

Abstract
Objective
Persistent immune activation is thought to contribute to increased CVD risk in HIV and statins may help modulate systemic immune activation. We aimed to compare the effects of two key statins on markers of systemic immune activation and arterial inflammation in the HIV population.
Design
Double-blind, active-controlled, parallel-group comparative trial performed in 45 sites
Methods
252 ART-treated HIV-infected participants with dyslipidemia were randomized (1:1) to pitavastatin 4mg daily vs. pravastatin 40mg daily in the INTREPID Trial. In this analysis of the INTREPID Trial, we assessed markers of immune activation and arterial inflammation using a modified intent to treat population. This trial is registered with ClinicalTrials.gov (NCT01301066).
Results
126 participants were randomized to receive pitavastatin and 126 to pravastatin. 99 participants in the pitavastatin group and 91 participants in the pravastatin group completed the study. Median age was 50(45,56) years [median(IQR)]. Baseline, LDL-C was 153(135,171) mg/dL, log HIV-1 viral load was 1.1±0.2 copies/mL and CD4 count was 580(439,794) cells/mm3. At Week 52, the pitavastatin group had a significantly greater reduction (% change) compared to pravastatin in sCD14 (−10.0 vs. 0.6%, P=0.02), oxLDL (−26.9 vs. −17.5%, P=0.02), and Lp-PLA2 (−26.6 vs. −15.5%, P=0.005) (pitavastatin vs. pravastatin).
Conclusions
52 weeks of pitavastatin 4mg daily (vs. pravastatin 40mg daily) led to a greater reduction in select markers of immune activation and arterial inflammation (sCD14, oxLDL and LpPLA2) among HIV-infected participants. Further work is needed to assess whether immune-modulatory effects of pitavastatin reduce CVD risk in HIV.
Keywords: HIV, statin, atherosclerosis, inflammation, cardiovascular disease
Introduction
Cardiovascular disease (CVD) risk is increased among HIV-infected individuals, including those with viral suppression on anti-retroviral therapy (ART).[1] Atherosclerosis, in particular, is a highly inflammatory process that is characterized by endothelial cell dysfunction, cytokine production, and the recruitment of monocytes to the intima of arteries. Among HIV-infected subjects on ART, arterial inflammation has been found to be increased compared to non-HIV-infected controls and the mechanisms underlying this difference remain to be elucidated.[2] Persistent immune activation, even after effective ART, has been postulated as a possible contributor.[3, 4] Development of successful strategies to reduce immune activation and arterial inflammation is a critical goal to reduce potential co-morbidities in HIV. However, relatively little is known regarding effective treatment strategies for residual immune activation among individuals living with HIV on ART.
Statins have been studied as a possible therapy to reduce CVD risk and immune activation in HIV-infected subjects on ART and have been shown to affect several indices of immune activation such as the proportion of activated T-cells and tissue factor positive patrolling monocytes.[5–9] Data are currently lacking for the effects of newer statins, such as pitavastatin, which have been found to have less drug-drug interactions with ART compared to older statins,[10] because of primary metabolism via glucuronidation as opposed to the cytochrome P450 system. Animal and in vitro data suggest potent effects of pitavastatin on inflammatory indices[11–13] but such effects have not been previously assessed in the HIV population. Currently, among individuals living with HIV, pravastatin is a recommended statin drug. As such we compared effects of pitavastatin to this commonly used statin on markers of arterial inflammation and immune activation in a large double-blind, active-controlled, parallel-group study in which pitavastatin 4mg had a greater effect in reducing low density lipoprotein cholesterol (LDL-C) after 52 weeks of statin therapy compared to pravastatin 40mg in HIV-infected participants.[14] In addition, the INTREPID study has shown that pitavastatin does not aggravate glucose parameters, an important consideration in choice of statins among HIV-infected patients.[15] We hypothesized that pitavastatin, at the recommended daily dose of 4mg, would have a greater effect in reducing key indices of arterial inflammation and immune activation, related to CVD in HIV, compared with pravastatin 40mg daily. No prior study has evaluated the effects of pitavastatin on immune indices and markers of arterial inflammation in HIV-infected patients.
Methods
Study design and participants
HIV-infected patieNts and TREatment with PItavastatin vs. pravastatin for Dyslipidemia (INTREPID) was a randomized, double-blind, double-dummy, active-controlled, parallel-group, superiority trial conducted at 45 sites in the United States and Puerto Rico. The study was performed from February 23, 2011 to March 29, 2013. The primary aim of the study was to compare the effect of pitavastatin 4mg vs. pravastatin 40mg on LDL-C reduction in individuals with HIV and dyslipidemia over 12 weeks, followed by a 40-week safety extension period and was approved by the Institutional Review Board at each participating center. Reporting of the primary results also included safety data. All participants provided written informed consent prior to enrollment. Males or females who were 18 to 70 years of age at the time of consent, had documented HIV infection, were receiving ART (except darunavir) for 6 months or greater, and had HIV-1 RNA <200 copies/mL and a CD4 count of >200 cells/mm3 for greater than 3 months were eligible. Statin-naïve participants and participants receiving statins at the initial screen, after a minimum 4-week washout period, were eligible for study participation. Participants were excluded for the following: conditions causing secondary dyslipidemia, history of coronary heart disease (CAD) or a CAD equivalent, active systemic infections, or prior or current muscular or neuromuscular disease of any type.
After the initial screening, all eligible participants underwent a 4-week dietary stabilization period, based on the Therapeutic Lifestyle Changes (TLC) diet[16], and were counseled on the TLC diet after the initial screen and for the remainder of the study period. After the wash-out and diet stabilization period, lipid eligibility was determined by a LDL-C value ≥130 mg/dL and ≤220 mg/dL and triglyceride (TG) value ≤400 mg/dL. Participants who met all eligibility criteria were then randomized 1:1 to receive active pitavastatin 4mg orally once daily and a matching pravastatin placebo versus active pravastatin 40mg orally once daily and a matching pitavastatin placebo for 52 weeks.
The study protocol called for exploratory analyses of future biomarkers. The present work represents an investigator-initiated biomarker analysis, conducted independently of the primary study, with specific markers chosen based on emerging research trends in the field related to an increasing appreciation of the role played by immune activation and arterial inflammation in the pathogenesis of CVD in HIV patients on ART, and the potential importance of statins to modulate these effects. In this context, percent change in oxidized LDL (oxLDL) as well as soluble CD163 (sCD163), high-sensitivity IL-6 (hsIL-6), monocyte chemoattractant protein-1 (MCP-1), soluble CD14 (sCD14), and lipoprotein-associated phospholipase 2 (Lp-PLA2) were assessed and changes from Baseline to Week 12 and Week 52 are reported.
Randomization and masking
Eligible participants were randomly assigned (1:1) using a central interactive voice response system. The randomization schedule was prepared by the biostatistics department of the contract research organization performing the trial. Treatment codes were not available to study investigators, site staff, or participants, and each participant received a matching placebo.
Procedures
Participants were seen every 4 weeks for the first 12 weeks of the study, with subsequent visits occurring quarterly through Week 52. EDTA plasma samples for markers of systemic immune activation and arterial inflammation were obtained at Baseline, Week 12, and Week 52 and were stored at −80°C. The specific numbers of specimens available for each analysis are listed in the data tables.
Measurements of the creatinine, thyroid stimulating hormone (TSH), lipid panel, CD4 count, and HIV-1 RNA were performed using standard techniques. Plasma sCD163 levels were quantified by ELISA according to manufacturer’s protocol (Trillium Diagnostics LLC, Bangor, ME). Plasma levels of hsIL-6, sCD14, MCP-1, and Lp-PLA2 were quantified by ELISA according to manufacturer’s protocol (R&D Systems, Minneapolis, MN). Plasma oxLDL levels were quantified by ELISA according to manufacturer’s protocol (Mercodia, Inc.) All ELISAs were performed in duplicate with appropriate quality control samples.
Statistical Analysis
The primary objective of INTREPID was to compare the between-group difference in the percent change in fasting serum LDL-C at Week 12 between the pitavastatin and pravastatin groups. In this current analysis, changes from Baseline to Week 12 and Baseline to Week 52 in oxLDL sCD163, hsIL-6, MCP-1, sCD14, and Lp-PLA2 are reported. Objectives of this current study included a comparison of the between-group absolute difference in these markers from Baseline to Week 12 and from Baseline to Week 52 and the within-group absolute difference in these markers from Baseline to Week 12 and from Baseline to Week 52. All available data within a modified intent-to-treat population (mITT) using a last observation carried forward method were included in the primary analyses. The mITT population included all randomized participants who had at least one post-baseline visit with blood samples available for the assessment of biomarkers. For those terminating prior to Week 12, the last post-baseline observation was carried forward to the Week 12 endpoint. Similarly, for those terminating after Week 12 but prior to Week 52, the last post-baseline observation was carried forward to the Week 52 endpoint. Sensitivity analyses using a per-protocol population (PPP) were performed and results shown in the Supplement. For the Week 12 endpoint, the PPP consisted of participants who had Baseline and Week 12 samples. For the Week 52 endpoint, the PPP consisted of participants who had Baseline and Week 52 samples. The study was powered for the primary endpoint of change in LDL. For the current analysis, with available data on change in biomarkers in 230 patients in the mITT analysis, the study had 90% power to detect a 0.43 SD difference between the groups.
Normality of the data was assessed using a Shapiro-Wilk test and normally distributed data are presented as mean ±SD and non-normally distributed data are presented by the median and interquartile range (IQR). The biomarkers were all non-normally distributed except for Lp-PLA2 and are all presented as median and IQR. Baseline comparisons between the pravastatin group and the pitavastatin group were made using the student’s t-test for normally distributed continuous variables, Wilcoxon rank sum test for non-normally distributed continuous variables, and χ2 test for categorical variables. Comparisons of the change in biomarkers over 12 weeks and 52 weeks between groups were assessed using the Wilcoxon rank sum test. To assess changes in the biomarkers over 12 weeks and 52 weeks within a group, a Wilcoxon signed rank test was used. Bivariate analyses between continuous variables were performed using a Spearman’s rank correlation coefficient if at least one variable was non-normally distributed. Sensitivity analyses comparing baseline levels of biomarkers, in a combined analysis of both statin groups, between males and females and the overall change in biomarkers at Week 52 stratified by statin group and sex were performed using the Wilcoxon rank sum test. Statistical analyses were performed using SAS JMP software (version 11.0; SAS Institute). The trial is registered on ClinicalTrials.gov (NCT01301066).
Results
Between February 23, 2011 to March 29, 2013, 594 participants were screened (Figure 1). Of these, a total of 342 were deemed ineligible, including 190 not meeting inclusion based on an LDL-C value ≥ 130 mg/dL and ≤ 220 mg/dL and triglyceride (TG) value ≤400 mg/dL. A total of 252 participants were randomly assigned to receive pitavastatin (126 participants) or pravastatin (126 participants). In the present analyses, all study participants with available blood samples for biomarkers at Baseline and at least one blood sample after the Baseline visit assessed for biomarkers were included (n=230; Figure 1).
Figure 1. Consort diagram.
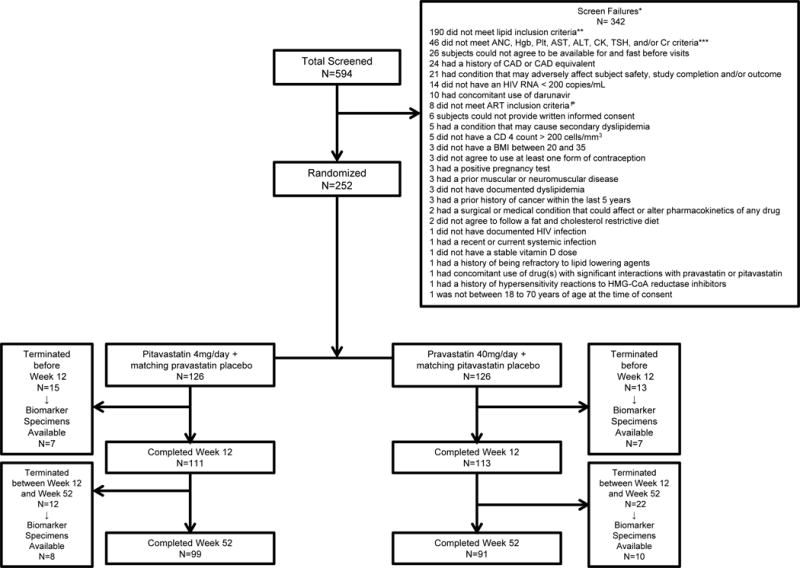
Five hundred and ninety-four participants were screened for enrollment; 342 participants were screen failures. Two-hundred and fifty-two participants were enrolled and randomized.
* Out of the 342 screen failures, 27 participants had at least 2 or more criteria for inclusion that were not met and/or exclusion criteria that were met
** Lipid inclusion criteria: elevated fasting plasma LDL-C≥130 mg/dL and ≤220 mg/dL, and TG levels ≤400 mg/dL, following statin washout and dietary lead-in period of a minimum of 4 weeks.
*** Additional lab inclusion criteria: absolute neutrophil count >750 cells/mm3, hemoglobin≥9.0 g/dL for female participants and ≥10.0 g/dL for male participants, platelets ≥100,000/mm3, ALT and AST ≤2.5 × upper limit of normal (ULN); note: participants co-infected with Hepatitis B or C were required to have ALT and AST ≤1.5 ULN, fasting serum glucose ≤125 mg/dL, CK ≤ × ULN. If a transient increase in CK level was suspected due to exercise or trauma, CK may have been repeated at screening after an “exercise washout” at the discretion of the Investigator, serum creatinine ≤1.3 × ULN and estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73m2 based on the modification of diet in renal disease (MDRD) equation at http://www.nephron.com/MDRD_GFR.cgi. if a creatinine level was suspected to be temporarily increased due to factors such as dehydration, creatinine testing may have been repeated and the eGFR may have been recalculated at screening at the discretion of the Investigator, and TSH <1.5 × ULN
₱ART inclusion criteria: On ART, except for darunavir, for at least 6 months prior to randomization and no change to the ART regimen within 3 months prior to randomization or anticipated need to change ART during the first 12 weeks of the study.
Abbreviations: ANC, absolute neutrophil count; hgb, hemoglobin; plt, platelet; AST, aspartate aminotransferase; ALT, alanine transferase; CK, creatinine kinase; TSH, thyroid stimulating hormone; Cr, creatinine; CAD, coronary heart disease; HIV RNA, Human Immunodeficiency Virus ribonucleic acid; ART, anti-retroviral therapy; BMI, body mass index; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; AE, adverse event; SAE, serious adverse event
Baseline Demographics
Overall, the median age for the study participants was 50(45, 56) years. Eighty-seven percent were male and 26% were Hispanic or Latino. The baseline LDL-C was 153(135, 171) mg/dL. The median Framingham Risk Score was 5(3, 9)%. With respect to HIV-specific parameters, baseline log HIV-1 viral load was 1.1±0.2 copies, and the CD4 count was 580(439, 794) cell/mm3 (Table 1; mITT population). Differences in baseline parameters, including immune activation markers were not seen other than for a small difference in CD4 count between groups. Baseline demographics for participants who did not have available blood samples for assessment of markers of immune activation and arterial inflammation (n=22) were not included in this analysis and were not significantly different from the demographics of those included (data not shown).
Table 1. Baseline Demographics and Markers of Immune Activation and Arterial Inflammation (mITT population).
Normally distributed data are reported as mean ± standard deviation and non-normally distributed data are reported as median (interquartile range), except for cardiac biomarkers which are all presented as median (interquartile range). Data are shown for mITT population.
| Pitavastatin | Pravastatin | |
|---|---|---|
| Age | 50 (46,56) N = 113 |
50 (45,56) N = 117 |
| Sex, % Male | 85% (96/113) | 89% (104/117) |
| Race White Black Asian Other |
84% (95/113) 13% (15/113) 1% (1/113) 2% (2/113) |
77% (90/117) 18% (21/117) 1% (1/117) 4% (5/117) |
| Hispanic | 25% (28/113) | 27% (32/117) |
| Hepatitis B/C | 8% (9/113) | 10% (12/117) |
| eGFR (ml/min/1.73m2) | 78 (70, 90) N = 113 |
78 (71, 90) N = 117 |
| TSH (mIU/L) | 1.6 (1.2, 2.3) N = 113 |
1.5 (1.1, 2.2) N = 117 |
| Total Cholesterol (mg/dL) | 236 (217, 260) N = 113 |
235 (214, 256) N = 117 |
| HDL-C (mg/dL) | 47 (39, 55) N = 113 |
47 (41, 56) N = 117 |
| LDL-C (mg/dL) | 155 (133, 172) N = 108 |
152 (136, 170) N = 116 |
| Triglycerides (mg/dL) | 149 (112, 220) N = 113 |
155 (113, 211) N = 117 |
| BMI (kg/m2) | 26 (24, 30) N = 113 |
28 (25, 31) N = 117 |
| Framingham Risk Score (%) | 5 (3, 10) N = 113 |
6 (3, 8) N = 117 |
| Log HIV-1 Viral Load (copies/ml) | 1.1±0.2 N = 112 |
1.1±0.2 N = 115 |
| CD4 count (cells/mm3)* | 650 (441, 859) N = 112 |
561 (433, 751) N = 117 |
| sCD163 (ng/mL) | 1,030 (731, 1,369) N = 112 |
949 (737, 1,139) N = 117 |
| hsIL-6 (pg/mL) | 0.9 (0.8, 1.4) N = 112 |
1.1 (0.7, 1.7) N = 117 |
| MCP-1 (pg/ml) | 142.2 (110.9,189.2) N = 112 |
142.6 (107.4, 180.7) N = 115 |
| sCD14 (ng/mL) | 1,801 (1,348, 2,276) N = 112 |
1,752 (1,382, 2,057) N = 117 |
| oxLDL (U/L) | 76.8 (60.6, 93.3) N = 112 |
77.9 (63.9, 88.6) N = 116 |
| Lp-PLA-2 (ng/mL) | 194 (157, 227) N = 112 |
183 (137, 240) N = 117 |
No differences between baseline values unless indicated,
P-value =0.03
Abbreviations: eGFR, estimated glomerular filtration rate; TSH, thyroid stimulating hormone; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein cholesterol; BMI, body mass index; HIV-1, Human Immunodeficiency Virus-1; sCD163, soluble CD163; hsIL-6, high sensitivity interleukin-6, MCP-1, monocyte chemoattractant protein-1; sCD14, soluble CD14; oxLDL, oxidized low density lipoprotein, Lp-PLA2, lipoprotein-associated phospholipase A2
Changes in Markers of Immune Activation and Arterial Inflammation
At Week 12, the pitavastatin group had a significantly greater reduction (% change) compared to pravastatin in oxLDL (−25.5 vs. −18.6%, P=0.001) and Lp-PLA2 (−23.4 vs. −17.4%, P=0.02, [pitavastatin vs. pravastatin]) (Table 2A). For absolute changes see Table 2A.
Table 2.
Data are shown as median (interquartile range); Significant between group P-values are shown in bold.
| A. Change in Markers of Immune Activation and Arterial Inflammation at Week 12 (mITT population) | ||||||
|---|---|---|---|---|---|---|
| Change in Pitavastatin | Change in Pravastatin | P-value* | Percent Change in Pitavastatin | Percent Change in Pravastatin | P-value* | |
| sCD163 (ng/mL) | −26.1 (−122.8, 130.8) N=112 |
−21.1 (−139.8, 110.3) N=116 |
0.88 | −3.8 (−14.5, 11.9) N=112 |
−2.1 (−14.9, 12.6) N=116 |
0.93 |
| hsIL-6 (pg/mL) | 0.0 (−0.3, 0.2) N=112 |
0.0 (−0.4, 0.1) N=116 |
0.49 | −2.5 (−27.7, 27.3) N=112 |
−2.2 (−30.9, 21.1) N=116 |
0.55 |
| MCP-1 (pg/ml) | −8.5 (−30.8, 18.0) N=110 |
−4.0 (−28.1, 17.5) N=114 |
0.62 | −5.3 (−18.5, 12.8) N=110 |
−2.9 (−17.0, 14.5) N=114 |
0.57 |
| sCD14 (ng/mL) | −37.1 (−425.8, 174.0)** N=112 |
−81.9 (−255.7, 106.8)** N=116 |
0.83 | −1.9 (−22.3, 10.6) N=112 |
−4.1 (−14.0, 8.1) N=116 |
0.90 |
| oxLDL (U/L) | −18.0 (−29.9, −10.8)** N=112 |
−13.4 (−23.6, −5.5) ** N=115 |
0.005 | −25.5 (−34.3, −16.4) N=112 |
−18.6 (−28.8, −8.0) N=115 |
0.001 |
| Lp-PLA2 (ng/mL) | −44.8 (−67.8, −19.8)** N=112 |
−30.2 (−52.7, −10.4)** N=116 |
0.01 | −23.4 (−36.2, −12.5) N=112 |
−17.4 (−29.8, −5.9) N=116 |
0.02 |
| B. Change in Markers of Immune Activation and Arterial Inflammation at Week 52 (mITT population) | ||||||
|---|---|---|---|---|---|---|
| Change in Pitavastatin | Change in Pravastatin | P-value* | Percent Change in Pitavastatin | Percent Change in Pravastatin | P-value* | |
| sCD163 (ng/mL) | 8.6 (−138.5, 122.2) N=98 |
−26.2 (−126.7, 128.6) N=98 |
0.92 | 1.1 (−12.3, 16.5) N=98 |
−2.2 (−13.9, 16.7) N=98 |
0.74 |
| hsIL-6 (pg/mL) | 0.0 (−0.3, 0.4) N=98 |
0.1 (−0.3, 0.4) N=98 |
0.57 | 1.9 (−28.2, 35.3) N=98 |
7.9 (−22.8, 40.9) N=98 |
0.41 |
| MCP-1 (pg/mL) | −2.2 (−31.2, 26.1) N=96 |
−2.2 (−21.7, 26.5) N=95 |
0.95 | −1.3 (−20.6, 21.9) N=96 |
−1.4 (−16.7, 18.5) N=95 |
0.91 |
| sCD14 (ng/mL) | −156.4 (−501.6, 114.5)** N=98 |
10.7 (−294.6, 307.3) N=98 |
0.01 | −10.0 (−23.2, 9.7) N=98 |
0.6 (−15.0, 19.2) N=98 |
0.02 |
| oxLDL (U/L) | −20.5 (−33.4, −8.8)** N=97 |
−13.2 (−25.2, −4.1)** N=95 |
0.02 | −26.9 (−36.0, −10.8) N=97 |
−17.5 (−30.0, −4.5) N=95 |
0.02 |
| Lp-PLA2 (ng/mL) | −44.3 (−77.8, −10.7)** N=97 |
−26.7 (−52.0, −5.0)** N=96 |
0.02 | −26.6 (−35.7, −8.4) N=97 |
−15.5 (−28.1, −3.2) N=96 |
0.005 |
P-value for between group difference
P<0.05 for within group difference
Abbreviations: sCD163, soluble CD163; hsIL-6, high sensitivity interleukin-6, MCP-1, monocyte chemoattractant protein-1; sCD14, soluble CD14; oxLDL, oxidized low density lipoprotein, Lp-PLA2, lipoprotein-associated phospholipase A2
A. Data are shown for mITT population with biomarker specimens available for Week 12.
B. Data are shown for mITT population with biomarker specimens available for Week 52.
At Week 52, the pitavastatin group had a significantly greater reduction (% change) compared to pravastatin in sCD14 (−10.0 vs. 0.6%, P=0.02), oxLDL (−26.9 vs. −17.5%, P=0.02), and Lp-PLA2 (−26.6 vs. −15.5%, P=0.005) (pitavastatin vs. pravastatin) (Table 2B; Figure 2). Within the pitavastatin group, the absolute changes in immune activation and inflammatory indices were significant for sCD14 [−156.4 (−501.6, 114.5)ng/ml (P=0.003)], oxLDL [−20.5(−33.4, −8.8)U/L (P<0.0001)], and Lp-PLA2 [−44.3 (−77.8, −10.7)ng/mL (P<0.0001). Within the pravastatin group, the absolute changes were significant for oxLDL [−13.2 (−25.2, −4.1)U/L (P<0.0001)] and Lp-PLA2 [−26.7 (−52.0, −5.0)ng/ml (P<0.0001)] (Table 2B).
Figure 2. Percent change in sCD14, oxLDL, and Lp-PLA2 at Week 52 in Pitavastatin group compared to Pravastatin group (mITT Population).
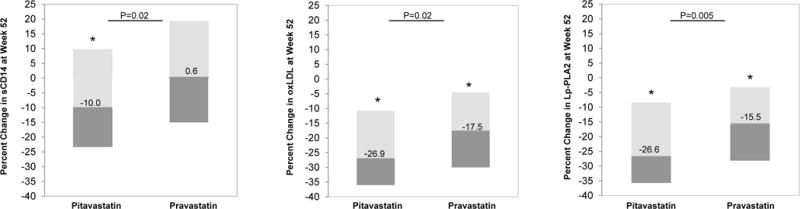
At Week 52, the pitavastatin group had a significantly greater reduction (% change) in sCD14, oxLDL, and Lp-PLA2 (sCD14: −10.0 vs. 0.6%, P=0.02; oxLDL −26.9 vs. −17.5%, P=0.02; Lp-PLA2 −26.6 vs. −15.5%, P=0.005). Data are median (IQR). P-values shown are for between group difference in percent change at Week 52.
Data are shown for mITT population with biomarker specimens available for Week 52.
Abbreviations: sCD14, soluble CD14; oxLDL, oxidized low density lipoprotein; Lp-PLA2, lipoprotein-associated phospholipase A2
* P<0.05 for within group difference
Changes in sCD14, oxLDL and Lp-PLA2 were compared at Week 52, and there was a high degree of correlation between change over 52 weeks in oxLDL and Lp-PLA2 (ρ=0.45, P<0.0001). The change in LDL-C at Week 52 was also strongly correlated with the changes in oxLDL (ρ=0.59, P<0.0001) and Lp-PLA2 at 52 weeks (ρ=0.64, P<0.0001) whereas the change in sCD14 was independent of the changes in LDL-C (ρ=0.04, P=0.63). The correlations between the change in LDL-C at Week 52 and the changes in sCD14, oxLDL and Lp-PLA2 were qualitatively similar between the pitavastatin and pravastatin group (Supplemental Table 1).
Predictive Markers of Change in Immune Activation and Arterial Inflammation in Response to Statins
Neither CD4 count nor viral load predicted change in sCD14, oxLDL or Lp-PLA2 at Week 52 (Table 3). Similarly, traditional risk markers including Framingham Risk Score and age did not predict changes in these markers. In contrast, baseline LDL-C was a significant predictor of changes in oxLDL (ρ=−0.17, P=0.02) and an even stronger predictor of change in Lp-PLA2 (ρ=−0.27, P=0.0002). In addition, baseline levels of each immune activation marker were significant and strong predictors of change in each marker, respectively (Table 3).
Table 3.
Correlation Analysis for Percent Change at Week 52 and Baseline Demographics (mITT population)
| Percent Change sCD14 | Percent Change oxLDL | Percent Change Lp-PLA2 | |
|---|---|---|---|
| Age | ρ= −0.07 P= 0.36 |
ρ= −0.07 P=0.32 |
ρ= −0.11 P=0.12 |
| Framingham Risk Score | ρ= −0.09 P=0.23 |
ρ= 0.05 P=0.48 |
ρ= −0.04 P=0.60 |
| CD4 count | ρ= 0.09 P=0.22 |
ρ= −0.01 P=0.87 |
ρ=0.02 P=0.74 |
| Log HIV-1 Viral Load | ρ= 0.04 P=0.54 |
ρ= 0.06 P=0.43 |
ρ=0.14 P=0.06 |
| LDL-C | ρ= −0.01 P=0.87 |
ρ= −0.17 P=0.02 |
ρ= −0.27 P=0.0002 |
| sCD14 | ρ= −0.48 P < 0.0001 |
ρ= −0.08 P=0.25 |
ρ= −0.03 P=0.67 |
| oxLDL | ρ= −0.02 P=0.75 |
ρ= −0.33 P < 0.0001 |
ρ= −0.03 P=0.66 |
| Lp-PLA2 | ρ= −0.04 P=0.56 |
ρ= 0.03 P=0.65 |
ρ= −0.27 P=0.0002 |
Significant P-values are shown in bold. Data are shown for mITT population with biomarker specimens available for Week 52.
Abbreviations: LDL-C, low density lipoprotein cholesterol; sCD14, soluble CD14; oxLDL, oxidized low density lipoprotein, Lp-PLA2, lipoprotein-associated phospholipase A
Stratification by Sex
For those variables that differed significantly between pitavastatin and pravastatin at Week 52, the changes among women were generally similar to those seen among men for oxLDL and Lp-PLA2 but qualitatively greater among women for sCD14 (−24.3 vs. 17.8% [pitavastatin vs. pravastatin, females], −6.0 vs. −2.0% [pitavastatin vs. pravastatin, males]) (Supplemental Table 2).
Sensitivity Analyses within the Per Protocol Population
Baseline demographics for the PPP are shown in Supplemental Table 3. Within the PPP, the pitavastatin group had a significantly greater reduction (% change) compared to the pravastatin group in oxLDL and Lp-PLA2 at Week 12 (Supplemental Table 4) and a significantly greater reduction (% change) in sCD14, oxLDL, and Lp-PLA2 at Week 52 (Supplemental Table 5). Similar to the mITT population, baseline LDL-C was a predictor of changes in oxLDL (ρ=˗0.17, P=0.03) and Lp-PLA2 (ρ=˗0.26, P=0.0007) (Supplemental Table 6). Changes in LDL-C at Week 52 strongly correlated with changes in oxLDL (ρ=0.58, P<0.0001) and changes in Lp-PLA2 at Week 52 (ρ=0.65, P<0.0001); and there was also a high degree of correlation between change over 52 weeks in oxLDL and Lp-PLA2 (ρ=0.44, P<0.0001). The correlations between the change in LDL-C at Week 52 and the changes in sCD14, oxLDL and Lp-PLA2 were qualitatively similar between the pitavastatin and pravastatin group as was seen in the mITT population (Supplemental Table 7). Sensitivity analyses by sex within the PPP revealed similar results to those of the mITT population (Supplemental Table 8).
Discussion
This study is the first and largest, prospective, active-controlled randomized trial evaluating the effect of two different statins on markers of systemic immune activation and arterial inflammation in HIV-infected subjects. The two statins compared in this study – pitavastatin and pravastatin – have in common a favorable pharmacokinetic (PK) profile when used together with ART, though there is a clinically significant interaction between pravastatin and darunavir that is not seen with pitavastatin.[17] We find that pitavastatin (vs. pravastatin) results in a greater percent and absolute reduction in sCD14, oxLDL, and LpPLA-2. Additionally, within each treatment group, levels of sCD14, oxLDL, and Lp-PLA2 were significantly reduced by Week 52 in the pitavastatin group while only oxLDL and Lp-PLA2 were significantly reduced by Week 52 in the pravastatin group.
Unlike other statins, such as atorvastatin and simvastatin, which are metabolized by the CYP450 3A4 iso-enzyme, pitavastatin is predominately metabolized by glucuronidation and only minimally by CYP450, marginally by CYP2C9 and to a lesser extent by CYP2C8 and has minimal interactions with anti-retroviral medications. In contrast, the known interaction between pravastatin and darunavir necessitated exclusion of patients receiving darunavir from the study.[17] Given the significant need to identify treatment strategies to reduce CVD risk in HIV-infected subjects on ART, it is highly desirable to identify statins such as pitavastatin that, not only demonstrate strong immune-modulatory effects, but also have a favorable PK profile when used together with ART.
In the current study, pitavastatin, but not pravastatin, led to a significant, reduction of 10% in levels of sCD14 at Week 52. Soluble CD14 has been found to be elevated in HIV-infected subjects as compared to non-HIV-infected subjects and has been found to be a predictor of mortality and progression of subclinical atherosclerosis in HIV-infected subjects. [4, 15, 18] In the SATURN-HIV study, a placebo-controlled study of rosuvastatin 10 mg/day, rosuvastatin similarly reduced sCD14 by approximately 10% at 48 weeks.[5] This reduction in monocyte activation, in light of the central role of monocytes and macrophages in atherogenesis, provides a possible mechanism by which statins could modulate CVD risk in the HIV population in addition to its lipid lowering effects.
In this study, pitavastatin also led to a greater reduction in oxLDL compared to pravastatin. Oxidized LDL is a pro-inflammatory form of LDL-C that has been found to be elevated in HIV-infected subjects compared to controls.[19] Notably, uptake of oxLDL by macrophages in the arterial wall is thought to contribute to foam cell generation; and oxLDL has also been shown to induce inflammatory changes in vascular smooth muscle cells and induce apoptosis of macrophages.[20, 21] Thus, the reduction of oxLDL in our study population provides another possible mechanism whereby statins could modulate CVD risk in the HIV-infected population. The percent reduction in oxLDL with pitavastatin in this current study was higher compared to that seen in a recent study by Nou et al., which reported a median reduction of oxLDL of −14.9% with atorvastatin in HIV-infected subjects over a 12 month period compared to a median reduction of −26.9% over 52 weeks in this analysis. Their study also found significant associations between changes in oxLDL and changes in non-calcified plaque, total plaque volume, positively remodeled plaque, and low attenuation plaque.[21] In SATURN-HIV, oxLDL was significantly reduced with rosuvastatin compared to placebo; and changes in oxLDL were associated with changes in sCD14 and changes in the percentage CD14dimCD16+ monocytes.[22] Together, these studies point to the possible effect of statins, through the reduction of oxLDL, to modulate the inflammation that characterizes accelerated atherosclerotic plaque formation and progression in HIV-infected subjects.
Pitavastatin as compared to pravastatin also led to a greater reduction in Lp-PLA2. Lp-PLA2 is a calcium-independent lipase that catalyzes a reaction on the surface of LDL that leads to the release of lysophosphatidylcholine, which has pro-oxidative effects, and oxidized fatty acids, which have pro-inflammatory effects. [23] HIV-infected subjects have also been shown to have abnormally elevated Lp-PLA2, a biomarker that has been associated with increased cardiovascular disease in both the general population and HIV-infected population.[24, 25] Statins have been found to decrease Lp-PLA2 in both HIV and non-HIV-infected subjects.[26] [27] [28] In this study, we demonstrate a larger percent reduction in Lp-PLA2 than was seen with rosuvastatin 10mg daily in SATURN-HIV.[26] Given both the pro-inflammatory and pro-oxidative effects of Lp-PLA2, it is plausible that the reduction of Lp-PLA2 with statin therapy could lead to a decrease in cardiovascular events among HIV-infected individuals.
We performed association analyses which suggested that higher levels of immune activation markers strongly predicted changes with statin therapy, suggesting a greater effect of statins among those with the highest degree of immune activation. In contrast, baseline immune function and HIV-1 viral load were not predictors of change over time with statin therapy. Baseline demographics did not predict change over time, but interestingly baseline LDL-C was a highly significant predictor of change in Lp-PLA2, suggesting a connection between dyslipidemia and arterial inflammation that may be statin amenable. In addition, changes in oxLDL and Lp-PLA2 were highly associated, suggesting a putative role for oxLDL in the promotion of arterial inflammation among HIV patients.
Similar to other studies evaluating the effects of statins (such as rosuvastatin [6, 26] and atorvastatin [21]) on immune activation in HIV-infected individuals, there was no significant change in the levels of sCD163 or hsIL-6 with either pitavastatin or pravastatin in our study. Levels of both sCD163 and IL-6 have been associated with atherosclerotic plaque in HIV-infected[3, 29] and non-HIV-infected subjects[30] and more recently with overall mortality in HIV-infected subjects[29, 31]. While the underlying mechanism of these effects remains unclear, the lack of a significant change in these biomarkers in our study, as in other statin studies, could relate to effects of the virus on specific immune pathways which are not modifiable by statins. Further research is needed to understand the clinical implications and mechanisms of the observed statin effects on immune function in this study.
Limitations of this study include the lack of data evaluating whether or not the changes seen in markers of immune activation and arterial inflammation related to changes in atherosclerotic plaque and/or cardiovascular events. Additionally, our study population included a specific group of HIV-infected participants with baseline dyslipidemia, i.e. LDL-C ≥130mg/dL to ≤220mg/dL; but our data on immune activation and arterial inflammation remain highly relevant, and we further explore relationships to change in LDL, not previously performed. The study was not specifically powered to assess changes in biomarkers, but was sufficiently large to detect clinically significant differences between the treatment groups. Strengths of this study include the randomization of two statins with favorable PK profiles in the context of ART and the relatively large sample size compared to other studies evaluating the effect of statins on these biomarkers. Importantly, participants were assessed early but also up to one year after initiation of therapy, permitting an investigation of the durability of the responses observed. Results were robust and similar in the mITT population and the per protocol population.
In conclusion, this large study is the first prospective, active-controlled, randomized trial to compare the effects of two statins, which are known to have minimal drug-drug interactions with ART, on markers of immune activation and arterial inflammation. This study provides novel data demonstrating that this new statin, pitavastatin, leads to a greater reduction than pravastatin in sCD14, oxLDL, and Lp-PLA2; and this effect is durable over 52 weeks. Analysis of data from this cohort additionally demonstrates that pitavastatin has a superior LDL lowering effect than pravastatin and that pitavastatin has net neutral effects on parameters of glucose homeostasis and insulin resistance.[15] This is important as other statins in HIV, such as rosuvastatin, have been shown to increase glucose and worsen insulin resistance.[32] Therefore, pitavastatin, through the reduction of indices of immune activation, its lipid-lowering effects, and its favorable PK profile when used in combination with ART, may be an effective treatment strategy in chronically treated HIV-infected subjects. Studies are now needed to evaluate whether the reduction of cardiovascular biomarkers by statins in HIV will translate into a reduction in the morbidity and mortality of the HIV-infected population. The effects of pitavastatin 4mg on primary prevention of CVD events among HIV-infected subjects are currently being assessed in the REPRIEVE trial, a large multicenter randomized trial to prevent vascular events in HIV-infected subjects.
Supplementary Material
Acknowledgments
This study was supported by an investigator-initiated grant to SKG from KOWA Pharmaceuticals America, Inc. and NIH P30 DK040561 the Nutrition Obesity Research Center at Harvard. Dr. Toribio was supported by the National Institutes of Health [T32DK007028-41].
Role of Sponsors:
The Sponsor funded the study but had no role in the analysis of the data nor in the decision to publish the data.
Footnotes
Contributors
MT was involved in reference search, figure preparation, data analysis, data interpretation, and writing; KVF was involved in data analysis and writing; LS was involved in data collection, data analysis, and preparation of figures; THB was involved in data collection and data interpretation; KCW was involved in data interpretation; MMP was involved in data collection; CAS was involved in study design, data collection, data interpretation, and writing; JAA was involved in parent study design, served as an investigator, and was involved in reference search, data interpretation, and writing; MVZ was involved in reference search, study design, and data interpretation. SKG was involved in reference search, figure preparation, study design, data collection, data analysis, data interpretation, and writing. All authors were involved in critical revision of the manuscript.
Declaration of interests:
KCW served on the scientific advisory board and was paid by Macrophage Therapeutics LLC, unrelated to this study. CAS and MMP are employees of KOWA Pharmaceuticals America, Inc. JAA received grants from KOWA Pharmaceuticals America, Inc during the conduct of the study, served in scientific advisory board personal from Janssen, Merck, and ViiV Healthcare, and received grants from Bristol-Myers Squibb and Gilead Sciences. MVZ participated in a scientific advisory board meeting for Roche Diagnostics and received grant support from Gilead Sciences, both unrelated to this study. SKG served as a paid consultant to Gilead Sciences, Theratechnologies, BMS, NovoNordisk, Merck, Navidea, and AstraZeneca and received grant support from Amgen, BMS, Gilead Sciences, KOWA Pharmaceuticals America, Inc and Theratechnologies unrelated to this study. SKG also received a grant to perform this project from KOWA Pharmaceuticals America, Inc. No other competing interests were reported.
Disclaimer: N/A
Previous Presentations: Results for this study were included in a poster presentation during the American Society of Microbiology and The Interscience Conference on Antimicrobial Agents and Chemotherapy 2016 on June 20, 2016 in Boston, MA and a poster presentation during the Massachusetts General Hospital Clinical Research Day on October 6, 2016 in Boston, MA.
References
- 1.Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV Infection and the Risk of Acute Myocardial Infarction. JAMA Intern Med. 2013:1–9. doi: 10.1001/jamainternmed.2013.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Subramanian S, Tawakol A, Burdo TH, Abbara S, Wei J, Vijayakumar J, et al. Arterial inflammation in patients with HIV. JAMA. 2012;308:379–386. doi: 10.1001/jama.2012.6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burdo TH, Lo J, Abbara S, Wei J, DeLelys ME, Preffer F, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J Infect Dis. 2011;204:1227–1236. doi: 10.1093/infdis/jir520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandler NG, Wand H, Roque A, Law M, Nason MC, Nixon DE, et al. Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis. 2011;203:780–790. doi: 10.1093/infdis/jiq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Funderburg NT, Jiang Y, Debanne SM, Labbato D, Juchnowski S, Ferrari B, et al. Rosuvastatin reduces vascular inflammation and T-cell and monocyte activation in HIV-infected subjects on antiretroviral therapy. J Acquir Immune Defic Syndr. 2015;68:396–404. doi: 10.1097/QAI.0000000000000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funderburg NT, Jiang Y, Debanne SM, Storer N, Labbato D, Clagett B, et al. Rosuvastatin treatment reduces markers of monocyte activation in HIV-infected subjects on antiretroviral therapy. Clin Infect Dis. 2014;58:588–595. doi: 10.1093/cid/cit748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganesan A, Crum-Cianflone N, Higgins J, Qin J, Rehm C, Metcalf J, et al. High dose atorvastatin decreases cellular markers of immune activation without affecting HIV-1 RNA levels: results of a double-blind randomized placebo controlled clinical trial. J Infect Dis. 2011;203:756–764. doi: 10.1093/infdis/jiq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss L, Chevalier MF, Assoumou L, Paul JL, Alhenc-Gelas M, Didier C, et al. Rosuvastatin Is Effective to Decrease CD8 T-Cell Activation Only in HIV-Infected Patients With High Residual T-Cell Activation Under Antiretroviral Therapy. J Acquir Immune Defic Syndr. 2016;71:390–398. doi: 10.1097/QAI.0000000000000879. [DOI] [PubMed] [Google Scholar]
- 9.Overton ET, Sterrett S, Westfall AO, Kahan SM, Burkholder G, Zajac AJ, et al. Effects of atorvastatin and pravastatin on immune activation and T-cell function in antiretroviral therapy-suppressed HIV-1-infected patients. AIDS. 2014;28:2627–2631. doi: 10.1097/QAD.0000000000000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chauvin B, Drouot S, Barrail-Tran A, Taburet AM. Drug-drug interactions between HMG-CoA reductase inhibitors (statins) and antiviral protease inhibitors. Clin Pharmacokinet. 2013;52:815–831. doi: 10.1007/s40262-013-0075-4. [DOI] [PubMed] [Google Scholar]
- 11.Qadir F, Alam SM, Siddiqi AQ, Kamran A. Pitavastatin is a potent anti-inflammatory agent in the rat paw model of acute inflammation. Pak J Pharm Sci. 2014;27:2169–2175. [PubMed] [Google Scholar]
- 12.Kaneyuki U, Ueda S, Yamagishi S, Kato S, Fujimura T, Shibata R, et al. Pitavastatin inhibits lysophosphatidic acid-induced proliferation and monocyte chemoattractant protein-1 expression in aortic smooth muscle cells by suppressing Rac-1-mediated reactive oxygen species generation. Vascul Pharmacol. 2007;46:286–292. doi: 10.1016/j.vph.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Abe M, Matsuda M, Kobayashi H, Miyata Y, Nakayama Y, Komuro R, et al. Effects of statins on adipose tissue inflammation: their inhibitory effect on MyD88-independent IRF3/IFN-beta pathway in macrophages. Arterioscler Thromb Vasc Biol. 2008;28:871–877. doi: 10.1161/ATVBAHA.107.160663. [DOI] [PubMed] [Google Scholar]
- 14.Sponseller C, Aberg J, Team I . Conference on Retroviruses and Opportunistic Infections. Boston: 2014. After 52 Weeks, Pitavastatin is Superior to Pravastatin for LDL-C Lowering in Patients with HIV. [Google Scholar]
- 15.Aberg J, Sponseller CA, Kryzhanovski VA, Kartman CE, Thompson MA. Endocrine Society Conference. San Francisco: 2013. Neutral effects of pitavastatin 4 mg and pravastatin 40 mg on blood glucose and HbA1c levels over 12 weeks: prespecified safety analysis from INTREPID (HIV-infected patieNts and TREatment with PItavastatin vs pravastatin for Dyslipidemia), a phase 4 trial. [Google Scholar]
- 16.Department of Health and Human Services (US) NIoH, National Heart, Lung, and Blood Institute. Your guide to lowering your cholesterol with TLC: therapeutic lifestyle changes. 2005 In. [Google Scholar]
- 17.Malvestutto CD, Ma Q, Morse GD, Underberg JA, Aberg JA. Lack of pharmacokinetic interactions between pitavastatin and efavirenz or darunavir/ritonavir. J Acquir Immune Defic Syndr. 2014;67:390–396. doi: 10.1097/QAI.0000000000000333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKibben RA, Margolick JB, Grinspoon S, Li X, Palella FJ, Jr, Kingsley LA, et al. Elevated Levels of Monocyte Activation Markers Are Associated With Subclinical Atherosclerosis in Men With and Those Without HIV Infection. J Infect Dis. 2015;211:1219–1228. doi: 10.1093/infdis/jiu594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zidar DA, Juchnowski S, Ferrari B, Clagett B, Pilch-Cooper HA, Rose S, et al. Oxidized LDL levels are increased in HIV infection and may drive monocyte activation. J Acquir Immune Defic Syndr. 2015 doi: 10.1097/QAI.0000000000000566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hardwick SJ, Hegyi L, Clare K, Law NS, Carpenter KL, Mitchinson MJ, et al. Apoptosis in human monocyte-macrophages exposed to oxidized low density lipoprotein. J Pathol. 1996;179:294–302. doi: 10.1002/(SICI)1096-9896(199607)179:3<294::AID-PATH590>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 21.Nou E, Lu MT, Looby SE, Fitch KV, Kim EA, Lee H, et al. Serum oxidized low-density lipoprotein decreases in response to statin therapy and relates independently to reductions in coronary plaque in patients with HIV. AIDS. 2015 doi: 10.1097/QAD.0000000000000946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hileman CO, Semba RD, Turner R, Labbato DE, Storer NJ, McComsey GA. Rosuvastatin Lowers Oxidative LDL in HIV-Infected Persons On Antiretroviral Therapy: SATURN-HIV. CROI. 2014 [Google Scholar]
- 23.Maiolino G, Bisogni V, Rossitto G, Rossi GP. Lipoprotein-associated phospholipase A2 prognostic role in atherosclerotic complications. World J Cardiol. 2015;7:609–620. doi: 10.4330/wjc.v7.i10.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, et al. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangili A, Ahmad R, Wolfert RL, Kuvin J, Polak JF, Karas RH, et al. Lipoprotein-associated phospholipase A2, a novel cardiovascular inflammatory marker, in HIV-infected patients. Clin Infect Dis. 2014;58:893–900. doi: 10.1093/cid/cit815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eckard AR, Jiang Y, Debanne SM, Funderburg NT, McComsey GA. Effect of 24 weeks of statin therapy on systemic and vascular inflammation in HIV-infected subjects receiving antiretroviral therapy. J Infect Dis. 2014;209:1156–1164. doi: 10.1093/infdis/jiu012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agouridis AP, Tsimihodimos V, Filippatos TD, Dimitriou AA, Tellis CC, Elisaf MS, et al. The effects of rosuvastatin alone or in combination with fenofibrate or omega 3 fatty acids on inflammation and oxidative stress in patients with mixed dyslipidemia. Expert Opin Pharmacother. 2011;12:2605–2611. doi: 10.1517/14656566.2011.591383. [DOI] [PubMed] [Google Scholar]
- 28.Schaefer EJ, McNamara JR, Asztalos BF, Tayler T, Daly JA, Gleason JL, et al. Effects of atorvastatin versus other statins on fasting and postprandial C-reactive protein and lipoprotein-associated phospholipase A2 in patients with coronary heart disease versus control subjects. Am J Cardiol. 2005;95:1025–1032. doi: 10.1016/j.amjcard.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 29.Hsu DC, Ma YF, Hur S, Li D, Rupert A, Scherzer R, et al. Plasma IL-6 levels are independently associated with atherosclerosis and mortality in HIV-infected individuals on suppressive antiretroviral therapy. AIDS. 2016;30:2065–2074. doi: 10.1097/QAD.0000000000001149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aristoteli LP, Moller HJ, Bailey B, Moestrup SK, Kritharides L. The monocytic lineage specific soluble CD163 is a plasma marker of coronary atherosclerosis. Atherosclerosis. 2006;184:342–347. doi: 10.1016/j.atherosclerosis.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 31.Knudsen TB, Ertner G, Petersen J, Moller HJ, Moestrup SK, Eugen-Olsen J, et al. Plasma CD163 independently predicts all-cause mortality from HIV-1 infection. J Infect Dis. 2016 doi: 10.1093/infdis/jiw263. [DOI] [PubMed] [Google Scholar]
- 32.Erlandson KM, Jiang Y, Debanne SM, McComsey GA. Rosuvastatin Worsens Insulin Resistance in HIV-Infected Adults on Antiretroviral Therapy. Clin Infect Dis. 2015;61:1566–1572. doi: 10.1093/cid/civ554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.