

Abstract
In recent years it has been recognized that clinical translation of novel therapeutic strategies for patients with adrenocortical carcinoma (ACC) often fails. These disappointing results indicate that the currently utilized tumor models only poorly reflect relevant pathophysiology and, thereby, do not predict clinical applicability of novel pharmacological approaches. However, also the development of new preclinical ACC models has remained a challenge with only one human cell line (NCI-H295R) and one recently established human pediatric xenograft model (SJ-ACC3) being available for this highly heterogeneous malignancy. Our current study furthermore reveals a poor reproducibility of therapeutic action between different clones of the most commonly used tumor model NCI-H295R. In an attempt to broaden the current preclinical armamentarium, we aimed at the development of patient-individual tumor models. During these studies, one xenograft (MUC-1) displayed marked engraftment and sustained tumor growth. MUC-1 tumor analysis revealed highly vascularized, proliferating and SF-1 positive xenografts. In a next step, we characterized all currently available human tumor models for ACC for Ki67, SF-1 and EGF-receptor status in comparison with MUC-1-xenografts. In addition, we established a primary culture, which is now viable over 31 passages with sustained nuclear SF-1 and cytoplasmic 3βHSD immuno-positivity. Subsequent investigation of therapeutic responsiveness upon treatment with the current systemic gold standard EDP-M (etoposide, doxorubicin, cisplatin and mitotane) demonstrated maintenance of the clinically observed drug resistance for MUC-1 exclusively. In summary, we provide evidence for a novel patient-derived tumor model with the potential to improve clinical prediction of novel therapeutic strategies for patients with ACC.
Keywords: adrenocortical carcinoma, patient-derived tumor-xenograft, preclinical animal model, cell line, clinical translation
Keywords: Abbreviations: ACC: adrenocortical carcinoma, STR: short tandem repeat, PDTX: patient derived tumor xenografts
INTRODUCTION
Adrenocortical carcinomas (ACC) are rare and highly malignant tumors with a poor prognosis. In recent years genetic and molecular profiling of surgical tumor specimens have led to the identification of novel biomarkers for ACC with potential prognostic impact [1]. However, the exact contribution of underlying pathways for adrenal tumorigenesis and their relevance for individualized therapeutic decisions is still largely unknown. In recent years new targeted therapies have been successfully introduced for various cancer types, but initial clinical evaluation of such therapies in patients with ACC have been disappointing [2–4]. Consequently, the classical multi-chemotherapeutic EDP-M scheme consisting of etoposide, doxorubicin, cisplatin and mitotane remains the gold standard therapy for patients not amendable for surgical resection [5]. Despite this therapeutic option, the overall survival of patients is still poor. Moreover, dose limiting site effects and therapy associated severe adverse events are commonly encountered [5]. Thus, novel treatment options for patients with extended disease are urgently needed.
Tumor models are important tools for preclinical therapeutic studies, but translation of novel therapeutic strategies in patients often fails indicating that prediction of clinical success solely on the basis of currently utilized models is not reliable. One reason for this shortcoming is that only two human cell lines, NCI-H295R and SW-13, are available for ACC, which do not reflect tumor heterogeneity and specific therapeutic responses of individual patients [6, 7]. In addition, SW-13 had originally been established from surgical material of a small cell carcinoma of the adrenal gland [7]. Thereby, its adrenocortical origin has been repeatedly questioned. Due to the lack of other ACC tumor models SW-13 cells have been used to complement preclinical experiments, but its predictive clinical value is highly debated as it lacks features of adrenocortical differentiation.
Even though these two commonly available cell lines can be utilized as xenografts in immunodeficient mice [8, 9], they originate from cell suspensions following long-term in vitro culture. There is good evidence that selection during multiple cell culture passages grossly changes biological properties compared to the original patient tumor [10]. To overcome this limitation, patient-derived tumor xenografts (PDTX) engrafted in immunodeficient mice have been established and tested for a variety of cancer types [11]. Following the same approach the establishment of a novel tissue-based xenograft model for pediatric ACC (SJ-ACC3) was recently reported [12]. Unfortunately, no cell line for complementary in vitro experiments could yet be derived from this xenograft.
Here we report on the limitations in clinical prediction of classical human tumor models for ACC as well as on the establishment of a new tumor model with the potential to improve the current unsatisfactory situation.
RESULTS
Clone dependent functional heterogeneity of NCI-H295R derived xenografts
During recent in vivo experiments on novel chemotherapeutic treatment schemes for ACC, our workgroup incidentally detected marked differences during tumor development of two clones of NCI-H295R (denoted as clone 1 and clone 2). While clone 1 was originally obtained from ATCC and utilized over a long period in our laboratory, clone 2 was again purchased from ATCC in 2012. Of note, both clones were recently analyzed by short-tandem repeat analyses confirming their authentication as NCI-H295R cells.
Macroscopically, xenografts derived from clone 2 (Figure 1C) developed large blood vessels, while this phenomenon was not noticed for clone 1 (Figure 1A). Increased tumor vascularization was confirmed by a detected higher number of blood vessel cross sections [μm2] of CD31 stained tumor slides (clone 1: 1258.6±209.1 vs. clone 2: 2228.5±293.7; p<0.05; Figure 1G). Moreover, we observed less effective engraftment of clone 2 in comparison to clone 1. Furthermore, subsequent histological and immunohistochemical analyses revealed highly necrotic xenografts of clone 1, while the proliferation rate was significantly higher for tumors derived from clone 2 (75.8±1.6%) compared to clone 1 (50.3±1.3%;p<0.001; Figure 1J). Interestingly, similarly extensive heterogeneities of in vivo properties were noticed independently by two different european laboratories (munich workgroup clones 1 and 2 in Figure 1, and Florence workgroup clones 3 and 4 in Supplementary Figure S1).
Figure 1. Pictures of athymic nude mice bearing NCI-H295R xenografts and H&E sections of clones 1.
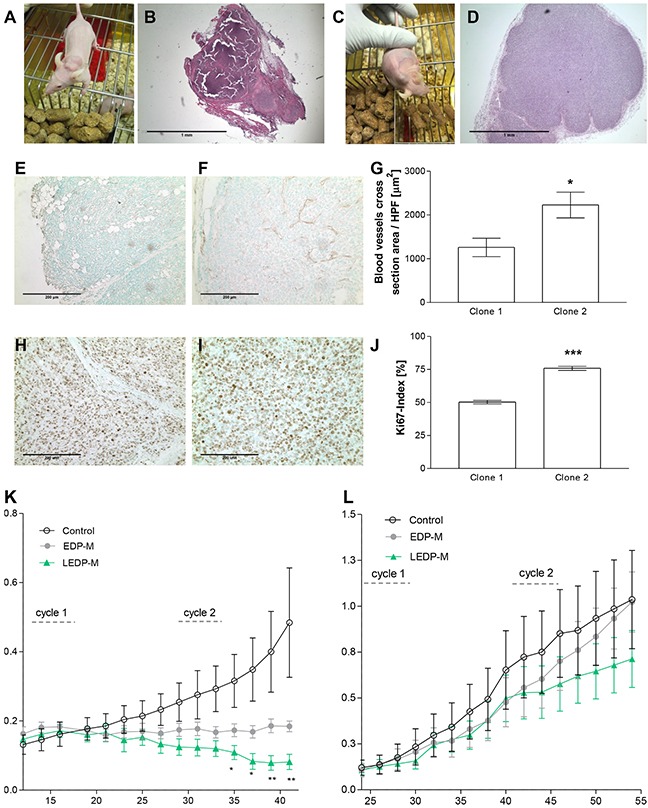
A, B. and 2 C, D. Representative CD31 E, F. and Ki67 H, I. stainings as well as the quantification of blood vessels cross sections G. and proliferation index J. of clone 1 and 2 tumors. Effects on tumor size of NCI-H295R xenografts of clone 1 K. and clone 2 L. after two therapeutic cycles with EDP-M (etoposide, doxorubicin, cisplatin and mitotane) and LEDP-M (etoposide, liposomal doxorubicin, liposomal cisplatin and mitotane). Stars denote significant differences compared with EDP-M.
To investigate whether these differences in biological behavior might have an impact on therapeutic prediction, we performed an intervention study with xenografts of clone 2 including the clinical gold standard treatment for ACC (etoposide, doxorubicin, cisplatin and mitotane, EDP-M) as well as a novel liposomal variant LEDP-M (etoposide, liposomal doxorubicin, liposomal cisplatin and mitotane). Previous studies with clone 1 had revealed significant differences between controls and LEDPM-treated tumors. Moreover, we detected significantly reduced tumor sizes in LEDP-M-treated tumors in comparison with EDP-M-treated tumors following the second therapeutic cycle as depicted by stars (Figure 1K, [13]). Analogous chemotherapeutic treatments on xenografts obtained from clone 2 did not reveal any significant reduction in tumor sizes upon EDP-M or LEDP-M treatment (Figure 1L). Thus, this comparative study revealed a marked and relevant spread of results based on the classical and most commonly used ACC tumor model.
Establishment and characterization of MUC-1 xenografts
In an attempt, to establish patient-individual endocrine tumor models, our working group initiated in a next step the implantation of ACC derived patient tumor specimen. During these studies, one xenograft (MUC-1) showed particular engraftment properties and sustained tumor growth. The respective tumor tissue was obtained from a 24-year-old male patient with a primary diagnosis of a left adrenal mass of 22 cm. While the tumor was initially diagnosed because of abdominal discomfort, no clinical symptoms of overt Cushing syndrome were reported. However, urinary steroid metabolome analysis [14] revealed a malignant secretory profile indicative of hormonal activity. The patient underwent adrenalectomy, nephrectomy and lymphadenectomy, and pathological examination revealed a Ki67 index of 30-40%. Despite extended radical resection, the patient developed abdominal metastatic spread, which was treated with four cycles of etoposide, doxorubicin, cisplatin and mitotane. The tumor pieces utilized for the preclinical study were derived from a subsequently developed subcutaneous metastasis at the neck.
Characterization of MUC-1 xenografts
Upon tumor inoculation of ten athymic nude mice aggressive tumor growth of two of the xenografts was noted. Immunohistochemical analyses revealed a highly vascularized and SF-1 positive (27.8 ± 1.6 cells/high power field)) xenograft in the murine host (Figure 2A–2D). The Ki67 index (9.5 ± 0.4 %) was comparable with the original surgical sample (p = n.s.; Figure 2E-H). Moreover, immunohistochemical analysis revealed β-catenin and p53 positivity for MUC-1 xenografts (Figure 2I-L) with staining patterns different from that obtained for the classical NCI-H295R tumor model (Figure 2M-P).
Figure 2. H&E, Ki67, SF-1 and CD-31 tumor analysis from the original patient tumor.
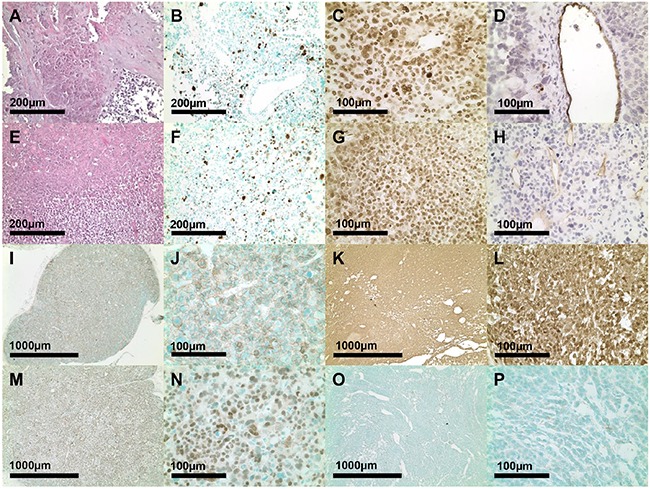
A-D. and of MUC-1 xenograft E-H. derived tumor slides from passage 2. Immunohistochemical β-catenin I-L. and p53 stainings M-P. of MUC-1 (I, J. M, N) and NCI-H295R (K, L, O, P) tumor slides.
The established xenograft was passed over into another group of animals. This procedure was repeated several times up to passage 5 (n= 10 (P1), 4 (P2), 10 (P3), 20 (P4) and 19 (P5), respectively) with sustained tumor growth. Remarkably, with the number of passages also the effective take-on rate increased from 20% in passage 1 up to approximately 70% in passages 4 and 5 (represented as number of implants showing large increase in tumor size per number of total implanted in %; Figure 3A). Simultaneously, the effective take-on time (defined as time until the first tumor of the passage reached a tumor size of 1.5 cm) decreased from 198 days in passage 2 down to 71 days in passage 5.
Figure 3. Schematic illustration of increasing engraftment rates (A, the white bars represent the total number of implanted mice per each passage while the grey area depicts the effective take on rates).
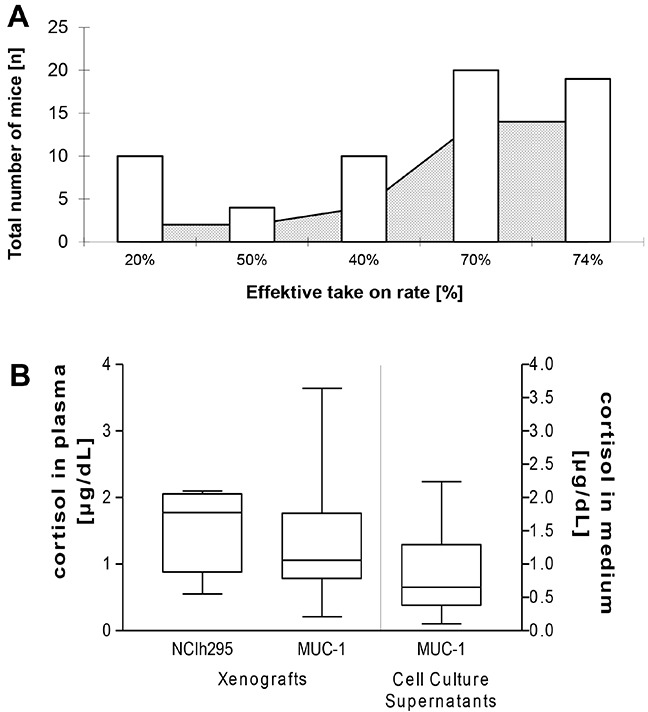
Cortisol measurements of plasma from NCI-H295R (n=6) and MUC-1 (n=17) tumor-bearing mice in vivo and of cell culture supernatants (n=14 of passages 3, 4, 5, 6, 8, 9, 10, 12 and 15 which were offset against a medium blank in triplicate) of MUC-1 cells in vitro B.
Comparison of MUC-1 with commonly available ACC xenografts
We further investigated the Ki67 indices [%] of all commonly available xenograft models for ACC by immunohistochemistry. While SW-13 tumors showed significantly higher proliferation rates (60.8±10.3%, p<0.05 versus MUC-1), Ki67 indices of NCI-H295R (50.3±1.3%) and SJ-ACC3 (33.7±3.6%) were comparable to those of MUC-1 xenografts (30.3±3.3%; p= n.s, Figures 4B and 4C).
Figure 4. Pictures from NCI-H295R, SW-13, SJ-ACC3 and MUC-1 xenografts in athymic nude mice.
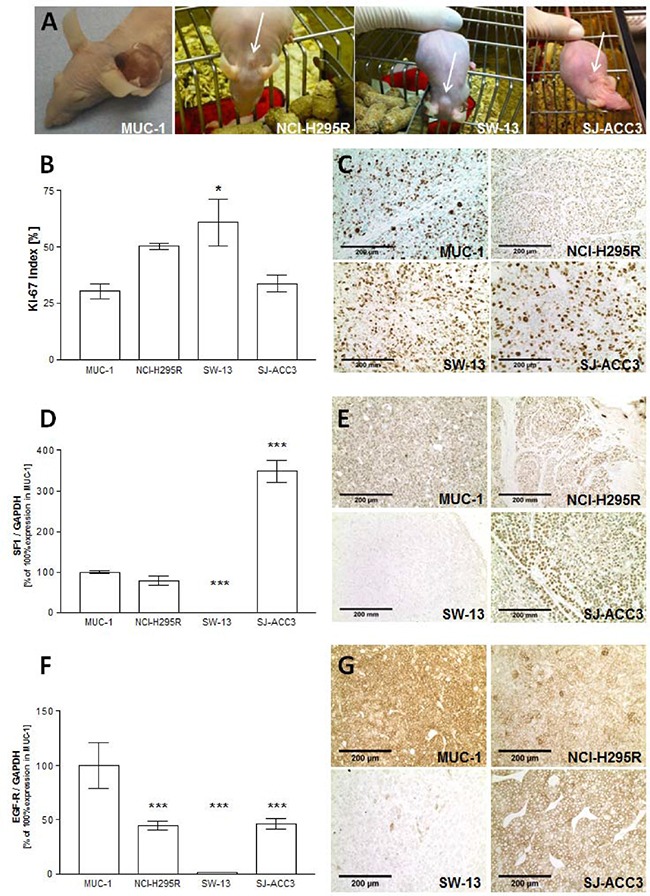
A. Quantification of Ki67indices B. as well as representative immunohistochemical stainings from all tumor models C. Real-Time PCR analysis and immunohistochemical stainings of SF-1 D, E. and EGF-receptor F, G. for all tumor xenografts. Stars represent significance vs. MUC-1 (*, p<0.05; **, p<0.01; ***, p<0.001).
In a next step, we analyzed SF-1 RNA (Figure 4D) and protein (Figure 4E) levels in all currently available ACC tumor models by quantitative real time PCR and immunohistochemistry, respectively. While SF-1 expression in MUC-1 (100 ± 0.0%) and NCI-H295R (78.3± 12.3%) was comparable (p=n.s.), we detected a highly significant SF-1 overexpression in the pediatric ACC tumor model SJ-ACC3 (348.2± 27.4%, p < 0.001 vs. MUC-1) and almost no detectable SF-1 expression in SW-13 xenografts (0.03± 0.0%, p < 0.001vs. MUC-1). Immunohistochemical analyses of representative tumor slides confirmed these findings (Figure 4E).
Regarding hormonal status, ELISA measurement revealed cortisol levels of 1.11 ± 0.2 in plasma samples of MUC-1 tumor bearing mice (n=17) which were statistically comparable with cortisol levels measured in plasma samples of mice bearing the commonly utilized hormonal active NCI-H295R-xenografts (1.57 ± 0.2; p = 0.5, n=6; Figure 3B).
In addition, we choose the EGF-receptor as one example to characterize potential differences in cellular signaling pathways between MUC-1 and the other currently existing in vivo models. While EGF-receptor expression was almost abrogated in the SW-13 tumor model (1.2 ± 0.2%; p < 0.001 vs. MUC-1) a significant higher expression was detectable in MUC-1 tumors (100 ± 0%) compared with all other tumor models (NCI-H295R: 44.5 ± 4.1% and SJACC-3: 45.9 ± 4.9%; both p < 0.001vs. MUC-1; (Figure 4F). Immunohistochemical analysis in tumor slides of the different xenografts also confirmed these as well as membranous receptor staining specifically for MUC-1, NCI-H295R and SJ-ACC3 xenografts (Figure 4G).
Establishment of a MUC-1 cell line
For the establishment of a novel in vitro ACC model we used explanted MUC-1 xenograft pieces for in vitro culturing. A first attempt failed after several passages due to massive contamination by murine fibroblasts. Thus, we initiated a second round of culturing involving continuous and highly specific murine and human fibroblast removal. The resulting multi-clonal cell suspension is now viable in passage 31. In these cultures, cross-contamination by murine fibroblasts could be excluded based on a universal primer probe assay using ApoE as specific target gene (Supplementary pdf 1). Representative pictures of passages 4, 7, 10 and 13 furthermore demonstrate specific stainings for Ki67, SF-1 as well as specific cytoplasmic 3βHSD expression for MUC-1 cells (Figure 5). Moreover, ELISA measurements revealed median cortisol levels of 0.8 ± 0.2 in cell culture supernatants MUC-1 cells during ongoing passaging (Figure 3B).
Figure 5. Bright-field pictures.
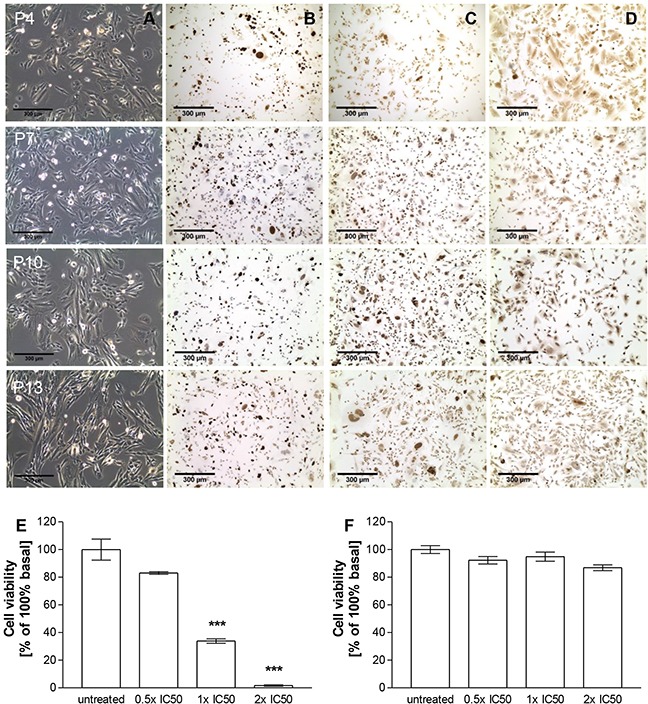
A. as well as Ki67 B., SF-1 C. and 3βHSD D. stainings of MUC-1 cells in passages 4, 7, 10 and 13 in vitro. Treatment dependent inhibition of cell viability of NCI-H295R E. and MUC-1 F. upon addition of different concentrations of EDP-M in vitro. Stars represent statistical significance over untreated controls.
Genetic characterization and comparison with commonly available ACC cell lines
Genetic characteristics of MUC-1 cells were investigated by PCR-Single-Locus Technology. These revealed a distinct short-tandem repeat (STR) profile for MUC-1 cells different from that of NCI-H295R and SW-13 cells (Table 1). Moreover, a sample with such STR profile is not reported in the online database of the DSMZ (German Collection of Microorganisms and Cell Cultures GmbH, https://www.dsmz.de). Thereby, MUC-1 cells were recently authenticated as a novel cell line of human origin (Supplementary pdf 2). Further genetic markers are summarized in Supplementary Table S1.
Table 1. Genetic characteristics of MUC-1 cells determined by PCR-Single-Locus Technology in comparison to the short tandem-repeat (STR) profiles of NCI-H295R and SW-13 as listed by ATCC.
| DNA marker | MUC-1 | NCI-H295R | SW-13 |
|---|---|---|---|
| AM | X | X | X |
| CSF1PO | 12, 12 | 10, 12 | 11, 12 |
| D13S317 | 9, 9 | 13 | 9 |
| D16S539 | 11, 14 | 11 | 12 |
| D5S818 | 11, 11 | 12 | 12 |
| D7S820 | 8, 10 | 9, 12 | 8, 10 |
| TH01 | 9.3 9.3 | 9, 3 | 7, 8 |
| TPOX | 8, 8 | 8 | 8 |
| vWA | 16, 17 | 17, 18 | 17, 19 |
Evaluation of responsiveness to EDP-M treatment
As the patient had been treated with EDP-M followed by metastatic spread including the tumor sample, which had been used for the MUC-1 xenograft we sought to investigate the therapeutic responsiveness in comparison to NCI-H295R cells. These experiments identified NCI-H295R cells (Clone 1, Figure 5E) as sensitive to EDP-M (% of 100% basal; untreated: 100.0±7.6% vs. 0.5 × IC50: 83.1±1.0%, p<0.001; 1 × IC50: 33.8±1.6%, p<0.001; 2 × IC50: 1.7±0.5%, p<0.001), while MUC-1 cells (Figure 5F) were resistant against treatment with all concentrations (% of 100% basal; untreated: 100.0±2.8% vs. 0.5 × IC50: 92.2±2.7%, p>0.05; 1 × IC50: 94.8±3.3%, p>0.05; 2 × IC50: 86.8±2.1%, p>0.05).
DISCUSSION
Only a few tumor models are available for ACC which can furthermore not reflect the high heterogeneity clinically observed for this tumor entity. However, even in those instances where different tumor models are available for a certain cancer type, it has been observed that cell lines diverge with extensive passaging in vitro, leading to a lack of clear correlation between therapeutic efficacy in cell-line based xenografts and clinical effectiveness. Key factors as invasive capabilities, genetic content, maintenance of a heterogeneous cell population, and the reliance on specific growth and survival pathways differ from the host and this process is until now only incompletely understood [15–17].
Our findings support this notion by demonstrating that different clones of the so far most commonly used tumor model for ACC significantly differ regarding tumor development, vascularization, proliferation, a marked decrease in engraftment potential following extensive passaging and most importantly in this context therapeutic responsiveness. This observation is endorsed by three independent working groups (reflected in Figure 1, Supplementary Figure S1 and by personal communication, Pierre Val, INSERM U1103, CNRS, Clermont Université, Clermont-Ferrand, France).
Keeping in vivo studies on the basis of patient-derived tissue-xenografts, separately from much more extensive passaging in vitro, has been already demonstrated to keep xenograft variability at bay [10]. Thus, to improve the predictive reliability of in vivo models patient-derived tumor xenografts (PDTXs) have been established for a wide range of cancer types and have been shown to maintain the original tumor architecture, histology, and gene expression in many instances [11, 18]. Accordingly, our workgroup aimed at the development of PDTX-models for endocrine tumors. Overall, some implanted and subsequently analyzed pieces remained vital and proliferating and retained specific patient characteristics in the murine host. However, in most instances these tumors lacked relevant growth properties (data not shown), limiting the applicability of PDTXs for preclinical therapeutic trials. In contrast to the majority of the established endocrine PDTXs, tissue-derived MUC-1 xenografts were characterized by the engraftment of large solid tumors and the maintenance of pathological and endocrine features comparable to that of the original patient tumor.
One of the most frequently cited reasons for the high failure rate of new therapeutic regimens in oncology is the lack of preclinical models reflecting patient heterogeneity [11, 15]. Initial characterization of MUC-1 xenografts and subsequent comparison with the commonly available tumor models for ACC revealed a molecular profile distinct from that of NCI-H295R, SW-13 and SJ-ACC3. In general, the histopathological diagnosis of adrenal tumors relies on a panel of different parameters: For the determination of the adrenocortical origin, the expression of SF-1 has emerged as the most valid marker [19] while for the discrimination of benign from malignant adrenocortical tumors the Ki67 index is of high importance. In addition to its value as prognostic marker, the Ki67 Index has been recently integrated in treatment flow charts for ACC patients and represents thereby an important determinant for ACC tumors [20, 21]. Moreover, EGF-R expression and furthermore membranous EGFR localization in immunohistochemical stainings has been reported to indicate a malignant phenotype of ACC [22].
Ki67 analyses of the different tumor models were mostly similar with slightly elevated indices for SW-13. SW-13-xenografts furthermore demonstrated an abrogation of both important markers, SF-1 and EGF-R, underlining the questionable value of SW-13 as tumor model for ACC. In contrast, NCI-H295R, SJ-ACC3 and MUC-1 derived tumors were positive for SF-1 and EGF-R. Interestingly, SJ-ACC3 displayed a highly significant overexpression of SF-1 RNA and protein, which is in accordance with previous reports on childhood adrenocortical tumors [23]. In contrast, MUC-1 tumors showed significantly elevated levels of EGF-R in comparison to the other tumor models.
Genetic analyses of known driver genes (CTNNB1, TP53, CDKN2A, RB1 and MEN1) and of genes recently reported to be of importance in ACC (ZNRF3, DAXX, TERT and MED12) revealed in the original patient tumor (also representing passage 1) a somatic mutation in TP53 (a frameshift deletion: Hg19 positions: 7574003 on Chr 17: G is deleted) while the tumor was devoid of mutations in any of the other investigated genes (T91/L91 in [1]). SW-13 cells carry a homozygous TP53 point mutation, NCI-H295R cells harbor a large deletion in the TP53 locus and SJ-ACC3 cells display a TP53 haplotype with G245C mutation as previously described for Li-Fraumeni syndrome [7, 8, 12]. NCI-H295R cells are furthermore known to have acquired an activating CTNNB1 mutation [24], which has not been described for SW-13 and SJ-ACC3 yet. Our immunohistochemical analyses are in accordance with the underlying genetic phenotype for p53 and β-catenin. While we detected strong nucleo-cytoplasmic distribution of β-catenin in the NCI-H295R tumor model, the staining appeared weaker and of cytoplasmic localization in MUC-1. In contrast, while strong and nuclear p53 staining was evident in MUC-1, no specific immunopositivity was detectable in NCI-H295R. Overall, these findings indicate that MUC-1 and NCI-H295R might represent different patient clusters accordingly to recent classifications of adult ACC tumors [1, 25].
Of note, also MUC-1 cells maintained in vitro hormonal activity and specific phenotypical characteristics for ACC, which was furthermore proven stable over extensive passages. With regard to therapeutic responsiveness, MUC-1 cells demonstrated drug resistance against the clinical gold standard EDP-M, which was not observed for the commonly utilized tumor model NCI-H295R. This finding was not unexpected as the patient, from which MUC-1 was obtained, had received several cycles of EDP-M before development of metastases, while such treatment was not reported for NCI-H295R [8]. In this context it has to be mentioned, that even though superior over other chemotherapies, the overall response- rate of EDP-M is poor ranging from 20-50% with a median progression free survival of only five months [5, 26]. Consequently, to improve the identification of novel and/or second line therapies, preclinical experiments should include a tumor model reflecting the clinically low response rates to EDP-M. Following the same line, we were recently able to include MUC-1-xenografts for the first time in a therapeutic study with two different anti-IGF-1R inhibiting approaches. These experiments also indicated sub-group dependent differences in the expression of components of the IGF-system, as well as in therapeutic outcome versus NCI-H295R and SJ-ACC3, thereby also better reflecting clinical observations than with NCI-H295R alone [20, 27].
In summary, we herein report on the establishment of the first tumor model for ACC, which provides both a human cell line as well as a tissue-derived xenograft model. Moreover, we provide evidence that the implementation of a panel of NCI-H295R, SJ-ACC3 and MUC-1 might be helpful for a more successful clinical translation of novel therapeutic regimens for ACC in the future.
MATERIALS AND METHODS
Animals and tumor models
Female athymic NMRI nu/nu mice were purchased from Harlan Winkelmann (Borchen, Germany) and housed under pathogen-free conditions. Patient tumor derived pieces (approximately 2 × 2 mm) were implanted subcutaneously into the neck of individual mice. The number of mice for initial establishment and characterization was n=10 and n=4-20 for MUC-1-xenografts in different passages as outlined in detail below.
For NCI-H295 15×106 and for SW-13 xenografts 13×106 tumor cells in a volume of 200 μl PBS were inoculated while SJ-ACC3 tumors [12] were induced by direct tumor implantation of small tumor pieces into athymic nude mice. Before implantation of these 2 × 2 mm measuring tumor pieces appropriate samples of SJ-ACC3 tumors were transferred from liquid nitrogen to a 37°C water bath and tumor tissue was rinsed several times in medium 199 (Gibco Invitrogen, Darmstadt, Germany) supplemented with 1% penicillin/streptomycin. Therapeutic experiments were performed as previously described [13].
All experiments were carried out following protocols approved by the Regierung von Oberbayern and in accordance with the German guidelines for animal studies. Furthermore, studies including patient biomaterial were approved by the local ethical committee and patients provided written consent. Furthermore, experiments including patient biomaterial were approved by the local ethical committee.
Pathological and immunohistochemical examination
Paraffin-embedded sections were rehydrated and incubated with blocking buffer containing 3 % BSA (Roche Diagnostics, Mannheim, Germany), 5 % goat or rabbit serum (Jackson ImmunoResearch Laboratories, West Grove, PA), and 0.5 % Tween 20 for 15 min. Immunohistochemical staining was performed using either monoclonal rabbit anti-human Ki67 (DCS, Hamburg, Germany; 1:200 in BB; Figures 1H and 1I, 2B and 2F, 4C and 5B), purified rat anti-mouse CD31 (Pharmingen, NJ, USA; 1:100 in BB; Figures 1E and 1F, 2D and 2H), monoclonal mouse anti-human SF-1 (Perseus Proteomics Inc., Tokyo, Japan; 1:100; Figures 2G, 4E, 5C), polyclonal rabbit anti-human SF-1, Novus Biological, Littleton, Colorado, USA; 1:200; Figure 2C), monoclonal rabbit anti-human EGF-receptor (Cell Signaling Technology, Danvers, USA; 1:100 in BB; Figure 4G) or polyclonal rabbit anti-mouse 3βHSD (provided by Anita Payne, University of Stanford, CA; Figure 5D), monoclonal mouse anti-human p53 (clone DO-7, Dako, Hamburg, Germany; 1:50 in BB; Figure 2I-L) and monoclonal mouse anti-human beta-Catenin (BD Transduction Laboratories, CA, USA; 1:500 in BB; Figure 2M-P) antibodies were used and incubated overnight at 4°C. After rinsing for 15 min in PBS, secondary antibody (for 3βHSD, Ki67, EGF-receptor and rabbit SF-1: goat anti-rabbit biotinylated IgG, Vector Laboratories, Burlingame, CA; for CD31: biotin-SP-conjugated AffiniPure goat anti-rat, Jackson Immuno Research Lab, CA; for mouse SF-1: ImmPRESS™ HRP Anti-Mouse IgG (Peroxidase) Polymer Detection Kit, Vector Laboratories; for p53 and beta-Catenin: biotinylated polyclonal rabbit anti-mouse, Dako, Hamburg, Germany) was applied for 30 min at room temperature. With the exception of a direct detection for the utilized SF-1 antibody complex, bound antibodies were visualized using the Vectastain ABC Kit (Vector Laboratories, Burlingame, CA, USA) followed by 3,3′-diaminobenzidine staining. For quantification 6 high power fields (HPF, 0.391 mm2, 400x magnification)/tumor were investigated and quantified for proliferation index (% Ki67 positive to negative cells).
The numbers of investigated samples for the different tumor models were n= 5-8 for Ki67, n=4-6 for SF-1, n=4 for EGF-R immunohistochemistry and n=4-7 for EGF-R and n=3-5 for SF-1 Real Time PCRs. The numbers of investigated samples for clones 1 and 2 of NCI-H295R were n= 5-7.
In vitro, 100 000 MUC-1 cells were seeded and PFA fixed on Falcon 4 well culture slides and incubated with the different antibodies as described for the tumor slides above.
Cortisol measurements
Cortisol was measured in plasma samples of tumor bearing mice using the human Cortisol ELISA (n=6 for NCI-H295R and n=17 for MUC-1). Cortisol levels of cell culture medium from MUC-1 cells supernatants of passages 3, 4, 5, 6, 8, 9, 10, 12 and15 were analyzed and offset against a medium blank (in triplicate). Cortisol concentration was measured by a routine competitive automated chemiluminescence immunoassay (Liaison, Diasorin, Sallugia, Italy). According to the manufacturer, cross-reactivity is highest for prednisolone (12.6%), 11-desoxycortisol (3.0%) and corticosterone (3.5%) and negligible for other structurally related steroids. In the media used in this study, we confirmed LoQ at 0.5 μg/dL and within and between- assay variability at 2 μg/dL at 4.0% and 9.2%, respectively. For this study, all samples were analyzed on one day in one analytical run.
Molecular investigation
SW-13, SJ-ACC3, NCI-H295R and MUC-1 tumors samples (n=4-7) were used for EGF-receptor Real-Time PCR analyses after RNA extraction (SV Total RNA Isolation system, Promega) and reverse transcription (RevertAid™ H Minus First Strand cDNA Synthesis Kit, Fermentas). For Real-Time PCR analyses we utilized the EvaGreen® reaction mix (Bio-Rad, Munich, Germany) in the Stratagene Mx3000PTM Cycler (Agilent Technologies, Waldbronn, Germany). Human primer catalogue numbers was #VHPS-10346 purchased from Biomol, Hamburg, Germany. For SF-1 Real-Time PCR analyses (n=3-5) human SF-1 Primer (forward: 5′-CAGCCTGGATTTGAAGTTCCT, reverse: 5′-CAGCATTTCGATGAGCAGGT) were used. Quantification was adjusted using the housekeeping gene GAPDH for human samples forward: 5′-AGC CTC CCG CTT CGC TCT CT-3′ and reverse: 5′-CCA GGC GCC CAA TAC GAC CA-3′.
Cell culture
Both NCI-H295R and SW-13 cell lines were originally obtained from ATCC and again authenticated in February 2015. SW-13 cells were cultured in DMEM/F-12 medium (Dulbecco's Modified Eagle Medium, Gibco Invitrogen, Darmstadt, Germany) in a 5% CO2-95% air atmosphere at 37°C. Cell culture medium was supplemented with 1% penicillin/streptomycin and 10% FBS. NCI-H295R cells were cultured in a 1:1 mixture of DMEM and Ham's F-12 medium (DMEM-F12 and supplements, Gibco Invitrogen). The medium was supplemented with insulin (10 mg/ml), transferrin (5.5 mg/ml), and selenium (5 ng/ml) (ITS), penicillin (100 U/ml), streptomycin (100 μg/ml) and 2% ultroser G (Cytogen, Sinn, Germany).
For the establishment of a MUC-1 cell line a piece of the xenograft was minced into pieces smaller than 0.5 mm using a razor blade. The resulting suspension was transferred into a 50 ml Falcon tube and centrifuged for 5 minutes by 1000 rpm (Universal 30RF, Hettich, Tuttlingen, Germany). The supernatant was discarded followed by an incubation of the pellet with 1mg sterile collagenase II (Biochrom, Berlin, Germany) dissolved in PBS. After 50 minutes, FCS (Life Technologies, Carlsbad, California) to a concentration of 10% was added to inactivate the collagenase. Upon another centrifugation step at 1000 rpm for 5 minutes, the supernatant was discarded and the pellet re-suspended for seven minutes at room temperature in erythrocyte lysis buffer (2 volumes of lysis buffer onto 1 volume of cells). After a final centrifugation step, the cells were re-suspended, filtered through 70μm cell strainer and cultured in Advanced DMEM/F12 Medium (containing 10% FBS and 1% penicillin/streptomycin. The cells were split every 2 weeks and directly after cell culture establishment as well as after passages 2, 3, 4, 6, 9, 11, 13 and 14 a mouse and human fibroblast removal step was performed using the Anti-Fibroblast MicroBeads and Feeder Removal MicroBeads from (Miltenyi Biotec, Bergisch Gladbach, Germany) following the manufacturers instruction.
Successful clearance of murine fibroblasts from the target cell line was demonstrated by investigating genomic DNA by an in-house assay established by Eurofins (Ebersberg, Germany) and a Universal-Primer Probe-Assay using ApoE as specific target gene.
For cell viability assay (MTT) 40 000 NCI-H295R and 14 000 MUC-1 cells were seeded on a 96 well plate and cultivated for 24 hours either with EDP-M. For quantification of cell viability, a MTT assay (Sigma-Aldrich, Steinheim, Germany) was used following the manufacturer's protocols. Measurements were made in a SPECTRA microplate reader from Tecan (Crailsheim, Germany).
The tested concentrations were based on the individual IC50 (half-maximum inhibitory concentration) of each drug regarding cell proliferation: etoposide: 1.2μM; doxorubicin: 11μM; cisplatin: 9.6μM and mitotane: 15.9μM. In each case drugs were tested from their lowest to highest concentration (0.5x IC50, 1x IC50, 2x IC50) as a combination of etoposide, doxorubicin, cisplatin and mitotane (EDP-M).
Genetic characterization
For detailed genetic characterization, genomic DNA of MUC-1 cells and the original patient tumor were extracted and a cell line authentication test was performed by Eurofins. Specifically, genetic characteristics were determined by PCR-single-locus technology. 21 independent PCR loci (Amelogenin, D3S1358, D1S1656, D6S1043, D13S317, Penta E, D16S539, D18S51, D2S1338, CSF1PO, Penta D, TH01, vWA, D21S11, D7S820, D5S818, TPOX, D8S1179, D12S391, D19S433 and FGA) were investigated (Promega, PowerPlex 21 PCR Kit). In parallel, positive and negative controls were carried out yielding correct results. The resulting sample with the DSMZ name CL151006_001 could not be verified in the online database of the DSMZ (German Collection of Microorganisms and Cell Cultures GmbH, http://www.dsmz.de/de/service/services-human-and-animal-cell-lines/online-stranalysis).
In a next step, genomic DNA of MUC-1 cells was investigated in more detail by Eurofins using the Argus-X12 PCR Kit for the analyses of nine independent PCR loci (Amelogenin, DXS8378, HPRTB, DXS7423, DXS7132, DXS10134, DXS10074, DXS10101, DXS10103, DXS10146, DXS10179, DXS10148 and DXS10135) including positive and negative controls.
Statistical analysis
All results are expressed as mean ± SEM. Statistical significance was determined using one-way ANOVA with Bonferroni's multiple comparison test or the paired t-test (Prizm software, Houston, TX). Statistical significance was defined as p<0.05 and is denoted as stars (*, p<0.05; **, p<0.01; ***, p<0.001) in the Figures if not stated otherwise.
SUPPLEMENTARY FIGURE AND TABLE
Acknowledgments
The authors are indebted to Susanne Mentz for her excellent technical support. The authors further thank Gerard Zambetti and Chris Morton (St. Jude Children's Research Hospital, Memphis, Tennessee) for providing the SJ-ACC3 tumor model and Guillaume Assie and Jérôme Bertherat (INSERM U1016, Institut Cochin, Paris, France) for providing the information about the mutational status of MUC-1.
Footnotes
CONFLICTS OF INTEREST
The authors have nothing to disclose
GRANT SUPPORT
This work was supported by the Seventh Framework Programme (FP7/2007-2013) under grant agreement n° 259735 to F.B. Furthermore, research leading to these results has received funding from the Wilhelm Sander Stiftung (2011.003.1/2), Mildred-Scheel-Stiftung (110190) to C.H. and F.B. and Associazione Italiana Ricerca sul Cancro (AIRC) Investigator Grant to M. L. (grant # IG2015-17691).
REFERENCES
- 1.Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, Elarouci N, Sbiera S, Kroiss M, Allolio B, Waldmann J, Quinkler M, et al. Integrated genomic characterization of adrenocortical carcinoma. Nature genetics. 2014;46:607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 2.Fassnacht M, Berruti A, Baudin E, Demeure MJ, Gilbert J, Haak H, Kroiss M, Quinn DI, Hesseltine E, Ronchi CL, Terzolo M, Choueiri TK, Poondru S, Fleege T, Rorig R, Chen J, et al. Linsitinib (OSI-906) versus placebo for patients with locally advanced or metastatic adrenocortical carcinoma: a double-blind, randomised, phase 3 study. The Lancet Oncology. 2015;16:426–435. doi: 10.1016/S1470-2045(15)70081-1. [DOI] [PubMed] [Google Scholar]
- 3.Kroiss M, Quinkler M, Johanssen S, van Erp NP, Lankheet N, Pollinger A, Laubner K, Strasburger CJ, Hahner S, Muller HH, Allolio B, Fassnacht M. Sunitinib in refractory adrenocortical carcinoma: a phase II, single-arm, open-label trial. The Journal of clinical endocrinology and metabolism. 2012;97:3495–3503. doi: 10.1210/jc.2012-1419. [DOI] [PubMed] [Google Scholar]
- 4.Wortmann S, Quinkler M, Ritter C, Kroiss M, Johanssen S, Hahner S, Allolio B, Fassnacht M. Bevacizumab plus capecitabine as a salvage therapy in advanced adrenocortical carcinoma. European journal of endocrinology / European Federation of Endocrine Societies. 2010;162:349–356. doi: 10.1530/EJE-09-0804. [DOI] [PubMed] [Google Scholar]
- 5.Fassnacht M, Terzolo M, Allolio B, Baudin E, Haak H, Berruti A, Welin S, Schade-Brittinger C, Lacroix A, Jarzab B, Sorbye H, Torpy DJ, Stepan V, Schteingart DE, Arlt W, Kroiss M, et al. Combination chemotherapy in advanced adrenocortical carcinoma. The New England journal of medicine. 2012;366:2189–2197. doi: 10.1056/NEJMoa1200966. [DOI] [PubMed] [Google Scholar]
- 6.Logie A, Boudou P, Boccon-Gibod L, Baudin E, Vassal G, Schlumberger M, Le Bouc Y, Gicquel C. Establishment and characterization of a human adrenocortical carcinoma xenograft model. Endocrinology. 2000;141:3165–3171. doi: 10.1210/endo.141.9.7668. [DOI] [PubMed] [Google Scholar]
- 7.Leibovitz A, McCombs WM, 3rd, Johnston D, McCoy CE, Stinson JC. New human cancer cell culture lines. I. SW-13, small-cell carcinoma of the adrenal cortex. Journal of the National Cancer Institute. 1973;51:691–697. [PubMed] [Google Scholar]
- 8.Gazdar AF, Oie HK, Shackleton CH, Chen TR, Triche TJ, Myers CE, Chrousos GP, Brennan MF, Stein CA, La Rocca RV. Establishment and characterization of a human adrenocortical carcinoma cell line that expresses multiple pathways of steroid biosynthesis. Cancer research. 1990;50:5488–5496. [PubMed] [Google Scholar]
- 9.Wolkersdorfer GW, Bornstein SR, Higginbotham JN, Hiroi N, Vaquero JJ, Green MV, Blaese RM, Aguilera G, Chrousos GP, Ramsey WJ. A novel approach using transcomplementing adenoviral vectors for gene therapy of adrenocortical cancer. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2002;34:279–287. doi: 10.1055/s-2002-33255. [DOI] [PubMed] [Google Scholar]
- 10.Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer research. 2013;73:5315–5319. doi: 10.1158/0008-5472.CAN-13-1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nature reviews Clinical oncology. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pinto EM, Morton C, Rodriguez-Galindo C, McGregor L, Davidoff AM, Mercer K, Debelenko LV, Billups C, Ribeiro RC, Zambetti GP. Establishment and characterization of the first pediatric adrenocortical carcinoma xenograft model identifies topotecan as a potential chemotherapeutic agent. Clinical cancer research. 2013;19:1740–1747. doi: 10.1158/1078-0432.CCR-12-3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hantel C, Jung S, Mussack T, Reincke M, Beuschlein F. Liposomal polychemotherapy improves adrenocortical carcinoma treatment in a preclinical rodent model. Endocrine-related cancer. 2014;21:383–394. doi: 10.1530/ERC-13-0439. [DOI] [PubMed] [Google Scholar]
- 14.Chortis V, Taylor AE, Schneider P, Tomlinson JW, Hughes BA, O'Neil DM, Libe R, Allolio B, Bertagna X, Bertherat J, Beuschlein F, Fassnacht M, Karavitaki N, Mannelli M, Mantero F, Opocher G, et al. Mitotane therapy in adrenocortical cancer induces CYP3A4 and inhibits 5alpha-reductase, explaining the need for personalized glucocorticoid and androgen replacement. The Journal of clinical endocrinology and metabolism. 2013;98:161–171. doi: 10.1210/jc.2012-2851. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JI, Decker S, Zaharevitz D, Rubinstein LV, Venditti JM, Schepartz S, Kalyandrug S, Christian M, Arbuck S, Hollingshead M, Sausville EA. Relationships between drug activity in NCI preclinical in vitro and in vivo models and early clinical trials. British journal of cancer. 2001;84:1424–1431. doi: 10.1054/bjoc.2001.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lum DH, Matsen C, Welm AL, Welm BE. Overview of human primary tumorgraft models: comparisons with traditional oncology preclinical models and the clinical relevance and utility of primary tumorgrafts in basic and translational oncology research. Current protocols in pharmacology / editorial board, SJ Enna. 2012 doi: 10.1002/0471141755.ph1422s59. Chapter 14:Unit 14 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidalgo M, Amant F, Biankin AV, Budinska E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Maelandsmo GM, Roman-Roman S, Seoane J, Trusolino L, Villanueva A. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer discovery. 2014;4:998–1013. doi: 10.1158/2159-8290.CD-14-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SY, Lin D, Gout PW, Collins CC, Xu Y, Wang Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Advanced drug delivery reviews. 2014;79-80C:222–237. doi: 10.1016/j.addr.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 19.Sbiera S, Schmull S, Assie G, Voelker HU, Kraus L, Beyer M, Ragazzon B, Beuschlein F, Willenberg HS, Hahner S, Saeger W, Bertherat J, Allolio B, Fassnacht M. High diagnostic and prognostic value of steroidogenic factor-1 expression in adrenal tumors. The Journal of clinical endocrinology and metabolism. 2010;95:E161–171. doi: 10.1210/jc.2010-0653. [DOI] [PubMed] [Google Scholar]
- 20.Fassnacht M, Kroiss M, Allolio B. Update in adrenocortical carcinoma. The Journal of clinical endocrinology and metabolism. 2013;98:4551–4564. doi: 10.1210/jc.2013-3020. [DOI] [PubMed] [Google Scholar]
- 21.Beuschlein F, Weigel J, Saeger W, Kroiss M, Wild V, Daffara F, Libe R, Ardito A, Al Ghuzlan A, Quinkler M, Osswald A, Ronchi CL, de Krijger R, Feelders RA, Waldmann J, Willenberg HS, et al. Major prognostic role of Ki67 in localized adrenocortical carcinoma after complete resection. The Journal of clinical endocrinology and metabolism. 2015;100:841–849. doi: 10.1210/jc.2014-3182. [DOI] [PubMed] [Google Scholar]
- 22.Adam P, Hahner S, Hartmann M, Heinrich B, Quinkler M, Willenberg HS, Saeger W, Sbiera S, Schmull S, Voelker HU, Strobel P, Allolio B, Fassnacht M. Epidermal growth factor receptor in adrenocortical tumors: analysis of gene sequence, protein expression and correlation with clinical outcome. Modern pathology. 2010;23:1596–1604. doi: 10.1038/modpathol.2010.153. [DOI] [PubMed] [Google Scholar]
- 23.Pianovski MA, Cavalli LR, Figueiredo BC, Santos SC, Doghman M, Ribeiro RC, Oliveira AG, Michalkiewicz E, Rodrigues GA, Zambetti G, Haddad BR, Lalli E. SF-1 overexpression in childhood adrenocortical tumours. European journal of cancer. 2006;42:1040–1043. doi: 10.1016/j.ejca.2006.01.022. [DOI] [PubMed] [Google Scholar]
- 24.Gaujoux S, Hantel C, Launay P, Bonnet S, Perlemoine K, Lefevre L, Guillaud-Bataille M, Beuschlein F, Tissier F, Bertherat J, Rizk-Rabin M, Ragazzon B. Silencing mutated beta-catenin inhibits cell proliferation and stimulates apoptosis in the adrenocortical cancer cell line H295R. PloS one. 2013;8:e55743. doi: 10.1371/journal.pone.0055743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faillot S, Assie G. ENDOCRINE TUMOURS: The genomics of adrenocortical tumors. European journal of endocrinology / European Federation of Endocrine Societies. 2016;174:R249–265. doi: 10.1530/EJE-15-1118. [DOI] [PubMed] [Google Scholar]
- 26.Else T, Kim AC, Sabolch A, Raymond VM, Kandathil A, Caoili EM, Jolly S, Miller BS, Giordano TJ, Hammer GD. Adrenocortical carcinoma. Endocrine reviews. 2014;35:282–326. doi: 10.1210/er.2013-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beuschlein F, Jakoby J, Mentz S, Zambetti G, Jung S, Reincke M, Suss R, Hantel C. IGF1-R inhibition and liposomal doxorubicin: Progress in preclinical evaluation for the treatment of adrenocortical carcinoma. Molecular and cellular endocrinology. 2016 doi: 10.1016/j.mce.2016.03.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.