

Abstract
Brain ischemia is a major cause of adult disability and death, and it represents a worldwide health problem with significant economic burden for modern society. The identification of the molecular pathways activated after brain ischemia, together with efficient technologies of gene delivery to the CNS, may lead to novel treatments based on gene therapy. Recombinant adeno-associated virus (rAAV) is an effective platform for gene transfer to the CNS. Here, we used a serotype 8 rAAV bearing the Y733F mutation (rAAV8-733) to overexpress co-chaperone E3 ligase CHIP (also known as Stub-1) in rat hippocampal neurons, both in an oxygen and glucose deprivation model in vitro and in a four-vessel occlusion model of ischemia in vivo. We show that CHIP overexpression prevented neuronal degeneration in both cases and led to a decrease of both eIF2α (serine 51) and AKT (serine 473) phosphorylation, as well as reduced amounts of ubiquitinated proteins following hypoxia or ischemia. These data add to current knowledge of ischemia-related signaling in the brain and suggest that gene therapy based on the role of CHIP in proteostasis may provide a new venue for brain ischemia treatment.
Keywords: brain ischemia, hypoxia, neuronal death, CHIP, ubiquitin-proteasome system, eIF2α, &Agr, &Kgr, &Tgr, rAAV
Graphical Abstract
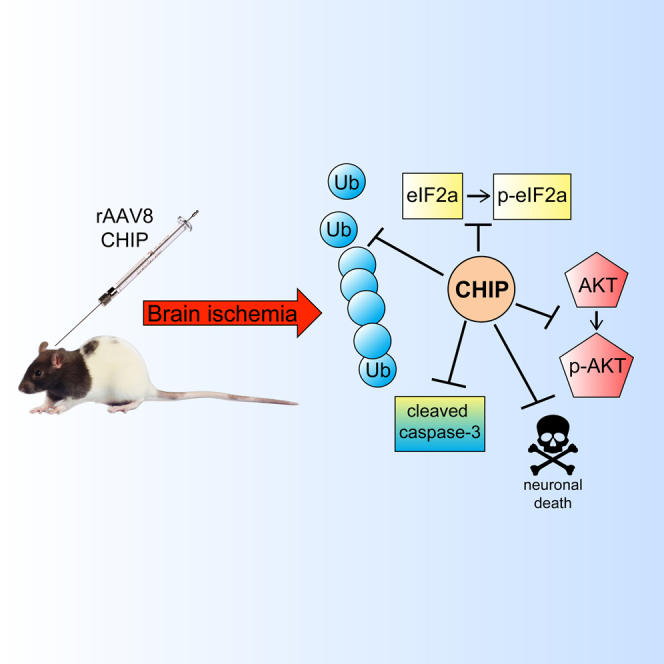
This work explored the potential use of CHIP as a gene therapy candidate. It provided the proof of principle that overexpression of CHIP promotes neuroprotection against ischemic injury in vitro and in vivo. It also explored some potential venues to explain how CHIP is working.
Introduction
Transient cerebral ischemia induces a cascade of cellular events leading to neuronal death, especially in vulnerable areas such as the hippocampus.1 Cerebral hypoxic and ischemic injuries are involved in many CNS diseases that can be acute, as promoted by occlusion of large arteries2 and intrapartum,3 or chronic, as promoted by sleep apnea4 and rare vascular conditions like Moyamoya disease.5 In addition, silent chronic brain infarction, which exhibits no apparent symptoms, is directly associated to increased morbidity and mortality in aged populations.6 Therefore, hypoxic and ischemic brain injuries are a major cause of acute mortality and chronic neurological morbidity, and understanding molecular pathways leading to cell death activation will contribute to the development of new therapeutic venues for treatment and risk factor identification.7 The phosphatidylinositol 3-kinase (PI3K)-protein kinase B (AKT) pathway has been implicated in protection from late onset apoptosis initiated by global brain ischemia (GBI) in the hippocampus.8, 9 Phosphorylation of AKT at the C-terminal mTORC2 phosphorylation site serine 473 (Ser473) starts 30 min after ischemia and endures for several hours following reperfusion.8 However, activation of AKT by oxidative stress may facilitate cell death depending on context,9 indicating that dynamic phosphorylation of AKT may affect several mechanisms related to cell fate. Proteostasis defects after brain ischemia are well documented, including disruption of the ubiquitin-proteasome system (UPS),10 endoplasmic reticulum (ER) stress,11, 12 and inhibition of global protein synthesis mediated by the phosphorylation of eukaryotic initiation factor-2α (eIF2α) at serine 51 (Ser51).13 These findings suggest that modulation of key components related to integrity of the proteome may promote neuroprotection after an ischemic insult. The C terminus Hsc70-interacting protein (CHIP), also known as STIP-1 homology and U-Box containing protein 1 (Stub-1), is a major component of the proteostasis machinery and has been implicated in the regulation of neurodegeneration.14 The function of CHIP in connecting the chaperone system with the ubiquitin-proteasome system (UPS) relies on three tandem tetratricopeptide repeat (TPR) motifs; through these, it interacts with chaperones Hsp70 and Hsp90, as well as on a U-BOX domain that induces E3 ligase activity, thus promoting the poli-ubiquitylation and proteasome-dependent degradation of chaperone-bound substrates.15 We have previously shown that overexpression of CHIP mediated by a recombinant adeno-associated viral vector prevented neurodegeneration in a model of acute endoplasmic reticulum stress induced in hippocampal slices by tunicamycin.16 Here we investigated a possible role for CHIP upon neuronal degeneration induced in both in vitro and in vivo models of global brain ischemia. To overexpress CHIP, we used recombinant adeno-associated virus, serotype 8, bearing the Y733F mutation (rAAV8-733), a vector that promotes highly efficient transduction of the targeted structure. Overall, we found that overexpression of CHIP prevented neuronal death, blocked phosphorylation of both eIF2α at Ser51 and AKT at Ser473, and prevented a decrease in the proportion of ubiquitinated proteins in the hippocampus following reperfusion.
Results
Overexpression of CHIP Prevents Neurodegeneration Induced by Either Oxygen and Glucose Deprivation In Vitro or Global Cerebral Ischemia In Vivo
To test for a neuroprotective effect of CHIP, we first used a well-established in vitro model of ischemic damage to the brain.17 Successful overexpression of transgenes by rAAV8-733 in hippocampal slices was shown in our previous publication, and neither GFP or CHIP overexpression altered propidium iodide (PI) uptake by itself.16 Hippocampal slices were subject to oxygen and glucose deprivation followed by 24 hr of reperfusion (OGD). Figure 1 shows representative photomicrographs of slices stained with PI in normoxia (Figure 1A), slices transduced by rAAV8-733-GFP and exposed to OGD (Figure 1B), and slices transduced by rAAV8-733-CHIP subject to OGD (Figure 1C). The results showed increased uptake of PI in the CA1 hippocampal region following OGD compared with controls (p = 0.0017). Transduction of GFP led to further increase in PI uptake (p < 0.0001), whereas transduction of CHIP prevented cell death (p = 0.7006) after OGD. Such increased PI uptake in rAAV-GFP-infected slices when compared to non-infected slices submitted to OGD might be due to GFP-mediated toxicity and increasing susceptibility to cell death.18 We then examined whether the neuroprotective effect of CHIP extended to an in vivo model of acute cerebral ischemia. Four-vessel occlusion (4VO) was done in adult rats transduced via intrahippocampal injections with either rAAV8-733-CHIP or rAAV8-733-GFP. The extent of hippocampal transduction was mapped in pilot experiments by examining immunolabeling for GFP in brain slices taken from animals transduced with rAAV8-733-GFP 3 weeks after stereotaxic surgery (Figure S1A). Increased CHIP content was confirmed in similar pilot experiments by comparing western blots from whole hippocampus samples taken from rats infected with rAAV8-733-CHIP compared with control tissue 3 weeks after transduction (Figure S1B). Vector-treated animals, together with untreated controls, were then subject to GBI, followed by a 7-day survival time,19 and neuronal degeneration was estimated in brain sections stained with FluoroJade C (FJ-C). Figure 2 illustrates the results of these experiments, in which FJ-C labeling was increased in the CA1 region following GBI in both untreated rats (p < 0.0001) (Figure 2B) and those injected with rAAV8-733-GFP (p < 0.0001) (Figure 2C) compared with controls (Figure 2A). There is no significant difference between uninfected mice (Figure 2B) and rAAV8-733-GFP-infected mice (Figure 2C) submitted to GBI. Treatment with rAAV8-733-CHIP significantly reduced cell degeneration following GBI (p = 0.0002) (Figures 2D and 2E). Consistent with these results, treatment with rAAV8-733-CHIP (n = 3) (Figure 2H), but not rAAV8-733-GFP (n = 3) (Figure 2G), nearly abolished immunolabeling of CA1 cells with an antibody to cleaved caspase-3 compared with sham-operated rats (n = 2) (Figure 2F). The findings showed that overexpression of CHIP prevented neurodegeneration in both in vitro and in vivo models of global brain ischemia.
Figure 1.

CHIP Overexpression Prevents Cellular Death Induced by Oxygen and Glucose Deprivation, Followed by 24 hr of Reperfusion in Hippocampal Slices In Vitro
(A–C) Representative photomicrographs (5× magnification) of propidium iodide uptake of organotypic hippocampal slices in normoxia (A), slices infected with rAAV8-733-GFP and submitted to oxygen and glucose deprivation followed by 24 hr of reperfusion (GFP+OGD) (B), or rAAV8-733-CHIP-infected slices submitted to OGD (C). (D) Quantification of the percentage of PI uptake in the CA1 region for each experimental condition compared to normoxia. n = 3 independent experiments from a pool of hippocampal slices taken from ten rats. 95% confidence interval (CI); **p < 0.01; ***p < 0.0001; NS, non-significant (p > 0.05). Error bars indicate ± SEM. Scale bar indicates 500 μm. N, normoxia; OGD, oxygen and glucose deprivation for 90 min followed by 24 hr of reperfusion; GFP+OGD, rAAV8-733-GFP infected + OGD; CHIP+OGD, rAAV8-733-CHIP infected + OGD.
Figure 2.

CHIP Overexpression Diminishes Neuronal Degeneration in the CA1 Region after Global Brain Ischemia, Followed by 7 Days of Reperfusion in the Hippocampus
(A–D) Representative photomicrographs of FluoroJade C-positive cells in the CA1 region of sham-operated rats (A), rats submitted to global brain ischemia followed by 7 days of reperfusion (GBI) (B), rAAV8-733-GFP-infected rats submitted to GBI (C), or rAAV8-733-CHIP-infected rats submitted to GBI (D). (E) Quantification of FluoroJade C-positive cells in the CA1 region per millimeter in different experimental conditions. Tukey’s post-test was performed for this analysis. 95% confidence interval (CI); **p < 0.001; NS, non-significant (p > 0.5). Error bars indicate ± SEM. (F–H) Representative photomicrographs of immunofluorescence for cleaved caspase-3-positive cells in the CA1 region of sham-operated rats (F), rAAV8-733-GFP-infected rats submitted to GBI (G), and rAAV8-733-CHIP rats submitted to GBI (H). n = 3–5 animals per group; 40× magnification. Scale bar indicates 50 μm.
Effects of the Overexpression of CHIP upon Phosphorylation of eIF2α in Serine 51 and AKT in Serine 473 Induced by Hypoxia or Ischemia
Global protein synthesis blockade mediated by phosphorylation of eIF2α at serine 51 is an important intracellular response to hypoxia and other forms of cellular stress.13, 19, 20 Previous results from our group confirmed that overexpression of CHIP blocks increased eIF2α phosphorylation induced by ER stress.16 We tested whether the overexpression of CHIP modulates OGD-induced phosphorylation of eIF2α (Figure 3). OGD in vitro also induced phosphorylation of eIF2α (p = 0.0319), and transduction of GFP led to further increase in the phosphorylation of Ser51 (p = 0.0091). In contrast, hippocampal slices transduced with CHIP and exposed to OGD showed levels of phosphorylated eIF2α (p-eIF2α) similar to controls (p = 0.9374). In keeping with the results of the OGD experiments, GBI in vivo also led to increased eIF2α phosphorylation at Ser51 in both untreated rats (p = 0.0062) and those treated with rAAV8-733-GFP (p = 0.0418), but not in animals treated with rAAV8-733-CHIP (p = 0.9979) (Figure 4B). Following GBI in vivo, the level of phosphorylation of AKT Ser473 was increased in whole hippocampus homogenates (p = 0.0075) (Figure 4A). Similar increased levels were detected in animals treated with rAAV8-733-GFP (p = 0.0234), whereas treatment with rAAV8-733-CHIP prevented the increase in phosphorylated AKT (p-AKT) Ser473 after 3 hr of reperfusion (p = 0.9996) (Figure 4A). These results indicate that overexpression of CHIP blocks downstream events associated with neurodegenerative signaling in the hippocampus.
Figure 3.

CHIP Overexpression Prevents eIF2α Phosphorylation Induced by Oxygen and Glucose Deprivation after 24 hr of Reperfusion In Vitro
(A) Western blots for p-eIF2α (Ser51) and total eIF2α from homogenates of organotypic hippocampal slices under different experimental conditions. (B) Quantification of normalized optical density for p-eIF2α and total eIF2α bands. α-tubulin was used as loading control. n = 3 independent experiments from a pool of hippocampal slices taken from nine rats. 95% confidence interval (CI); *p < 0.05; **p < 0.01; NS, non-significant. Error bars indicate ± SEM. N, normoxia; OGD, oxygen and glucose deprivation for 90 min followed by 24 hr of reperfusion; GFP+OGD, rAAV8-733-GFP-infected slices submitted to OGD; CHIP+OGD, rAAV8-733-CHIP-infected slices submitted to OGD.
Figure 4.

CHIP Overexpression Prevents Both AKT Phosphorylation in Serine 473 and eIF2α Phosphorylation in Serine 51 Induced by Global Brain Ischemia, Followed by 3 hr of Reperfusion in the Hippocampus
(A) Left: western blots for p-AKT (Ser473) and total AKT of whole hippocampus homogenates under different experimental conditions. Right: quantification of normalized optical density for p-AKT and total AKT bands. (B) Left: western blots for p-eIF2α (Ser51) and total eIF2α of whole hippocampus homogenates under different experimental conditions. Right: quantification of normalized optical density for p-eIF2α and total eIF2α bands. β-actin was used as a loading control. n = 4 animals per group; 95% confidence interval (CI); *p < 0.05; **p < 0.01; NS, non-significant. Error bars indicate ± SEM. S, sham operated; GBI, global brain ischemia followed by 3 hr of reperfusion; GFP+GBI, rAAV8-733-GFP infected + GBI; CHIP+GBI, rAAV8-733-CHIP infected + GBI.
Overexpression of CHIP Attenuates Increased Protein Ubiquitination Induced by Either Oxygen and Glucose Deprivation In Vitro or Global Cerebral Ischemia In Vivo
Various research groups have shown that brain ischemia leads to the accumulation of ubiquitin-containing protein aggregates.10 Following OGD, ubiquitin content was significantly increased in the hippocampal slices (p = 0.0046), while a trend toward increased ubiquitination in tissue transduced with GFP was not statistically significant (p = 0.0722) (Figure 5). However, the level of ubiquitinated protein in CHIP-transduced slices was comparable to controls (p = 0.9885). The levels of ubiquitin-containing aggregates were also estimated in whole hippocampus homogenates after 3 hr of reperfusion following in vivo brain ischemia. Treatment with rAAV8-733-CHIP significantly reduced the levels of protein ubiquitination compared with rAAV8-733-GFP-treated rats (p = 0.0028) (Figure 6). In the absence of brain ischemia, there were no significant differences among the levels of protein ubiquitination in the hippocampus of sham-operated rats, rAAV8-733-GFP-injected rats (p = 0.2240), or rAAV8-733-CHIP-injected rats (p = 0.0529) (Figure S2).
Figure 5.

CHIP Overexpression Attenuates Increased Ubiquitinated Proteins Induced by Oxygen and Glucose Deprivation Followed by 24 hr of Reperfusion In Vitro
Western blot for ubiquitin from homogenates of organotypic hippocampal slices submitted to different experimental conditions. N, normoxia; OGD, oxygen and glucose deprivation for 90 min followed by 24 hr of reperfusion; GFP+OGD, rAAV8-733-GFP-infected slices submitted to OGD; CHIP+OGD, rAAV8-733-CHIP-infected slices submitted to OGD. 95% confidence interval (CI); **p < 0.01; NS, non-significant. Error bars indicate ± SEM. Molecular weights are shown on the right.
Figure 6.

CHIP Overexpression Attenuates the Increase in Ubiquitinated Proteins Induced by Transient Global Brain Ischemia Followed by 3 hr of Reperfusion
(A) Western blot for ubiquitin from whole homogenates of hippocampal samples taken from sham-operated animals (S), rAAV8-733-GFP-infected animals after 3 hr of reperfusion (GFP 3HR), or rAAV8-733-CHIP-infected animals after 3 hr of reperfusion (CHIP 3HR). (B) Quantification of optical density of each vertical lane normalized by the loading control (actin). n = 3 animals per group. 95% confidence interval (CI); *p < 0.01. Error bars indicate ± SEM. Molecular weights are represented on the right.
Discussion
In this study, we showed that overexpression of CHIP protected hippocampal neurons from both oxygen and glucose deprivation in vitro and brain ischemia in vivo, consistent with our previous finding that CHIP protects organotypic hippocampal cultures from neurodegeneration triggered by endoplasmic reticulum stress.16 Our results contrast those of Stankowski et al.,21 who reported that primary neuronal cultures transfected with a small interfering RNA (siRNA) against CHIP resulted in improved survival following oxidative stress. Such a discrepancy is likely due to their use of dissociated cell cultures, whereas we examined both in vitro and in vivo models of hypoxic responses in structured brain tissue. It has long been recognized that cellular responses to hypoxic insult vary among distinct brain regions, as well as between in vitro and in vivo preparations and among differing cell types.22, 23, 24 Thus, our organotypic cultures and global brain ischemia models likely represent more closely the pathophysiological changes following ischemic insult in hippocampal tissue. Furthermore, the same group has reported the role of CHIP in the maintenance of mitochondria homeostasis, hence regulating neuronal survival following OGD.25
Results of experiments in both in vivo and in vitro models of brain ischemia have disclosed an increased level of clustered ubiquitinated proteins that may lead to protein aggregates in neurons of the hippocampal CA1 region and ensuing cell death.10 It has been suggested that irreversible aggregation of translational complex components, chaperones, and protein folding enzymes following brain ischemia lead to inhibition of translation and subsequent neuron death.26 Consistent with this view, we detected augmented levels of ubiquitinated proteins after either OGD followed by 24 hr of reperfusion or GBI followed by 3 hr of reperfusion, particularly among high molecular weight protein bands in western blots. This is corrected upon rAAV8-733-CHIP treatment in the hippocampus, thus suggesting that increased E3 ligase activity promoted by CHIP overexpression is sufficient to decrease protein ubiquitination and neuronal degeneration in ischemic conditions. After global brain ischemia, phosphorylation of eIF2α has been pointed out as being responsible for the shutdown of protein synthesis in vulnerable regions of the CNS, such as the hippocampus.13 Accordingly, we detected increased phosphorylation of eIF2α after both in vitro and in vivo ischemia, which was prevented by overexpression of CHIP. Early work suggested a detrimental role for the phosphorylation of eIF2α after GBI. However, compelling results suggested the opposite in a focal cerebral ischemia model, in which salubrinal, an inhibitor of eIF2α dephosphorylation, enhanced neuronal survival.27 It is possible that these differing results are related to region-specific mechanisms that operate after local ischemic conditions.22, 23, 28 Thus, the role of eIF2α in the outcome of stressful conditions that lead to neuron death is still debatable, and a better understanding of its modulation and crosstalk with other mechanisms of proteostasis may offer novel strategies for stroke therapy.29 The PI3K-AKT pathway has been attributed a major role in the protection of neurons from caspase-mediated cell death after brain ischemia.30 Phosphorylation of AKT at serine 473 was increased after several minutes of reperfusion and was sustained for days following ischemia.8 In turn, it was shown that CHIP promotes downregulation and degradation of phosphatase and tensin homolog (PTEN), a negative modulator of AKT phosphorylation, which increases the levels of p-AKT.31 We found that the overexpression of CHIP abrogated the phosphorylation of AKT at serine 473 in the ischemic hippocampus. Although these results seem to contradict a well-established neuroprotective role for the activation of AKT in tissue exposed to oxidative stress, reports suggest that the phosphorylation of AKT at Ser473 may also increase the levels of reactive oxygen species (ROS) and induce cell death.9, 32 Of particular interest, a report suggests that phosphorylation of eIF2α at serine 51 may control the activation of AKT promoting either cell death or survival in response to oxidative stress.33 The latter studies are, therefore, compatible with our finding that the overexpression of CHIP inhibited the phosphorylation of eIF2α and of AKT. Altogether, the present data from both in vitro and in vivo models of global cerebral ischemia are consistent with the hypothesis that the overexpression of CHIP is sufficient to prevent neurodegeneration in the hippocampus. Our experiments were designed to evaluate the effect of CHIP overexpression upon neuronal degeneration induced by brain ischemia in the hippocampus, providing evidence that CHIP overexpression was sufficient to block neuronal death, rather than rescue neuronal degeneration after ischemic insult. The central assumption of this work is to highlight CHIP as a potential therapeutic target for brain ischemia. Notwithstanding the need for an evaluation of the CHIP transgene delivery effect after ischemia, mostly for acute conditions, our data confirmed that rAAV8-733 efficiently delivers the CHIP transgene to the CNS. In addition, our data suggest that an rAAV8-733-CHIP vector qualifies as a potential candidate for neuroprotective gene therapy for chronic ischemic conditions in the brain, such as brain obstructive chronic vascular diseases.34
Materials and Methods
Animals
Animals were housed in microisolators in the controlled conditions of a temperature and light cycle, with an ad libitum diet. Male Lister Hooded rats were used for both in vitro and in vivo approaches. All procedures were approved by the University Ethics Committee under protocol IBCCF 172 and 054/16.
Production of Recombinant Adeno-Associated Viral Vector
Vector preparation was produced by a two-plasmid co-transfection method as previously described.35 A crude iodixanol fraction with rAAV vectors was further purified and concentrated by chromatography on a 5 mL HiTrap Q Sepharose column using an AKTA fast protein liquid chromatography (FPLC) system (Amersham Biosciences). Vectors were eluted from the column using 215 mM NaCl (pH 8.0), and the vector-containing fractions were collected, pooled, concentrated, and buffer exchanged into Alcon BSS PLUS irrigating solution with 0.014% Tween 20, using a Biomax 100 K concentrator (Millipore). The titer of DNase-resistant vector genomes was measured by real-time PCR relative to a standard. Finally, the purity of the vector was validated by silver-stained SDS-PAGE, assayed for sterility and lack of endotoxin, and then aliquoted and stored at −80°C. Vectors contained the sequences encoding either GFP or human CHIP under the control of the ubiquitous chicken beta-actin (CBA) promoter. Recombinant adeno-associated viral vector serotype 8 with capsid Y733F mutant (rAAV8-733) was used to express CHIP or GFP in hippocampal tissue. GFP was used as a control both in vitro and in vivo.
Hippocampal Slice Cultures
Hippocampal slices were produced as described previously, with slight modifications.17 Briefly, male Lister Hooded rats at postnatal day 6–7 were decapitated, and the hippocampi were dissected under sterile conditions in ice-cold Hank’s balanced salt solution (HBSS, Gibco). The tissue was sliced transversally at 400 μm with a McIlwain Tissue Chopper (Mickle Laboratories). Slices were then transferred to 30-mm-diameter membrane inserts (Millicell, Millipore) and cultured for 14 days in 6-well culture trays with 1 mL of medium per well. Culture medium included 50% minimum essential medium (MEM), 25% HBSS, 25% heat inactivated horse serum, 1% penicillin, and 1% streptomycin, in addition to 25 mM HEPES, 36 mM D-glucose, and 4 mM Na2HCO3 (pH 7.3). Cultures were kept in a humidified incubator at 37°C and 5% CO2. Medium was changed every 3 days.
rAAV8-733 Transduction
A total of 1 μL of vector solution at 1.3E12 vetor genomes (VGs)/mL was applied in both in vitro and in vivo models. Isolated hippocampi were infected by laying the vector suspension on top of the tissue slices after 30 min of culture (day 0). For the in vivo approach, male rats at postnatal day 50–60 were deeply anesthetized using a mixture of xylazine and ketamine (1:3). A stereotaxic apparatus coupled to a microinjection pump (Harvard Systems) and a Hamilton microsyringe was used for the procedure. The cranium was exposed though a skin incision, and bilaterally symmetrical holes were opened using a dental drill (Beltec LB-100). The tip of the microsyringe needles were placed at −3.9 mm antero-posterior, ±2.0 mm lateral, and −2.5 mm dorso-ventral relative to the bregma, and injections were done at 0.1 μL/min. Withdrawal of the needle was done 5 min after the end of each injection to avoid reflux of the vector suspension. Dental cement was used to close the incision, and then the rats were returned to their home cages and kept under close monitoring until they were awake.
Oxygen and Glucose Deprivation
The procedure was conducted as previously described with slight modifications.36 After 13 days of culture, hippocampal slices previously infected with rAAV (day in vitro 0) and non-infected controls were rinsed three times with glucose-free medium (1.26 mM CaCl2, 5.36 mM KCl, 136.9 mM NaCl, 0.34 mM H2PO4, 0.49 mM MgCl2, 0.44 mM MgSO4, 25 mM HEPES, and 4 mM NaHCO3 [pH 7.3]) and left in 1 mL of this medium for 30 min. The medium was then replaced by fresh medium previously bubbled with nitrogen for 30 min. The cultures were then transferred to an anaerobic chamber (ProOx 110/ProCO2, BioSpherix) at 0.1% O2, 5% CO2, and 94.9% N2 at 37°C for 90 min, after which the slices were returned to initial conditions of culture for 24 hr before analysis.
Global Brain Ischemia
We used the four-vessel occlusion model to induce transient global brain ischemia in rats as previously described,37, 38 with slight modifications. Briefly, both rAAV-infected male rats (injected at postnatal day 60) and non-infected controls at postnatal day 90 were anesthetized with a mixture of oxygen and isoflurane (model 72-6474, Harvard Systems) and had anesthesia sustained by a mask attached to the system. An incision was made caudal to the occipital bone, directly overlying the first two cervical vertebrae. Paraspinal muscles were separated from the midline to gain access to the right and left alar foramina of the first cervical vertebrae. Using an electrocautery (Medcir Vet120), the vertebral arteries were permanently occluded. Twenty-four hours later, animals were again anesthetized and had their common carotid arteries exposed through a ventral midline neck incision. Using aneurism clips (Fine Science Tools), arteries were occluded for 5 min and anesthesia was immediately discontinued. After releasing the arteries, anesthesia was applied again as the neck skin was sutured and animals were returned to their home boxes after recovery from surgery. Tramadol (50 mg/kg) was injected intramuscularly twice a day for 3 consecutive days to diminish animal suffering. Sham-operated controls were subject to the same procedures except for the occlusion of carotids. Neurodegeneration was evaluated after 7 days, and molecular changes were studied 3 hr after surgery. Animals were excluded from the study and euthanized if they failed to show pupillary dilation during occlusion and failed to present bilateral paw extension during or after the cerebral ischemia. They were also excluded from the study if they showed abnormal vocalization, generalized convulsions, or excessive hypoactivity.
Assessment of Necrotic Cell Death In Vitro
Propidium iodide (PI) at 5 μg/mL was added to the medium of organotypic hippocampal slice cultures after the experiment. Cell death by necrosis was identified by the uptake of PI.15 Images were obtained with an inverted epi-illumination fluorescence microscope (MRMm Rev3, Zeiss) with a cold charge-coupled device (CCD) camera system (Axiocam) at 4× magnification. The same exposure time was used for all independent experiments. PI fluorescence in the CA1 region was quantitatively analyzed using ImageJ Software and was expressed as a percentage of the maximum fluorescence (Ff), obtained after tissue fixation with 4% paraformaldehyde (4% PFA). Cell death (%) = (F − F0)/(Ff − F0) × 100, where F is the PI fluorescence of slices measured at 24 hr of drug exposure and F0 is the background fluorescence before treatment.
Assessment of Neurodegeneration In Vivo
Adult rats subject to global cerebral ischemia survived for 7 days after surgery (n = 3–5 animals per group). Animals were euthanized and perfused through the heart with 0.9% saline followed by 4% paraformaldehyde in 0.1 M PBS. Then, their brains were removed and immersed in 30% sucrose in 0.1 M PBS for cryoprotection. Brains were frozen in dry ice, and coronal sections were cut in a cryostat at a 16 μm width. Sections of the brains from distinct experimental groups were collected on the same slide to minimize experimental bias in the comparisons. Sections were collected starting 3.6 mm from the bregma and, after dehydration, were oxidized with KMgO4 0.06% solution for 10 min and then stained with FluoroJade C (FJ-C Histo-Chem), following the manufacturer’s instructions.39 For immunolabeling of cleaved caspase-3, sections were incubated with 10% normal goat serum, 5% bovine serum albumin, and 1% Triton X for 1 hr and then incubated with an antibody to cleaved caspase-3 (AB 3623, 1:200, Millipore) at 37°C overnight, followed by an Alexa Fluor 555-conjugated goat antibody anti-rabbit (1:200, Invitrogen) for 2 hr at room temperature. To assess neural degeneration, sections −3.5 to −5 mm distant from the bregma were stained with FluoroJade C or immunostained for cleaved caspase-3. FluoroJade C-positive cells were counted in the CA1 layer of the hippocampus under 20× magnification and plotted as cells per millimeter. Images were obtained with either an Apotome microscope (MRMm Rev3, Zeiss) or a confocal microscope (LSM 510META, Zeiss).
Western Blot
Samples from hippocampal slices were rinsed with PBS; homogenized on ice in radioimmunoprecipitation assay (RIPA) lysis buffer containing 1% Triton X-100, 1% sodium deoxycholate (DOC), 1% nonidet P-40 (NP-40), 150 mM NaCl, 10 mM Tris-HCl, 5 mM EDTA, 0.1% SDS, PMSF (10 mg/mL), pepstatin (1 mg/mL), aprotinin (2 mg/mL), leupeptin (2 mg/mL), NaF (22 mg/mL), and sodium orthovanadate (92 mg/mL); and then subject to vigorous vortex shaking to be homogenized. Samples of whole hippocampus homogenates were taken from adult animals after euthanasia and quick dissection of whole hippocampus on an ice-cold surface. The same solution used for hippocampal cultures was used for the homogenization of the whole hippocampus. Protein concentration was determined by the Lowry assay. Per lane, 15 μg of protein were applied for electrophoresis in 10% SDS-PAGE and then transferred to nitrocellulose membranes (Bio-Rad) and processed for western blotting. The membranes were blocked with 5% milk in Tween 20-Tris-buffered saline (T-TBS) buffer (0.1% Tween in 20 mM Tris-HCl/137 mM NaCl [pH 7.3]) and then incubated overnight with primary antibodies: anti-rabbit phosphorylated eIF2α (1:500, Bioscience), anti-rabbit eIF2α (1:1,000, Santa Cruz), anti-rabbit phosphorylated AKT (1:500, Cell Signaling Technology), anti-rabbit AKT (1:1,000, Cell Signal), anti-rabbit ubiquitin (1:500, Cell Signal), anti-mouse β-actin (1:15,000, Abcam 6276-100), and anti-mouse α-tubulin (1:30,000, Sigma). Washed membranes were incubated with a horseradish peroxidase (HRP)-conjugated secondary anti-antibody for 2 hr and visualized with Luminata western blotting analysis reagent (Amersham Biosciences). Optical density was measured with ImageJ software. Respective loading controls are indicated in each figure.
Statistics
Plotted values are expressed as means ± SEM. The p values were considered statistically significant when p ≤ 0.05. Statistical significance was assessed with one-way ANOVA followed by Dunnet’s multiple comparison post-test or Tukey’s post-test. For in vitro experiments, four to six slices obtained from nine or ten rats were used for each experimental condition, in three independent experiments. For in vivo experiments, we analyzed three to five animals per experimental group for each condition.
Author Contributions
F.C.-M., E.N.-S., and J.A.-N. designed and executed all experiments and analyzed and interpreted the data. V.C. was responsible for the rAAV productions. F.C.-M., W.W.H., R.L., H.P.-S., and L.B.C. contributed to the design of the experiments and provided funding for the acquisition of data.
Acknowledgments
This work was funded by CNPq and FAPERJ. We thank Seok-Hong Min, from the University of Florida, for providing the CHIP construction.
Footnotes
Supplemental Information includes two figures and can be found with this article online at http://dx.doi.org/10.1016/j.ymthe.2016.11.017.
Supplemental Information
References
- 1.Chen J., Nagayama T., Jin K., Stetler R.A., Zhu R.L., Graham S.H., Simon R.P. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J. Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beumer D., Mulder M.J., Saiedie G., Fonville S., van Oostenbrugge R.J., van Zwam W.H. Occurrence of intracranial large vessel occlusion in consecutive, non-referred patients with acute ischemic stroke. Neurovascular Imaging. 2016;2:11. [Google Scholar]
- 3.Martinez-Biarge M., Cheong J.L., Diez-Sebastian J., Mercuri E., Dubowitz L.M., Cowan F.M. Risk factors for neonatal arterial ischemic stroke: the importance of the intrapartum period. J. Pediatr. 2016;173:62–68.e1. doi: 10.1016/j.jpeds.2016.02.064. [DOI] [PubMed] [Google Scholar]
- 4.Gami A.S., Olson E.J., Shen W.K., Wright R.S., Ballman K.V., Hodge D.O., Herges R.M., Howard D.E., Somers V.K. Obstructive sleep apnea and the risk of sudden cardiac death: a longitudinal study of 10,701 adults. J. Am. Coll. Cardiol. 2013;62:610–616. doi: 10.1016/j.jacc.2013.04.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derdeyn C.P. Moyamoya disease and moyamoya syndrome. N. Engl. J. Med. 2009;361:97. doi: 10.1056/NEJMc090780. author reply 98. [DOI] [PubMed] [Google Scholar]
- 6.Fanning J.P., Wong A.A., Fraser J.F. The epidemiology of silent brain infarction: a systematic review of population-based cohorts. BMC Med. 2014;12:119. doi: 10.1186/s12916-014-0119-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurinami H., Shimamura M., Ma T., Qian L., Koizumi K., Park L., Klann E., Manfredi G., Iadecola C., Zhou P. Prohibitin viral gene transfer protects hippocampal CA1 neurons from ischemia and ameliorates postischemic hippocampal dysfunction. Stroke. 2014;45:1131–1138. doi: 10.1161/STROKEAHA.113.003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Endo H., Nito C., Kamada H., Nishi T., Chan P.H. Activation of the Akt/GSK3beta signaling pathway mediates survival of vulnerable hippocampal neurons after transient global cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2006;26:1479–1489. doi: 10.1038/sj.jcbfm.9600303. [DOI] [PubMed] [Google Scholar]
- 9.Los M., Maddika S., Erb B., Schulze-Osthoff K. Switching Akt: from survival signaling to deadly response. BioEssays. 2009;31:492–495. doi: 10.1002/bies.200900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caldeira M.V., Salazar I.L., Curcio M., Canzoniero L.M., Duarte C.B. Role of the ubiquitin-proteasome system in brain ischemia: friend or foe? Prog. Neurobiol. 2014;112:50–69. doi: 10.1016/j.pneurobio.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Badiola N., Penas C., Miñano-Molina A., Barneda-Zahonero B., Fadó R., Sánchez-Opazo G., Comella J.X., Sabriá J., Zhu C., Blomgren K. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tajiri S., Oyadomari S., Yano S., Morioka M., Gotoh T., Hamada J.I., Ushio Y., Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 13.Page A.B., Owen C.R., Kumar R., Miller J.M., Rafols J.A., White B.C., DeGracia D.J., Krause G.S. Persistent eIF2alpha(P) is colocalized with cytoplasmic cytochrome c in vulnerable hippocampal neurons after 4 hours of reperfusion following 10-minute complete brain ischemia. Acta Neuropathol. 2003;106:8–16. doi: 10.1007/s00401-003-0693-2. [DOI] [PubMed] [Google Scholar]
- 14.Paul I., Ghosh M.K. A CHIPotle in physiology and disease. Int. J. Biochem. Cell Biol. 2015;58:37–52. doi: 10.1016/j.biocel.2014.10.027. [DOI] [PubMed] [Google Scholar]
- 15.Matsumura Y., Sakai J., Skach W.R. Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J. Biol. Chem. 2013;288:31,069–31,079. doi: 10.1074/jbc.M113.479345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cabral Miranda F., Adão-Novaes J., Hauswirth W.W., Linden R., Petrs-Silva H., Chiarini L.B. CHIP, a carboxy terminus HSP-70 interacting protein, prevents cell death induced by endoplasmic reticulum stress in the central nervous system. Front. Cell. Neurosci. 2015;8:438. doi: 10.3389/fncel.2014.00438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoppini L., Buchs P.-A., Muller D. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 18.Ansari A.M., Ahmed A.K., Matsangos A.E., Lay F., Born L.J., Marti G., Harmon J.W., Sun Z. Cellular GFP toxicity and immunogenicity: potential confounders in in vivo cell tracking experiments. Stem Cell Rev. 2016;12:553–559. doi: 10.1007/s12015-016-9670-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanderson T.H., Deogracias M.P., Nangia K.K., Wang J., Krause G.S., Kumar R. PKR-like endoplasmic reticulum kinase (PERK) activation following brain ischemia is independent of unfolded nascent proteins. Neuroscience. 2010;169:1307–1314. doi: 10.1016/j.neuroscience.2010.05.076. [DOI] [PubMed] [Google Scholar]
- 20.Owen C.R., Kumar R., Zhang P., McGrath B.C., Cavener D.R., Krause G.S. PERK is responsible for the increased phosphorylation of eIF2alpha and the severe inhibition of protein synthesis after transient global brain ischemia. J. Neurochem. 2005;94:1235–1242. doi: 10.1111/j.1471-4159.2005.03276.x. [DOI] [PubMed] [Google Scholar]
- 21.Stankowski J.N., Zeiger S.L., Cohen E.L., DeFranco D.B., Cai J., McLaughlin B. C-terminus of heat shock cognate 70 interacting protein increases following stroke and impairs survival against acute oxidative stress. Antioxid. Redox Signal. 2011;14:1787–1801. doi: 10.1089/ars.2010.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pérez-Rodríguez D., Anuncibay-Soto B., Llorente I.L., Pérez-García C.C., Fernández-López A. Hippocampus and cerebral cortex present a different autophagic response after oxygen and glucose deprivation in an ex vivo rat brain slice model. Neuropathol. Appl. Neurobiol. 2015;41:e68–e79. doi: 10.1111/nan.12152. [DOI] [PubMed] [Google Scholar]
- 23.Skelding K.A., Spratt N.J., Fluechter L., Dickson P.W., Rostas J.A. αCaMKII is differentially regulated in brain regions that exhibit differing sensitivities to ischemia and excitotoxicity. J. Cereb. Blood Flow Metab. 2012;32:2181–2192. doi: 10.1038/jcbfm.2012.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Birse-Archbold J.L., Kerr L.E., Jones P.A., McCulloch J., Sharkey J. Differential profile of Nix upregulation and translocation during hypoxia/ischaemia in vivo versus in vitro. J. Cereb. Blood Flow Metab. 2005;25:1356–1365. doi: 10.1038/sj.jcbfm.9600133. [DOI] [PubMed] [Google Scholar]
- 25.Palubinsky A.M., Stankowski J.N., Kale A.C., Codreanu S.G., Singer R.J., Liebler D.C., Stanwood G.D., McLaughlin B. CHIP is an essential determinant of neuronal mitochondrial stress signaling. Antioxid. Redox Signal. 2015;23:535–549. doi: 10.1089/ars.2014.6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge P., Luo Y., Liu C.L., Hu B. Protein aggregation and proteasome dysfunction after brain ischemia. Stroke. 2007;38:3230–3236. doi: 10.1161/STROKEAHA.107.487108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakka V.P., Gusain A., Raghubir R. Endoplasmic reticulum stress plays critical role in brain damage after cerebral ischemia/reperfusion in rats. Neurotox. Res. 2010;17:189–202. doi: 10.1007/s12640-009-9110-5. [DOI] [PubMed] [Google Scholar]
- 28.Kreisman N.R., Soliman S., Gozal D. Regional differences in hypoxic depolarization and swelling in hippocampal slices. J. Neurophysiol. 2000;83:1031–1038. doi: 10.1152/jn.2000.83.2.1031. [DOI] [PubMed] [Google Scholar]
- 29.Nakka V.P., Prakash-babu P., Vemuganti R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: potential therapeutic targets for acute CNS injuries. Mol. Neurobiol. 2016;53:532–544. doi: 10.1007/s12035-014-9029-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouyang Y.B., Tan Y., Comb M., Liu C.L., Martone M.E., Siesjö B.K., Hu B.R. Survival- and death-promoting events after transient cerebral ischemia: phosphorylation of Akt, release of cytochrome c and activation of caspase-like proteases. J. Cereb. Blood Flow Metab. 1999;19:1126–1135. doi: 10.1097/00004647-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Lv Y., Song S., Zhang K., Gao H., Ma R. CHIP regulates AKT/FoxO/Bim signaling in MCF7 and MCF10A cells. PLoS ONE. 2013;8:e83312. doi: 10.1371/journal.pone.0083312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nogueira V., Park Y., Chen C.C., Xu P.Z., Chen M.L., Tonic I., Unterman T., Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajesh K., Krishnamoorthy J., Kazimierczak U., Tenkerian C., Papadakis A.I., Wang S., Huang S., Koromilas A.E. Phosphorylation of the translation initiation factor eIF2α at serine 51 determines the cell fate decisions of Akt in response to oxidative stress. Cell Death Dis. 2015;6:e1591. doi: 10.1038/cddis.2014.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes J.W., Wyckoff J.A., Hollander A.S., Derdeyn C.P., McGill J.B. Moyamoya syndrome causing stroke in young women with type 1 diabetes. J. Diabetes Complications. 2016;30:1640–1642. doi: 10.1016/j.jdiacomp.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petrs-Silva H., Dinculescu A., Li Q., Deng W.T., Pang J.J., Min S.H., Chiodo V., Neeley A.W., Govindasamy L., Bennett A. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol. Ther. 2011;19:293–301. doi: 10.1038/mt.2010.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simões Pires E.N., Frozza R.L., Hoppe J.B., Menezes Bde.M., Salbego C.G. Berberine was neuroprotective against an in vitro model of brain ischemia: survival and apoptosis pathways involved. Brain Res. 2014;1557:26–33. doi: 10.1016/j.brainres.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 37.Schmidt-Kastner R., Paschen W., Ophoff B.G., Hossmann K.A. A modified four-vessel occlusion model for inducing incomplete forebrain ischemia in rats. Stroke. 1989;20:938–946. doi: 10.1161/01.str.20.7.938. [DOI] [PubMed] [Google Scholar]
- 38.Ramos A.B., Vasconcelos-Dos-Santos A., Lopes de Souza S.A., Rosado-de-Castro P.H., Barbosa da Fonseca L.M., Gutfilen B., Cintra W.M., Mendez-Otero R. Bone-marrow mononuclear cells reduce neurodegeneration in hippocampal CA1 layer after transient global ischemia in rats. Brain Res. 2013;1522:1–11. doi: 10.1016/j.brainres.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 39.Schmued L.C., Stowers C.C., Scallet A.C., Xu L. Fluoro-Jade C results in ultra high resolution and contrast labeling of degenerating neurons. Brain Res. 2005;1035:24–31. doi: 10.1016/j.brainres.2004.11.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.