

Abstract
In order to maintain a stable genome, cells need to detect and repair DNA damage before they complete the division cycle. To this end, cell cycle checkpoints prevent entry into the next cell cycle phase until the damage is fully repaired. Proper reentry into the cell cycle, known as checkpoint recovery, requires that a cell retains its original cell cycle state during the arrest. Here, we have identified Tousled‐like kinase 2 (Tlk2) as an important regulator of recovery after DNA damage in G2. We show that Tlk2 regulates the Asf1A histone chaperone in response to DNA damage and that depletion of Asf1A also produces a recovery defect. Both Tlk2 and Asf1A are required to restore histone H3 incorporation into damaged chromatin. Failure to do so affects expression of pro‐mitotic genes and compromises the cellular competence to recover from damage‐induced cell cycle arrests. Our results demonstrate that Tlk2 promotes Asf1A function during the DNA damage response in G2 to allow for proper restoration of chromatin structure at the break site and subsequent recovery from the arrest.
Keywords: Asf1A, cell cycle, checkpoint recovery, DNA damage, Tlk2
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
DNA damage poses a continuous threat to the integrity of our genome. To deal with this threat, cells are equipped with DNA damage checkpoints that promote repair of the damage while delaying cell cycle progression 1. Once the DNA damage has been repaired and the checkpoint response is shut down, cells need to resume the cell cycle to be able to divide. This is particularly challenging in G2 cells, because the entry into mitosis critically depends on the presence of a variety of mitotic regulators, whose expression and activity is inhibited upon activation of the DNA damage checkpoint. These include key mitotic entry regulators such as Plk1 and cyclin B1/Cdk1 1, 2, 3, 4. In addition to inhibition of their kinase activity, transcription and protein stability are also repressed in damaged cells 3, 5, 6. Regulation of their expression is a delicate process, since too high levels of expression can compromise the DNA damage response (DDR), resulting in premature cell cycle reentry, while excessive reduction of expression pushes cells in a state of permanent arrest 6, 7.
Therefore, maintenance of G2‐specific gene expression is crucial to maintain cells competent to recover from a DNA damage‐induced G2 arrest 8, 9. This recovery competence is controlled by a balance of transcriptional repression via p53 on the one hand and transcriptional activation by transcription factors such as FoxM1, B‐Myb, and Lin‐9 on the other 9, 10. As the DDR progresses, the function of p53 is repressed by the p53‐induced phosphatase Wip1 11, 12, while the function of FoxM1 is maintained through Cdk‐dependent phosphorylation 9, 13. However, additional regulators of recovery competence are likely to exist 14. For instance, the contribution of chromatin remodeling at the break site to checkpoint recovery remains elusive. Chromatin remodeling is essential for the processing and repair of the break site, and for the restoration of normal chromatin function 15. Recovery of transcription after DNA damage has been shown to depend on histone exchange, indicating that chromatin restoration could possibly play a role in the control of recovery competence 16.
Here, we identified Tousled‐like kinase 2 (Tlk2) as a key regulator of checkpoint recovery. Tlk2 is a homolog of Tousled, first identified in Arabidopsis thaliana 17. Humans contain 2 homologs of Tousled, Tlk1 and Tlk2, whose activities are cell cycle regulated and peak during S phase 18. Tlk1 and 2 phosphorylate Asf1A and Asf1B and regulate their stability 19, 20. Asf1A and Asf1B are histone chaperones that are required to insert histone H3‐H4 dimers into newly replicated DNA 21, 22. In addition to their function in S phase, Tlks have also been implicated in the DDR 23, 24, 25, 26. Tlks are a target of the DDR suggesting a possible role during checkpoint recovery 24.
Here, we report that Tlk2 is a kinase whose activity is responsible for recovery from a DNA damage‐induced arrest in the G2 phase of the cell cycle. Absence of Tlk2 activity results in loss of recovery competence. We further show that Tlk2 is required for Asf1A‐dependent chromatin restoration after DNA damage and thereby retains expression of pro‐mitotic genes required for efficient recovery.
Results
An siRNA‐based screen identifies potential regulators of recovery
To identify kinases that are specifically required for recovery, we performed a screen in U2OS osteosarcoma cells using an siRNA kinome library. Each gene was targeted by a pool of 4 independent siRNAs targeting different sequences in the gene. We analyzed whether siRNA‐mediated protein depletion would result in reduced recovery from DNA damage. For comparison, we also monitored the effect of protein depletion on unperturbed cycling cells (Fig EV1A). To analyze recovery after DNA damage, we treated DNA damage‐arrested U2OS cells with caffeine for 8 h and monitored mitotic entry by staining with antibodies directed at phosphorylated‐S10 on histone H3 (Fig EV1A and B). Caffeine inhibits the DDR and efficiently promotes recovery in G2‐arrested cells 27, 28, 29.
Figure EV1. Primary screen details.
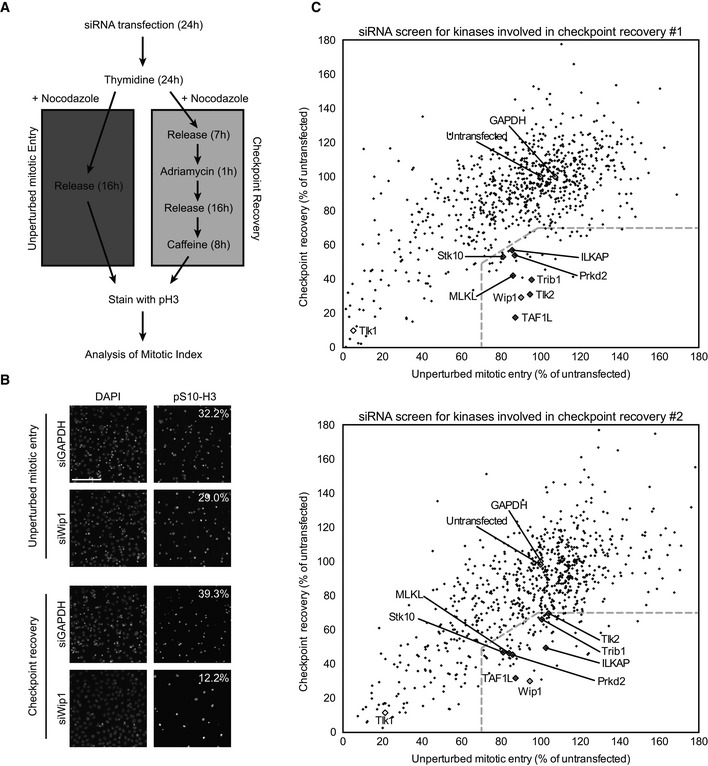
- Protocol used for the siRNA‐based kinome screen. U2OS cells were transfected for 24 h, synchronized at the G1/S border by thymidine for 24 h, and then either released into nocodazole for 16 h (unperturbed mitotic entry) or released for 7 h, treated with 0.5 μM adriamycin for 1 h, and after 16 h, cells were induced to recover by addition of caffeine for 8 h (checkpoint recovery). Cells were fixed and stained with DAPI and antibodies directed at histone H3‐pS10. Finally, automated analysis of the percentage of histone H3‐pS10‐positive DAPI‐stained nuclei was carried out.
- Representative images of positive control (siWip1)‐ and negative control (siGAPDH)‐transfected cells treated as in (A). Calculated percentages are indicated. Scale bar indicates 100 μm.
- Plots of duplicate screens. U2OS cells were treated as in (A). Black diamonds indicate individual siRNA‐targeted genes from the library, light gray diamonds indicate positive (Wip1‐depleted) and negative (untransfected and GAPDH‐depleted) controls, dark gray diamond indicates the genes that met the criteria in both screens, and white diamonds indicate Tlk1‐depleted conditions. Dotted lines indicate selection criteria for recovery‐specific genes.
We were primarily interested in genes that would affect recovery after a DNA damage arrest but would not affect unperturbed mitotic entry. As a positive control, we used knockdown of Wip1, a phosphatase known to control checkpoint recovery (Fig EV1B, 8). We performed the kinome‐wide screen in duplicate and considered genes that would show < 70% recovering cells relative to untransfected controls, but > 70% of cells entering mitosis in the unperturbed mitotic entry relative to untransfected controls. In addition, the ratio of recovering cells over cells entering mitosis in the non‐damaged cultures had to be < 0.7. Using this approach, we were able to identify seven genes that potentially affect checkpoint recovery (Figs 1 and EV1C, Dataset EV1, Table EV1).
Figure 1. An siRNA‐based screening approach identifies potential regulators of recovery.
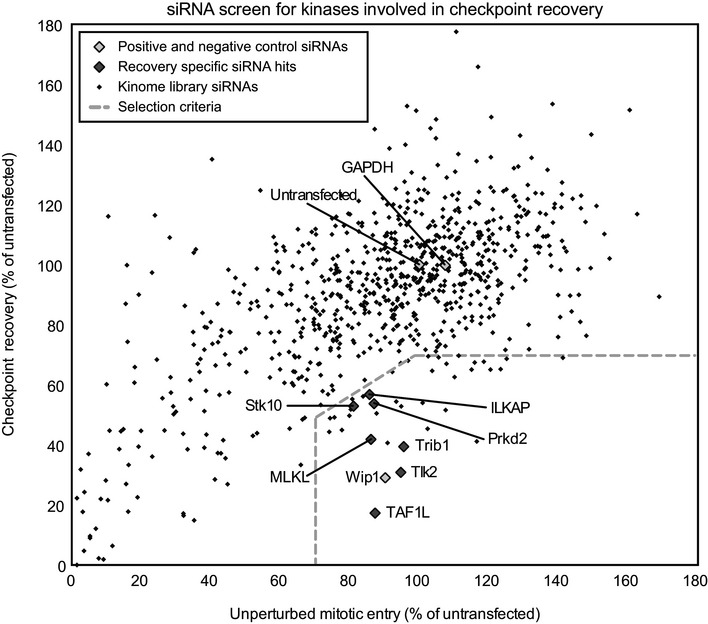
Plot of one of the screens (the duplicates are shown in Fig EV1C). U2OS cells were transfected, synchronized at the G1/S border by thymidine for 24 h, and then either released into nocodazole for 16 h (unperturbed mitotic entry) or for 7 h followed by treatment with 0.5 ?M adriamycin for 1 h, and after a 16‐h G2 arrest, cells were induced to recover by addition of caffeine (checkpoint recovery). Mitotic index was scored based on the percentage of histone H3‐pS10‐positive DAPI‐nuclei and normalized to the untransfected controls. Black diamonds indicate individual siRNA‐targeted genes from the library, light gray diamonds indicate positive (Wip1‐depleted) and negative (untransfected and GAPDH‐depleted) controls, and dark gray diamonds indicate the hits based on the two screens. Dotted lines indicate selection criteria for recovery‐specific genes.
Tlk2 is important for checkpoint recovery
We next performed a secondary screen in which we deconvoluted the single siRNAs from the pools used in our primary screen and considered only those genes that met our criteria for at least three out of the four single siRNAs as bona fide hits. For Tlk2, all four different targeting sequences displayed a defect in our checkpoint recovery assay, but not in the unperturbed situation (Fig 2A). The other 6 genes identified in the primary screen did not meet the strict criteria we set for the secondary screen, making Tlk2 our sole hit (Fig EV2A–C). There was a slight variation in the extent of the recovery defect observed with the different Tlk2 siRNAs, which correlated well with the extent of protein depletion achieved with the independent siRNAs (Fig 2B).
Figure 2. Tlk2 kinase activity is required for recovery from a DNA damage‐induced arrest.
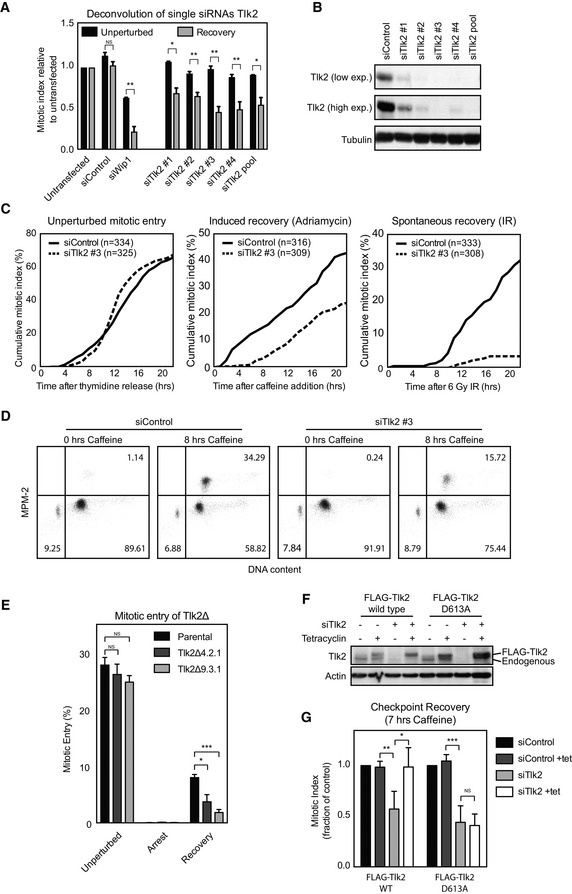
- U2OS cells were transfected with four independent siRNAs from the pools used in the screen, treated as in Fig 1, and analyzed for mitotic index. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, *P ≤ 0.05, **P ≤ 0.01).
- U2OS cells were synchronized with a single thymidine block, released into G2, and damaged with 0.5 μM adriamycin for 1 h. After a 16‐h G2 arrest, cells were harvested for Western blot analysis and analyzed for Tlk2 protein levels.
- Live cell imaging of thymidine‐synchronized unperturbed or damaged G2 cells. Cumulative percentage of cells entering mitosis were scored and plotted.
- U2OS cells were transfected with either a control siRNA or Tlk2 siRNA #3, synchronized, and damaged in G2. Cells were either harvested or treated with caffeine for 8 h before harvest, and cell cycle distribution was analyzed by FACS. Percentages of cells in each quadrant are indicated.
- Tlk2Δ cells were generated using CRISPR/Cas9 genome editing. Cells were synchronized in G2 by thymidine release and damaged with 0.5 μM adriamycin for 1 h. After a 16‐h G2 arrest, cells were induced to recover by addition of caffeine for 8 h and analyzed by FACS. Error bars represent SD, n = 4. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, *P ≤ 0.05, ***P ≤ 0.001).
- U2TR cells stably expressing Tlk2 siRNA #3‐insensitive tetracycline‐inducible FLAG‐Tlk2‐wt or FLAG‐Tlk2‐D613A were thymidine‐synchronized and damaged in G2. Tetracycline was present form the start of the experiment where indicated.
- Caffeine‐induced recovery assay of cell lines shown in (F). Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001).
Figure EV2. Secondary screening of identified kinases.
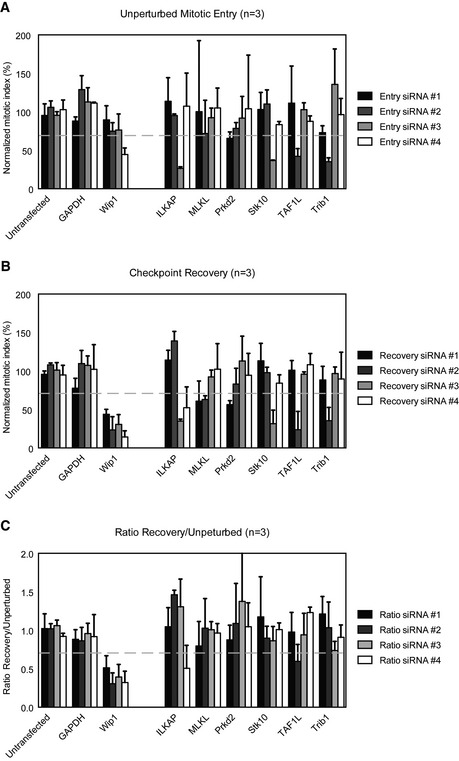
-
A–CSingle siRNAs which were pooled in the primary screen were deconvolved. U2OS cells were transfected with the siRNAs, and mitotic index is determined as in Fig 1. For each gene identified in the screen, the mitotic index of the single siRNAs is shown in both the unperturbed setup (A), recovery setup (B) and a ratio between the two (C). The gray‐dotted line indicates the cutoff criteria. Error bars represent SD, n = 3.
To more carefully determine the kinetics of cells entering mitosis in our assays, we followed the cells by time‐lapse microscopy and plotted the cumulative mitotic index. The timing of mitotic entry in control and Tlk2‐depleted cells was very similar (Fig 2C). However, cumulative mitotic entry after DNA damage showed a clear defect in mitotic entry in Tlk2 depletion upon the addition of caffeine (Fig 2C). To rule out that the recovery defect was specific to caffeine‐induced recovery, we also monitored spontaneous recovery. While G2‐arrested U2OS cells could spontaneously recover after irradiation with 6 Gy of ionizing radiation (IR), we found that Tlk2‐depleted cells were severely impaired (Fig 2C).
In order to confirm that the defect in recovery is not caused by a general defect in DNA replication, we performed FACS analysis of control and Tlk2‐depleted cells at the moment of damage and 8 h after induction of recovery. Indeed, we could clearly see that in all cases, more than 90% of the cells have a 4n DNA content and that the only difference is the distribution of these cells between G2 and mitosis (Fig 2D). In addition, Y15 phosphorylation, an inhibitory phosphorylation site on Cdk1 remained high in Tlk2‐depleted cells, indicating a potent arrest (Fig EV4A). In contrast, normal cell proliferation and EdU incorporation in unperturbed cultures were not significantly altered upon depletion of Tlk2 (Fig EV3A–D), compared to a low dose of 1 mM hydroxyurea (Fig EV3A–D). To definitively rule out that Tlk2 depletion might compromise cell viability under normal conditions, we used CRISPR/Cas9 genome editing 30 to generate two Tlk2 knockout cell lines, Tlk2Δ 4.2.1 and Tlk2Δ 9.3.1, respectively (Fig EV3E–G). Both knockout cell lines displayed normal growth kinetics (Fig EV3H–J) and mitotic entry after release from a thymidine block was unaffected (Fig 2E). Importantly, recovery after a DNA damage‐induced arrest was greatly reduced (Figs 2E and EV3K), providing conclusive evidence that the depletion of Tlk2 selectively affects cell cycle reentry after DNA damage and does not affect normal cell division. This is consistent with the recent finding that Tlk2 is not essential for cell viability 31. As a final validation of Tlk2 as a regulator of recovery, we generated cell lines expressing tetracycline‐inducible siRNA‐resistant FLAG‐tagged versions of Tlk2 to perform a rescue experiment. Using this setup, we could express either wild‐type Tlk2 or a kinase‐dead mutant (D613A) in cells depleted of endogenous Tlk2 (Fig 2F). We could clearly observe the recovery defect after depletion of endogenous Tlk2. More importantly, we were able to rescue this defect by inducing wild‐type FLAG‐Tlk2 (Fig 2G). However, reconstitution with the D613A mutant did not result in a rescue, showing that cells need Tlk2 kinase activity to properly recover from a DNA damage‐induced arrest. While Tlk2 might not be at its peak activity at this moment, cells could rely on a minimal general level of Tlk2 activity or localized elevation of its activity at the sites of damage. Interestingly, DNA damage‐induced inhibition of a kinase that plays an essential role during the DNA damage response is not without precedent, as we have previously shown that minimal Cdk activity is essential during the DDR, even though it is actively repressed 9. Also, the activity of other DNA damage‐sensitive kinases, such as Plk1, is required to start repair during a highly active DDR 32. Taken together, these observations show that during checkpoint recovery, cells require active Tlk2 to recover from a DNA damage‐induced cell cycle arrest in G2.
Figure EV4. Analysis of Tlk1 and Asf1A during DNA damage and recovery.
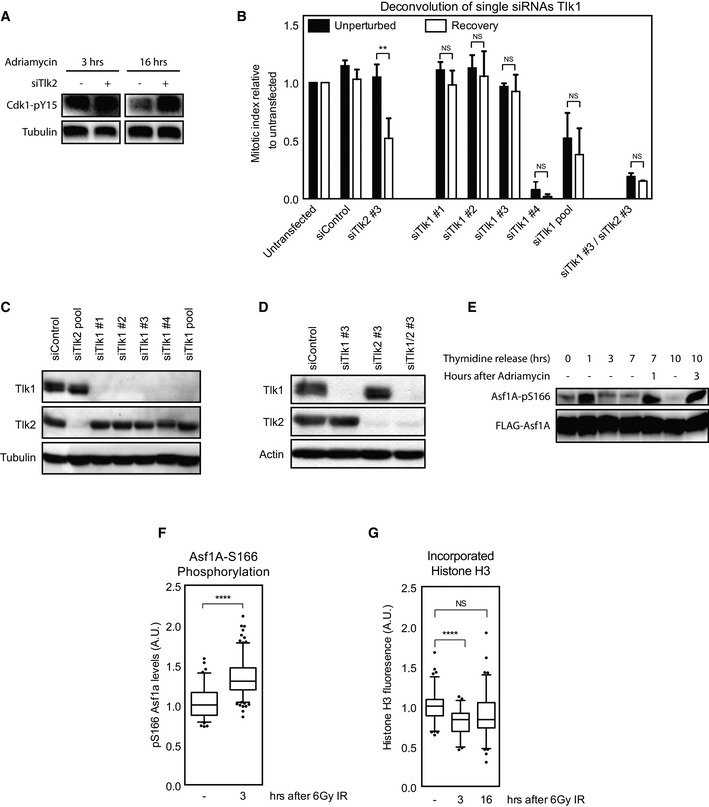
-
AU2OS cells were transfected with control or Tlk2 siRNAs and synchronized in G2 by a single thymidine block. Cells were subsequently damaged in G2 by 0.5 μM adriamycin for 1 h and harvested at the indicated times after damage. Samples were analyzed by Western blot with the indicated antibodies.
-
BCells were treated as in Fig 2A. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, **P ≤ 0.01).
-
C, DU2OS cells were synchronized with a single thymidine block, released into G2, and damaged with 0.5 μM adriamycin for 1 h. After a 16‐h G2 arrest, cells were harvested for Western blot analysis and analyzed for Tlk1 and Tlk2 protein levels.
-
EInducible FLAG‐Asf1A‐wt cell lines were synchronized by a single thymidine block and released for the hours indicated. Adriamycin was added for 1 h at 0.5 μM and washed out for the indicated time. FLAG‐Asf1A was immunoprecipitated and blotted with the indicated antibodies.
-
FCells were synchronized in G2 phase by release from a single thymidine block. Cells were left untreated or were irradiated with 6 Gy of IR. Samples were fixed 3 h afterward and stained for Asf1A‐pS166. Nuclear fluorescence intensity was measured relative to DAPI. Whiskers represent 5–95% of data points. Statistical significance was tested using an unpaired two‐tailed t‐test (****P ≤ 0.0001).
-
GU2OS cells were synchronized in G2 by a single thymidine block and were left untreated or were irradiated with 6 Gy of IR. Samples were pre‐extracted at the indicated time points and were fixed for analysis. Cells were stained for total levels of histone H3 and quantified relative to DAPI. Whiskers represent 5–95% of data points. Statistical significance was tested using an unpaired two‐tailed t‐test (NS P > 0.05, ****P ≤ 0.0001).
Figure EV3. Tlk2 is not essential for replication and cell viability.
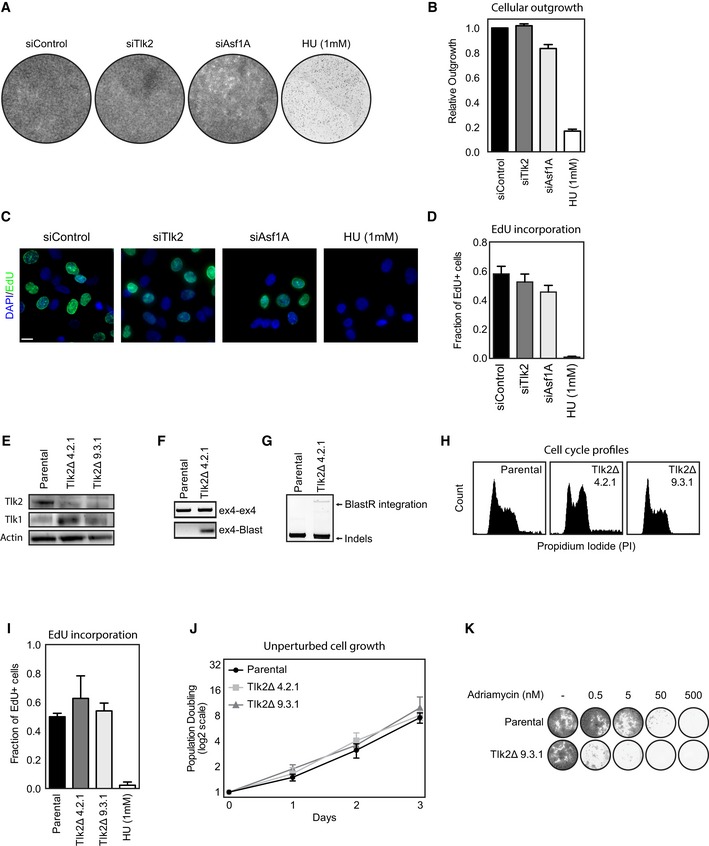
-
AAsynchronously cycling U2OS cells were transfected with the indicated siRNAs and analyzed for outgrowth after 7 days using crystal violet staining. Hydroxyurea was added where indicated at a concentration of 1 mM.
-
BQuantification of the experiment in (A). Error bars represent SD, n = 3.
-
CAsynchronously cycling U2OS cells were treated as in (A). Cells were treated with EdU for 2 h, fixed, and analyzed for Edu incorporation. Scale bar indicates 10 μm.
-
DQuantification of the experiment in (C). Error bars represent SD, n = 3.
-
EWestern blot analysis of Tlk2 knockout cells.
-
F, GIncorporation of the BlastR gene as measured by PCR amplification of genomic DNA.
-
HCell cycle analysis of asynchronous parental and Tlk2Δ cell lines.
-
IAsynchronously cycling parental and Tlk2Δ cell lines were treated with EdU for 4 h and analyzed for EdU incorporation similar to (D). Error bars represent SD, n = 3.
-
JPopulation doubling during 3 consecutive days of asynchronously cycling parental and Tlk2Δ cell lines. Error bars represent SD, n = 3.
-
KAdriamycin sensitivity in parental and Tlk2Δ cells. Cells were treated with the indicated amounts of adriamycin and analyzed for outgrowth using crystal violet staining after 7 days.
Evidence thus far suggests that both Tlk1 and Tlk2 display similar functions 18, 19. Therefore, we were curious to know why we did not pick up Tlk1 in our screen. In our screen, we found that Tlk1 depletion resulted in very clear general mitotic entry defect (Fig EV1C). We deconvoluted the pooled siRNAs from the library and assessed the effects on normal mitotic entry and recovery. Interestingly, only siRNA #4 produced a clear mitotic entry defect, whereas the other three siRNAs did not seem to affect mitotic entry or recovery in any way (Fig EV4B). All four siRNAs depleted Tlk1 very efficiently (Fig EV4C). Therefore, we have to conclude that siRNA #4 produces an off‐target effect and Tlk1 has no essential role in mitotic entry either with or without DNA damage. This suggests that Tlk2 performs a unique function after DNA damage, while Tlk1 and 2 might act redundantly in non‐damaged cells. To corroborate this notion, we co‐depleted Tlk1 and 2 and found a general failure to enter mitosis in either condition (Fig EV4B and D), indicating that Tlk1 and Tlk2 indeed perform redundant roles in non‐damaged cells. However, when cells are challenged with insults to their DNA, they rely on Tlk2 for efficient recovery.
Tlk2 controls Asf1A to promote recovery
Tlks have been implicated in the DDR previously 23, 24, 25, 26, and therefore, we asked whether depletion of Tlk2 would affect repair of the DNA damage. To this end, we synchronized cells in G2, damaged them using 0.5 μM adriamycin, and monitored the kinetics of γH2AX foci over time. In both the control and Tlk2‐depleted conditions, we saw efficient induction of γH2AX foci shortly after damage and this signal disappeared over time (Fig 3A and B). However, we noted a slightly higher induction and a faster decrease of γH2AX in Tlk2‐depleted cells. At the earlier time points, this difference was not significant, but at 24 h after damage we observed a significantly lower level of γH2AX in the Tlk2‐depleted cells. We next performed a more extensive time course to analyze the induction of p53 and p21, as well as the activation of Chk1, as visualized by the phosphorylation at S317 (Fig 3C). Similar to the altered timing of γH2AX, we again observed a slightly faster induction of Chk1 and p53 in the Tlk2‐depleted cells (Fig 3C). In addition, we observed that the phosphorylation of S317 on Chk1 decreased faster in the Tlk2‐depleted cells. While these data suggest that Tlk2 depletion leads to a minor shift in the kinetics of the DDR, they also demonstrate that Tlk2‐depleted cells are able to efficiently activate and silence a functional DDR in response to genotoxic insults, and therefore, we believe the impact of these alterations to be negligible.
Figure 3. Depletion of Tlk2 leads to altered kinetics of the DNA damage response.
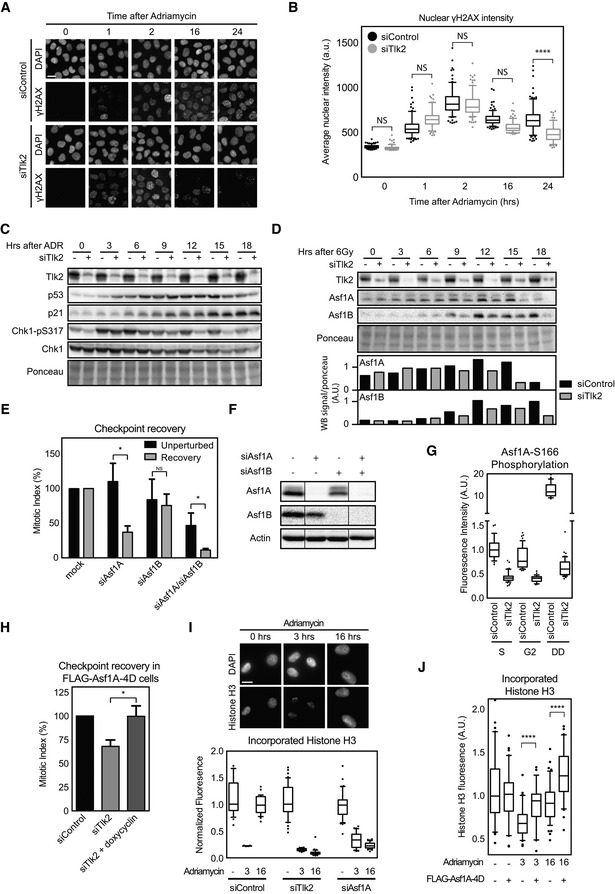
- U2OS cells were grown on glass coverslips and synchronized in G2 after thymidine release. Cells were fixed at the indicated times after adriamycin‐induced DNA damage and stained for DAPI and γH2AX. Scale bar indicates 10 μm.
- Quantification of cells in (A). Each dot represents the total signal of γH2AX in the nucleus of a single cell. Bars indicate the mean and error bars indicate the SD of the data points in each condition. Whiskers represent 5–95% of data points. Statistical significance was tested using an unpaired two‐tailed t‐test (NS P > 0.05, ****P ≤ 0.0001).
- U2OS cells were synchronized in G2 and harvested at the indicated times after induction of adriamycin‐induced DNA damage with or without Tlk2 depletion. Samples were analyzed by Western blotting for the indicated proteins.
- U2OS cells were synchronized in G2 and damaged with 6 Gy of IR. Samples were taken at the indicated times after induction of damage with or without Tlk2 depletion. Samples were analyzed by Western blot for the indicated proteins. Band signal intensity for Asf1A and Asf1B was measured and corrected for Ponceau S staining.
- U2OS cells were transfected with the indicated siRNAs and treated as in Fig 2A. Mitotic index was analyzed by FACS. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, *P ≤ 0.05).
- U2OS cells were synchronized with a single thymidine block, released into G2, and damaged with 0.5 μM adriamycin for 1 h. After a 16‐h G2 arrest, cells were harvested for Western blot analysis and analyzed for the indicated protein levels. Lanes were all present on the same blot but pasted together for comparison as indicated by the lines.
- Cells were synchronized in S or G2 phase by a single thymidine block. Damage was induced by 0.5 μM adriamycin for 1 h, and cells were allowed to incubate for 16 h afterward. Samples were fixed at the indicated cell cycle stages and stained for Asf1A‐pS166. Nuclear fluorescence intensity was measured relative to DAPI. Whiskers represent 5–95% of data points.
- Cells were treated and analyzed as in Fig 2E. Induction of FLAG‐Asf1A‐4D was induced using doxycycline. Error bars represent SD, n = 5. Statistical significance was tested using a paired two‐tailed t‐test (*P ≤ 0.05).
- U2OS cells were synchronized in G2 by a single thymidine block and where indicated treated with 0.5 μM adriamycin for 1 h and after the indicated time points were pre‐extracted and fixed. Cells were stained for total levels of histone H3 and quantified relative to DAPI. Scale bar indicates 10 μm. Whiskers represent 5–95% of data points.
- FLAG‐Asf1A‐4D‐inducible cells were treated as in (I), and expression was induced using doxycycline where indicated. Whiskers represent 5–95% of data points. Statistical significance was tested using an unpaired two‐tailed t‐test (****P ≤ 0.0001).
Given the minor difference in checkpoint activation, we next decided to investigate events triggered by Tlk2 itself. Known substrates of Tlks include Asf1A and Asf1B 19, 20, 33. These histone chaperones are required for proper chromatin assembly during S phase and after DNA damage, although the latter is mostly established in yeast 19, 34. In mammalian cells, Asf1A and Asf1B have redundant roles in S phase 35, 36, and therefore, we wondered whether these histone chaperones could be the targets of Tlk2 to promote recovery after DNA damage and might share isoform specificity during checkpoint recovery similar to Tlks. To test this, we first analyzed whether expression of Asf1A or Asf1B was affected by IR. Protein samples were taken at several time points after IR and blotted for both histone chaperones (Fig 3D). Interestingly, we observe a clear induction of Asf1A and Asf1B at 9 to 12 h after IR, but this induction was absent in Tlk2‐depleted cells. This observation indicates that Tlk2 is needed for proper induction of Asf1A and Asf1B after DNA damage, possibly through regulation of Asf1A and Asf1B protein stability 20. Therefore, we asked whether depletion of Asf1A and/or Asf1B also results in a recovery defect. Asf1A depletion clearly reduced the amount of cells that were able to enter mitosis during checkpoint recovery but showed no effect in cells that were allowed to enter mitosis in the absence of DNA damage (Fig 3E and F). In contrast, Asf1B produced a relatively mild defect in both unperturbed mitotic entry and checkpoint recovery (Fig 3E and F). Compared to the induction of replication stress by low dose of hydroxyurea, depletion of Asf1A had a very minimal effect on cell proliferation and EdU incorporation (Fig EV3A–D), indicating that replication is not affected by depletion of Asf1A only. In contrast, co‐depletion of Asf1A and Asf1B resulted in clear reduction in mitotic entry in otherwise unperturbed cells (Fig 3E). This synergistic effect is a reflection of the essential role of the Asf1 histone chaperones during replication 35, 36.
Tlks have been shown to phosphorylate the C‐terminal tail of Asf1A and are thought to be important for opening up its histone‐binding pocket 37. The best‐described phosphorylation site is Asf1A‐S166, and we wondered whether this site is affected during the DNA damage‐induced arrest in a Tlk2‐dependent manner. We synchronized cells in either S phase or G2 phase and quantified the total amount of S166 phosphorylation. We observed that Tlk2 depletion caused a significant decrease in the amount of S166 phosphorylation both in S phase and in G2 phase (Fig 3G). Strikingly, induction of DNA damage in G2 resulted in an extensive increase in S166 phosphorylation, far beyond the level observed in S phase cells (Figs 3G and EV4E). Moreover, Asf1A is also readily phosphorylated after IR albeit to a lesser extent (Fig EV4F). S166 phosphorylation appears to be largely dependent on Tlk2 (Fig 3G), suggesting that activation of Asf1A by Tlk2 promotes recovery. To further corroborate this notion, we investigated whether a phosphomimetic mutant of Asf1A could overcome the recovery defect instigated by Tlk2 depletion. To this end, we made use of a cell line that inducibly expresses a Flag‐tagged mutant of Asf1A (FLAG‐Asf1A‐4D) with phosphomimetic mutations at the four most prominent Tlk‐dependent phosphorylation sites 37. Indeed, expression of the FLAG‐Asf1A‐4D mutant overcame the defects caused by depletion of Tlk2 and restored full recovery (Fig 3H), indicating that phosphorylation of Asf1A by Tlk2 is an essential step to promote recovery.
Asf1A is required to incorporate H3‐H4 histone dimers into DNA, and a recent report showed that H3 histones are evicted from the DNA following treatment with adriamycin 38. Therefore, we examined the amount of histone H3 that was incorporated in the DNA following DNA damage and during recovery. To this end, we permeabilized cells, washed out the freely diffusible proteins, and stained for chromatin‐bound histone H3 after fixation. Indeed, we observed that chromatin‐bound histone H3 levels dramatically drop shortly after induction of DNA damage and levels were restored 16 h after treatment (Fig 3I). Depletion of Tlk2 or Asf1A both resulted in an inability to reincorporate histone H3 at 16 h after damage, showing that Asf1A and Tlk2 are both essential to reincorporate histone H3 into chromatin after DNA damage in G2 (Fig 3I). In addition, chromatin‐bound histone H3 decreased similarly after IR (Fig EV4G). Interestingly, expression of FLAG‐Asf1A‐4D significantly increased the amount of H3 incorporation shortly after DNA damage and during recovery (Fig 3J). These observations indicate that Asf1A, like Tlk2, plays a unique role during the DDR in G2 cells, promoting Asf1A phosphorylation and chromatin restoration to allow for checkpoint recovery.
In yeast, Asf1 and K56 acetylation of H3 have been shown to be important for chromatin re‐assembly at the newly repaired break site 34. Asf1 stimulates the acetylation of H3 at K56 via recruitment of CBP/p300 39. Interestingly, while repair occurs normal in Asf1 deletion mutants in yeast, they are unable to deposit new histones after repair of the DSB and fail to recover from the DDR 34. There are several reports suggesting a role for acetylated H3K56 during DNA damage in mammalian cells 39, 40. However, its role is controversial as there seems to be very little acetylated H3K56 bound to Asf1 complexes 41 and acetylated H3K56 on newly incorporated histones is negligible 42. More in line with the latter, we could not obtain conclusive results of a role for Tlk2 or Asf1A on K56 acetylation after DNA damage (data not shown). Therefore, it remains elusive which modifications are important for deposition of histones in newly repaired DNA and how this affects the essential role of Tlk2 and Asf1A in checkpoint recovery.
Furthermore, the role of Tlk2 in maintaining chromatin structure might also offer an explanation for the Tlk2‐dependent changes in the kinetics of the DDR (Fig 3A–C). Several reports implicate DDR kinetics when altering chromatin structure. Chromatin compaction is required for restricting the DDR 43, 44, and reducing chromatin compaction produces a hypersensitive response 45. The loss of histone H3‐dependent chromatin structure after Tlk2 depletion might explain the effects on DDR kinetics.
Tlk2 depletion leads to a loss of recovery competence
During S phase, stability of Asf1 controls repression of transcription and restoration of chromatin is an essential step for recovery of transcription following UV‐induced genotoxic lesions 16, 46. Regulation of transcription plays a vital role during the process of checkpoint recovery in G2, as it determines the competence to recover 8, 9, 10, 47. Recovery competence in G2 is regulated by controlling the expression levels of a subset of pro‐mitotic genes, such as Plk1 and cyclin B1. To address whether Tlk2‐depleted cells also lose recovery competence via reduced expression of pro‐mitotic genes, we monitored the expression of their respective mRNAs by qPCR. When comparing an unperturbed and a DNA damage‐arrested population of G2 cells, we observed that depletion of Tlk2 significantly reduced the expression of mRNAs encoding for Plk1 and cyclin B1 in the damaged cells (Fig 4A). Reduction of Plk1 and cyclin B1 mRNA was also observed in Tlk2 knockout cells that were damaged in G2 (Fig 4B). To monitor the effects on protein expression, we immunoblotted for Plk1 and cyclin B1 protein levels right before and 16 h after the induction of damage. We found that both Plk1 and cyclin B1 levels were severely reduced after DNA damage in Tlk2‐depleted cells, indicating that Tlk2 depletion can lead to loss of expression of pro‐mitotic proteins (Fig 4C). The repression of these genes during the DDR is controlled by p53, and therefore, we tested whether the recovery defect we observed in these cells could be alleviated by removing p53. In agreement with the literature, we could overcome the defect in checkpoint recovery induced by Wip1 depletion by co‐depleting p53 (Fig 4D) 8. In addition, we also observed a clear increase in recovering cells when we compared Tlk2 depletion with co‐depletion of Tlk2 and p53 (Fig 4D). These results clearly show that p53 has an important role in the loss of recovery competence observed in Tlk2‐depleted cells. Next we asked whether we could see the same difference in p53‐proficient and p53‐deficient HCT116 colon carcinoma cell lines. Similar to the phenotypes observed in U2OS cells, we see a clear reduction in recovery after depletion of Tlk2 in p53‐proficient HCT116 cells, whereas the defect in recovery was not observed in cells that do not express p53 (Fig EV5A and B). In addition, Tlk2 depletion did not significantly affect mitotic entry in unperturbed HCT116 cells (Fig EV5C). These data show that the effect of Tlk2 on recovery is not limited to U2OS cells. In addition, they demonstrate that the effect of Tlk2 on recovery can be rescued by p53. We hypothesize that p53 is not directly involved in the defect, but removal of p53 rather alleviates the pressure of the DDR on transcriptional activity.
Figure 4. Depletion of Tlk2 leads to a loss of recovery competence.
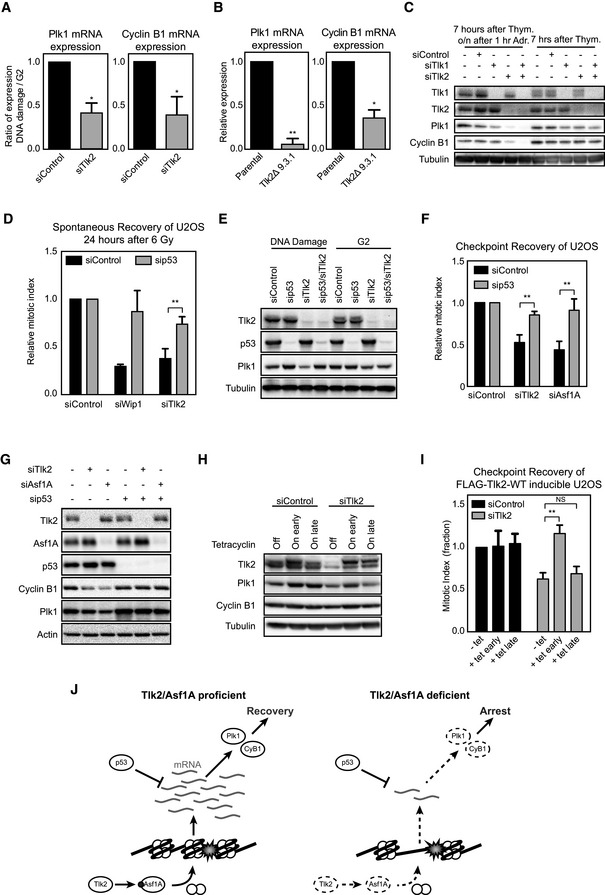
- U2OS cells were treated as in Fig 2B after which mRNA was isolated and analyzed by qPCR. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (*P ≤ 0.05).
- Tlk2Δ 9.3.1 cells were treated and analyzed as in Fig 3A. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (*P ≤ 0.05, **P ≤ 0.01).
- U2OS cells were synchronized in G2 and harvested or synchronized in G2 and harvested 16 h after adriamycin‐induced DNA damage. Samples were analyzed by Western blot for the indicated proteins.
- U2OS cells were transfected with the indicated siRNAs, synchronized in G2, and damaged with 6 Gy of IR. Cells were harvested 24 h later, and the mitotic index was analyzed by FACS. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (**P ≤ 0.01).
- Cells transfected with the indicated siRNAs were treated and analyzed as in (C).
- U2OS cells were transfected with the indicated siRNAs, arrested in G2 with adriamycin, and induced to recover by addition of caffeine. Mitotic index was analyzed by FACS. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (**P ≤ 0.01).
- U2OS cells were synchronized in G2 and harvested 16 h after adriamycin‐induced DNA damage. Samples were analyzed by Western blot for the indicated proteins.
- U2TR cells stably expressing Tlk2 siRNA #3‐insensitive tetracycline‐inducible FLAG‐Tlk2‐wt were thymidine‐synchronized, damaged in G2, and harvested 24 h afterward. Tetracycline was either absent (off), added from the start of the experiment (early) or 16 h after induction of DNA damage (late). Samples were analyzed by Western blot for the indicated proteins.
- Cells were treated as in (G) and recovery was induced by addition of caffeine for 8 h. Mitotic index was analyzed by FACS and normalized to the control. Error bars represent SD of three independent experiments. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, **P ≤ 0.01).
- Model of Tlk2/Asf1A‐dependent recovery competence. Red circle denotes phosphorylation event at S166, and white circles denote histones. See text for further details.
Figure EV5. Analysis of recovery competence.
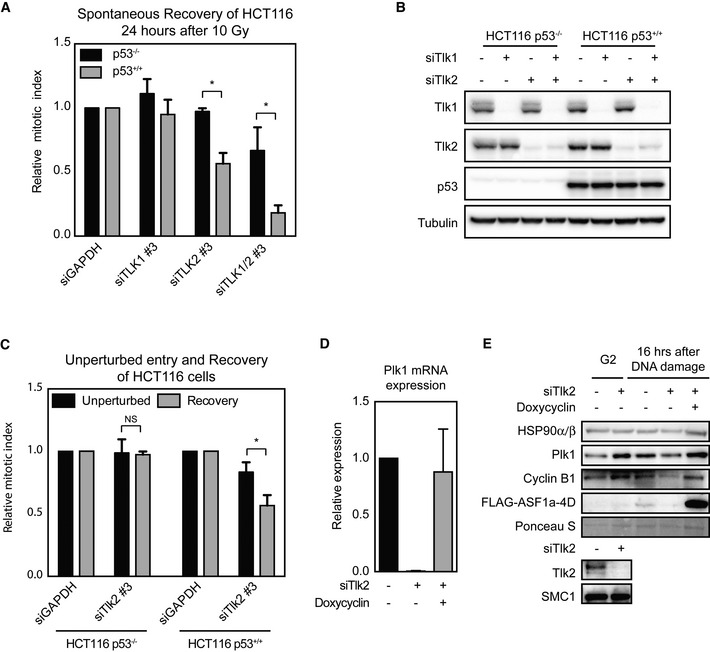
- HCT116‐p53+/+ and HCT116‐p53−/− cells were transfected with the indicated siRNAs, synchronized in G2, and damaged with 10 Gy of IR. Cells were harvested 24 h later, and the mitotic index was analyzed by FACS. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (*P ≤ 0.05).
- HCT116‐p53+/+ and HCT116‐p53−/− cells were treated as in (A) and analyzed by Western blot to determine the knockdown efficiency of the indicated siRNAs and levels of p53.
- HCT116‐p53−/− and HCT116‐p53+/+ cells were either released for 24 h from a 24‐h thymidine block into medium containing nocodazole or released for 7 h into G2, irradiated with 10 Gy IR, and released for 24 h into medium containing nocodazole. Mitotic index was analyzed by FACS and normalized to the control‐treated cells. Error bars represent SD, n = 3. Statistical significance was tested using a paired two‐tailed t‐test (NS P > 0.05, *P ≤ 0.05).
- FLAG‐Asf1A‐4D‐inducible U2OS cells were treated, synchronized in G2 by a single thymidine block and damaged in G2 by 0.5 μM adriamycin for 1 h, and were released for 16 h. Expression was induced by addition of doxycycline. mRNA was isolated and analyzed by qPCR. Error bars represent the SD of three independent experiments, n = 2.
- Cells were treated as in (D), and samples were analyzed by Western blot with the indicated antibodies.
In accordance with the phenotypes observed in Fig 4D, excessive suppression of Plk1 was not observed in cells co‐depleted of p53, indicating that p53 is responsible for the repression of pro‐mitotic proteins in Tlk2‐depleted cells and thus drives loss of recovery competence (Fig 4E). Similar results were obtained when co‐depleting p53 with Asf1A (Fig 4F). In both Tlk2‐ and Asf1A‐depleted cells, Plk1 and cyclin B1 expression levels during the DDR were reduced while p53 depletion resulted in a restoration of Plk1 and cyclin B1 expression (Fig 4G). In addition, induction of FLAG‐Asf1A‐4D in this setting resulted in restoration of Plk1 mRNA levels as well as protein levels of Plk1 and cyclin B1 (Fig EV5D and E).
Finally, we reasoned that if Tlk2 depletion leads to loss of recovery competence, the timing of the Tlk2 rescue by our RNAi‐insensitive inducible Tlk2 could be crucial. Indeed, when we restored Tlk2 expression before DNA damage was inflicted, cells were able to retain recovery competence. However, when we restored Tlk2 only after the loss of Plk1 and cyclin B1 had already taken place (i.e., 8 h after induction of DNA damage), we were unable to rescue the recovery defect of Tlk2‐depleted cells (Fig 4H and I). In addition, late induction of FLAG‐Tlk2 also failed to restore normal levels of both Plk1 and cyclin B1 in the DNA damage cultures (Fig 4H). Taken together, these results clearly demonstrate that Tlk2, through Asf1A, controls chromatin restoration after DNA damage. Failure to properly restore chromatin results in loss of pro‐mitotic gene transcription. The resulting loss of sufficient amounts of Plk1 and cyclin B1 to restart the cell cycle renders cells incapable of recovering from a DNA damage‐induced arrest in G2 (Fig 4J).
Materials and Methods
Antibodies, reagents, Western blotting, and qPCR
Antibodies directed against the following proteins were used: Tlk2, SMC1 (all Bethyl), MPM‐2 (Millipore), α‐tubulin (Sigma), γH2AX, histone H3‐pS10 (Upstate), actin, Chk1, Plk1, cyclin B, p53, HSP90α/β (all Santa Cruz), Tlk1, Asf1A, Asf1B, Chk1‐pS317, Cdk1‐pY15 (all Cell Signaling), FLAG (Sigma), and histone H3 (Abcam). The antibody directed at Asf1A‐pS66 was a gift from A. Groth 37. Secondary antibodies Alexa‐488, Alexa‐568, and Alexa‐633 were from Molecular Probes and horseradish peroxidase‐coupled secondary antibodies from Dako. The following chemicals were used: thymidine (2.5 mM, Sigma), caffeine (5 mM, Sigma), adriamycin (0.5 μM, Sigma), nocodazole (250 ng/ml, Sigma), DAPI (Sigma), PI (Sigma), Ponceau S (Sigma), doxycycline (1 μg/ml, Sigma), and tetracycline (1 μg/ml, Sigma). For Western blot analysis, cells were lysed in Laemmli sample buffer normalized for total protein content and analyzed by immunoblotting. For qPCR, samples were harvested and mRNA was isolated with RNeasy kit (Qiagen). cDNA synthesis was performed by means of SuperScript III reverse transcriptase kit (Invitrogen) with oligo‐dT primers according to the manufacturer's protocols. cDNA was analyzed by qPCR using SensiMix SYBR Low‐ROX (Bioline) on an real‐time PCR system (Applied Biosystems 7500). The fold enrichment of Plk1 and cyclin B1 was calculated using the 2−ΔΔCt method after normalization by GAPDH mRNA levels. Primer sequences were described previously 48.
Cell culture and generation of cell lines
Human osteosarcoma U2OS cells HCT116p53+/+ and HCT116p53−/− human colon carcinoma cells were grown in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% FCS (Lonza), 2 mM L‐glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin. Tlk2 sequence‐verified cDNA was obtained from Open Biosystems. Tlk2 was amplified and N‐terminally tagged with FLAG by PCR with the following forward primer: 5′‐ATGGATATCACCATGGATTATAAAGATGATGATGATAAAATGATGGAAGAATTGCATAG‐3′ and reverse primer: 5′‐ATGCTCGAGTCAATTAGAAGAACTGTTAT‐3′. PCR products were then cloned into pCDNA4/TO with EcoRV and XhoI (New England Biolabs). FLAG‐Tlk2‐wt was made resistant to siRNA #3 (targeting sequence: 3′‐GAUAGAAAGACAACGGAAA‐5′) by site‐directed mutagenesis PCR using the following primers: 5′‐TAAATTCACAGAGGGAAGAGATCGAAAGGCAACGAAAAATGTTAGCAAAGCGGAAAC‐3′ and 5′‐GTTTCCGCTTTGCTAACATTTTTCGTTGCCTTTCGATCTCTTCCCTCTGTGAATTTA‐3′.
FLAG‐Tlk2‐D613A was generated by mutagenesis PCR using the following primers: 5′‐GTGTGGAGAGATAAAAATTACAGCTTTTGGTCTTTCGAAGATCATG‐3′ and 5′‐CATGATCTTCGAAAGACCAAAAGCTGTAATTTTTATCTCTCCACAC‐3′. U2OS‐derived U2TR cells stably expressing FLAG‐Tlk2‐wt and FLAG‐Tlk2‐D613A were generated by calcium phosphate transfection of the constructs, selection of stable clones by zeocin (400 mg/ml, Invitrogen) treatment for 2 weeks followed by clonal selection. Stable clones were grown in media containing tetracycline system‐approved fetal bovine serum (Lonza). Clones were further selected for 2 weeks with tetracycline present in the medium to generate clones that tolerate expression of the exogenous Tlk2 clones. For induction of expression, cells were treated for the indicated times with tetracycline (1 mg/ml).
Tlk2 knockout cell lines were generated using CRISPR/Cas9 genome editing. CRISPR sequences were designed targeting Tlk2 (exon 4, 5′‐GAACCATATGAAACTAGCCA ‐3′; exon 9, 5′‐TCTGACTTAGAGAAGAAGGA‐3′) and cloned into pX330 30. U2OS cells were transfected with the exon‐specific pX330 plasmids in addition to a plasmid containing a guide RNA to the zebrafish TIA gene (5′‐ ggtatgtcgggaacctctcc‐3′) and a cassette of a 2A sequence followed by a BlastR gene, flanked by two TIA target sites. Co‐transfection results in infrequent integration of the BlastR gene at the targeted genomic locus by NHEJ, as previously described 31. Successful integration of the cassette renders cells resistant to blasticidin. Two days following transfection, the culture medium was supplemented with blasticidin (10 μg/ml). Surviving colonies were clonally expanded, screened for cassette integration and indels into the query gene by PCR (using primer sets 5′‐GAAGGTTTTGTAGCAGCTCTGA‐3′ and 5′‐AACAAAAGAGTCCCCGAAGAA‐3′ and 5′‐ACATTGTTCAGTGCACAGTAGTGA‐3′ and 5′‐GGTTCCTACTCAGATTCCCTCA‐3′), and checked on Western Blot for expression of Tlk2.
Cell synchronization
For analysis of checkpoint recovery, cells were synchronized at the G1/S border by thymidine (2.5 mM) for 24 h followed by a 7‐h release and 1‐h incubation with adriamycin (0.5 μM) or irradiation with 6 Gy with a 137Cs source. Afterward, cells were kept for 16 h in nocodazole (250 ng/ml). Recovery was induced by adding caffeine (5 mM) for 8 h. Unperturbed mitotic entry was assayed by a 24‐h thymidine block followed by a 16‐h release into nocodazole. For reconstitution assays, expression was induced by addition of tetracycline (1 mg/ml) at the indicated times.
siRNA transfection and automated image analysis
For siRNA experiments, cells were grown in 96‐well plates (Viewplate‐96, PerkinElmer) and transfected with 20 nM siRNA using HiPerFect (Qiagen) or RNAiMAX (Life Technologies) according to the manufacturer's recommendations. The human ON‐TARGETplus kinome library (GU‐103500) and individual siRNAs targeting Wip1, Tlk1, Tlk2, Asf1A, and Asf1B were obtained from Dharmacon. Cells were fixed by addition of equal volume of an 8% formaldehyde solution to the medium to prevent loss of mitotic cells, permeabilized with methanol, and stained with DAPI and anti‐histone H3‐pS10 antibodies. Image acquisition was performed using a Cellomics ArrayScan VTI (Thermo Scientific) using a 20× 0.40 NA objective. Image analysis was performed using Cellomics ArrayScan HCS Reader (Thermo Scientific). In short, cells were identified based on DAPI staining, and they were scored as mitotic if the intensity of histone H3‐pS10 staining reached a preset threshold. All images and automated image quantifications were subsequently checked manually. Image analysis was performed on at least 500 cells per condition.
Fluorescence‐activated cell sorting
Cells were harvested by trypsinization and fixed with ice‐cold 70% ethanol. Cells were stained using the MPM‐2 antibody (Millipore) and Alexa‐488‐conjugated secondary antibodies (Molecular Probes). DNA was stained using propidium iodide, and samples were analyzed on a FACSCalibur flow cytometer (BD Biosciences). Cell cycle distribution was determined by flow cytometry counting 104 events as described.
Immunofluorescence, real‐time imaging, and cumulative index
For immunofluorescence, cells were fixed/permeabilized in 3.7% PFA with 0.2% Triton X‐100 or fixed in 3.7% PFA and permeabilized by ice‐cold methanol. Cells were blocked with 4% BSA and stained with the indicated primary antibodies. Samples were subsequently incubated with Alexa Fluor secondary antibodies and DAPI and mounted with Prolong Gold Antifade. For pre‐extraction, cells were permeabilized at the indicated time points with 0.2% Triton X‐100 in PBS and were rigorously washed twice with PBS. Subsequently, cells were fixed with 3.7% paraformaldehyde and stained as described. Images were acquired on a Zeiss Axiovert 200 M using NA 0.75 objectives or on a DeltaVision Elite imaging system using NA 1.4 objectives. Images were processed and analyzed with FIJI as described. For time‐lapse microscopy, cells were grown on LabTek II chambered cover glasses in Leibovitz's L‐15 medium (Gibco) supplemented with 10% FCS (Lonza), 2 mM L‐glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin and were imaged with DIC on a Zeiss Axiovert 200 M using 20× NA 0.75 objectives or on a Deltavision imaging system using 20× 0.75 NA objectives. Images were taken every 15 min, and movies were manually scored for cumulative mitotic index.
Author contributions
WB, JvdB, and RHM conceived and designed the experiments and analyzed the data. WB and RHM wrote the manuscript. WB, JvdB, and MA performed the experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Dataset EV1
Review Process File
Acknowledgements
We thank the Medema, Kops, Lens, and Wolthuis laboratories for input and useful discussion. We thank David A. Egan for technical assistance with the automated screening, Anja Groth for providing us with the inducible Asf1A cell lines and the α‐Asf1A‐pS166 antibody, and Thijn Brummelkamp for sharing the pTIA‐Blast vector. We thank Anja Groth and Travis H. Stracker for critical reading of the manuscript. RM is supported by the Netherlands Genomics Initiative of NWO (91210065) and by the NWO Gravitation program CancerGenomics.nl (024.001.0281). WB, JvdB and MA are supported by the Netherlands Genomics Initiative of NWO (91210065).
EMBO Reports (2016) 17: 659–670
References
- 1. Shaltiel IA, Krenning L, Bruinsma W, Medema RH (2015) The same, only different – DNA damage checkpoints and their reversal throughout the cell cycle. J Cell Sci 128: 607–620 [DOI] [PubMed] [Google Scholar]
- 2. Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH (2000) Polo‐like kinase‐1 is a target of the DNA damage checkpoint. Nat Cell Biol 2: 672–676 [DOI] [PubMed] [Google Scholar]
- 3. Wiebusch L, Hagemeier C (2010) p53‐ and p21‐dependent premature APC/C‐Cdh1 activation in G2 is part of the long‐term response to genotoxic stress. Oncogene 29: 3477–3489 [DOI] [PubMed] [Google Scholar]
- 4. Lee J, Kim JA, Barbier V, Fotedar A, Fotedar R (2009) DNA damage triggers p21WAF1‐dependent Emi1 down‐regulation that maintains G2 arrest. Mol Biol Cell 20: 1891–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, Logothetis CJ, McDonnell TJ (2006) Identification of cell cycle regulatory genes as principal targets of p53‐mediated transcriptional repression. J Biol Chem 281: 25134–25142 [DOI] [PubMed] [Google Scholar]
- 6. Riley T, Sontag E, Chen P, Levine A (2008) Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol 9: 402–412 [DOI] [PubMed] [Google Scholar]
- 7. Lindqvist A, Rodríguez‐Bravo V, Medema RH (2009) The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 185: 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindqvist A, de Bruijn M, Macurek L, Brás A, Mensinga A, Bruinsma W, Voets O, Kranenburg O, Medema RH (2009) Wip1 confers G2 checkpoint recovery competence by counteracting p53‐dependent transcriptional repression. EMBO J 28: 3196–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alvarez‐Fernández M, Halim VA, Krenning L, Aprelia M, Mohammed S, Heck AJ, Medema RH (2010) Recovery from a DNA‐damage‐induced G2 arrest requires Cdk‐dependent activation of FoxM1. EMBO Rep 11: 452–458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mannefeld M, Klassen E, Gaubatz S (2009) B‐MYB is required for recovery from the DNA damage‐induced G2 checkpoint in p53 mutant cells. Cancer Res 69: 4073–4080 [DOI] [PubMed] [Google Scholar]
- 11. Fiscella M, Zhang H, Fan S, Sakaguchi K, Shen S, Mercer WE, Vande Woude GF, O'Connor PM, Appella E (1997) Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53‐dependent manner. Proc Natl Acad Sci USA 94: 6048–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu X, Nannenga B, Donehower LA (2005) PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev 19: 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Laoukili J, Alvarez M, Meijer LAT, Stahl M, Mohammed S, Kleij L, Heck AJR, Medema RH (2008) Activation of FoxM1 during G2 requires cyclin A/Cdk‐dependent relief of autorepression by the FoxM1 N‐terminal domain. Mol Cell Biol 28: 3076–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Halim VA, Alvarez‐Fernández M, Xu YJ, Aprelia M, van den Toorn HWP, Heck AJR, Mohammed S, Medema RH (2013) Comparative phosphoproteomic analysis of checkpoint recovery identifies new regulators of the DNA damage response. Sci Signal 6: rs9 [DOI] [PubMed] [Google Scholar]
- 15. Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734 [DOI] [PubMed] [Google Scholar]
- 16. Adam S, Polo SE, Almouzni G (2013) Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 155: 94–106 [DOI] [PubMed] [Google Scholar]
- 17. Roe JL, Rivin CJ, Sessions RA, Feldmann KA, Zambryski PC (1993) The Tousled gene in A. thaliana encodes a protein kinase homolog that is required for leaf and flower development. Cell 75: 939–950 [DOI] [PubMed] [Google Scholar]
- 18. Silljé HH, Takahashi K, Tanaka K, Van Houwe G, Nigg EA (1999) Mammalian homologues of the plant Tousled gene code for cell‐cycle‐regulated kinases with maximal activities linked to ongoing DNA replication. EMBO J 18: 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silljé HH, Nigg EA (2001) Identification of human Asf1 chromatin assembly factors as substrates of Tousled‐like kinases. Curr Biol 11: 1068–1073 [DOI] [PubMed] [Google Scholar]
- 20. Pilyugin M, Demmers J, Verrijzer CP, Karch F, Moshkin YM (2009) Phosphorylation‐mediated control of histone chaperone ASF1 levels by Tousled‐like kinases. PLoS One 4: e8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Recht J, Tsubota T, Tanny JC, Diaz RL, Berger JM, Zhang X, Garcia BA, Shabanowitz J, Burlingame AL, Hunt DF et al (2006) Histone chaperone Asf1 is required for histone H3 lysine 56 acetylation, a modification associated with S phase in mitosis and meiosis. Proc Natl Acad Sci USA 103: 6988–6993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsubota T, Berndsen CE, Erkmann JA, Smith CL, Yang L, Freitas MA, Denu JM, Kaufman PD (2007) Histone H3‐K56 acetylation is catalyzed by histone chaperone‐dependent complexes. Mol Cell 25: 703–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Canfield C, Rains J, De Benedetti A (2009) TLK1B promotes repair of DSBs via its interaction with Rad9 and Asf1. BMC Mol Biol 10: 110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Groth A, Lukas J, Nigg EA, Sillje HHW, Wernstedt C, Bartek J, Hansen K (2003) Human Tousled like kinases are targeted by an ATM‐ and Chk1‐dependent DNA damage checkpoint. EMBO J 22: 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kelly R, Davey SK (2013) Tousled‐like kinase‐dependent phosphorylation of Rad9 plays a role in cell cycle progression and G2/M checkpoint exit. PLoS One 8: e85859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Krause DR, Jonnalagadda JC, Gatei MH, Sillje HHW, Zhou B‐B, Nigg EA, Khanna K (2003) Suppression of Tousled‐like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene 22: 5927–5937 [DOI] [PubMed] [Google Scholar]
- 27. Hall‐Jackson CA, Cross DA, Morrice N, Smythe C (1999) ATR is a caffeine‐sensitive, DNA‐activated protein kinase with a substrate specificity distinct from DNA‐PK. Oncogene 18: 6707–6713 [DOI] [PubMed] [Google Scholar]
- 28. Lau CC, Pardee AB (1982) Mechanism by which caffeine potentiates lethality of nitrogen mustard. Proc Natl Acad Sci USA 79: 2942–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van Vugt MATM, Brás A, Medema RH (2004) Polo‐like kinase‐1 controls recovery from a G2 DNA damage‐induced arrest in mammalian cells. Mol Cell 15: 799–811 [DOI] [PubMed] [Google Scholar]
- 30. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A et al (2015) Gene essentiality and synthetic lethality in haploid human cells. Science 350: 1092–1096 [DOI] [PubMed] [Google Scholar]
- 32. Yata K, Lloyd J, Maslen S, Bleuyard J‐Y, Skehel M, Smerdon SJ, Esashi F (2012) Plk1 and CK2 Act in Concert to Regulate Rad51 during DNA Double Strand Break Repair. Mol Cell 45: 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carrera P, Moshkin YM, Gronke S, Sillje HHW, Nigg EA, Jackle H, Karch F (2003) Tousled‐like kinase functions with the chromatin assembly pathway regulating nuclear divisions. Genes Dev 17: 2578–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen C‐C, Carson JJ, Feser J, Tamburini B, Zabaronick S, Linger J, Tyler JK (2008) Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 134: 231–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Groth A, Ray‐Gallet D, Quivy J‐P, Lukas J, Bartek J, Almouzni G (2005) Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 17: 301–311 [DOI] [PubMed] [Google Scholar]
- 36. Corpet A, De Koning L, Toedling J, Savignoni A, Berger F, Lemaître C, O'Sullivan RJ, Karlseder J, Barillot E, Asselain B et al (2011) Asf1b, the necessary Asf1 isoform for proliferation, is predictive of outcome in breast cancer. EMBO J 30: 480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Klimovskaia IM, Young C, Strømme CB, Menard P, Jasencakova Z, Mejlvang J, Ask K, Ploug M, Nielsen ML, Jensen ON et al (2014) Tousled‐like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat Commun 5: 3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O et al (2013) Drug‐induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun 4: 1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das C, Lucia MS, Hansen KC, Tyler JK (2009) CBP/p300‐mediated acetylation of histone H3 on lysine 56. Nature 459: 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Battu A, Ray A, Wani AA (2011) ASF1A and ATM regulate H3K56‐mediated cell‐cycle checkpoint recovery in response to UV irradiation. Nucleic Acids Res 39: 7931–7945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jasencakova Z, Scharf AND, Ask K, Corpet A, Imhof A, Almouzni G, Groth A (2010) Replication Stress Interferes with Histone Recycling and Predeposition Marking of New Histones. Mol Cell 37: 736–743 [DOI] [PubMed] [Google Scholar]
- 42. Alabert C, Barth TK, Reverón‐Gómez N, Sidoli S, Schmidt A, Jensen ON, Imhof A, Groth A (2015) Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29: 585–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, Goodarzi AA (2010) 53BP1‐dependent robust localized KAP‐1 phosphorylation is essential for heterochromatic DNA double‐strand break repair. Nat Cell Biol 12: 177–184 [DOI] [PubMed] [Google Scholar]
- 44. Floyd SR, Pacold ME, Huang Q, Clarke SM, Lam FC, Cannell IG, Bryson BD, Rameseder J, Lee MJ, Blake EJ et al (2013) The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 498: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murga M, Jaco I, Fan Y, Soria R, Martinez‐Pastor B, Cuadrado M, Yang S‐M, Blasco MA, Skoultchi AI, Fernandez‐Capetillo O (2007) Global chromatin compaction limits the strength of the DNA damage response. J Cell Biol 178: 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Im J‐S, Keaton M, Lee KY, Kumar P, Park J, Dutta A (2014) ATR checkpoint kinase and CRL1βTRCP collaborate to degrade ASF1a and thus repress genes overlapping with clusters of stalled replication forks. Genes Dev 28: 875–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Krenning L, Feringa FM, Shaltiel IA, van den Berg J, Medema RH (2014) Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol Cell 55: 59–72 [DOI] [PubMed] [Google Scholar]
- 48. Laoukili J, Kooistra MRH, Brás A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH (2005) FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol 7: 126–136 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Dataset EV1
Review Process File