

Abstract
The coding sequence of each human pre-mRNA is interrupted, on average, by 11 introns that must be spliced out for proper gene expression. Each intron contains three obligate signals: a 5′ splice site, a branch site, and a 3′ splice site. Splice site usage has been mapped exhaustively across different species, cell types, and cellular states. In contrast, only a small fraction of branch sites have been identified even once. The few reported annotations of branch site are imprecise as reverse transcriptase skips several nucleotides while traversing a 2–5 linkage. Here, we report large-scale mapping of the branchpoints from deep sequencing data in three different species and in the SF3B1 K700E oncogenic mutant background. We have developed a novel method whereby raw lariat reads are refined by U2snRNP/pre-mRNA base-pairing models to return the largest current data set of branchpoint sequences with quality metrics. This analysis discovers novel modes of U2snRNA:pre-mRNA base-pairing conserved in yeast and provides insight into the biogenesis of intron circles. Finally, matching branch site usage with isoform selection across the extensive panel of ENCODE RNA-seq data sets offers insight into the mechanisms by which branchpoint usage drives alternative splicing.
Splicing is a remarkable process whereby large intervening sequences (introns) from the primary transcript are removed and the flanking sequences (exons) are ligated together to make mRNA. Splicing occurs through a two-step mechanism. The 2′ hydroxyl group of the branchpoint nucleotide first attacks the first phosphate of the intron (i.e., the 5′ splice site or 5′ss) to yield a branched RNA lariat. In the second step, the 3′ hydroxyl group of the 5′ splice site attacks the 3′ splice site, ligating the exons and releasing the excised RNA lariat. In higher eukaryotes, this chemistry is catalyzed by the spliceosome—a catalytic complex comprised of five distinct small ribonucleic protein particles (snRNPs) and numerous other proteins (Hoskins and Moore 2012). One of these particles, U2snRNP, recognizes the surrounding branch site sequence (TRYTRAY motif) in the pre-mRNA by an imperfect RNA:RNA base-pairing interaction with the branch site recognition sequence (GUAGUA). This imperfect duplex is characterized by a bulged nucleotide in the pre-mRNA that is activated as a branchpoint nucleophile. Previous studies have indicated that while Saccharomyces cerevisiae branch site sequence has a strong consensus sequence of TACTAAC, the mammalian sequence is more degenerate. However, recent studies have demonstrated that even in S. cerevisiae there exist branch sites with a more degenerate branch site motif (Gould et al. 2016). Experiments with an orthogonal U2snRNA:branch site system systematically tested the catalytic viability of alternate bulged duplexes in the U2snRNA:pre-mRNA interaction and demonstrated flexibility in the position of the bulged nucleotide (Smith et al. 2009; Taggart et al. 2012).
U2snRNP is recruited to the branch site by U2 auxiliary factor, which is comprised of two proteins, U2AF2 and U2AF1. U2AF2 binds the polypyrimidine track, which along with the branch site, the 5′ss, and the 3′ss AG, compose the core splicing elements. Most branch sites are between 19 and 35 nt away from the 3′ss (Taggart et al. 2012; Mercer et al. 2015). Prior analysis of AG selection in the second step of splicing indicates a general distance limitation of at least 8 nt between the branch site and the AG that is used as the 3′ss (which can be modified by competing AG dinucleotides) (Taggart et al. 2012). Bound U2snRNP and other splicing factors sterically interfere with the usage of competitor AGs closer than 12–18 nt downstream from the branchpoint (BP) (Smith et al. 1993; Chua and Reed 2001). The arrangement of these elements can affect splice site recognition.
Distal branch sites located beyond the normal distance to the 3′ss are associated with exon skipping whereas proximal branchpoints may affect 3′ss AG selection (Corvelo et al. 2010; Taggart et al. 2012). Interestingly, recurrent mutations in U2AF1 and components of U2snRNP SF3B1 have been found in several cancers (Yoshida et al. 2011; Bonnal et al. 2012; The Cancer Genome Atlas Network 2012; Imielinski et al. 2012; Harbour et al. 2013). Chemical inhibition and oncogenic mutations of SF3B1, including K700E, have been associated with the activation of cryptic intronic 3′ss presumably through a reduction of branch site selection fidelity (Corrionero et al. 2011; Buonamici et al. 2014; Darman et al. 2015; DeBoever et al. 2015; Papasaikas et al. 2015; Alsafadi et al. 2016). It was proposed that alternative upstream branch sites were selected in introns that spliced via SF3B1-induced cryptic 3′ss usage (Alsafadi et al. 2016).
Branchpoints were originally identified in vitro by RNase mapping/thin layer chromatography (TLC) and later by primer extension (Padgett et al. 1984; Cellini et al. 1986; Jacquier and Rosbash 1986). Branchpoints have been mapped in vivo by lariat PCR which exploited reverse transcriptase's (RT) ability to traverse the 2′-5′ linkage (Vogel et al. 1997; Gao et al. 2008). Our group developed a method to search deep sequencing databases for these lariat reads (Taggart et al. 2012). Recently, this approach has been applied to other data sets, combined with 2-D gel purification or enriched for intron-containing reads by capture (Awan et al. 2013; Mercer et al. 2015). While these approaches have expanded both the number and species of branch sites discovered, the use of enrichment technologies introduces biases related to intron length, variations in hybridization efficiency, and capture probe binding location. Furthermore, reverse transcriptase often skips several nucleotides while traversing a 2′-5′ linkage, leaving the precise location of the branchpoint unknown. Also, due to the chemistry of the 2′-5′ linkage, the RT is more likely to fall off at this position and undergo template switching, leading to the calling of false branchpoints.
In order to survey branch site usage in all regions of human introns, we have analyzed a series of internally generated and public RNA-seq data sets (ENCODE) for lariat reads (The ENCODE Project Consortium 2012; Bitton et al. 2014; Yue et al. 2014). U2snRNP/pre-mRNA base-pairing models were applied to lariat read data to ascertain the most likely branch site at each location. In this manner, we create the largest data set of branchpoints in human introns reported to date (Supplemental Fig. S1; Supplemental Tables S1–S8), accounting for 16.8% of all introns. The extensive collection of ENCODE RNA-seq data allows for a side-by-side comparison of branch site usage with alternative splice site usage. The following study describes the contribution of branch site location to mechanisms of splice site selection in exon skipping, cryptic splice site usage, and oncogenic SF3B1 usage.
Results
To study the full range of branchpoints and profile the steady-state levels of all lariats in the cell, we developed an analysis pipeline that inferred branchpoint location from gapped, inverted lariat reads (Fig. 1A; Taggart et al. 2012). A fine-grain map was drawn to identify precisely where the branchpoint occurs in the branch site sequence. Novel modes of U2snRNA:pre-mRNA base-pairing were discovered in human introns, and true branchpoints were called after correcting for RT skipping (Fig. 1B). A coarse-grain map was used to create a framework for relating branchpoint position to splicing outcome (Fig. 1C). Comparing the coarse-grain map from three different species confirms a preferred branch site location in a window upstream of the 3′ss. In humans, the expected region is 10–60 nt upstream of the 3′ss AG. The branchpoint distribution in yeast (Schizosaccharomyces pombe) was shifted significantly closer to the 3′ss than their mammalian counterparts. Similarly, 255 branchpoints that correspond to U12 minor introns were positioned relatively closer to the 3′ss (n = 255, P-value < 0.001) as reported previously (Supplemental Fig. S2; Mertins and Gallwitz 1987; Dietrich et al. 2001). Approximately 9% of human U2 branchpoints were found <10 nt from the 3′ss AG. A subset of these apparent branchpoints (∼3% of all branchpoints) appears to be forming at the 3′ss, creating a circular intronic product. The remaining 8% of branchpoints form distally (>60 nt from AG), beyond the region presumed to be optimal for the second step of splicing (Fig. 1C).
Figure 1.
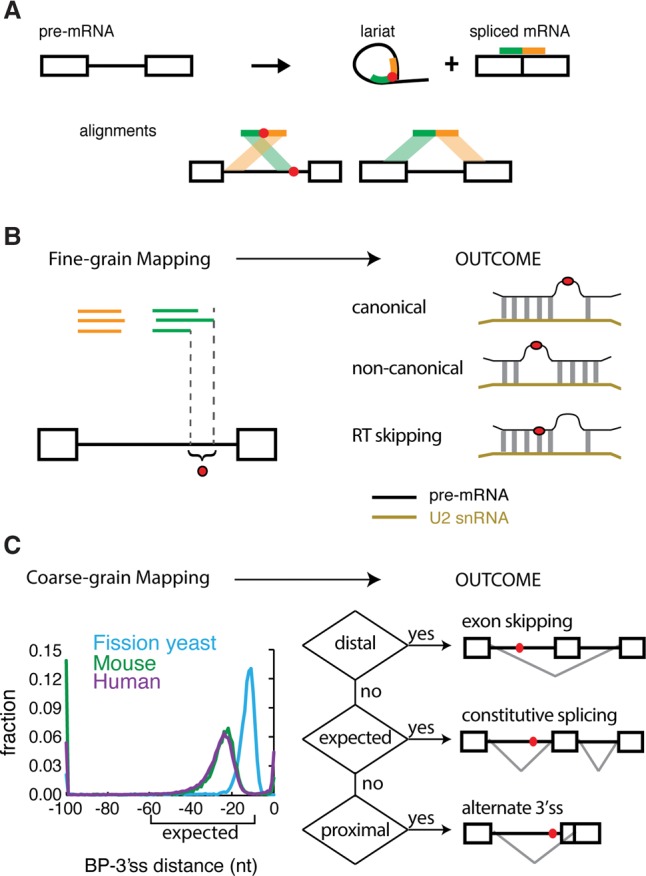
The technique and purpose of mapping branchpoints. (A) Overview of branchpoint mapping (adapted by permission from Macmillan Publishers Ltd: [Nature Structural and Molecular Biology] (Taggart et al. 2012), © 2012). Inverted fragmental cDNA reads across the 2′-5′ linkage of the lariat RNAs were collected to map the branchpoint. (B) Fine-grain mapping. Discovering the precise locations of branch sites enables the discovery of canonical and noncanonical modes of U2snRNA pairing and the characterization of RT ‘skipping’ behavior around 2′-5′ linkages. (C) Coarse-grain mapping. General branchpoint location was analyzed in three species: fission yeast, mouse, and human. The human and mouse branchpoints were largely located within the expected region (between 10 and 60 nt upstream of the 3′ss AG), but the fission yeast branchpoints were more proximal. The ENCODE RNA-seq data sets were used to test the relationship between branchpoint location and alternative splicing outcomes.
Fine-grain map of human branchpoints suggests alternate modes of U2snRNA:pre-mRNA base-pairing
To determine the manner in which U2snRNA can productively base-pair with pre-mRNA, we aligned the branchpoint recognition sequence of U2snRNA with the pre-mRNA. The alignment was performed to allow G:U base pairs and bulges of length 0–3 occurring in the pre-mRNA at any position in the U2-premRNA alignment (Fig. 1B). The enrichment of each possible mode of alignment was determined using an iterative, masking strategy that (1) avoided over counting degenerate alignments,(2) controlled for variable skipping of the RT (Fig. 1B; Supplemental Figs. S3, S4), and (3) enabled mismatches to occur within the motif. The application of this method to lariat data from S. pombe indicates predominant usage (63.56%) of the canonical U2snRNA:pre-mRNA model, with an overwhelming preference for A as the branch site (i.e., TRYTRAY), while 23.0% utilize an alternate mode defined by a 3-nt pre-mRNA loop located between position 2 and 3 in the U2:branch site duplex (i.e., TRANNYTRY) and 13.4% match no model at all (Fig. 2A).
Figure 2.

Alternate modes of U2snRNA:pre-mRNA base-pairing emerged from lariat mapping. (A) U2snRNA:pre-mRNA base-pairing models found in Schizosaccharomyces pombe. Twenty-nucleotide windows around unprocessed branchpoint reads were iteratively aligned to U2snRNA in a manner that allowed branchpoint location, loop location, and the loop length to vary. Significantly enriched modes of base-pairing were identified and recorded. Left column: schematics of significantly enriched modes of base-pairing, middle column: percent of lariat reads that fit model, right column: motif of lariat reads that fit model. (B) U2snRNA:pre-mRNA base-pairing models found in human. Left column: schematics of significantly enriched modes of base-pairing, middle column: percent of lariat reads that fit model, right column: motif of lariat reads that fit model. (C) Branch site motif distribution relative to the 3′ss. (D) The skipping behavior and mutational profile of reverse transcriptase (RT) was inferred from the human canonical branchpoint (TRYTRAY) mapping. The histogram describes the frequency of each size of RT-induced skip (x-axis). The striped portion of bars represents the fraction of misincorporation events at the apparent branchpoint. (E) Exemplars of branch site motifs were validated in a strong (APRT, upper) and weak (SHMT2, lower) branch site reporter minigene. Branch sites of the first intron (between the blue and red exons) were first deleted (ΔBP, lanes 3,17) or replaced by branch site motifs identified in B to test for their ability to restore splicing. Inclusion level of the middle exon is indicated under the gel image.
Applying an identical approach to human branch sites also returned canonical binding as a dominant motif (Fig. 2B). The alternative mode of U2snRNA pairing observed in yeast was also returned as an enriched motif. The four remaining modes were permutations of these classes. In other words, the canonical TRYTRAY was also observed to utilize a C branchpoint (i.e., TRYTRCY) and to permit a bulge length > 1(i.e., TRYTRNAY). Similarly, the TRANNYTRY was also observed with small bulge lengths (TRANYTRY, TRAYTRY). In the case of the canonical TRYTRAY motif (where R = “A”), there exist 12 examples of either A being used as the nucleophile (Supplemental Fig. S5), in agreement with previous studies (Smith et al. 2009). Eight and one-half percent of motifs arose through template switching RT artifacts. Template switching events are clear false positives and lack the signature distribution downstream from the 3′ss (Fig. 2C). The degree of template switching varies between human samples and is completely absent from S. pombe, which is presumably due to the smaller intron size and the near absence of nonfunctional matches to 5′ss motifs in yeast introns which are required for false priming (Supplemental Fig. S6). In addition to template switching, RT will frequently skip nucleotides as it traverses the 2′-5′ linkage of the branched site, making simple questions such as the base preference for the branchpoint nucleophile ambiguous. It is difficult to determine if a closely spaced cluster of branchpoints detected from lariat reads (Fig. 1A) arose through biological promiscuous branch site selection or RT skipping. Studies with an artificial U2snRNA sequence establish that the −2 position (i.e., the base paired to the second T in the TRYTRAY motif) was not used as the branchpoint nucleophile (Smith et al. 2009). Presumably the more than 3000 branchpoints that occur at the −2 position in the ENCODE matches to the canonical TRYTRAY motif represent the highest confidence examples of RT skipping (Fig. 2D). By restricting analysis to these high confidence branch sites, it becomes obvious that RT skipping behavior is highly dependent on the sequence of the branchpoint and −1 positions. Branchpoints and sites with a purine at −1 can result in skipping in up to 50% of the reads. The smaller pyrimidine bases induce RT skipping at a much lower level (Supplemental Fig. S4). Fitting lariat read data into the best fitting U2snRNA base-pairing modes and calling the branchpoint at the bulged positions adjusts 21.9% of lariat reads to correct for RT skipping and allows many of the 46,724 small clusters of observed sites to coalesce into 36,078 BP coordinates. This approach can also be used to refine raw lariat sequence reads, avoiding the biases of relying on mutated A nucleotides. While the model does not specify the nucleotide identity at the bulged position, the fraction of the canonical “A” BP called in this manner increases from 40% in the raw data to 55%, whereas the fraction of C branchpoints remains stable (i.e., 31% to 33%). The resulting database of branchpoints and its supporting set of observed lariat reads has been made available with quality measures (see fairbrother.biomed.brown.edu/Lariat2016 and Supplemental Tables S1–S8).
We observe roughly equal spatial distribution (Fig. 2C) and read validation between canonical and alternate U2snRNA pairing modes (Supplemental Fig. S7A). If the alternate U2snRNA pairing modes arose from false positives in the data set and were nonfunctional, we would expect that they would be more likely to co-occur in the same introns with a second, functional branchpoint. However, when we compare the set of introns that contain canonical branchpoints to the set of introns that contain branchpoints with an alternate U2snRNA pairing mode, we observe no major differences in the co-occurrence with additional branchpoints (Supplemental Fig. S7B). The novel branch site motifs have never been validated in a biochemical assay. In order to test whether these modes of U2snRNA:pre-mRNA base-pairing could support splicing in vivo, minigene reporter constructs were prepared to assay branch site function. Each reporter was constructed from a genomic three-exon, two-intron fragment. Branch sites in the upstream intron were mapped and then mutated to completely eliminate splicing (∼90% exon skipping) (Fig. 2E, lanes 3,17). Substituting the canonical branch site resulted in the full or majority restoration of splicing in both constructs as expected (Fig. 2E, lanes 4,5,18,19). The addition of an extra nucleotide into the predicted bulge restored splicing in one construct but had little effect in the other. The TRAYTRY and the TRANNYTRY spliced robustly in both constructs. TRANNYTRY was the only alternate mode enriched in both the human and yeast data. However, TRYTRCY is clearly a weaker branch site motif in both reporters (Fig. 2E). As the recognition of the 5′ss, the 3′ss, and branch site are coordinated events, it is possible a strong 3′ss could compensate for a weak branch site through enhanced recruitment of U2AF2. Consistent with this hypothesis, the weakest branch site motif, TRYTRCY, was associated with the strongest average 3′ss scores, the strongest average polypyrimidine tract scores, and higher 5′ss scores (Supplemental Fig. S8). All full branch site sequences inserted into the construct are listed in Supplemental Table S9.
Intron circles arise from conserved post-splicing events
Despite the evidence that most introns require a minimal distance between the branch site and AG, there exists a subset of introns where branchpoints can appear to form within a few nucleotides of the AG (Fig. 1C). The most extreme case of proximal branch site usage occurs at position zero (∼3% of all branchpoints)—a complete circularization of the intron. Mechanistically, these cases were hypothesized to represent (1) true branchpoints created at the first step of splicing, or (2) a secondary product generated from a conventional lariat, or (3) an artifact of the branchpoint mapping protocol. To explore whether circularization events were functional, lariat PCR was performed on introns that underwent circularization (Fig. 3A). Of 12 circularization events validated in human, seven circularized in human, mouse, and rat. Additionally, introns that circularize tend to, on average, have higher conservation scores than introns that do not circularize (Supplemental Fig. S9A). These results suggest that circularization either creates a functional intronic product or is a byproduct splicing.
Figure 3.

Intron circularization via 3′-5′ linkage is conserved and arises in introns that also contain branchpoints in the expected region. (A) Conservation of the intron circles was assessed by inverted nested RT-PCR in multiple species (primer location indicated by arrows) and Sanger sequencing. The PCR results inconsistent with conserved distal branchpoints are boxed in red. ‘N/A’ indicates certain introns were not tested. (B) Mutational profile of branchpoints from expected region (left) compared with mutational profile from full intron circularization events (right). (C) Location of additional branchpoints in introns which circularize (red) was compared to introns with no observed evidence for circularization (blue). (D) Model for circles implicates a 3′-5′ circular linkage arising from a conventional lariat.
It could be hypothesized that these junctions represent true branchpoints that splice to an alternate downstream 3′ss. However, this is unlikely because we observe no enrichment for alternate 3′ss in a 60-nt window downstream from the circularization junction. Analysis of U2AF2 binding at the polypyrimidine tract, which is located downstream from the branchpoint and upstream of the 3′ss, using CLIP binding studies (Shao et al. 2014), shows a peak immediately upstream of the circularization junction point at these loci (Supplemental Fig. S9B). Additionally, the circle reads do not match the distinct mutational profile associated with reverse transcription through a 2′-5′ linkage (P-value < 0.0001) (Fig. 3B). In contrast, the mutational rate and profile at circularized introns more closely resembles the fidelity of reverse transcriptase observed at normal 3′-5′ linkages (Fig. 3B, right panel). While it is beyond the scope of this study to definitively characterize each circularization event, the data suggest circularization occurs post-splicing. Consistent with this model, many introns that circularize also form conventional lariats that branch in the expected range (Fig. 3C; Supplemental Fig. S7B). The resulting model suggests circularization occurs either as a third nucleophilic attack on the 2′-5′ phosphate or as a two-step debranching/ligation reaction (Fig. 3D). These results suggest that intron circles are a chemically distinct RNA species from the lariat intermediate. Furthermore, the introns that undergo circularization in humans tend to undergo circularization in rodents (Fig. 3A). It is not yet evident why intron circularization is conserved, but there are examples of circular RNAs that serve a function outside of splicing (i.e., miRNA sponges) (Hansen et al. 2013; Memczak et al. 2013; Salzman et al. 2013).
Distal branch site usage is conserved, coincident with U2AF2 binding, and associated with exon skipping
In agreement with our and other previous studies (Corvelo et al. 2010; Taggart et al. 2012; Bitton et al. 2014), there is significantly more distal branchpoint usage upstream of conditionally skipped exons than constitutively used exons (P < 0.001). Prior biochemical analysis has focused on branchpoint usage in tissues that are permissive to inclusion. The ENCODE data set also allows distal branchpoint usage to also be measured in cell lines that skip the exon in question. Using 16 different cell lines, alternative splicing maps were drawn connecting branchpoint usage in the intron to the splicing outcome of the message. Briefly, each lariat read was traced back to the cell line context from which it was discovered in the ENCODE poly(A) depleted RNA-seq data set. The branchpoint was then classified according to the degree of exon inclusion in that cell line context using the mRNA splice junction usage patterns observed in the poly(A) selected data. Extreme categories were designated “includes” for introns with >95% exon inclusion in that cell line and “skip” for introns that skip (<5% inclusion). Branchpoints mapped in conditions that favor inclusion were binned and depicted in the top panel, and branchpoints mapped in conditions favoring skipping were binned and depicted in the bottom panel (Fig. 4A). Interestingly, distal branchpoint usage occurs at ∼12% upstream of conditionally skipped exons in both permissive and restrictive cell lines. However, the aggregate data suggests that in permissive cell lines an additional branchpoint is usually recognized in the expected zone at that locus (Fig. 4A). In restricted cell lines, this additional branchpoint is located in the expected zone of the downstream, used exon.
Figure 4.

Distal branchpoints are conserved and associated with alternative exon usage. (A) The location of branchpoints was mapped in exon inclusion (top) and exon skipping (bottom) transcripts. Distal branchpoints more than 100 nt upstream of the 3′ss were binned at −100 nt. y-axes indicate the fraction of the bars. All branchpoints depicted use the same donor site (marked by *). (B) Distal branchpoints were validated by nested lariat PCR in human, mouse, rat, and zebrafish. Sequencing was used to distinguish conserved branch site usage (black box) from nonconserved (red). (C) The CLIP signal of U2AF2 binding was mapped relative to distal branchpoints (top) and all branchpoints (bottom). (D) Conservation of four introns that contain distal branchpoints. Thick bars indicate exons, and the red dots are distal branchpoints.
While distal branchpoints have been previously associated with exon skipping (Corvelo et al. 2010), it is possible that these species arise from template switching or represent errors made by the splicing machinery. To determine if these distal lariat reads represent errors, candidate distal branchpoints were selected for RT-PCR validation, first in human and then in additional vertebrate species (Fig. 4B, left column). To provide more definitive evidence of branchpoint function, this validation was repeated in orthologous introns using RNA extracted from multiple vertebrates (Fig. 4B, right lanes). These experiments suggest that distal branchpoint usage is highly conserved. All eight distal branchpoints utilized in human are conserved and splice using branch sites that map within 4 nt of the orthologous position in rodent. The majority (five of eight) of these distal branch sites are also utilized in zebrafish, suggesting the use of these branch sites has been preserved by natural selection (Fig. 4B). In a typical intron, a polypyrimidine tract located upstream of the 3′ss AG is bound by U2AF2, which recruits U2snRNP to the branch site. Evidence of the generality of U2AF2 function was found by intersecting prior U2AF2 CLIP binding studies (Shao et al. 2014) with the global branchpoint map generated here (Fig. 4C, bottom). Repeating this approach with the distal subset of branch sites demonstrates a similar location of peak occupancy centered at distal branch sites (Fig. 4C, top). The data suggest that distal branchpoints are conserved functional elements that share with canonical branchpoints the same requirement for U2AF2 binding. Furthermore, many distal branch sites are associated with highly conserved intronic regions that extend well beyond the canonical 3′ss region (Fig. 4D).
Distal branchpoints have a modest effect on splice site selection and influence the kinetics of splicing
To explore a potential role for distal branchpoints in splicing, three minigenes were constructed to model three separate introns that utilized distal branchpoints (i.e., intron 15, LONP1; intron 2, SHMT2; intron 2, APRT). For each locus, the entire three-exon, two-intron subregion involved in exon skipping was cloned into a mammalian expression vector and transfected into HEK293T cells (schematic of subregion tested in Fig. 5A). The introns upstream of the skipped exon were selected to include two mapped branchpoints—one located in the distal and one in the expected zone. The deletion of the distal branchpoints had a modest negative effect on exon inclusion, and the magnitude of that effect was inversely correlated to their distance to 3′ss (Fig. 5A). In contrast, the deletion of the branch site in the expected range reduced exon inclusion in all three cases. Double deletion of branch sites in both distal and expected regions (ΔBP1ΔBP2) caused similar levels of inclusion as the single deletion of the branch site in the expected region (ΔBP1) (Fig. 5A). One prediction of distal branchpoint usage would be loss of end product through a dead end complex. However, in two of the three cases tested (SHMT2 and LONP1), total transcript level was reduced in ΔBP1, arguing against the usage of distal branch sites resulting in a dead end pathway. To further test the functionality of distal branchpoints, distal branch sites were substituted for a disabled branchpoint located in the expected region and tested for their ability to rescue splicing (BP2→BP1) (Fig. 5B). While the uninserted construct failed to include the middle exon, the relocation of the distal branch site into the expected zone demonstrated the branch site was catalytically active.
Figure 5.

Distal branchpoints are functional elements that are associated with delayed splicing. (A) Both or either one of the distal (BP1) and expected (BP2) branchpoints was mutated in a splicing minigene as depicted at the top. Minigenes were transfected into HEK293T cells and inclusion of the middle exon was measured by RT-PCR as a readout of branchpoint activity (two replicates displayed for each construct). The splicing inclusion level was estimated by ImageQuant software across ≥3 replicates and recorded below each panel and in the histogram to the right. (B) Expected branch site of APRT (i.e., BP2) was replaced by the sequence of the distal branch site (BP2→BP1) and the functionality of branch sites was assayed by RT-PCR as in A. (C) Scheme for determining relative order of intron removal from paired-end reads, using all human introns. The bar plot indicates the degree to which the upstream (blue) and downstream (orange) introns splice first. (D) Introns that contain distal branch sites (orange) were analyzed for order of intron removal. “Upstream first” contains loci where the upstream intron reliably spliced first (i.e., >90%) and “downstream first” contains loci where the distal intron spliced before the upstream intron (left). Similar comparisons were performed on the downstream intron (right).
One requirement of a regulated exon skipping event is for the spliceosome to have the opportunity to select between two alternate splice choices. Splicing is a reversible, cotranscriptional reaction that occurs at splice sites that can be separated by hundreds of thousands of nucleotides. Without a mechanism to delay splicing, the upstream intron could be spliced before the downstream intron has been synthesized. To test the ability of distal branch sites to delay upstream splicing in vivo, paired-end deep sequencing was analyzed to determine the relative order of the splicing of introns (see scheme; Fig. 5C). The ENCODE data set was searched for alignments that defined partially spliced transcripts (i.e., one read aligns across an intron while its mate pair spans an exon/exon junction). Each of these read pairs was counted and interpreted as an unbiased sampling of the order of splicing (i.e., a measurement of the fraction of time the upstream intron spliced first). The distribution of 102,000 comparisons suggests that, in about half of all comparisons, there is a clear preference (i.e., more than 90%) of the reads for a particular order, i.e., for either the upstream or downstream intron to splice first. Examining cases which exhibit this clear preference for order reveals a bias for the upstream intron to be processed first (55%) in all introns. If the upstream intron contains a distal branch site, it is usually spliced after the downstream intron, splicing first only 41% of the time (Fig. 5D). Additionally, if the downstream intron contains a distal branch site, it is only spliced first 28% of the time, compared to the background of 45% of downstream introns splicing first (Fig. 5D). Together, our data suggest that distal branch sites contribute only moderately to the productive splicing compared to branch sites in the expected region, and their main role may be to delay splicing kinetics to enable for alternative splicing events. We propose that these distal branchpoints may act as a holding state, allowing time for downstream alternate splice sites to be transcribed. A more complete exploration of potential mechanisms for how these distal branchpoints affect splicing kinetics and pre-mRNA processing is outlined in the discussion.
Proximal branch site location associates with alternate 3′ss use
In addition to the association of distal branchpoints with exon skipping, branch site location can influence alternate 3′ss usage. Prior applications of machine learning to branch site data found that minimal distance restrictions between AGs and the branch sites explain 95% of the AG selection data (Taggart et al. 2012). These rules suggest that alternate utilization of a more proximal branch site can drive the selection of a downstream AG. To test the relationship between branch site selection and AG usage in genomic data, ENCODE cell lines were classified according to their degree of alternate 3′ss usage at each alternately processed transcript. As with distal branch sites, extreme categories were designated upstream for introns with >95% proximal 3′ss usage and distal for introns with >95% distal 3′ss usage. Mapping an intron's branchpoints in cell lines that conform to these criteria at that locus suggest a role for branch site selection in alternate 3′ss selection. Downstream AG usage is associated with branch sites that would appear to preclude upstream AG selection because the branchpoint's location is too close or downstream from the upstream AG (Fig. 6A).
Figure 6.

Branchpoint location predetermines the 3′ss choice in wild-type and SF3B1 mutant background. (A) Branchpoint location relative to the 3′ss analyzed in transcripts undergoing alternative 3′ss usage across ENCODE cell lines. Transcripts in cell lines using predominantly the upstream 3′ss (top) are compared to branchpoint usage in cases where the downstream 3′ss is used (bottom). (B) Branchpoint to 3′ss distance analyzed in wild type or the oncogenic mutant SF3B1 K700E NALM-6 cell line.
To further explore the mechanism that may underlie alternate branch site usage, a specific subset of cryptic and alternate 3′ss AG selection was considered. It has been observed that certain driver mutations in the SF3B1 protein cause a shift toward upstream 3′ss AG selection in a variety of cancers (Buonamici et al. 2014; DeBoever et al. 2015). As SF3B1 is a component of U2snRNP, recent studies have suggested that the SF3B1 K700E mutation acts by reducing the fidelity of branch site utilization (Alsafadi et al. 2016). Mapping branch sites in RNA extracted from isogenic cell lines that are heterozygous for SF3B1 K700E revealed no significant difference between the global distributions of branch sites in wild-type versus mutant backgrounds (P = 0.93, Kolmogorov-Smirnov test) (Fig. 6B). However, it is possible that most loci are unaffected, and loss of branchpoint fidelity is only observed at loci with SF3B1-dependent alternate 3′ss usage. To explore further the idea of altered branch site specificity, targeted branchpoint mapping was performed by RT-PCR in 10 substrates thought to undergo SF3B1-dependent alternate 3′ss usage. Differences in branchpoint usage were initially observed by Sanger sequencing, in which lariat reads amplified by RT-PCR from four of the introns tested in an SF3B1 K700E mutant background exhibited differential branch site usage (Supplemental Fig. S10). However, deep sequencing of these PCR products revealed the alternate branchpoints associated with mutant SF3B1 were also used at some level in the wild type (Supplemental Fig. S11).
Discussion
This study seeks to map branchpoints, an understudied class of functional elements, in human, mouse, and yeast introns. This branchpoint map suggests important insights into the mechanism of splicing. We build upon our prior work and present the most comprehensive map to date. A branch site list drawn from a compilation of all human studies performed to date covers 44,332 introns, ∼16.8% of annotated introns in the genome (Supplemental Fig. S1). There are numerous benefits of such a map, including a better understanding of expression quantitative trait loci (eQTL) and noncoding disease-causing variants (Ward and Kellis 2012), mechanisms of U2snRNA:pre-mRNA recognition, and the role of branch site selection in alternative splicing.
The branch site is an obligate signal for intron splicing, yet in higher eukaryotes it lacks the strong sequence motifs associated with the 5′ss and 3′ss. A general region of branchpoint formation is observed in an expected range of −18 to −35 nt upstream of the 3′ss (Fig. 1C). Experiments with orthogonal U2snRNA demonstrated flexibility in the U2snRNA:pre-mRNA duplex. The canonical duplex contains a trinucleotide repeat TRYTRY with a bulged A at position 5 (i.e., TRYTRAY). The orthogonal U2 experiment demonstrated that the duplex was insensitive to sequence and even tolerated bulges at several alternate positions (Smith et al. 2009). This flexibility is reflected in the branch sites identified by lariat reads (Fig. 2). While prior observations identified a low information TRAY motif for branch sites in humans, it can be seen that this motif corresponds to the common region of two equally represented populations of longer motifs (Lim and Burge 2001; Padgett 2012). It is also possible that an extended mode of binding could be being utilized, similar to what was observed in S. cerevisiae (Garrey et al. 2006).
Despite the increased noise in distal branch site mapping, distal branch site reads are real branch sites known to be important in alternatively spliced exons. The first identified case was the mutually exclusive splicing of tropomyosin (Helfman and Ricci 1989) and additional examples have been subsequently characterized (Smith and Nadal-Ginard 1989; Goux-Pelletan et al. 1990; Southby et al. 1999; Wollerton et al. 2004). As AG selection can occur through a downstream scanning mechanism that initiates at the branch site (Smith et al. 1993; Taggart et al. 2012), introns depleted of AG in their 3′ termini have been interpreted as an indication of distal branch sites (Gooding et al. 2006). In agreement with this study, these introns with extended AG exclusion zones are more likely to flank skipped exons (Corvelo et al. 2010; Hallegger et al. 2010). Here, we report direct evidence of widespread distal branchpoint usage and profile branch site selection in permissive and restricted tissues. Distal branch sites are functional elements maintained by purifying selection. Distal branch sites match U2snRNA binding models, co-occur with U2AF2 binding, are utilized in additional vertebrate orthologs, and are associated with regions of strong sequence conservation (Fig. 4B–D). Increased distance from the branchpoint clearly weakens 3′ss AG recognition conferring dependence of the substrate on the activity of Prp22, Prp18, and Slu7 (Smith et al. 2008). In some cases, this weakness is utilized by evolution to block the second step of splicing. In other words, the natural product of the gene is the first step intermediate. Such an example can be seen in the 3′ end processing of Ter1 in S. pombe, which is performed by the spliceosome at a donor site to a branch site separated by an excessive distance from an acceptor 3′ss (Kannan et al. 2013). Despite this, there is unequivocal evidence that some distally branched introns can progress through the second step of splicing (Smith and Nadal-Ginard 1989; Southby et al. 1999; Hallegger et al. 2010). It is not clear what factors affect the maximum distance between a branchpoint and the 3′ss AG, but some data suggest secondary structure can bridge excessive distance between the two elements (Hall et al. 1988; Deshler and Rossi 1991; Estes et al. 1992; Gahura et al. 2011).
It has been demonstrated in vitro that the spliceosome can revert a fully spliced message and excised lariats back to unspliced pre-mRNA (Tseng and Cheng 2008). We suggest that some distally branched lariat intermediates could be a holding state where the excessive distance to the 3′ss greatly reduces the forward reaction such that the reverse reaction is more likely. Unfortunately, technical limits on substrate size in in vitro splicing prevent a similar type of biochemical validation. However, if distally branched lariat RNAs are reversed by the spliceosome, distal branchpoint usage should only be observed at the site of splicing, in the chromatin associated with transcribing polymerase. In agreement with this hypothesis, distally branched lariats are 2.9 times more abundant in the chromatin fraction than in total RNA (Supplemental Fig. S12).
A role for proximal branch sites in splicing has not been widely studied. Prior analysis of AG selection in the second step of splicing indicates a general distance limitation of at least 8 nt between the branch site and 3′ss (Taggart et al. 2012). Bound U2snRNP and other splicing factors sterically interfere with the use of other AGs closer than 12–18 nt downstream from the BP (Smith et al. 1993; Chua and Reed 2001). This concept of a minimum distance is reinforced by mapping branchpoints around alternative 3′ss processing events. Examining branch site usage in cell lines where a proximal 3′ss is used detects more upstream usage of branch sites than in cell lines where the distal 3′ss is used (Fig. 6A). Alternate cryptic 3′ss usage is also seen in disease states such as multiple types of cancer. SF3B1 is commonly mutated in chronic lymphocytic leukemia at K700E (Yoshida et al. 2011; The Cancer Genome Atlas Network 2012; Imielinski et al. 2012; Harbour et al. 2013). Prior studies reported a loss of fidelity in specifying 3′ss, with upstream AG frequently being included in the transcript (Chua and Reed 2001; Gooding et al. 2006). Profiling isogenic cells carrying the K700E mutation did not reveal significant differences in type or distribution of branch site usage in the samples tested (Fig. 6B). Testing particular loci that were found to undergo alternate 3′ss usage revealed apparent shifts in branch site usage; however, deep sequence analysis discovered that all SF3B1 mutant branchpoints were also used at a detectable level in the SF3B1 wild-type background (Supplemental Figs. S10, S11). While such evidence does not rule out relaxed branchpoint selection in the SF3B1 K700E background, the effect is not large enough to detect with the technology and approach employed by this study.
Methods
Lariat read identification
Human RNA-seq data were obtained from the ENCODE Project (GSE30567) (The ENCODE Project Consortium 2012). Additional human RNA-seq data were analyzed from GSE53328 (Mercer et al. 2015). Mouse and fission yeast RNA-seq data were obtained from accessions GSE39619 (Yue et al. 2014) and GSE50246 (Bitton et al. 2014), respectively. In-house Perl scripts were used to identify lariat reads, using a previously described algorithm (see Supplemental Methods; Taggart et al. 2012). Perl scripts used in identifying lariat reads are made publicly available on the lab website (http://fairbrother.biomed.brown.edu/data/Lariat2016/), on GitHub (https://github.com/allisontaggart/lariat), and in the Supplemental Material. Alignment is performed using the Bowtie aligner (Langmead et al. 2009). Annotations used were hg19 UCSC genes, mm9 UCSC genes, and Ensembl EF2 for human, mouse, and fission yeast, respectively. U12 introns were determined using the U12DB (Alioto 2007). Branchpoint counts and motif statistics stratified by sample can be found in Supplemental Table S10.
Mapping modes of U2snRNA:pre-mRNA interactions
Modes of U2snRNA:branch site interaction were discovered using the set of expected branchpoints 18–35 nt upstream of the 3′ splice site. Background control sets were generated using positions 18–35 nt upstream of the annotated 3′ splice sites. Sequences were aligned to the compliment of the U2 sequence (TRYTRY). This alignment was allowed within a 20-nt window centered around the branchpoint and allowed for wobble base-pairing, a bulge anywhere within the aligned sequence, and a bulge of size 0–3 nt. The real set and a background control set were both sampled 1000 times, and the most statistically significant enriched alignment in the real set was identified. The sequences that fit that alignment were then removed, and the sampling was repeated until there were no more statistically significant U2 alignments (P < 0.02). After the novel modes were identified from perfect matches to the U2 sequence, the remaining reads were aligned to these modes allowing for mismatches using the paster package (Hertz and Stormo 1999). Template switching artifacts were filtered based on sequence complementarity to the 5′ss. See Supplemental Methods for full details.
Minigene splicing reporter
To test the effect of branchpoint mutation on splicing, genomic fragments spanning three exons and two introns from SHMT2, APRT, and LONP1 were cloned into the pcDNA3.1/zeo(+) vector (Invitrogen) between the EcoR1 and Nhe1 sites. Subsequently, ±3 nt (total 7 nt) around the distal branchpoints in the upstream intron and/or ±3 nt around the expected branchpoints were deleted. To test branchpoint motifs generated from the U2 alignment experiment, various motifs were inserted at the expected branchpoint position into the double branchpoint mutants of SHMT2 or APRT backbone (Supplemental Table S9). Alternatively, 10 nt of the distal branch site (±5 nt around the branchpoint) were inserted at the expected branchpoint position of APRT to test the functionality of the distal branch site sequence. These constructs were transfected into HEK293T or HeLa cells, and the RNA was harvested, reverse transcribed, and subjected to PCR to assay the exon inclusion. See Supplemental Methods for full details.
Alternative splicing maps
Alternative splicing for 16 ENCODE cell lines (GSE30567) was determined using the MISO software package (Katz et al. 2010) and alternative splicing events index. For both exon skipping and alternate 3′ splice site usage, events were binned into >95% inclusion or exclusion, and the remaining events were discarded. If branchpoint information from a particular cell line was mapped for a 95% inclusion or exclusion event from that cell line, it was included in the distribution. For exon skipping, branchpoints were mapped to the closest downstream 3′ splice site. For alternate 3′ splice sites, branchpoints were mapped using the more proximal 3′ splice site as position 0.
Determining the order of intron removal in a transcript
ENCODE RNA-seq paired-end sequencing reads (GSE30567) covering partially spliced transcripts were used to infer splicing order. An informative read contained one end mapping across an intronic or exon-intron pre-mRNA region and the other end mapping across a spliced exon-exon junction. Reads across intron pairs were counted and interpreted to infer the fraction of time one intron spliced before the other. Intron pairs that had at least 10 informative reads and at least 90% unidirectional order were determined to have a clear order preference and were included in this analysis.
Lariat enrichment and library preparation
RNA from SF3B1 K700K or SF3B1 K700E knocked-in NALM-6 cell lines (gift from H3 Biomedicine) was treated with Ribominus to remove the ribosomal RNA. The resulting RNA was homogenized, followed by phenol/chloroform extraction and ethanol precipitation to remove the RNase. RNA was reverse transcribed to make cDNA. cDNA was subjected to second-strand cDNA synthesis, end-repair, dA-tailing, ligation, and amplification. To enrich the cDNA for lariat reads, two methods were used. The cDNA libraries of SF3B1 K700K and K700E NALM-6 cells were hybridized with biotinylated RNA probes targeting the 5′ end of introns. Alternatively, the cDNA library was treated by duplex-specific nuclease to enrich for rare species. Lariat reads were enriched by various rounds of treatments: DSN only, capture only, capture+DSN, capture+capture, capture+DSN+capture, and capture+DSN+capture+DSN. The enriched cDNA libraries were sequenced using a HiSeq paired-end 100-bp protocol at BGI HongKong. See Supplemental Methods for full details.
Branchpoint validation by nested RT-PCR
RNA from cell lines was extracted from HEK293T cells or HeLa cells of SF3B1 wild type or K700E background (gift from H3 Biomedicine). RNA from multiple vertebrate tissues was extracted from total brains of mouse or rat, or 24-h zebrafish whole embryos. Total RNA was isolated and digested with DNase I to remove DNA contamination. cDNA was synthesized using random 9-mer primers and reverse transcribed. PCR was then performed on the cDNA first using the outer primer pair (Supplemental Table S11), followed by a second PCR using “nested” primers (Supplemental Table S11), with the initial PCR product used as the template. The product of the second PCR was then examined by electrophoresis, and the appropriate bands were excised and purified. PCR products were then cloned into pCR2.1 using a TOPO TA Cloning kit (Invitrogen, Cat.# K4500) and transformed into TOP10 Escherichia coli cells. Individual colonies were grown, and plasmid DNA was isolated and was subsequently sequenced. See Supplemental Methods for full details.
Data access
All raw sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP094107 and SRP094058. Perl scripts used in identifying lariat reads are available on the lab website (http://fairbrother.biomed.brown.edu/data/Lariat2016/), on GitHub (https://github.com/allisontaggart/lariat), and in the Supplemental Material. Branchpoint data tables are available in Supplemental Tables S1–S8, on the lab website (http://fairbrother.biomed.brown.edu/data/Lariat2016/), and in a searchable, public session on the UCSC Genome Browser (https://genome.ucsc.edu/cgi-bin/hgTracks?hgS_doOtherUser=submit&hgS_ otherUserName=allisontaggart&hgS_otherUserSessionName= hg19_BPs).
Supplementary Material
Acknowledgments
We thank Silvia Buonamici (H3 Biomedicine) for the kind gift of SF3B1 engineered HeLa and NALM-6 cell lines and the sharing of alternative 3′ss data of SF3B1 engineered NALM-6 cells. We also thank the Mouse Transgenic and Gene Targeting Facility, the Richard Freiman lab and the Robbert Creton lab at Brown University for their gift of animal tissues. We thank Tom Mayo for valuable discussions and his assistance with understanding the kinetics of alternate steady state pathways. Part of this research was conducted using computational resources and services at the Center for Computation and Visualization and the Genomic Core Facility at Brown University, supported by the National Institutes of Health, National Institute of General Medical Sciences (NIGMS grant number P30GM103410, and NCRR grant numbers P30RR031153, P20RR018728, and S10RR02763), National Science Foundation (EPSCoR grant number 0554548), Lifespan Rhode Island Hospital, and the Division of Biology and Medicine, Brown University. This work was supported by National Institutes of Health under award number R01GR527276 and by NIGMS under grant number R01GM105681.
Author contributions: A.J.T., C.-L.L., B.S., S.K., and C.H. performed the experiments. W.G.F., C.-L.L., and A.J.T. prepared the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.202820.115.
References
- Alioto TS. 2007. U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic Acids Res 35: D110–D115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awan AR, Manfredo A, Pleiss JA. 2013. Lariat sequencing in a unicellular yeast identifies regulated alternative splicing of exons that are evolutionarily conserved with humans. Proc Natl Acad Sci 110: 12762–12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitton DA, Rallis C, Jeffares DC, Smith GC, Chen YY, Codlin S, Marguerat S, Bahler J. 2014. LaSSO, a strategy for genome-wide mapping of intronic lariats and branch points using RNA-seq. Genome Res 24: 1169–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnal S, Vigevani L, Valcarcel J. 2012. The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 11: 847–859. [DOI] [PubMed] [Google Scholar]
- Buonamici S, Lim KH, Feala J, Park E, Corson L, Aicher M, Aird D, Chan B, Corcoran E, Darman R, et al. 2014. SF3B1 mutations induce aberrant mRNA splicing in cancer and confer sensitivity to spliceosome inhibition. In Proceedings of the American Association of Cancer Research annual meeting, p. 2932, San Diego, CA. [Google Scholar]
- The Cancer Genome Atlas Network. 2012. Comprehensive molecular portraits of human breast tumours. Nature 490: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellini A, Felder E, Rossi JJ. 1986. Yeast pre-messenger RNA splicing efficiency depends on critical spacing requirements between the branch point and 3′ splice site. EMBO J 5: 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua K, Reed R. 2001. An upstream AG determines whether a downstream AG is selected during catalytic step II of splicing. Mol Cell Biol 21: 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrionero A, Minana B, Valcarcel J. 2011. Reduced fidelity of branch point recognition and alternative splicing induced by the anti-tumor drug spliceostatin A. Genes Dev 25: 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvelo A, Hallegger M, Smith CW, Eyras E. 2010. Genome-wide association between branch point properties and alternative splicing. PLoS Comput Biol 6: e1001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshler JO, Rossi JJ. 1991. Unexpected point mutations activate cryptic 3′ splice sites by perturbing a natural secondary structure within a yeast intron. Genes Dev 5: 1252–1263. [DOI] [PubMed] [Google Scholar]
- Dietrich RC, Peris MJ, Seyboldt AS, Padgett RA. 2001. Role of the 3′ splice site in U12-dependent intron splicing. Mol Cell Biol 21: 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes PA, Cooke NE, Liebhaber SA. 1992. A native RNA secondary structure controls alternative splice-site selection and generates two human growth hormone isoforms. J Biol Chem 267: 14902–14908. [PubMed] [Google Scholar]
- Gahura O, Hammann C, Valentova A, Puta F, Folk P. 2011. Secondary structure is required for 3′ splice site recognition in yeast. Nucleic Acids Res 39: 9759–9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao K, Masuda A, Matsuura T, Ohno K. 2008. Human branch point consensus sequence is yUnAy. Nucleic Acids Res 36: 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrey SM, Voelker R, Berglund JA. 2006. An extended RNA binding site for the yeast branch point-binding protein and the role of its zinc knuckle domains in RNA binding. J Biol Chem 281: 27443–27453. [DOI] [PubMed] [Google Scholar]
- Gooding C, Clark F, Wollerton MC, Grellscheid SN, Groom H, Smith CW. 2006. A class of human exons with predicted distant branch points revealed by analysis of AG dinucleotide exclusion zones. Genome Biol 7: R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould GM, Paggi JM, Guo Y, Phizicky DV, Zinshteyn B, Wang ET, Gilbert WV, Gifford DK, Burge CB. 2016. Identification of new branch points and unconventional introns in Saccharomyces cerevisiae. RNA 22: 1522–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goux-Pelletan M, Libri D, d'Aubenton-Carafa Y, Fiszman M, Brody E, Marie J. 1990. In vitro splicing of mutually exclusive exons from the chicken β-tropomyosin gene: role of the branch point location and very long pyrimidine stretch. EMBO J 9: 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall KB, Green MR, Redfield AG. 1988. Structure of a pre-mRNA branch point/3′ splice site region. Proc Natl Acad Sci 85: 704–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallegger M, Sobala A, Smith CW. 2010. Four exons of the serotonin receptor 4 gene are associated with multiple distant branch points. RNA 16: 839–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. 2013. Natural RNA circles function as efficient microRNA sponges. Nature 495: 384–388. [DOI] [PubMed] [Google Scholar]
- Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, Bowcock AM. 2013. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet 45: 133–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfman DM, Ricci WM. 1989. Branch point selection in alternative splicing of tropomyosin pre-mRNAs. Nucleic Acids Res 17: 5633–5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz GZ, Stormo GD. 1999. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics 15: 563–577. [DOI] [PubMed] [Google Scholar]
- Hoskins AA, Moore MJ. 2012. The spliceosome: a flexible, reversible macromolecular machine. Trends Biochem Sci 37: 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, et al. 2012. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150: 1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquier A, Rosbash M. 1986. RNA splicing and intron turnover are greatly diminished by a mutant yeast branch point. Proc Natl Acad Sci 83: 5835–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan R, Hartnett S, Voelker RB, Berglund JA, Staley JP, Baumann P. 2013. Intronic sequence elements impede exon ligation and trigger a discard pathway that yields functional telomerase RNA in fission yeast. Genes Dev 27: 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz Y, Wang ET, Airoldi EM, Burge CB. 2010. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Burge CB. 2001. A computational analysis of sequence features involved in recognition of short introns. Proc Natl Acad Sci 98: 11193–11198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. 2013. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495: 333–338. [DOI] [PubMed] [Google Scholar]
- Mercer TR, Clark MB, Andersen SB, Brunck ME, Haerty W, Crawford J, Taft RJ, Nielsen LK, Dinger ME, Mattick JS. 2015. Genome-wide discovery of human splicing branchpoints. Genome Res 25: 290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Gallwitz D. 1987. Nuclear pre-mRNA splicing in the fission yeast Schizosaccharomyces pombe strictly requires an intron-contained, conserved sequence element. EMBO J 6: 1757–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA. 2012. New connections between splicing and human disease. Trends Genet 28: 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA, Konarska MM, Grabowski PJ, Hardy SF, Sharp PA. 1984. Lariat RNA's as intermediates and products in the splicing of messenger RNA precursors. Science 225: 898–903. [DOI] [PubMed] [Google Scholar]
- Papasaikas P, Tejedor JR, Vigevani L, Valcarcel J. 2015. Functional splicing network reveals extensive regulatory potential of the core spliceosomal machinery. Mol Cell 57: 7–22. [DOI] [PubMed] [Google Scholar]
- Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. 2013. Cell-type specific features of circular RNA expression. PLoS Genet 9: e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Yang B, Wu T, Huang J, Tang P, Zhou Y, Zhou J, Qiu J, Jiang L, Li H, et al. 2014. Mechanisms for U2AF to define 3′ splice sites and regulate alternative splicing in the human genome. Nat Struct Mol Biol 21: 997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CW, Nadal-Ginard B. 1989. Mutually exclusive splicing of α-tropomyosin exons enforced by an unusual lariat branch point location: implications for constitutive splicing. Cell 56: 749–758. [DOI] [PubMed] [Google Scholar]
- Smith CW, Chu TT, Nadal-Ginard B. 1993. Scanning and competition between AGs are involved in 3′ splice site selection in mammalian introns. Mol Cell Biol 13: 4939–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Query CC, Konarska MM. 2008. “Nought may endure but mutability”: spliceosome dynamics and the regulation of splicing. Mol Cell 30: 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DJ, Konarska MM, Query CC. 2009. Insights into branch nucleophile positioning and activation from an orthogonal pre-mRNA splicing system in yeast. Mol Cell 34: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southby J, Gooding C, Smith CW. 1999. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of α-actinin mutally exclusive exons. Mol Cell Biol 19: 2699–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart AJ, DeSimone AM, Shih JS, Filloux ME, Fairbrother WG. 2012. Large-scale mapping of branchpoints in human pre-mRNA transcripts in vivo. Nat Struct Mol Biol 19: 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng CK, Cheng SC. 2008. Both catalytic steps of nuclear pre-mRNA splicing are reversible. Science 320: 1782–1784. [DOI] [PubMed] [Google Scholar]
- Vogel J, Hess WR, Borner T. 1997. Precise branch point mapping and quantification of splicing intermediates. Nucleic Acids Res 25: 2030–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward LD, Kellis M. 2012. Interpreting noncoding genetic variation in complex traits and human disease. Nat Biotechnol 30: 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell 13: 91–100. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. [DOI] [PubMed] [Google Scholar]
- Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, et al. 2014. A comparative encyclopedia of DNA elements in the mouse genome. Nature 515: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.