

Abstract
The endoplasmic reticulum (ER) serves as a cellular storehouse for Ca2+, and Ca2+ released from the ER plays a role in a host of critical signaling reactions, including exocytosis, contraction, metabolism, regulation of transcription, fertilization, and apoptosis. Given the central role played by the ER, our understanding of these signaling processes could be greatly enhanced by the ability to image [Ca2+]ER directly in individual cells. We created a genetically encoded Ca2+ indicator by redesigning the binding interface of calmodulin and a calmodulin-binding peptide. The sensor has improved reaction kinetics and a Kd ideal for imaging Ca2+ in the ER and is no longer perturbed by large excesses of native calmodulin. Importantly, it provides a significant improvement over all previous methods for monitoring [Ca2+]ER and has been used to directly show that, in MCF-7 breast cancer cells, the antiapoptotic protein B cell lymphoma 2 (Bcl-2) (i) lowers [Ca2+]ER by increasing Ca2+ leakage under resting conditions and (ii) alters Ca2+ oscillations induced by ATP, and that acute inhibition of Bcl-2 by the green tea compound epigallocatechin gallate results in an increase in [Ca2+]ER due to inhibition of Bcl-2-mediated Ca2+ leakage.
Keywords: calcium, cameleon
To date, researchers have attempted to use a number of strategies to deduce the role of [Ca2+]ER in signaling processes, but all possess shortcomings (1, 2). Conventional methods use cytosolic small-molecule Ca2+ indicators to infer that an observed signal likely originated from the endoplasmic reticulum (ER). However, as more studies identify the complex interplay among levels of Ca2+ in the ER and Ca2+ influx (3), as well as a close interaction network between the ER and mitochondria (4, 5), it seems increasingly important for our understanding of signaling processes that organelle Ca2+ be observed directly to differentiate Ca2+ signals from different organelles. Ca2+ indicator dyes can be loaded at 37°C rather than 25°C, thus favoring the loading of internal compartments. However, this method loads all internal compartments (not just the ER), and the remaining cytosolic dye must be effectively removed, or quenched. ER-targeted aequorin probes (6) can be genetically targeted to the ER to measure [Ca2+]ER directly, but the aequorin probes are luminescent and therefore not bright enough for single-cell imaging without specialized equipment. Perhaps more problematic is that for aequorin to be active it must be reconstituted with the coelenterazine cofactor, and to do so the ER must initially be completely depleted of Ca2+. Finally, the low-affinity FRET-based cameleon (7) YC4.3ER can also be genetically targeted and enables single-cell imaging, but its Kd for Ca2+ is not well suited to monitor changes in [Ca2+]ER, and therefore it has a low sensitivity.
Given the critical role of apoptosis in human diseases as well as normal physiology and the increasing evidence that the balance of B cell lymphoma 2 (Bcl-2) family proteins plays a role in the tumorigenesis of a number of cancers (8), a fundamental need exists to better understand how these proteins interact and the multiple mechanisms by which they influence apoptotic cascades. An important aspect of this is identifying inhibitors of different Bcl-2 family members that could serve as tools to perturb and thereby better our understanding of the signaling cascades, as well as to act as potential therapeutic agents. We set out to generate an indicator for [Ca2+]ER so that we could examine the role of [Ca2+]ER in the regulation of Bcl-2 family-mediated apoptosis. Although most studies have focused on the role of the mitochondria in apoptosis, recently it has been shown that Bcl-2 family members can localize to the ER and alter [Ca2+]ER, thus adding another dimension to the role of Bcl-2 family members in apoptosis (9–13). Importantly, it has been shown that changes in [Ca2+]ER can directly influence the propensity of a given stimulus to lead to apoptosis (12, 14), although there have been a number of conflicting reports (15) as to exactly how the antiapoptotic protein Bcl-2 influences [Ca2+]ER. Of particular interest is a recent report that Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) in WEHI7.2 T cells, suggesting that this interaction might be the mechanism by which Bcl-2 alters ER Ca2+ homeostasis (16). The creation of a robust and reliable sensor to directly monitor [Ca2+]ER could help alleviate some of the uncertainties associated with indirect Ca2+ measurements and would provide a significant advantage in probing questions related to Ca2+ homeostasis in the ER.
Materials and Methods
Peptide synthesis and Biacore (Neuchâtel, Switzerland) experiments were performed as described in Supporting Text, which is published as supporting information on the PNAS web site. The mutant peptide and calmodulins (CaMs) were cloned between a truncated enhanced cyan fluorescent protein (CFP) and citrine fluorescent protein (17), as described (7). The constructs were cloned between the BamHI/EcoRI sites in pRSETB or pBAD for protein purification and pcDNA3 for expression in mammalian cells. To generate an ER-targeted cameleon, the calreticulin signal sequence MLLPVLLLGLLGAAAD was added 5′ to CFP, and an ER retention sequence, KDEL, was added to the 3′ end of citrine.
For protein purification, cameleons were expressed in either JM109 (Stratagene) or LMG194 (Invitrogen) and grown over-night at 25°C. Constructs in pBAD/LMG194 were induced with 0.2% arabinose. Protein was extracted with Bacterial Protein Extraction Reagent (Pierce), purified via an N-terminal 6×Histag by using Ni-NTA agarose, and buffer-exchanged into 10 mM 4-morpholinepropanesulfonic acid/100 mM NaCl, pH 7.4, by using Amicon Centricon-30 columns (Millipore). Absorbance measurements were conducted on a Cary 3E UV-Visible spectrometer (Varian). Fluorescence measurements were conducted on a Spex (Spex Industries, Metuchen, NJ) Fluorolog-3 Fluorimeter (Horiba group) at a cameleon concentration of 0.4 μM. Ca2+/EGTA, and Ca2+/N-(2-hydroxyethyl)ethylene dinitrilo-N,N′,N′-triacetic acid (HEEDTA) buffers were prepared as described (18, 19) and were used in Ca2+ titrations to achieve concentrations of <50 μM. All solutions of >50 μM were unbuffered.
Stopped-flow experiments were performed on an Applied Photophysics (Surrey, U.K.) Stopped Flow. Equal volumes of cameleon (final concentration of 0.8 μM) and Ca2+ (various concentrations) were rapidly mixed, and the fluorescence was monitored by using an excitation wavelength of 420 nm and an emission cutoff filter of 500 nm. The observed first-order rate constant (kobs) was calculated from each averaged (n > 4) data set by nonlinear regression analysis.
Cell-culturing details are provided in Supporting Text. For imaging, cells were rinsed twice and then maintained in Hanks' balanced salt solution (HBSS) with 20 mM Hepes and 2 g/liter d-glucose at pH 7.4. Ca2+-free solutions were generated by adding 0.5 mM EGTA, 1 mM Mg2+, 1g/liter glucose, and 20 mM Hepes to Ca2+ and Mg2+-free HBSS (GIBCO). For experiments in which both the ER and cytosolic Ca2+ levels were monitored, cells were treated with 4 μM fura-2-AM (Molecular Probes) with 0.04% Pleuronic F-127 in HBSS for 30 min at room temperature. Cells were then washed with HBSS and allowed to incubate at room temperature for 15 min to ensure cleavage of the acetoxymethyl ester. (—)Epigallocatechin gallate (EGCG) and (—)epicatechin (EC) were purchased from Sigma, dissolved in 100% DMSO, and stored at –80°C until use.
To calibrate the cameleon probe in the ER, Rmin was obtained by treating the cells with 3 mM EGTA and 2 μM ionomycin, and Rmax was determined by treating cells with 25 μM digitonin, followed by 5–10 mM Ca2+, 1 mM ATP, and 1 mM Mg2+. The Rmin and Rmax values were used to convert cell data to % FRET (of maximum). Finally, the apparent Kd1 (0.58), Kd2 (56.46), Rmax1 (0.28), Rmax2 (0.72), n1 (1.18), and n2 (1.67) values obtained from fitting the in vitro data with 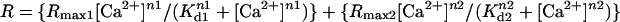 were used in the calibration. To calibrate fura-2, cells were treated with 8 μM ionomycin and 10 mM EGTA in Ca2+-free HBSS to obtain Rmin, followed by 2 μM ionomycin and 20 mM Ca2+ in HBSS to obtain Rmax. The in situ Rmin and Rmax values were adjusted (multiplied by 0.85) to account for the minimum viscosity effect (20). The standard equation:
were used in the calibration. To calibrate fura-2, cells were treated with 8 μM ionomycin and 10 mM EGTA in Ca2+-free HBSS to obtain Rmin, followed by 2 μM ionomycin and 20 mM Ca2+ in HBSS to obtain Rmax. The in situ Rmin and Rmax values were adjusted (multiplied by 0.85) to account for the minimum viscosity effect (20). The standard equation: 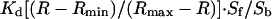 was used to convert the Fura-2 350:380 ratio to [Ca2+]cyt, where Sf and Sb are the emission intensity at 380 nm for Ca2+-free and Ca2+-bound fura-2, respectively.
was used to convert the Fura-2 350:380 ratio to [Ca2+]cyt, where Sf and Sb are the emission intensity at 380 nm for Ca2+-free and Ca2+-bound fura-2, respectively.
Cells were imaged on a Zeiss Axiovert 200M microscope with a cooled charge-coupled device camera (Roper Scientific, Trenton, NJ), controlled by metafluor 6.1 software (Universal Imaging, Downington, PA). Emission ratio imaging of the cameleon was accomplished by using a 436DF20 excitation filter, 450-nm dichroic mirror, and two emission filters (475/40 for enhanced CFP and 535/25 for citrine) controlled by a Lambda 10-2 filter changer (Sutter Instruments, Novato, CA). Excitation ratio imaging for fura-2 was accomplished by using 350/10 and 380/10 excitation filters, a 450-nm dichroic mirror, and a 535/45 emission filter. Fluorescence images were background corrected. Exposure times were typically 100–1,000 ms, and images were collected every 8–20 s.
Results
To create a sensor that would be appropriate for monitoring [Ca2+]ER in single living cells, we started with the original cameleon construct, comprised of two fluorescent proteins (CFP and citrine) and two sensing proteins [CaM and a CaM-binding peptide derived from skeletal muscle myosin light chain kinase (skMLCK)] that undergo a conformational change upon binding (7). Our goal was to redesign the binding interface between CaM and the peptide to generate highly specific protein/peptide pairs that would display a range of Ca2+ affinities and that would not be perturbed by endogenous proteins, such as WT CaM. This was particularly important, given that previous cameleons have suffered from perturbation by endogenous proteins (21, 22). Toward this end, we initially generated peptides that would not bind to WT CaM and then sought to reengineer CaM to reconstitute binding to the mutant peptides.
As a starting point for redesign, we targeted the six possible salt-bridge interactions identified in the NMR solution structure between CaM and the skMLCK peptide (23). A series of biotinylated peptides was synthesized in which a basic residue (either K or R) at the N and C termini of the peptide was replaced with an acidic E residue. Charge reversals were made sequentially, generating peptides with two, four, and six charge reversals until all of the salt-bridge interactions were destroyed. Binding of CaM to the peptides was assayed by using surface plasmon resonance (Biacore 3000), in which binding events are optically detected as a change in the refractive index at the surface. To accomplish this, biotinylated peptides were coupled to a streptavidin-coated chip, and CaM was injected over the surface in the presence of Ca2+ (Fig. 6, which is published as supporting information on the PNAS web site). Initially, we verified that binding of CaM to the WT skMLCK was ∼10 nM, comparable to previously reported values obtained in solution (Fig. 7, which is published as supporting information on the PNAS web site). The mutant peptide with four charge reversals displayed a dramatically lower affinity for CaM than the WT skMLCK peptide (Fig. 1a). Injection of increasing concentrations of CaM enabled determination of the Kd > 700 μM (Fig. 8, which is published as supporting information on the PNAS web site). Thus, four charge reversals were sufficient to decrease the affinity for WT CaM by >104.
Fig. 1.

In vitro characterization of the mutant CaM and peptide and the resulting redesigned cameleon. (a) Biacore sensorgram of WT CaM binding to skMLCK (solid line) and four-charge reversal skMLCK (dashed line). CaM was injected over the surface in the presence of saturating Ca2+ starting at time t = 0 for 360 s. (b) Binding of WT (squares, dashed line) and mutant (circles, solid line) CaM to mutant skMLCK with four-charge reversals. The binding of CaM reached a steady state during the association phase, and therefore a Scatchard analysis was used to determine the dissociation constants (Fig. 8). (c) In vitro calcium titration curves of YC2.1 (diamonds, dashed line), YC3.3 (squares, dashed line), YC4.3 (triangles, dashed line), and D1 (circles, solid line), along with corresponding fits of the data. The gray box represents the typical range of [Ca2+] in the ER, from resting to the depleted state. (d) Percent of the maximum FRET response of YC3.3 (squares) and D1 (circles) with increasing concentrations of WT CaM.
A small library of CaM mutants in which the four complementary positions in CaM (E11, E84, E87, and E127) were changed to E, K, or R was then assayed for binding to both WT skMLCK and the mutant peptide. The best hit had a Kd of 270 μM, and therefore a higher affinity than WT CaM for the mutant peptide (Fig. 1b). Upon sequencing, this mutant was identified as E11K, E84R, E87K, or E127E. Mutation of E127 to R did not increase the affinity of CaM for the peptide (data not shown).
The mutant CaM/peptide pair, designated as Design 1 (D1), was cloned between CFP and citrine to yield a reengineered cameleon. Under saturating conditions of Ca2+, the cameleon showed a decrease in CFP emission and an increase in citrine emission, indicative of increased FRET. The magnitude of the FRET change was comparable to that observed for the original cameleons. Fig. 1c shows the Ca2+ titration curves of the design along with previous cameleons (YC2, YC3, and YC4). D1 has a biphasic Ca2+ response, and a two-site saturation fit yielded Kd values of 0.81 and 60 μM. The two dissociation constants likely result from the different affinities of the N- and C-terminal domains of CaM for Ca2+, as observed for YC2.1 and YC4.3 (7). Fig. 1c shows that the cameleon fills a gap in Ca2+ sensitivity, falling directly between YC3.3 and YC4.3. Because the ER is expected to range from hundreds of micromolar (resting state) to low micromolar (depleted state), this sensor is ideally suited for imaging Ca2+ in the ER.
An important part of our design strategy was to develop a sensor that would not be perturbed by endogenous cellular proteins, such as WT CaM, because this has posed a problem for previous cameleons (21). Fig. 1d shows the FRET response of YC3.3 and the D1 in the presence of excess CaM. For YC3.3, the FRET response under saturating Ca2+ conditions decreases with the addition of increasing concentrations of CaM, because excess CaM in solution binds to the skMLCK portion of the cameleon, forming an intermolecular complex and preventing the sensor from registering a FRET increase. Importantly, in the charge reversal redesign (D1), addition of increasing CaM has almost no effect on the FRET response, indicating that this cameleon is not perturbed by large excesses of CaM.
Stopped-flow fluorescence measurements were undertaken to determine the binding kinetics of the cameleon. Rapid mixing of the cameleon protein with various concentrations of Ca2+ resulted in a rapid increase in FRET. The initial responses were fitted with a single exponential to obtain the observed rate (kobs). A fit to the data (Fig. 9, which is published as supporting information on the PNAS web site) yields kon of 3.6 × 106 M-1·s—1, koff of 256 s—1, and Kd of 69 μM for D1, where the observed Kd corresponds well to the results from the Ca2+ titration curve. Therefore, although this cameleon has a kon similar to previous cameleons, it has a much faster koff (250 s—1 instead of 10 s—1), making it more appropriate to monitor rapidly changing Ca2+ dynamics.
Addition of the calreticulin signal sequence and a KDEL ER-retention tag led to effective and specific localization of the cameleon to the ER in mammalian cells (Fig. 2a). Fig. 2 b and c show the dramatic improvement in [Ca2+]ER sensitivity of D1ER over YC4.3ER, which was previously the best indicator for directly monitoring [Ca2+]ER at the single-cell level. Treatment of HeLa cells with pharmacological blockers such as thapsigargin, the irreversible sarcoendoplasmic reticulum Ca2+ ATPase (SERCA) inhibitor (Fig. 2b), or with agonists such as ATP, which results in IP3 production and activation of IP3R (Fig. 2c), causes a depletion in [Ca2+]ER that is robust and readily visible by using D1ER. Additionally, upon ATP treatment, oscillations in [Ca2+]ER were directly observed with D1ER, whereas YC4.3ER was not sensitive enough to detect oscillations. The D1ER cameleon is not saturated under resting conditions, because treatment with 25 μM digitonin and 5 mM Ca2+ led to a ratio increase (data not shown); thus, D1ER is poised to detect both increases and decreases in [Ca2+]ER.
Fig. 2.

Comparison of the response of ER-targeted YC4.3 and D1 in HeLa cells. (a) Fluorescence image of D1ER in HeLa cells showing effective ER localization. (b) Emission ratio of D1 (red) and YC4.3 (blue) in the ER upon treatment with thapsigargin. (c) Emission ratio of D1 (red lines, representing two different cells) and YC4.3 (blue) in the ER upon treatment with ATP. (d) Simultaneous imaging of Ca2+ in the cytosol (using fura-2, green) and ER (D1, red). The left axis represents the excitation ratio of fura-2 (350/380 nm), and the right axis represents the emission ratio of the cameleon (citrine/CFP). (e) Same experimental conditions as in d but for an extended amount of time. In the continued presence of 10 μM ATP, the ER begins to refill with Ca2+ after ∼1,000 s.
Because CFP and citrine are spectrally distinct from the excitation and emission of fura-2, D1ER can be used in conjunction with this high-affinity Ca2+ indicator in the cytosol to simultaneously monitor both [Ca2+]ER and [Ca2+]cyt (Fig. 2 d and e). As can be seen from Fig. 2d, in HeLa cells upon stimulation with 10 μM ATP, there is a rise in [Ca2+]cyt followed by rapid oscillations. These oscillations are also observed in the ER, and, as evidenced by the data in Fig. 2d Inset, the [Ca2+]ER oscillations are directly anticorrelated with those in the cytosol. Dampening out of the oscillations coincides with a large decrease in [Ca2+]ER. At longer time points (Fig. 2e), once the [Ca2+]cyt has returned to essentially basal levels, lower-frequency sustained oscillations are observed in the cytosol, as described (24). We can now observe that these oscillations occur only when the ER starts to refill, presumably due to capacitative Ca2+ entry.
Given the dramatic improvement in our ability to directly measure [Ca2+]ER at the single-cell level with D1ER and the potential to monitor Ca2+ simultaneously in the ER and the cytosol, we turned our focus to the effect of the antiapoptotic Bcl-2 protein on [Ca2+]ER in MCF-7 breast cancer epithelial cell lines stably expressing either a control plasmid (neo) or Bcl-2 (25). As reported (25), overexpression of Bcl-2 in MCF-7 cells protected against apoptosis induced by thapsigargin, staurosporine, and H2O2. Fig. 3a shows that in MCF-7 Bcl-2 cells, [Ca2+]ER is decreased by ∼40% [from an average of 103 ± 12 μM in neo cells (n = 45 cells) to 66 ± 7 μM in Bcl-2 cells (n = 79 cells)]. Overexpression of SERCA2b in the Bcl-2 cells restores [Ca2+]ER to control levels. Blocking SERCA pumps with thapsigargin causes depletion of [Ca2+]ER due to leak of Ca2+ across the ER membrane (Fig. 3a). Taking the derivative of the change in [Ca2+]ER upon thapsigargin treatment enables a comparison of the leakage rate for neo and Bcl-2 cells, and Bcl-2 cells overexpressing SERCA2b. As is evident from Fig. 3b, the leakage rate for Bcl-2 cells is greater than that for neo cells, indicating that under resting conditions, there is less [Ca2+]ER in Bcl-2 cells, because Bcl-2 increases the leak of Ca2+ out of the ER, as has been found in some other cell types (10, 11, 26). In cells overexpressing both Bcl-2 and SERCA2b, the leakage rate is still much greater than in control cells, indicating that the increase in resting [Ca2+]ER levels is due to increased pumping of Ca2+ into the ER rather than a decreased leak.
Fig. 3.

The effect of Bcl-2 overexpression on the [Ca2+]ER of MCF-7 cells. (a) Comparison of [Ca2+]ER in neo (red), Bcl-2 (blue), and Bcl-2 also overexpressing SERCA2b (green) cells. Each trace represents the average of more than five cells. Emission ratios were converted into [Ca2+], as described in Materials and Methods. Thapsigargin was added at time t = 0 in the absence of external Ca2+ to prevent Ca2+ influx due to capacitative Ca2+ entry. (b) Leakage rate of Ca2+ from the ER as a function of [Ca2+]ER for Bcl-2 (blue), neo (red), and Bcl-2 overexpressing SERCA2b (green) cells.
The molecular mechanism by which Bcl-2 alters Ca2+ homeostasis is still unclear. Bcl-2 may facilitate Ca2+ leakage from the ER by forming a channel, either by homooligomerizing or by interacting with another member of the Bcl-2 family, because it has been reported that in planar lipid bilayers, Bcl-2 can form a cation selective channel at physiological pH (27). Alternatively, Bcl-2 could interact with and alter the function of an endogenous release channel or pore-forming protein in the ER. Indeed, in a recent study, Bcl-2 was found to interact with the IP3R in WEHI7.2 T cells by coimmunoprecipitation, and this interaction appeared to influence the amount of Ca2+ released from the ER and the open probability of the IP3R in lipid bilayers (16). If Bcl-2 interacts with the IP3R in MCF-7 cells, it would potentially alter Ca2+ oscillations triggered by ATP. Any differences between Bcl-2 and neo cells could be detected by simultaneously monitoring the ER and the cytosol.
Treatment of MCF-7 cells with ATP results in a large release of Ca2+ from the ER into the cytosol, followed by Ca2+ oscillations in both the ER and the cytosol upon refilling of the ER (Fig. 4). Bcl-2 cells required a larger dose of ATP than neo cells for oscillations to be induced (100 vs. 10 μM), suggesting that they are less sensitive to ATP. Fig. 4 a and b show responses of representative single Bcl-2 and neo cells. It is evident that less Ca2+ is released into the cytosol in Bcl-2 cells, both in the initial dump and in subsequent oscillations. The average initial decrease in [Ca2+]ER is 51 ± 4 μM for Bcl-2 cells and 85 ± 9 μM for neo cells, resulting in average cytosolic increases of 890 ± 370 nM for Bcl-2 and 1580 ± 650 nM for neo (n = 12 for Bcl-2 cells and n = 11 for neo cells). Importantly, in both Bcl-2 and neo cells, [Ca2+]ER decreases to ∼ 5 μM after the initial release; therefore, the smaller Ca2+ release in Bcl-2 cells results from the lower Ca2+ level in the ER under resting conditions.
Fig. 4.

Comparison between ATP-induced Ca2+ oscillations in the cytosol and the ER for Bcl-2 and neo MCF-7 cells. The differences between Bcl-2 (a and c) and neo (b and d) cells are highlighted in c and d, which are on the same timescale and focus on the oscillations induced by ATP. The [Ca2+]cyt signal is in green, and the [Ca2+]ER signal is in blue for Bcl-2 and red for neo cells. The fura-2 excitation ratio (350/380 nm) and cameleon emission ratio (citrine/CFP) were converted to [Ca2+], as outlined in Materials and Methods.
Aside from the intensity of the Ca2+ oscillations, the most significant difference between the Bcl-2 and neo cells is that the oscillations are typically more rapid and the duration of ER Ca2+ release is shorter in Bcl-2 overexpressing cells (Fig. 4 c and d). As a consequence, Bcl-2 cells tend to oscillate at a higher frequency. Oscillation data from more individual cells are presented in Fig. 10, which is published as supporting information on the PNAS web site, demonstrating that, although there is some variability in the oscillations, the trends in oscillation frequency and duration are maintained.
Identifying novel inhibitors of Bcl-2 should provide insight into the role of Bcl-2 in endogenous signaling cascades leading to apoptosis, as well as lead to the potential development of therapeutic agents. Previously, Leone et al. (28) found that (—)EGCG, the most abundant component of green tea, could bind to the hydrophobic pocket of Bcl-2 in vitro and inhibit its interaction (Ki = 335 nM) with a peptide derived from the proapoptotic BH3-only protein Bad, whereas a compound lacking the gallate moiety, (—)EC, had no effect (see Fig. 11, which is published as supporting information on the PNAS web site, for structures). We treated both neo and Bcl-2 cells with EGCG and found that the compound could induce apoptosis in a dose-dependent manner, as determined by FACS analysis of propidium iodide- and annexin V-stained cells (Fig. 5a; details of assay given in Supporting Text). Importantly, EGCG was much more effective than other proapoptotic stimuli such as thapsigargin and H2O2 in inducing apoptosis in Bcl-2 cells, indicating that it was capable of overcoming protection by Bcl-2. Given that green tea polyphenols have been shown to inhibit tumor formation and growth in certain models (29), this raises the possibility that this antitumor activity could be partially related to overcoming Bcl-2 protection of cancer cells.
Fig. 5.

The effect of the green tea compound EGCG and a control compound EC on apoptosis and [Ca2+]ER of MCF-7 cells. (a) Cell viability (percent of the control), as determined by FACS analysis of propidium iodide and annexin V-stained neo (red) and Bcl-2 (blue) cells after 48-h treatment with nothing (control), EC, and increasing concentrations of EGCG. (b) Treatment of MCF-7 cells overexpressing Bcl-2 with EGCG and EC, as denoted by the green and blue boxes. The traces represent two different cells. (c) Treatment of MCF-7 neo cells with EGCG, as denoted by the green box. The traces represent two different cells. It should be noted that EC did not cause an increase in [Ca2+]ER in neo cells. (d) Bcl-2 cells treated with EGCG (green traces, each representing an individual cell) compared with neo (red) and Bcl-2 (blue) cells. At time t = 0, cells were treated with 2 μM thapsigargin in the absence of external Ca2+ to determine the ER Ca2+ leakage rate. (e) Comparison of the leakage rates of Ca2+ from the ER for Bcl-2 (blue circles), neo (red diamonds), and Bcl-2 cells pretreated with EGCG (green, upside-down triangles and squares), showing the decreased leakage rate upon EGCG treatment.
To gain more insight into how EGCG overcomes Bcl-2 protection, we examined the effect of EGCG on [Ca2+]ER. As can be shown from Fig. 5b, treatment of Bcl-2 cells with 150 μM EGCG results in an immediate increase in [Ca2+]ER, from an average of ∼60 to ∼125 μM. This effect can be reversed upon washing out the inhibitor. On the contrary, the control compound, EC, has no effect on [Ca2+]ER. Fig. 5c shows that in neo cells there is either no increase or a very modest increase in [Ca2+]ER, consistent with inhibition of the small amount of endogenous Bcl-2 in these cells. To determine the cause of the Ca2+ increase, we pretreated Bcl-2 cells with EGCG and then added thapsigargin (Fig. 5d) to monitor the leak rate. As can be seen from Fig. 5e, EGCG causes a dramatic decrease in the Ca2+ leak rate of Bcl-2 cells, such that the leak is the same or slower than that in neo cells. This suggests that the binding of EGCG to Bcl-2 prevents Bcl-2 from increasing the Ca2+ leak rate.
Discussion
The improved ER-targeted cameleon poses a number of significant advantages over all previous methods for monitoring [Ca2+]ER. Compared with the ER-targeted aequorin probes, the cameleon signal is ratiometric, permits single-cell resolution, and is compatible with fura-2 in the same cell. Importantly, these characteristics enable the monitoring of Ca2+ oscillations within the ER. Additionally, the cameleon is not consumed by Ca2+ as aequorin is, and therefore the instantaneous signal depends only on Ca2+ and not the entire past history of Ca2+ in the cell (1). Because the cameleons and aequorins have different Ca2+ response curves, they should yield different mean [Ca2+] whenever there is spatial heterogeneity in [Ca2+]. The cameleons would tend to give slightly lower mean [Ca2+] values than aequorins.
The improved ER-targeted cameleon enabled us to show that Bcl-2 plays an important role in governing [Ca2+]ER in breast cancer cells by altering the resting level of [Ca2+]ER as well as the amplitude, duration, and frequency of IP3-mediated Ca2+ oscillations. Our study does not directly address whether Bcl-2 interacts with the IP3R in MCF-7 cells, because Ca2+ oscillations are controlled by a host of factors, including the level of Ca2+ in the ER and capacitative calcium entry, both of which are altered in Bcl-2-overexpressing cells (9, 11, 26). However, our data show that, in addition to altering the resting level and leak rate of [Ca2+]ER, Bcl-2 overexpression can also modify fundamental Ca2+ signaling processes such as the nature of IP3-induced Ca2+ oscillations. This raises the intriguing possibility that Bcl-2 may have an effect on signaling pathways that are downstream of Ca2+ oscillations and therefore has the potential to influence processes such as exocytosis, mitochondrial redox state, and differential gene transcription (30–32). This disruption of Ca2+ homeostasis by Bcl-2 may be essential in determining the susceptibility of breast cancer cells to different apoptotic stimuli, because Scorrano et al. (12) have found that certain apoptotic stimuli (lipid second messengers and oxidative stress, staurosporine, etoposide, and brefeldin-A) depend either critically or partially on ER Ca2+.
We have also identified a small-molecule inhibitor (EGCG) of Bcl-2 that can overcome Bcl-2 protection and induce apoptosis in breast cancer cells. Interestingly, this inhibitor also reverses the effect of Bcl-2 on [Ca2+]ER. Because EGCG is known to bind in the hydrophobic groove and prevent the interaction of Bcl-2 with BH3-only peptides, this suggests that binding in this hydrophobic pocket inhibits the channel function of Bcl-2, either by preventing its interaction with other proteins or by causing a conformational change in Bcl-2 itself. Recently, Bassik et al. (33) found that phosphorylation within the unstructured loop region between the BH3 and BH4 domains of Bcl-2 inhibited the interaction between Bcl-2 and proapoptotic family members and caused an increase in Ca2+ leak rate (33). They also showed that the ability to bind BH3-only proteins, but not Bax or Bak, was critical for Bcl-2 to lower [Ca2+]ER. The combination of that study with the present one strongly suggests that Bcl-2 must interact with other proteins (either itself or other family members) through the hydrophobic groove to facilitate leakage of Ca2+ from the ER. It also implies that the interaction of other family members with Bcl-2 localized to the ER can have a dramatic effect on [Ca2+]ER and potentially apoptosis. We are currently examining whether a direct connection exists between blockage of the channel function, increase in [Ca2+]ER, and the increased apoptosis observed in the Bcl-2-overexpressing cells.
Supplementary Material
Acknowledgments
We thank L. Xu, S. R. Adams, L. Gross, P. Steinbach, and Q. Xiong for assistance and advice and Professor J. Adams for the use of his Applied Photophysics stopped-flow instrument. This work was supported by Ruth L. Kirschstein National Research Service Award Postdoctoral Fellowship F32-GM067488-01 (to A.E.P.), Department of Defense Postdoctoral Fellowship DAMD17-99-1-9096 (to C.J.), Department of Energy Grant DE-FG-01-ER63276 (to R.Y.T.), and National Institutes of Health Grants NS27177 (to R.Y.T.) and GM60554 (to J.C.R.).
Author contributions: A.E.P., C.J., J.C.R., and R.Y.T. designed research; A.E.P. and C.J. performed research; A.E.P., C.J., and J.C.R. contributed new reagents/analytic tools; A.E.P., C.J., J.C.R., and R.Y.T. analyzed data; and A.E.P. and R.Y.T. wrote the paper.
Abbreviations: ER, endoplasmic reticulum; SERCA, sarcoendoplasmic reticulum Ca2+ ATP-ase; CaM, calmodulin; skMLCK, skeletal muscle myosin light chain kinase; Bcl-2, B cell lymphoma 2; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; EGCG, epigallocatechin gallate; EC, epicatechin; CFP, cyan fluorescent protein; HBSS, Hanks' balanced salt solution; D1, Design 1.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AY796115).
References
- 1.Alvarez, J. & Montero, M. (2002) Cell Calcium 32, 251–260. [DOI] [PubMed] [Google Scholar]
- 2.Solovyova, N. & Verkhratsky, A. (2002) J. Neurosci. Methods 122, 1–12. [DOI] [PubMed] [Google Scholar]
- 3.Sneyd, J., Tsaneva-Atanasova, K., Yule, D. I., Thompson, J. L. & Shuttleworth, T. J. (2004) Proc. Natl. Acad. Sci. USA 101, 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnaudeau, S., Kelley, W. L., Walsh, J. V., Jr., & Demaurex, N. (2001) J. Biol. Chem. 276, 29430–29439. [DOI] [PubMed] [Google Scholar]
- 5.Filippin, L., Magalhaes, P. J., Benedetto, G. D., Colella, M. & Pozzan, T. (2003) J. Biol. Chem. 278, 39224–39234. [DOI] [PubMed] [Google Scholar]
- 6.Brini, M., Pinton, P., Pozzan, T. & Rizzuto, R. (1999) Microsc. Res. Tech. 46, 380–389. [DOI] [PubMed] [Google Scholar]
- 7.Miyawaki, A., Llopis, J., Heim, R., McCaffery, J. M., Adams, J. A., Ikura, M. & Tsien, R. Y. (1997) Nature 388, 882–887. [DOI] [PubMed] [Google Scholar]
- 8.Cory, S. & Adams, J. M. (2002) Nat. Rev. Cancer 2, 647–656. [DOI] [PubMed] [Google Scholar]
- 9.Lam, M., Dubyak, G., Chen, L., Nunez, G., Miesfeld, R. & Distelhorst, C. W. (1994) Proc. Natl. Acad. Sci. USA 91, 6569–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foyouzi-Youssefi, R., Arnaudeau, S., Borner, C., Kelly, W. L., Tschopp, J., Lew, D. P., Demaurex, N. & Krause, K.-H. (2000) Proc. Natl. Acad. Sci. USA 97, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinton, P., Ferrari, D., Magalhaes, P., Schulze-Osthoff, K., Virgilio, F. D., Pozzan, T. & Rizzuto, R. (2000) J. Cell Biol. 148, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scorrano, L., Oakes, S. A., Opferman, J. T., Chang, E. H., Sorcinelli, M. D., Pozzan, T. & Korsmeyer, S. J. (2003) Science 300, 135–139. [DOI] [PubMed] [Google Scholar]
- 13.Baffy, G., Miyashita, T., Williamson, J. R. & Reed, J. C. (1993) J. Biol. Chem. 268, 6511–6519. [PubMed] [Google Scholar]
- 14.Pinton, P., Ferrari, D., Rapizzi, E., Virgilio, F. D., Pozzan, T. & Rizzuto, R. (2001) EMBO J. 20, 2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Distelhorst, C. W. & Shore, G. C. (2004) Oncogene 23, 2875–2880. [DOI] [PubMed] [Google Scholar]
- 16.Chen, R., Valencia, I., Zhong, F., McColl, K. S., Roderick, H. L., Bootman, M. D., Berridge, M. J., Conway, S. J., Holmes, A. B., Mignery, G. A., et al. (2004) J. Cell Biol. 166, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A. & Tsien, R. Y. (2001) J. Biol. Chem. 276, 29188–29194. [DOI] [PubMed] [Google Scholar]
- 18.Tsien, R. Y. & Pozzan, T. (1989) Methods Enzymol. 172, 230–263. [DOI] [PubMed] [Google Scholar]
- 19.Tsien, R. Y. & Rink, T. J. (1983) in Current Methods in Cellular Neurobiology, ed. Barber, J. L. (Wiley, New York), Vol. 3, pp. 249–312. [Google Scholar]
- 20.Poenie, M. (1990) Cell Calcium 11, 85–91. [DOI] [PubMed] [Google Scholar]
- 21.Hasan, M. T., Friedrich, R. W., Larkum, M. E., Euler, T., Both, M., Duebel, J., Waters, J., Giese, G., Bujard, H., Griesbeck, O., et al. (2004) Pub. Lib. Sci. 2, 763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heim, N. & Griesbeck, O. (2004) J. Biol. Chem. 279, 14280–14286. [DOI] [PubMed] [Google Scholar]
- 23.Ikura, M., Clore, G. M., Gronenborn, A. M., Zhu, G., Klee, C. B. & Bax, A. (1992) Science 256, 632–638. [DOI] [PubMed] [Google Scholar]
- 24.Bootman, M. D., Cheek, T. R., Moreton, R. M., Bennett, D. L. & Berridge, M. J. (1994) J. Biol. Chem. 269, 24783–24791. [PubMed] [Google Scholar]
- 25.Froesch, B. A., Aime-Sempe, C., Leber, B., Andrews, D. & Reed, J. C. (1999) J. Biol. Chem. 274, 6469–6475. [DOI] [PubMed] [Google Scholar]
- 26.Abeele, F. V., Skryma, R., Shuba, Y., Coppenolle, F. V., Slomianny, C., Roudbaraki, M., Mauroy, B., Wuytack, F. & Prevarskaya, N. (2002) Cancer Cell 1, 169–179. [DOI] [PubMed] [Google Scholar]
- 27.Schendel, S. L., Xie, Z., Montall, M. O., Matsuyama, S., Montal, M. & Reed, J. C. (1997) Proc. Natl. Acad. Sci. USA 94, 5113–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leone, M., Zhai, D., Sareth, S., Kitada, S., Reed, J. C. & Pellecchia, M. (2003) Cancer Res. 63, 8118–8121. [PubMed] [Google Scholar]
- 29.Yang, C. S. & Wang, Z. Y. (1993) J. Natl. Cancer Inst. 85, 1038–1049. [DOI] [PubMed] [Google Scholar]
- 30.Berridge, M. J., Bootman, M. D. & Roderick, H. L. (2003) Nat. Mol. Cell Biol. 4, 517–529. [DOI] [PubMed] [Google Scholar]
- 31.Berridge, M. J., Lipp, P. & Bootman, M. D. (2000) Nat. Mol. Cell Biol. 1, 11–21. [DOI] [PubMed] [Google Scholar]
- 32.Blaustein, M. P. & Golovina, V. A. (2001) Trends Neurosci. 24, 602–608. [DOI] [PubMed] [Google Scholar]
- 33.Bassik, M. C., Scorrano, L., Oakes, S. A., Pozzan, T. & Korsmeyer, S. J. (2004) EMBO J. 23, 1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.