ) and mantle zone (MZ).
RESULTS
Conditional Deletion of Nkx2-1 in the Ventral Forebrain Results in Quantitative and Spatial Changes in MGE Gene Expression
To study the role of NKX2-1 in the developing forebrain, we crossed a conditional Nkx2-1 mouse (Kusakabe et al., 2006) with an Olig2-tva-Cre line as well as the Ai14-Cre reporter (Madisen et al., 2010; Schüller et al., 2008) to eliminate expression of Nkx2-1 in MGE progenitors. We refer to the conditional mutant as Nkx2-1cKO. Olig2-tva-Cre begins forebrain Cre expression at approximately embryonic day (E) 10.5 (Figures 1A–1C and 1G–1I). The mutant had reduced Nkx2-1 MGE expression by E11.5, with nearly complete elimination by E13.5 (Figures 1D–1F and 1J–1L). As expected, the mutant MGE failed to produce CINs (Sussel et al., 1999). Nkx2-1cKOs expressed Nkx2-1 in the preoptic area, a region lacking Cre-mediated recombination (Figure 1L).
Figure 1. Conditional Deletion of Nkx2-1 in the Embryonic MGE.
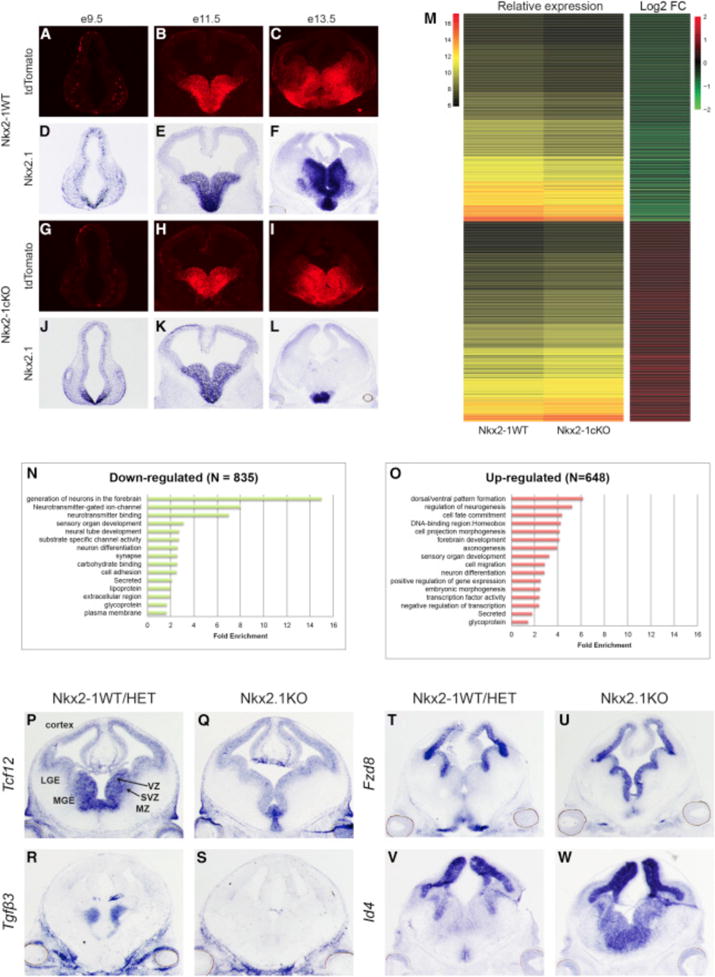
(A–L) AI14 immunofluorescence (A–C, G–I) and Nkx2-1exon-2 in situ (D–F, J–L) on WT and Nkx2-1cKO at E9.5, E11.5, and E13.5.
(M) Heatmaps showing relative expression and log2 fold change of dysregulated genes in the Nkx2-1cKO MGE at E13.5.
(N and O) Functional enrichment analysis on down- (N) and up-regulated (O) genes in the Nkx2-1cKO MGE at E13.5.
(P–S) In situ analysis using probes detecting the downregulated genes Tcf12 (P and Q) and Tgf3 (R and S) in WT and Nkx2-1cKO forebrain at E13.5.
(T–W) In situ analysis using probes detecting the upregulated genes Fzd8 (T and U) and Id4 (V and W) in WT and Nkx2-1cKO forebrain at E13.5.
We identified dysregulated genes in the Nkx2-1cKO via RNA expression microarray analysis from E13.5 MGE. We identified 1,483 dysregulated genes; 648 increased RNA expression in Nkx2-1cKO relative to WT tissue and 835 with reduced RNA levels (Data S1A). Unsupervised clustering was used to generate co-expression groups (Figure 1M). The upregulated genes were associated with gene ontology (GO) biological process annotations that included generation of forebrain neurons, neurotransmitter-gated ion channels, and neurotransmitter binding, while downregulated genes were associated with annotation terns such as dorsal/ventral patterning, regulation of neurogenesis, and cell-fate commitment (Figures 1N and 1O; Data S1B and S1C).
Differential expression was validated for 36 genes via in situ hybridization (ISH) (Figures 1P–1W; Figure S1). We observed a reduction of Lhx6 and Lhx8, two NKX2-1 target genes (Figure S1) (Butt et al., 2008; Du et al., 2008; Sussel et al., 1999). Overall, the SVZ and MZ showed substantial reduction expression across genes examined via ISH. The SVZ had reduced expression in 19 of 24 downregulated genes, including Tcf12 and Tgfβ3 (Figures 1P–1S). Four genes (AdarB2, Elfn1, Gbx1, and Gbx2) were reduced in the pallidal MZ (e.g., ventral pallidum [VP] and GP; Figure S1). These patterns suggest that NKX2-1, or genes regulated by NKX2-1, is required to promote gene transcription in the SVZ and maturing pallidal neurons.
Conversely, ISH of genes with increased RNA in the Nkx2-1cKO revealed altered levels in the VZ of the MGE (8 of 12 analyzed), including Fzd8 and Id4 (Figures 1T–1W; Figure S1). In addition to the increased transcription in the VZ, six genes had increased transcription in the SVZ of the MGE (e.g., Meis2, Oct6, and Zfp503), particularly in SVZ1, the layer closest to the VZ (Petryniak et al., 2007). As in previous studies, these transcriptional changes support the transformation of MGE progenitors into cells with a transcriptional profile resembling adjacent progenitor domains: the lateral and caudal ganglionic eminences (LGE and CGE) (Butt et al., 2008; Sussel et al., 1999). Overall, the ISH results support that NKX2-1 mediates transcriptional repression in the VZ and SVZ1, as well as transcriptional activation in the SVZ and MZ.
NKX2-1 Genomic Binding Characterized via ChIP-Seq
The transcriptional changes in Nkx2-1cKO MGE might be due to direct regulation of target genes by NKX2-1 binding to REs or might be mediated indirectly via the functions of NKX2-1 regulated genes. To define primary changes and characterize the binding and functional properties of NKX2-1, we performed ChIP-seq on E13.5 basal ganglia. There was strong concordance in NKX2-1 binding across replicates (Figures 2A and 2B). We derived a set of 4,997 binding regions based on the presence of significant enrichment in NKX2-1 ChIP libraries and no enrichment in the input and IgG control datasets.
Figure 2. Genomic Characterization of NKX2-1 Binding in the MGE.
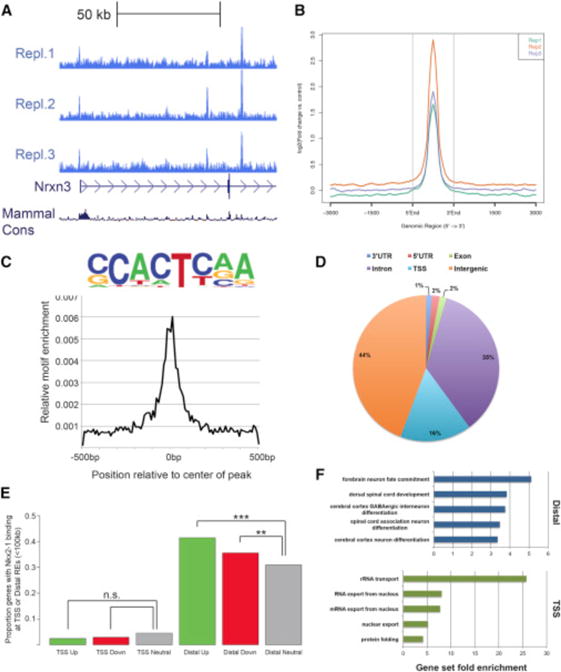
(A) Biological replicates of NKX2-1 ChIP-seq showing consistent NKX2-1 binding at the 5′ end of Nrxn3.
(B) Average profile of NKX2-1 binding at called ChIP-seq peaks comparing the three biological replicates.
(C) NKX2-1 consensus motif derived from de novo motif analysis of NKX2-1 ChIP-seq data.
(D) Genomic features of NKX2-1-bound REs.
(E) Proportion of differentially expressed genes in the Nkx2-1cKO with TSS and distal NKX2-1 binding. Fisher’s exact test was used to test significance between the groups: ***p < 0.001, **p < 0.01.
(F) Enriched gene ontology annotations of putative NKX2-1 binding target genes for distal REs (blue) and NKX2-1-bound TSSs (green).
De novo motif discovery using genomic sequences associated with NKX2-1 binding was performed using HOMER (Heinz et al., 2010). The most frequent de novo motif occurred at the center of NKX2-1-bound regions and likely represents the primary sequence motif recognized in E13.5 MGE (Figure 2C). This core domain is similar to the primary consensus motif derived from NKX2-1 ChIP-seq performed in NKX2-1-amplified lung adenocarcinomas (Watanabe et al., 2013). Earlier studies have shown that the Drosophila NK-2 HD and mouse NKX2-2 HD bind to parts of this sequence in vitro (Berger et al., 2008; Noyes et al., 2008). Together, this analysis provides strong evidence that (G|C)CACT(C|T)AA is the primary motif recognized by NKX2-1 in the developing MGE.
NKX2-1 binding was located in both intergenic (44%) and gene regions (56%; 3′ UTR, 5′ UTR, exon, transcriptional start site, and introns) (Figure 2D). We observed strong sequence conservation across vertebrate evolution at NKX2-1 binding regions (Data S1D). 16% of binding occurred at transcriptional start sites (TSSs), consistent with previous data (Snyder et al., 2013; Watanabe et al., 2013). We observed that NKX2-1 binding to distal loci, within 100 kb, was enriched near both up- and downregulated genes in the Nkx2-1cKOs, suggesting that NKX2-1 acts as both a transcriptional activator and repressor via direct binding at REs near target genes (Figure 2E). Surprisingly, NKX2-1 binding at the TSS was not enriched for either up- or downregulated gene sets, providing evidence that NKX2-1 primarily exerts its effect through distal REs in the E13.5 MGE (Figure 2E).
We next examined enrichment of annotation terms among NKX2-1 target genes via GREAT (McLean et al., 2010). For distal REs, the top-ranked GO terms were associated with CNS development and, in particular, forebrain and GABAergic cell development (Figure 2F; Data S1E). Genes annotated to TSS-enriched regions had a strong correlation with mRNA processing and basic cellular processes and lacked enrichment for CNS/developmental annotation terms (Figure 2F; Data S1E).
Epigenomic Profiling Reveals that NKX2-1 Regulates Activation and Repression
We next examined the relationship between NKX2-1 binding and chromatin state to assess how NKX2-1 acts as both an activator and repressor. Histone ChIP-seq was performed on MGE tissue at E13.5. We assessed histone modifications associated with proximal and distal gene activation (H3K4me3, H3K4me1, and H3K27ac) and repression (H3K27me3). Over 93% of NKX2-1 binding regions were flanked by at least one of these histone modifications, with the largest proportion exhibiting H3K4me1, supporting the idea that NKX2-1 binding occurs at REs. We classified all NKX2-1-bound TSSs and distal REs by the pattern of histone modifications as TSS activating (H3K4me3), TSS bivalent (H3K4me3 and H3K27me3), distal (strong) activating (H3K27ac and high H3K4me3), distal (weak) activating (H3K27ac and low/no H3K4me3), distal bivalent (H3K27me3 and H3K27ac), repressing (H3K27me3), latent (H3K4me1 only), or no histone modifications (Figures 3A and 3B; Data S1F). This general strategy has been used to classify REs (Attanasio et al., 2014; Bernstein et al., 2006; Ernst et al., 2011), and it effectively classified regulatory activity of NKX2-1 binding regions. The majority of NKX2-1-bound TSSs and distal REs exhibit activating or bivalent marks, with a smaller set of distal REs exhibiting repressive, latent, or no histone modifications (Figures 3A and 3B). We observed a strong correlation between NKX2-1-bound regions classified by chromatin state and expression of the nearest TSS as determined by micro-array experiments (Figure 3C).
Figure 3. Histone Profile of NKX2-1-Bound REs in the MGE.
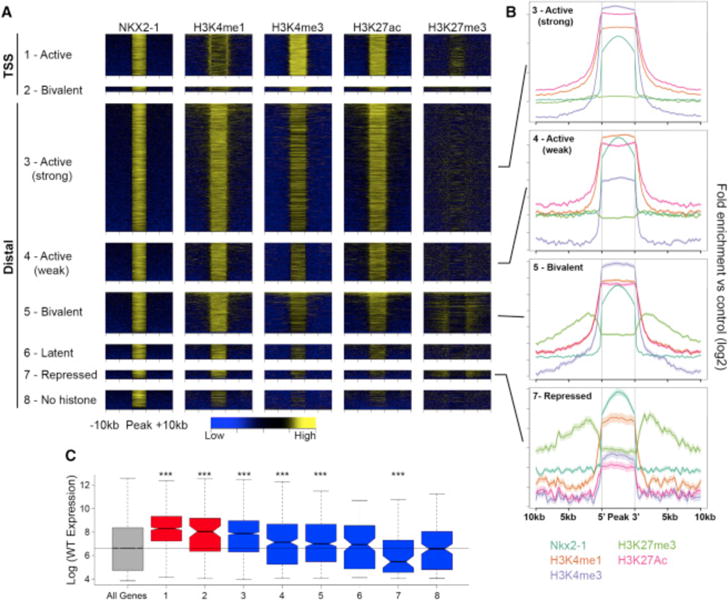
(A) Heatmaps showing H3K4me1, H3K4me3, H3K27ac, and H3K27me3 enrichment around NKX2-1-bound REs. REs divided into eight groups defined by their combined histone profile: (1) active TSS (1.1.1.0 = H3K4me1+, H3K4me3+, H3K27ac+, and H3K27me3−); (2) bivalent TSS (1.1.1.1); (3) active (strong) distal (1.1.1.0); (4) active (weak) distal (1.0.1.0); (5) bivalent distal (1.1.1.1); (6) latent distal (1.0.0.0); (7) repressed distal (1.0.0.1); and (8) no histone (0.0.0.0).
(B) Average profile of H3K4me1, H3K4me3, H3K27me3, and H3K27ac at (3) active (strong) distal, (4) active (weak) distal, (5) bivalent distal, and (7) repressed distal REs.
(C) Relative gene expression of closest TSS in relation to RE groups as defined in (A). Two-sample t tests were used to test significance compared to the control gene set: ***p < 0.001, **p < 0.01, and *p < 0.05.
While the epigenomic data show that chromatin state is a strong indicator of RE function, WT chromatin state at NKX2-1-bound REs is associated with weak or no trends in differential expression of the nearest gene in the Nkx2-1cKO (Figure S2), indicating that NKX2-1 function cannot be inferred based on WT RE chromatin state alone. However, enrichment of distal NKX2-1 binding with both up- and downregulation of gene expression in the Nkx2-1cKO suggests that NKX2-1 binding has context-dependent activity. For example, REs exhibiting a strong activation signature overall in the MGE may be activated or repressed by NKX2-1 binding depending on cell type. To directly test for NKX2-1-mediated activation or repression via genomic binding and to understand context-dependent function of NKX2-1 binding, we performed the same set of ChIP-seq experiments targeting histone modifications to characterize chromatin state in E13.5 MGEs from Nkx2-1cKO mice. We first examined genes that were differentially expressed in the microarray and ISH experiments, observing differential epigenomic signatures in Nkx2-1cKO mice in the region around differentially expressed genes. For example, Lhx8 had reduced expression in Nkx2-1cKOs, as well as NKX2-1 binding at distal REs that showed decreased H3K4me1 and H3K27ac in the Nkx2-1cKO (Figures 4A, 4C, and 4D). Conversely, Gli2, which showed increased MGE expression and distal NKX2-1 binding, exhibited an inverse pattern of histone marks with reduced H3K4me1, H3K4me3, and H3K27ac and increased H3K27me3 in the Nkx2-1cKO (Figures 4B, 4E, and 4F). The presence of differential epigenomic states at NKX2-1-bound REs suggests that we can indeed use these Nkx2-1cKO datasets to establish TF binding function.
Figure 4. Definition of aREs and rREs Based on Changes in the Histone Profile in the Nkx2-1cKO MGE.
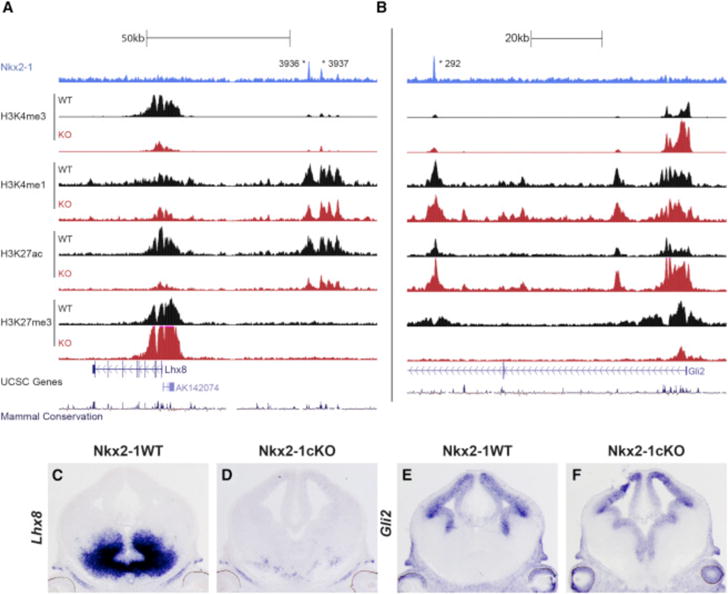
(A and B) Genomic regions of the Lhx8 (A) and Gli2 (B) loci with the ChIP-seq datasets and genomic features shown; NKX2-1 ChIP-seq, H3K4me3, H3K4me1, H3K27ac, H3K27me3, UCSC genes, and mammalian conservation. The H3 modification ChIP-seqs were performed on both WT and Nkx2-1cKO MGE at E13.5. Called TF binding peaks are labeled with an asterisk.
(C–F) In situ analysis of Lhx8 (C and D) and Gli2 (E and F) in WT and Nkx2-1cKO forebrain at E13.5.
Next, we assessed genome-wide changes in chromatin state at all NKX2-1-bound REs to identify differential modifications flanking NKX2-1-bound regions. ~25% of NKX2-1-bound REs exhibited differential enrichment for histone modifications in Nkx2-1cKOs at a stringent significant cutoff. It is likely that many remaining REs bound by NKX2-1 exhibited sub-significant changes in Nkx2-1cKOs or cell-type-specific changes that were masked by cellular heterogeneity in the MGE. We defined two types of REs that were functionally dependent on NKX2-1 binding, as they showed changed chromatin states in Nkx2-1cKO MGE. First, we defined NKX2-1 activating REs (aREs, n = 1,505; Data S1F); these exhibited loss of an activating mark (H3K27ac or H3K4me3) or gain of a repressing mark (H3K27me3) in Nkx2-1cKOs (Figure 5A; Figure S3A). Second, we defined NKX2-1 repressing REs (rREs, n = 664; Figure 5B; Data S1F); these exhibited a gain of activation marks or loss of repressing marks in the Nkx2-1cKO. H3K27ac and H3K27me3 decreases flanking NKX2-1 binding sites were the strongest histone modification changes at aREs and rREs, respectively (Figures 5A and 5B; Figures S3A–S3C), and changes to these marks correlated most strongly with differential gene expression. Reduction of H3K4me3 in the Nkx2-1cKO correlated with reduced gene expression, but gain of H3K4me3 was not associated with differential expression. Reductions in H3K4me1 were smaller in magnitude in Nkx2-1cKOs but were associated with loss of activation and of repression (Figures S3A–S3C and S4A). In the REs where H3K4me1 did change, other histone modifications were also observed, consistent with H3K4me1 being a general mark of enhancers and not having the specificity for activity/repression of H3K27ac, H3K27me3, or H3K4m3 (Bernstein et al., 2006; Creyghton et al., 2010).
Figure 5. Identification of NKX2-1-Bound REs Mediating Transcriptional Activation and Repression.
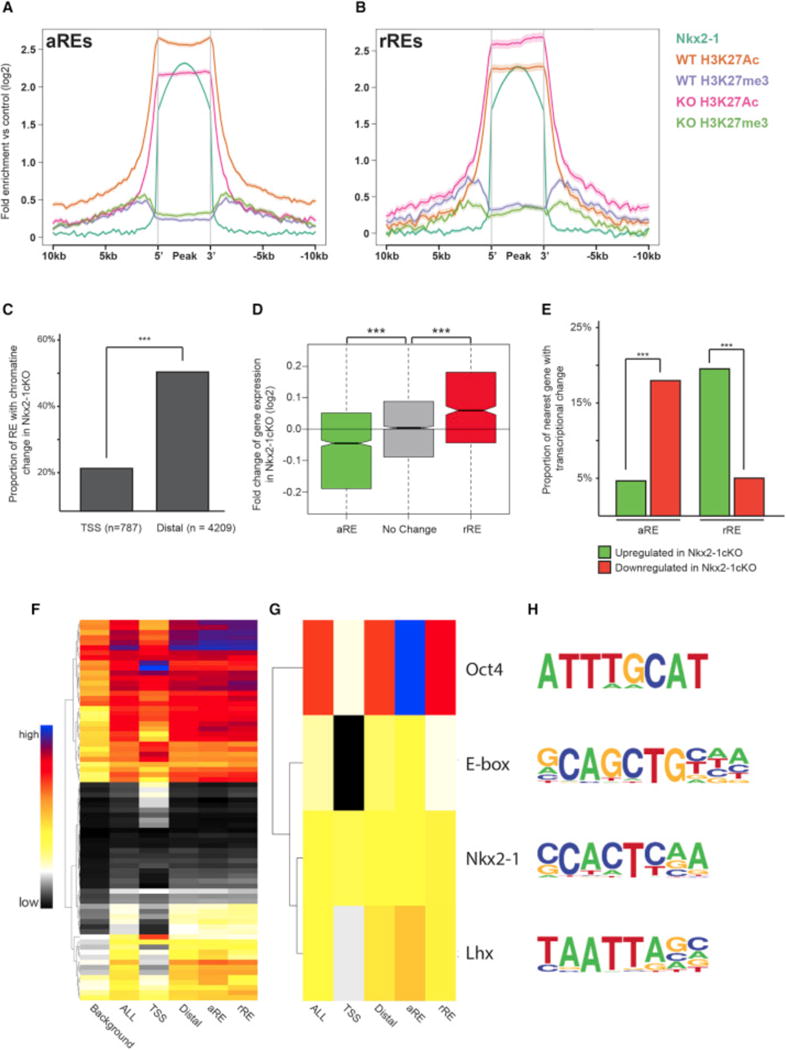
(A and B) Average profile of H3K27ac and H3K27me3 at aREs (A) and rREs (B) in WT and Nkx2-1cKO MGE at E13.5.
(C) Proportion of TSS and distal NKX2-1-bound REs with changes in the chromatin profile in the Nkx2-1cKO in the E13.5 MGE.
(D) Average fold change in expression of genes associated to distal aREs and rREs in the Nkx2-1cKO MGE at E13.5. Two-sample t test was used to test significance between the groups: ***p < 0.001.
(E) Proportion of distal aREs and rREs with an associated increase or decrease in expression of the gene with closest TSS.
(F–H) Proportion of background, TSS, all NKX2-1-bound Res, distal Res, distal aREs, and distal rREs with identified de novo motifs. All motifs (F) and manually curated list (G). Sequence of motifs and potential TF recognizing the motifs (H). Fisher’s exact test was used to test significance between the groups for (C) and (E). Wilcox test was used to compare expression distribution in (D): ***p < 0.001, **p < 0.01.
Both aREs and rREs occurred more frequently distally versus at gene promoters (Figure 5C), consistent with distal RE NKX2-1 binding mediating differential expression of genes in the Nkx2-1cKO MGE and with the lack of correlation between NKX2-1 TSS binding and transcriptional sensitivity to Nkx2-1cKO (Figures 2E and 5C). Only a small proportion of NKX2-1-bound TSSs were classified as an aRE (7%) or rRE (8%), with a subset of the promoter aREs (13/55) and rREs (4/63) also exhibiting change in expression in the Nkx2-1cKO. This suggests that most TSS epigenomic changes associated with loss of NKX2-1 binding are associated with subtle or no expression changes. By comparison, NKX2-1-sensitive distal aREs and rREs occurred more frequently (25% and 13% of all distal REs, respectively) and were strongly enriched within 100 kb of a differentially expressed gene TSS (Figure 5D). For 20% of the rREs, we observed increased transcription of the nearest gene in the Nkx2-1cKO, and for 18% of the aREs, we observed decreased transcription of the nearest gene. In comparison, cKO-sensitive REs showed background levels of enrichment near genes differentially expressed in the opposite direction (Figure 5E). While WT chromatin state alone was associated with weak or no trend in differential gene expression in the Nkx2-1cKO (Figure S2), separation of REs assigned by chromatin state into activating and repressing REs revealed strong association between predicted NKX2-1 activating or repressing activity and change of expression in the cKO (Figure 5E; Figure S4B). This indicates that epigenomic state and transcriptional expression are linked and integrated via NKX2-1 binding at distal REs.
OCT4, E-box, and LHX Motifs Are Enriched at NKX2-1-Bound aREs
We hypothesized that NKX2-1 activity as an activator or repressor is cell-type specific and that the direction of regulation is driven in part by transcriptional co-regulators in the same or parallel TF cascades that bind the same REs that NKX2-1 targets. To test this, we searched NKX2-1-bound RE sequences for enrichment of TF-binding motifs and then tested them for further enrichment within specific RE classes (TSS, distal, aRE, and rRE). Identified de novo motifs were present at different frequencies in the diverse RE classes (Figure 5F; Figure S5). We identified motifs that were generally correlated to NKX2-1 motif (Figures 5G and 5H) and tested whether these motifs were significantly associated with aRE and rRE. OCT4, E-box, and LHX motifs were present in both aRE and rRE sequences at levels above genomic background but were significantly enriched in aREs versus rREs. We did not observe any motifs that were enriched in rREs versus aREs after controlling for sequence content. The motif enrichment pattern suggests that binding of other relevant TFs at the same REs may bias whether NKX2-1-bound REs function to activate or repress transcription in specific cell types.
LHX6 and NKX2-1 Co-occupy aREs in the MGE
NKX2-1 is necessary for Lhx6 expression, which may promote transcriptional activation in the SVZ and MZ, as it regulates the differentiation of several MGE-derived neuronal cell types. We hypothesized that LHX6 binds to the LHX motif in tandem with NKX2-1 in the MGE. To test this and characterize LHX6 binding in the MGE, we performed LHX6 ChIP-seq on the E13.5 basal ganglia. All three biological replicates showed a consistent binding pattern, and after merging them together, we identified 1,064 significant LHX6-bound REs (Data S1G). De novo motif analysis identified an LHX6 consensus motif similar to the motif found in the NKX2-1 aREs (compare Figures 5H and 6A). Half (53%) of LHX6 REs were also significantly enriched for NKX2-1 binding (defined as +/+REs). 55% of these regions were aREs based on a significant loss of activated chromatin marks in the Nkx2-1cKO. The proportion of aREs was significantly higher for +/+REs when compared to the NKX2-1 RE alone and NKX2-1 REs without significant LHX6 binding (+/−REs; Figure 6B). 8% of the +/+REs were rREs, which is significantly lower compared to the proportion of rREs among all Nkx2-1 REs as well as among +/−REs (Figure 6C). Thus, REs bound by both LHX6 and NKX2-1 were considerably enriched for activating properties as defined by whole MGE ChIP-seq analysis, whereas LHX6 binding was present at far fewer rREs (Figures S3A–S3C).
Figure 6. NKX2-1 and LHX6 Co-occupy aREs in the MGE.
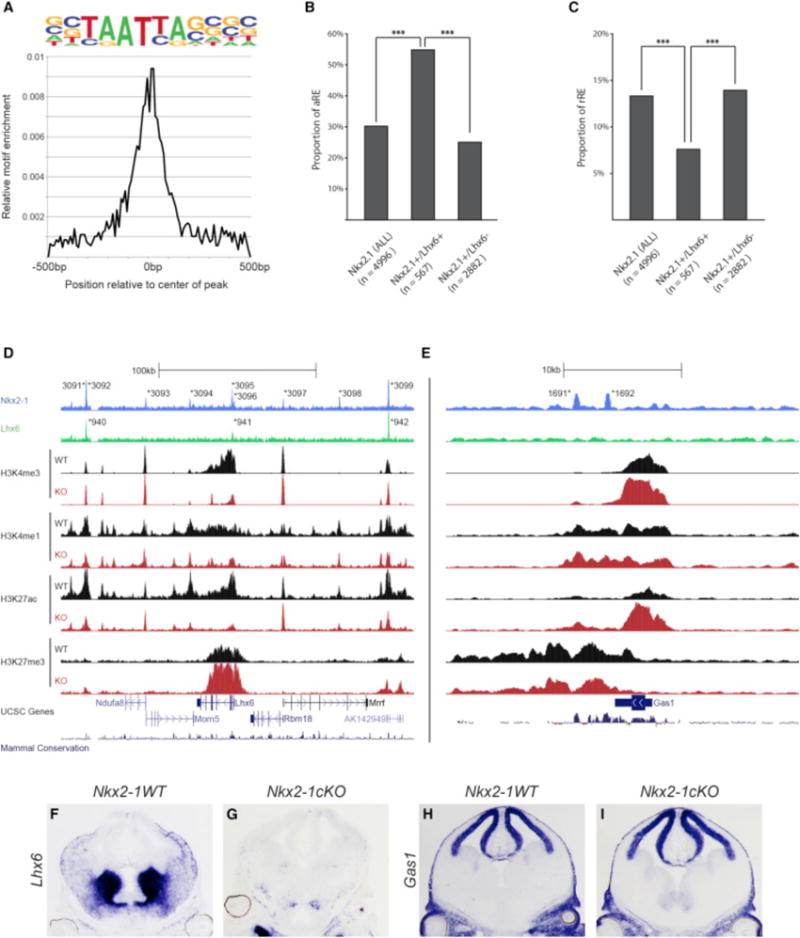
(A) Enrichment of primary LHX6 de novo motif in relation to the center of LHX6-bound REs.
(B) Proportion of aREs in NKX2-1+ (all), NKX2-1+/LHX6+, and Nkx2-1+/Lhx6− bound REs. Chi-square test was used to test significance between the groups: ***p < 0.001.
(C) Proportion of rREs in NKX2-1+ (all), NKX2-1+/LHX6+, and Nkx2-1+/Lhx6− bound REs. Fisher’s exact test was used to test significance between the groups: ***p < 0.001.
(D and E) Genomic region of the Lhx6 (D) and Gas1 (E) loci (+/−100 kb from TSS); NKX2-1 ChIP-seq, LHX6 ChIP-seq, H3K4me1, H3K4me3, H3K27Ac, H3K27me3, UCSC genes, and mammalian conservation. Both WT and Nkx2-1cKO data are shown for the H3 modifications. Called peaks are labeled with an asterisk.
(F–I) In situ analysis of Lhx6 (F and G) and Gas1 (H and I) transcription in WT and Nkx2-1cKO forebrain at E13.5.
Comparison of in situ expression patterns in the Allen Brain Atlas (http://www.brain-map.org/) of genes that are putative targets of +/+REs showed a significantly higher proportion of MGE expression compared to a randomized set of control genes (+/+:58% MGE expressed, Control: 2%; Fisher’s exact test, p value < 0.001), supporting the MGE-specific activity of the +/+REs (Data S1H). Altogether, these data show that a large proportion of the NKX2-1-bound REs are also bound by LHX6 and that these aREs likely activate gene expression in a region- or cell-type-specific manner in the MGE. To better understand the role of +/+REs and +/−REs (NKX2-1 REs without significant LHX6 binding) in the context of active and repressed loci, we looked at the genomic profile of the expressed Lhx6 and the repressed Gas1 genes (+/−100 kb from TSS; Figures 6D and 6E). The Lhx6 locus had three +/+REs (3,091/940, 3,095/941, and 3,099/942) and six +/−REs (3,092–3,094 and 3,096–3,098). All REs except for one (3,093) were classified as aREs based on their reduced H3K27ac in the Nkx2-1cKO MGE. Overall, in the Nkx2-1cKO, the Lhx6 locus showed an extensive reduction of H3K4me3 and H3K27ac, consistent with the reduction of Lhx6 transcription (Figures 6D, 6F, and 6G; Data S1D). The Gas1 locus had two +/−REs (1,691 and 1,692), but no +/+REs. The absence of +/+REs is consistent with a low level of Gas1 expression in the WT MGE. Both +/−REs had activating and repressing histone marks, suggesting that these regions were in a bivalent state. In the Nkx2-1cKO, we observed an increase of H3K4me3 and H3K27ac and a reduction of H3K27me3 at both Gas1 rREs, which is in line with the increase in Gas1 transcript in the VZ of the MGE in the Nkx2-1cKO. Together, these data suggest that NKX2-1 attenuates Gas1 transcription via establishing/maintaining epigenetic repression of the identified +/−REs (Figures 6E, 6H, and 6I; Data S1D). In conclusion, NKX2-1 and LHX6 have been shown to bind a common set of 325 REs, of which a large proportion mediates transcriptional activation (199 aREs versus 26 rREs, or 7.6-fold enrichment for aRE versus rRE). In comparison, for REs with NKX2-1 binding without LHX6 (+/−REs, n = 2,140), the enrichment of aRE versus rRE is 1.9-fold (482 aREs versus 255 rREs).
NKX2-1 and LHX6 Combinatorial Binding Predicts RE Sub-regional Activity in the MGE
Based on Nkx2-1cKO histone and ISH data, the majority of +/+REs likely activate transcription, whereas the +/−REs either are neutral or directly repress transcription. To analyze the activity of +/−RE and +/+RE in vivo, we examined transgenic enhancer assays using sectioned E11.5 forebrain enhancer activity patterns available in the VISTA database (see VISTA database; http://enhancer.lbl.gov) (Visel et al., 2013). We assessed RE enhancer activity in the cortex, LGE, and MGE for +/−REs and +/+REs (Figure 7A). Both +/−REs and +/+REs had high activity in cortex and LGE. The +/−REs showed a lower proportion of activity in the MGE compared to cortex and LGE, suggesting that NKX2-1 is repressing transcription via binding at +/−REs (Figure 7A). The Nkx2-1cKO histone data supports this, as illustrated by three VISTA regions (Figure 7C; Figure S6). All three elements showed an increase in H3K27ac and/or H3K4me3 in the mutant that correlated with increased transcription of their nearby genes (Data S1D, hs187 = FoxP1, hs271 = CoupTFI, and hs1336 = BMPER).
Figure 7. LHX6 Is Required for Activating Transcription in the SVZ and MZ of the MGE.
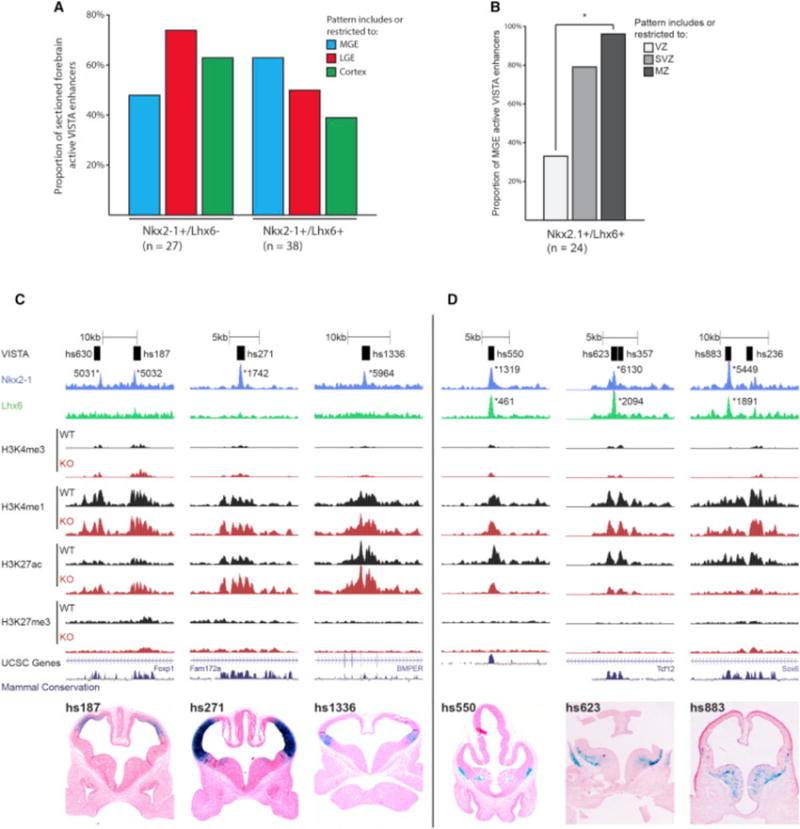
(A) Proportion of MGE, LGE, and cortex activity of +/−REs and +/+REs at E11.5.
(B) VZ, SVZ, and MZ activity of +/+REs in the MGE at E11.5. Chi-square test was used to test significance between the groups: *p < 0.05.
(C) Browser view of +/−REs with tracks; VISTA transgenic ID, NKX2-1 ChIP-seq, LHX6 ChIP-seq, H3K4me1, H3K4me3, H3K27ac, H3K27me3, UCSC genes, and mammalian conservation. Both WT and Nkx2-1cKO data are shown for H3 modifications. Called peaks are labeled with an asterisk. VISTA transgenics showing in vivo activity of +/−REs (hs187, hs271, and hs1336) at E11.5.
(D) Browser view of +/+REs with tracks; VISTA transgenic ID, NKX2-1 ChIP-seq, LHX6 ChIP-seq, H3K4me1, H3K4me3, H3K27ac, H3K27me3, UCSC genes, and mammalian conservation. Both WT and Nkx2-1cKO data are shown for H3 modifications. Called peaks are labeled with an asterisk. VISTA transgenics showing in vivo activity of +/+REs (hs550, hs623, and hs883) at E11.5.
We observed higher MGE enhancer activity for the +/+REs compared to the +/−REs (Figure 7A). This activity was specific to the LHX6-expressing SVZ and MZ of the MGE and exemplified by hs550, hs623, and hs883 (Figures 7B and 7D). In the Nkx2-1cKO, we found a significant decrease of H3K27ac at all three regions; thus, their activity requires the combined activity of NKX2-1 and LHX6 (Figure 7D; Figure S6). In addition, the nearby genes showed reduced transcription (Data S1D, hs550 = Etv1, hs623 = Tcf12, and hs883 = Sox6). These results support the idea that NKX2-1 represses transcription in the MGE and that NKX2-1 and LHX6 combined binding is associated with transcriptional activation specific to the SVZ and MZ in the MGE.
NKX2-1 Represses and LHX6 Activates Transcription in the MGE
We used three types of experiments to test the model that NKX2-1 binding is required for repression and LHX6 binding for activation. First, we generated a transgenic mouse line harboring an activating +/+RE reporter that is a potential Tgfβ3 enhancer (mm1429, Figure 8A). As predicted by the model, the mm1429 RE had a high enhancer activity specific to the SVZ of the MGE at E12.5 (Figure 8B). Next, we mutated two NKX2-1 and two LHX6 consensus motifs in mm1429 separately and in combination (Figure 8C). The activity of the WT and mutant enhancers were evaluated using a transcription reporter assay performed using E13.5 MGE primary cultures. Mutation of the LHX6 sites eliminated the majority of mm1429 activity, providing strong evidence that LHX6 is required for activating mm1429 in the SVZ of the MGE (Figure 8C). On the other hand, mutation of the NKX2-1 motifs increased mJR23 activity. In an enhancer with both NKX2-1 and LHX6 motifs mutated, activity levels were similar to the WT sequence (Figure 8C). To further test whether NKX2-1 represses transcription, we generated a stable transgenic mouse line for the VISTA element hs1336 (a repressing +/−RE. The hs1336 mice were crossed to Nkx2-1cKOs. In this KO, we observed a de-repression of hs1336 combined with an increased transcription of the supposed hs1336 target gene BMPER (Figures S7A–S7D). Together, these results provide experimental support for our model that NKX2-1 acts on REs to repress transcription in the MGE, whereas LHX6 activation of REs increases transcription in the SVZ.
Figure 8. In Vivo Activity of NKX2-1 in the Developing MGE.
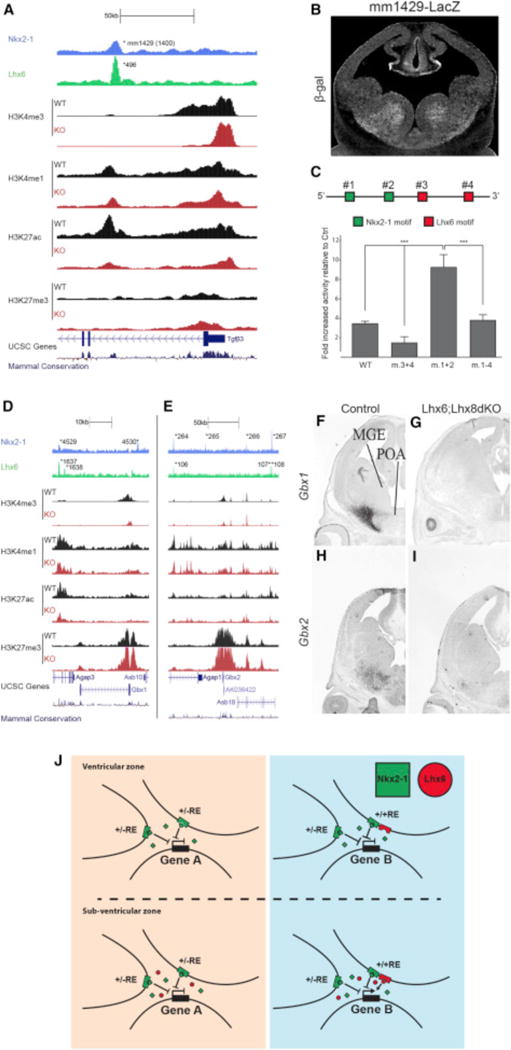
(A) mm1429 locus with the ChIP-seq datasets and genomic features; NKX2-1 ChIP-seq, LHX6 ChIP-seq, H3K4me1, H3K4me3, H3K27ac, H3K27me3, UCSC genes, and mammalian conservation. Both WT and Nkx2-1cKO data are shown for H3 modifications. Called peaks are labeled with an asterisk.
(B) mm1429 transgenic showing activity in the SVZ and MZ of the MGE at E12.5.
(C) Schematic of mm1429 with NKX2-1 and LHX6 motifs. Luciferase reporter assay showing the opposing effects of mutating NKX2-1 and LHX6 motifs in mm1429. Data are represented as mean ± SEM (n = 3).
(D–I) Genomic regions of the Gbx1 (D) and the Gbx2 (E) loci with the same ChIP-seq datasets and genomic features shown in Figure 5A. In situ analysis of Gbx1 (F and G) and Gbx2 (H and I) transcription in WT and Lhx6; Lhx8dKO forebrain at E13.5.
(J) Summary model showing the combined activity of NKX2-1 and LHX6 at REs in the VZ and SVZ.
LHX6 Is Required to Activate Gbx1 and Gbx2 Transcription
Many +/+REs are active in the SVZ and MZ of the MGE, likely due to LHX6 binding. Since NKX2-1 binds to +/+REs, we tested whether NKX2-1 is sufficient for maintaining transcription of genes through the +/+REs in the absence of LHX6. We examined transcriptional regulation of Gbx1 and Gbx2, two downregulated genes in the Nkx2-1cKO (Figure S1; Data S1A). The Gbx1 locus (+/−100 kb of TSS) contains one +/−RE (4,530), one +/+RE (4,529/1,637), and one −/+RE (1,638). The Gbx2 locus contains two +/+REs (264/106 and 267/108), two +/−REs (265 and 266), and one −/+RE (107 aREs; Figures 8D and 8E). All LHX6-bound Gbx1 and Gbx2 REs are classified as aREs based on changes in the histone profiles in the Nkx2-1cKO (Figures 8D and 8E; Data S1D). Since LHX6 is strongly reduced in the Nkx2-1cKO, it is impossible to separate the contribution of NKX2-1 and/or LHX6 to the loss of Gbx1 and Gbx2 transcription and the reduced activity at their associated REs in the mutant. To overcome this caveat, we looked at Gbx1 and Gbx2 transcription in a Lhx6; Lhx8 double KO, as LHX6 and LHX8 are known to have redundant activities in the basal ganglia (Flandin et al., 2011). In the Lhx6; Lhx8 double KO, we see a complete loss of Gbx1 and a near-complete loss of Gbx2 expression (Figures 8F–8I). Since NKX2-1 expression is normal in the Lhx6; Lhx8 double KO (Flandin et al., 2011), we conclude that LHX6 and LHX8 are required for Gbx1 and Gbx2 expression and that NKX2-1 binding is not sufficient to maintain Gbx1 and Gbx2 expression in the absence of LHX6 and LHX8. Examination of REs and target genes with significant binding of LHX6, but not NKX2-1, suggests that LHX6 can act independent of NKX2-1 to activate gene expression and that activation is the primary functional result of LHX6 binding both with and without NKX2-1 (Figure S7E; Data S1F).
DISCUSSION
TF and Epigenomic Analysis Reveal Genetically Encoded Neurodevelopmental Pathways
Studies applying genomic approaches to annotate REs and understand their function in vivo in the mouse brain have demonstrated the power of epigenomic assays. These studies have captured the specific regional enhancer activity patterns and highly dynamic nature of RE function during mouse brain development (Nord et al., 2013; Visel et al., 2009). However, these efforts leave unanswered the question of how RE activity is mediated by the TF binding. This represents a major barrier to our ability to predict RE function and understand transcriptional regulation at the mechanistic level. TF binding is known to be more predictive of tissue-specific RE enhancer activity than histone modifications (Dogan et al., 2015; Kwasnieski et al., 2014). Thus, genomic analysis of TF cascades will be critical toward unlocking RE function and understanding genomic regulatory wiring. Our findings enable comparison of binding properties, epigenomic state, and regional transcriptional activity, providing a model for expanding our understanding of TF-mediated RE activity mediating brain development. We showed that NKX2-1 and LHX6 exhibit combinatorial binding at distal REs that drives cell-type-specific activation and repression. Data from the MGE suggest that diverse neurodevelopmental activity patterns of REs can be predicted via integrating TF binding patterns with epigenomic profiling in relevant brain regions and cell types. Further, this analysis strategy provides a path toward reverse engineering genomic-encoded regulatory activity underlying neurodevelopment (Visel et al., 2013).
NKX2-1 is critical for basal ganglia development and interneuron specification. Previously, its genome-wide binding properties and activity in the developing brain were unknown. We found that NKX2-1 binding plays a direct role in transcriptional activation and repression of discrete gene sets. The presence of histone modifications at NKX2-1-bound REs that indicate active or repressed functionality in our results are in line with studies that have functionally classified REs using these signatures (Attanasio et al., 2014; Bernstein et al., 2006). In addition to H3K4me1 and H3K27ac as indicative of distal RE activity, we also find H3K4me3 at active distal REs near genes with a relatively high expression. This confirms that the H3K4me3 signal should be interpreted as a label of active distal REs, as well as active TSSs (Core et al., 2014; Pekowska et al., 2011). Furthermore, we identified NKX2-1-bound distal REs and TSSs that exhibit bivalent signatures with both activating (H3K4me3/H3K27ac) and repressive (H3K27me3) histone modifications, a feature described in earlier studies (Attanasio et al., 2014). Our findings suggest that these bivalent regions may be under tight developmental control, with many putative regulators functioning as neurodevelopmental TFs (e.g., Lhx6, Lhx8, Gli2, Gbx1, and Gbx2). Comparison of WT and Nkx2-1cKO epigenomic state and TF binding motifs highlight that other TFs, besides NKX2-1 (OCT, bHLH, and LHX), contribute to the epigenomic state of REs. Thus, neurodevelopmental REs may be targeted at various stages of development and RE activity is refined by region-specific TF binding. Further studies will be required to characterize specific features of bivalent REs and to dissect combinatorial binding and function of other neurodevelopmental TFs that bind the same pool of REs as NKX2-1.
NKX2-1 Controls MGE Regional Fate by Transcriptional Repression
Here we provide evidence that NKX2-1 promoted MGE identity through transcriptional repression in MGE progenitors (VZ). +/−REs show higher activity in cortex and LGE compared to the MGE, and alterations in the histone marks that occur on these REs in Nkx2-1cKOs show increased permissive chromatin and activity when there is no NKX2-1 binding. Many NKX2-1-repressed genes encode TFs and regulators of the sHH, WNT, and BMP signaling pathways (Figure 8J; Data S1A), which regulate cell differentiation and regional patterning. NKX2-1 repressive function is analogous to the activity of homeodomain TFs that regulate dorsoventral patterning of the neural tube (Briscoe et al., 2000; Muhr et al., 2001). Gro/TLE regulates transcription by reducing histone acetylation and inducing histone methylation, thereby promoting chromatin condensation and impairing activator recruitment. Local recruitment of Gro/TLE proteins by NKX2-1 could explain the changes in histone modifications associated with rREs in Nkx2-1cKOs (Chen et al., 1999; Patel et al., 2012). We used computational de novo motif analysis to identify candidate TFs that confer either activating or repressing properties to NKX2-1-bound REs (aREs and rREs). Here we find that aREs and rREs have an enrichment of OCT4 motifs. OCT4 can recruit other TFs and chromatin-modifying proteins to interaction hubs, REs that regulate ESC maintenance (Boyer et al., 2005; Chen et al., 2008; van den Berg et al., 2010). These data suggest that NKX2-1 functionally integrates a transcriptional network regulating local patterning and cell differentiation in the developing MGE.
NKX2-1 Is Required for the Initiation of Lhx6 Expression
After NKX2-1 represses the fates of regions adjacent to the MGE, it induces TFs that drive MGE-lineages. This is clearest in the SVZ, where Lhx6 and Lhx8 expression are induced by NKX2-1. Here we provide comprehensive evidence that NKX2-1 binds to and promotes the activity of REs in the Lhx6 and Lhx8 loci based on TF ChIP-seq and changes in the epigenome in the Nkx2-1cKO. Unlike its repressive function in the VZ, NKX2-1 promotes activation of Lhx6 and Lhx8 in the SVZ. Switching NKX2-1 from a repressor to an activator could involve differences in interacting proteins that are expressed in the VZ and SVZ. We found that binding motifs for LHX and E-box bHLH TFs are enriched in aREs and showed that LHX6 binding is enriched at aREs. We conclude that LHX6 is essential for mediating transcriptional activation downstream of NKX2-1 (Figure 8J). Furthermore, we predict that E-box proteins present in the MGE, such as ASCL1, OLIG1/2, and TCF3/4/12, also coordinate with NKX2-1 function together on aREs. Of note, bHLH and LIM/HD TFs coordinately regulate neural subtype specification in the ventral neural tube (Lee and Pfaff, 2003; Ma et al., 2008).
LHX6 and LHX8 Can Drive Gene Transcription Independent of NKX2-1
LHX6 and LHX8 regulate a common set of target genes in the developing MGE (Flandin et al., 2011). Here we show that LHX6 and LHX8 are required for activating Gbx1 and Gbx2 transcription in the MZ of the MGE. Even though NKX2-1 and LHX6 bind to the same aREs in the Gbx1 and Gbx2 loci, and NKX2-1 is expressed in the Lhx6; Lhx8 double mutant, NKX2-1 is insufficient to drive Gbx1 and Gbx2 transcription in the absence of LHX6 and LHX8. These data provide evidence that in the neurons developing in MZ, NKX2-1 is not required to activate transcription after the onset of LHX6 and LHX8 expression.
Combined Binding of NKX2-1 and LHX6 Identify Cell-Type-Specific Res
MGE-derived CINs initially express NKX2-1, LHX6, and LHX8 in the SVZ yet only express LHX6 as they leave the MGE (Sussel et al., 1999). Du et al. (2008) showed that LHX6 is sufficient to rescue the production of CINs in the Nkx2-1 KO. Thus, once Lhx6 and Lhx8 transcription is initiated, NKX2-1 is not required for the maturation of CINs. Consistent with this, one-third of LHX6-bound REs lack significant NKX2-1 binding. Indeed, −/+REs are associated with genes expressed in MGE SVZ and CINs, suggesting regulation by LHX6 during CIN development. Two-thirds of the LHX6-bound REs have robust NKX2-1 binding. These elements may regulate gene transcription in two types of cells: (1) SVZ cells that are fated to generate NKX2-1−; LHX6+ neurons (e.g., CINs in which NKX2-1 expression is eventually lost) and (2) SVZ and/or MZ cells that are fated to generate NKX2-1+; LHX6+ neurons (e.g., pallidal projection neurons [PPNs]). In this regard, we found +/+REs near genes expressed in CINs, including Arx, Cxcr7, Dlx1/2, Dlx5/6, Elmo1, Lhx6, Maf, Nrxn3, Nxph1, Robo1, Robo2, and Sox6; some of these genes are also expressed in the PPN (Dlx1, Lhx6, and Sox6). Thus, this approach efficiently identified known important regulators of MGE and CIN development as well as other unknown NKX2-1 and LHX6 target genes that are candidates for elucidating new molecular mechanisms regulating MGE development.
Transcriptional Circuits Specifying Cell Fate and Differentiation of MGE Lineages
Herein, we showed novel data that contribute to the elucidation of transcriptional circuits regulating cell-fate specification and differentiation of MGE lineages. Intersection of NKX2-1 and LHX6 ChIP-seq identified REs active in the MGE SVZ and/or MZ; proximity of these REs provides evidence for genes that regulate specification and differentiation of MGE-derived neurons that contribute to the GP, VP, and CINs. Comparison of MGE epigenomic state between WT and Nkx2-1cKO demonstrated that NKX2-1 binding is required to establish or maintain chromatin state and epigenetic control of expression. The MGE at E13.5 is a heterogeneous mix of cell types and states. While we cannot differentially dissect epigenomic state or NKX2-1 and LHX6 binding of cell populations within the MGE, intersecting regional transcription patterns and RE activity (via transgenic assays) increase our ability to predict the cell types where specific TF-mediated RE functions occur. Nonetheless, analysis of binding and epigenomic state of specific purified MGE cell types or regions (e.g., VZ versus SVZ) will be necessary to dissect complex epigenomic signatures, such as the presence of both activating and repressing histone modifications at bivalently marked REs.
TFs, Enhancers, and the Regulation of Gene Expression Underlying Brain Development
Our study provides a novel perspective of RE activity at levels of specificity and detail not previously examined in vivo in mammalian brain. The combination of genomic datasets, including TF binding, epigenomics, and global RNA expression, with in vivo regional RNA expression via in situ and in vivo RE activity via transgenic mouse assays enables us to examine how REs function to regulate expression in vivo. The intersection of TF binding to the same set of primarily distal REs in our study supports a model of REs as binding hubs that are critical for regulating spatiotemporal gene expression patterns. Further studies are necessary to increase the resolution of these analyses to separately define the transcriptional and epigenetic states of the MGE VZ and SVZ and their neuronal and glial products. Based on this initial examination of MGE TFs and activity patterns of bound REs, we propose that common REs direct gene expression patterns in the developing brain via complex activity mediated by cell-specific TF binding. These REs exhibit a remarkable level of evolutionarily sequence conservation, suggesting evolutionarily ancient genomic control of patterning and lineage specification in the developing brain guided by the coordinated binding of NKX2-1 and LHX6 in addition to many other linked TF pathways. REs identified here as bound by NKX2-1 are likely part of this set of common REs that exhibit differential activity depending on specific TF expression/binding and on RE epigenetic state. This initial effort at elucidating NKX2-1-mediated transcriptional pathways suggests that by dissecting neurodevelopmental transcription factor networks using the combination of genetics, genomics, and developmental neurobiology, we will gain further insight into the genetically encoded wiring diagram that ultimately gives rise to the magnificent functional capacity and complexity of the brain.
EXPERIMENTAL PROCEDURES
Mice
1336CT2IG was amplified from human genomic DNA and subcloned into Hsp68-CreERT2-IRES-GFP (Visel et al., 2013). Stable transgenic mice were generated by pronuclear injection at the UCSF Transgenic Core. 1336CT2IG was genotyped with forward primer ACACAGGTGCTCTGACTTTAC and reverse primer AGTGCTGCCTCTGACCTCAT, generating a product of 500 bp. All other mice strains have been previously reported: Nkx2-1f/f (Kusakabe et al., 2006), Olig2-tva-Cre (Schüller et al., 2008), Lhx6-PLAP (Choi et al., 2005), Lhx8KO (Zhao et al., 1999), and AI14 Cre-reporter (Madisen et al., 2010). All animal care and procedures were performed according to the University of California at San Francisco Laboratory Animal Research Center guidelines.
Dissection of Embryos
NKX2-1 and LHX6 ChIP-seq experiments were performed on micro-dissected basal ganglia from three litters of E13.5 CD1 mice. Histone ChIP-seq and RNA micro-array hybridization were performed on micro-dissected E13.5 MGE from Nkx2-1WT/HET and Nkx2-1cKO. All MGE dissections were performed as follows. The dorsal boundary was defined by the sulcus separating LGE and MGE. The caudal end of the sulcus defined the caudal boundary. Septum was removed.
TF ChIP
Dissected basal ganglia were disassociated and crosslinked at room temperature for 10 min in 1% formaldehyde. The crosslinked chromatin was sheared using a Bioruptor UCD-200 for 15 rounds (1 round = 30 s on/1 min off at high intensity) and incubated at 4°C overnight with 4 mg NKX2-1 polyclonal (Santa Cruz Biotechnology, Cat# sc-13040, RRID: AB_10015210) or LHX6 polyclonal antibody (GenScript) (Figure S8). 20× blocking peptide (LHX6 aa1-67, NKX2-1 aa1-190) was used as negative control. Protein/antibody complexes were collected using Dynabeads (20 μL protein A + 20 μL protein G) and processed as earlier described (Vokes et al., 2007).
Native Histone ChIP
Each ChIP was performed starting with ~300,000 nuclei from Nkx2-1WT/HET and Nkx2-1cKO MGE. The native ChIP was performed as earlier described (Magklara et al., 2011). Antibodies used were: H3K4me1 (Abcam, Cat# ab8895, RRID: AB_306847), H3K4me3 (Abcam, Cat# ab8580, RRID: AB_306649), H3K27me3 (Active Motif, Cat# 39155, RRID: AB_2561020), and H3K27ac (Abcam, Cat# ab4729, RRID: AB_2118291).
qPCR
All ChIP experiments were validated with ChIP-qPCR. This was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems) using SYBR GreenER qPCR SuperMix (Invitrogen, Cat. No. 11760-100). For primer sequences, see Supplemental Experimental Procedures. The qPCR data were analyzed as described in Vokes et al. (2007). For detail on primers used, see Supplemental Experimental Procedures.
ChIP-Seq Library Preparation and Sequencing
The ChIP-seq libraries were made using the Ovation Ultralow DR Multiplex System (Part no. 0330, NuGEN). Generated libraries were size selected (180–350 bp) and sequenced at the Center for Advanced Technology at UCSF (http://cat.ucsf.edu/) and the Genomics Core Facility (http://humangenetics.ucsf.edu/genomics-services/).
Histology
Immunofluorescence was performed on 16 mm cryosection as previously described (Zhao et al., 2008). In situ hybridization was performed as previously described (Schaeren-Wiemers and Gerfin-Moser, 1993). For details on how probes were generated, see Supplemental Experimental Procedures.
Differential Expression
RNA was isolated using RNeasy Mini Kit (QIAGEN). Quality control (QC) and RNA-micro array was performed at the UCSF SABRE Functional Genomics Facility (http://arrays.ucsf.edu/). For detailed protocols, see Supplemental Experimental Procedures.
ChIP-Seq Computational Analysis
Clustering, base calling, and quality metrics were performed using standard Illumina software. Sequencing libraries were analyzed for overall quality and were filtered, and reads were mapped to the mouse genome (mm9) using Burrows-Wheeler Alignment (BWA) (Li and Durbin, 2009). Peak calling on merged aligned bam files was performed using Macs2 considering both negative binding control and input DNA control. Coverage and heatmap diagrams were produced using the ngs.plot.r package (Shen et al., 2014). For NKX2-1 and LHX6, merged peak sets were generated across three replicates and annotated to genomic features. Motif analysis performed using HOMER (Heinz et al., 2010) with default settings and genomic background. Classification of chromatin state for NKX2-1 and LHX6 peaks performed based on presence of overlapping or flanking histone modification peak (H3K4me1, H3K4me3, H3K27ac, and H3K27me3). To identify NKX2-1-bound REs where chromatin state was sensitive to change in the Nkx2-1cKO, we compared differential ChIP enrichment by running the WT versus the cKO Macs2. Custom R scripts were used for computational analysis and are available at request. The data discussed in this publication have been deposited in NCBI’s GEO and are accessible through GEO Series accession number GSE85705 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE85705) and via a track hub which can be loaded for viewing in the UCSC genome browser (http://nordlab.genomecenter.ucdavis.edu/trackhubs/Nkx2.1/hub.txt). For details, see Supplemental Experimental Procedures.
Luciferase Reporter Assay
See Supplemental Experimental Procedures for details on primary culture preparation, transfection, motif mutagenesis, and luciferase assays.
Supplementary Material
Highlights.
Novel approach to dissect TF function by integrating genetic and genomic data
First comprehensive analysis of NKX2-1 TF function in the embryonic brain
NKX2-1 promotes regional identity by repressing genetic programs in the VZ of MGE
NKX2-1 and LHX6 co-occupy REs driving gene expression in the SVZ and MZ of the MGE
Acknowledgments
We thank members of the J.L.R.R. lab for advice and critical evaluation of the data. This project was supported by Vetenskapsrådet 2011-38865-83000-30 (M.S.), Svenska Sällskapet för Medicinsk Forskning (M.S.), R01HG003988 (A.V.), R24HL123879 (A.V.), U01DE024427 (A.V.), U54HG006997 (A.V.), and NIH grant MH081880 (J.L.R.R.). Research conducted at Lawrence Berkeley National Laboratory was performed under Department of Energy Contract DE-AC02- 05CH11231, University of California. J.L.R.R. is cofounder, stockholder, and currently on the scientific board of Neurona, a company studying the potential therapeutic use of interneuron transplantation.
Footnotes
ACCESSION NUMBERS
The accession number for the data reported in this paper is GEO: GSE85705.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, eight figures, and supplemental data and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2016.08.020.
AUTHOR CONTRIBUTIONS
Conceptualization, M.S., A.S.N., and J.L.R.R.; Methodology, M.S. and P.F.; Software, A.S.N. and L.S.-F.; Validation, M.S., P.F., A.S.N., J.D.P., and J.S.H.; Formal Analysis, M.S. and A.S.N.; Investigation, M.S.; Resources, S.S., C.K., and A.V.; Data Curation, A.N.; Writing – Original Draft, M.S., A.S.N., and J.L.R.R.; Writing – Review & Editing, M.S., A.S.N., and J.L.R.R.; Visualization, M.S. and A.S.N.; Supervision, M.S., A.S.N., and J.L.R.R.; Project Administration, M.S. and J.L.R.R; Funding Acquisition, M.S., J.L.R.R., and A.V.
References
- Attanasio C, Nord AS, Zhu Y, Blow MJ, Biddie SC, Mendenhall EM, Dixon J, Wright C, Hosseini R, Akiyama JA, et al. Tissue-specific SMARCA4 binding at active and repressed regulatory elements during embryogenesis. Genome Res. 2014;24:920–929. doi: 10.1101/gr.168930.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Peña-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–1276. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell. 2000;101:435–445. doi: 10.1016/s0092-8674(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Butt SJB, Sousa VH, Fuccillo MV, Hjerling-Leffler J, Miyoshi G, Kimura S, Fishell G. The requirement of Nkx2-1 in the temporal specification of cortical interneuron subtypes. Neuron. 2008;59:722–732. doi: 10.1016/j.neuron.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell K. Dorsal-ventral patterning in the mammalian telencephalon. Curr Opin Neurobiol. 2003;13:50–56. doi: 10.1016/s0959-4388(03)00009-6. [DOI] [PubMed] [Google Scholar]
- Chen G, Fernandez J, Mische S, Courey AJ. A functional interaction between the histone deacetylase Rpd3 and the corepressor groucho in Drosophila development. Genes Dev. 1999;13:2218–2230. doi: 10.1101/gad.13.17.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- Choi GB, Dong HW, Murphy AJ, Valenzuela DM, Yancopoulos GD, Swanson LW, Anderson DJ. Lhx6 delineates a pathway mediating innate reproductive behaviors from the amygdala to the hypothalamus. Neuron. 2005;46:647–660. doi: 10.1016/j.neuron.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet. 2014;46:1311–1320. doi: 10.1038/ng.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci USA. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogan N, Wu W, Morrissey CS, Chen KB, Stonestrom A, Long M, Keller CA, Cheng Y, Jain D, Visel A, et al. Occupancy by key transcription factors is a more accurate predictor of enhancer activity than histone modifications or chromatin accessibility. Epigenetics Chromatin. 2015;8:16. doi: 10.1186/s13072-015-0009-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Xu Q, Ocbina PJ, Anderson SA. NKX2.1 specifies cortical interneuron fate by activating Lhx6. Development. 2008;135:1559–1567. doi: 10.1242/dev.015123. [DOI] [PubMed] [Google Scholar]
- Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flandin P, Zhao Y, Vogt D, Jeong J, Long J, Potter G, Westphal H, Rubenstein JLR. Lhx6 and Lhx8 coordinately induce neuronal expression of Shh that controls the generation of interneuron progenitors. Neuron. 2011;70:939–950. doi: 10.1016/j.neuron.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkouli A, van Wijk NV, Lopes R, Kessaris N, Pachnis V. LIM homeodomain transcription factor-dependent specification of bipotential MGE progenitors into cholinergic and GABAergic striatal interneurons. Development. 2009;136:3841–3851. doi: 10.1242/dev.038083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Kawaguchi A, Hoshi N, Kawaguchi R, Hoshi S, Kimura S. Thyroid-specific enhancer-binding protein/NKX2.1 is required for the maintenance of ordered architecture and function of the differentiated thyroid. Mol Endocrinol. 2006;20:1796–1809. doi: 10.1210/me.2005-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA. High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 2014;24:1595–1602. doi: 10.1101/gr.173518.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Pfaff SL. Synchronization of neurogenesis and motor neuron specification by direct coupling of bHLH and homeodomain transcription factors. Neuron. 2003;38:731–745. doi: 10.1016/s0896-6273(03)00296-4. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YC, Song MR, Park JP, Henry Ho HY, Hu L, Kurtev MV, Zieg J, Ma Q, Pfaff SL, Greenberg ME. Regulation of motor neuron specification by phosphorylation of neurogenin 2. Neuron. 2008;58:65–77. doi: 10.1016/j.neuron.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–140. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magklara A, Yen A, Colquitt BM, Clowney EJ, Allen W, Markenscoff-Papadimitriou E, Evans ZA, Kheradpour P, Mountoufaris G, Carey C, et al. An epigenetic signature for monoallelic olfactory receptor expression. Cell. 2011;145:555–570. doi: 10.1016/j.cell.2011.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Rubenstein JLR. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhr J, Andersson E, Persson M, Jessell TM, Ericson J. Groucho-mediated transcriptional repression establishes progenitor cell pattern and neuronal fate in the ventral neural tube. Cell. 2001;104:861–873. doi: 10.1016/s0092-8674(01)00283-5. [DOI] [PubMed] [Google Scholar]
- Nord AS, Blow MJ, Attanasio C, Akiyama JA, Holt A, Hosseini R, Phouanenavong S, Plajzer-Frick I, Shoukry M, Afzal V, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155:1521–1531. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nord AS, Pattabiraman K, Visel A, Rubenstein JLR. Genomic perspectives of transcriptional regulation in forebrain development. Neuron. 2015;85:27–47. doi: 10.1016/j.neuron.2014.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–1289. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SR, Bhumbra SS, Paknikar RS, Dressler GR. Epigenetic mechanisms of Groucho/Grg/TLE mediated transcriptional repression. Mol Cell. 2012;45:185–195. doi: 10.1016/j.molcel.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekowska A, Benoukraf T, Zacarias-Cabeza J, Belhocine M, Koch F, Holota H, Imbert J, Andrau JC, Ferrier P, Spicuglia S. H3K4 tri-methylation provides an epigenetic signature of active enhancers. EMBO J. 2011;30:4198–4210. doi: 10.1038/emboj.2011.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petryniak MA, Potter GB, Rowitch DH, Rubenstein JLR. Dlx1 and Dlx2 control neuronal versus oligodendroglial cell fate acquisition in the developing forebrain. Neuron. 2007;55:417–433. doi: 10.1016/j.neuron.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeren-Wiemers N, Gerfin-Moser A. A single protocol to detect transcripts of various types and expression levels in neural tissue and cultured cells: in situ hybridization using digoxigenin-labelled cRNA probes. Histochemistry. 1993;100:431–440. doi: 10.1007/BF00267823. [DOI] [PubMed] [Google Scholar]
- Schüller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, Huillard E, Sun T, Ligon AH, Qian Y, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E. ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014;15:284. doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EL, Watanabe H, Magendantz M, Hoersch S, Chen TA, Wang DG, Crowley D, Whittaker CA, Meyerson M, Kimura S, Jacks T. Nkx2-1 represses a latent gastric differentiation program in lung adenocarcinoma. Mol Cell. 2013;50:185–199. doi: 10.1016/j.molcel.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- van den Berg DLC, Snoek T, Mullin NP, Yates A, Bezstarosti K, Demmers J, Chambers I, Poot RA. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell. 2010;6:369–381. doi: 10.1016/j.stem.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Taher L, Girgis H, May D, Golonzhka O, Hoch RV, McKinsey GL, Pattabiraman K, Silberberg SN, Blow MJ, et al. A high-resolution enhancer atlas of the developing telencephalon. Cell. 2013;152:895–908. doi: 10.1016/j.cell.2012.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt D, Hunt RF, Mandal S, Sandberg M, Silberberg SN, Nagasawa T, Yang Z, Baraban SC, Rubenstein JLR. Lhx6 directly regulates Arx and CXCR7 to determine cortical interneuron fate and laminar position. Neuron. 2014;82:350–364. doi: 10.1016/j.neuron.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vokes SA, Ji H, McCuine S, Tenzen T, Giles S, Zhong S, Longabaugh WJR, Davidson EH, Wong WH, McMahon AP. Genomic characterization of Gli-activator targets in sonic hedgehog-mediated neural patterning. Development. 2007;134:1977–1989. doi: 10.1242/dev.001966. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Francis JM, Woo MS, Etemad B, Lin W, Fries DF, Peng S, Snyder EL, Tata PR, Izzo F, et al. Integrated cistromic and expression analysis of amplified NKX2-1 in lung adenocarcinoma identifies LMO3 as a functional transcriptional target. Genes Dev. 2013;27:197–210. doi: 10.1101/gad.203208.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Guo YJ, Tomac AC, Taylor NR, Grinberg A, Lee EJ, Huang S, Westphal H. Isolated cleft palate in mice with a targeted mutation of the LIM homeobox gene lhx8. Proc Natl Acad Sci USA. 1999;96:15002–15006. doi: 10.1073/pnas.96.26.15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Marín O, Hermesz E, Powell A, Flames N, Palkovits M, Rubenstein JLR, Westphal H. The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci USA. 2003;100:9005–9010. doi: 10.1073/pnas.1537759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Flandin P, Long JE, Cuesta MD, Westphal H, Rubenstein JLR. Distinct molecular pathways for development of telencephalic interneuron subtypes revealed through analysis of Lhx6 mutants. J Comp Neurol. 2008;510:79–99. doi: 10.1002/cne.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.