

Abstract
Plants have evolved several strategies, including regulation of genes through epigenetic modifications, to cope with environmental stresses. DNA methylation is dynamically regulated through the methylation and demethylation of cytosine in response to environmental perturbations. High-affinity potassium transporters (HKTs) have accounted for the homeostasis of sodium and potassium ions in plants under salt stress. Wheat (Triticum aestivum L.) is sensitive to soil salinity, which impedes its growth and development, resulting in decreased productivity. The differential expression of HKTs has been reported to confer tolerance to salt stress in plants. In this study, we investigated variations in cytosine methylation and their effects on the expression of HKT genes in contrasting wheat genotypes under salt stress. We observed a genotype- and tissue-specific increase in cytosine methylation induced by NaCl stress that downregulated the expression of TaHKT2;1 and TaHKT2;3 in the shoot and root tissues of Kharchia-65, thereby contributing to its improved salt-tolerance ability. Although TaHKT1;4 was expressed only in roots and was downregulated under the stress in salt-tolerant genotypes, it was not regulated through variations in cytosine methylation. Thus, understanding epigenetic regulation and the function of HKTs would enable an improvement in salt tolerance and the development of salt-tolerant crops.
Keywords: : DNA methylation, epigenetics, gene regulation, salt stress, Triticum aestivum
Introduction
Plants, being sessile, have developed several strategies to cope with environmental stresses, including alterations in the expression level of genes through epigenetic modifications such as DNA methylation. DNA methylation plays a key role in gene expression through the RNA-directed DNA methylation (RdDM) of genes and the induction of histone modifications. Cytosine methylation has been reported to be involved in many vital biological processes, including transposon movement, genome imprinting, and regulation of gene expression (Yan et al., 2010). Abiotic stresses have direct, negative effects on the biochemical and physiological processes that are associated with plant growth and development, which results in a significant reduction in crop yield.
One of the detrimental effects of salinity is the accumulation of sodium ion (Na+) in plant tissues, which inhibits the uptake of the potassium ion (K+) from soil. Na+ and K+ have similar chemical properties and content ratio in nonsaline soils; however, the physiological effects of these ions on the metabolism and growth of plants are quite different. Maintaining a high K+/Na+ ratio has been suggested to be a major strategy for plants to cope with salt stress (Hamamoto et al., 2015).
At the cellular level, the mechanisms for salt tolerance function to reduce Na+ accumulation in the cytoplasm by limiting the entry of Na+ into cells, actively transporting Na+ out of cells, and compartmentalizing Na+ into vacuoles (Shi et al., 2003). K+ is preferred for uptake into roots from the soil, and most plants exhibit a high degree of K+/Na+ discrimination for uptake. High-affinity potassium transporters (HKTs) have been reported to be active at the plasma membrane level and function as Na+/K+ symporters as well as selective Na+ uniporters (Horie et al., 2009). HKTs may have two major functions, namely to take up Na+ from the soil to reduce the requirement of K+ when K+ is a limiting factor and to reduce the accumulation of Na+ in the leaf by removing Na+ from the xylem sap and loading Na+ into the phloem sap (Brini et al., 2009). HKTs belong to the HKT/Trk/Ktr-type K+ transporter superfamily that is found in microorganisms and plants (Yamaguchi et al., 2013).
Loss-of-function analysis of HKT1 in Arabidopsis and wheat established that the primary role of AtHKT1 is to retrieve Na+ from the xylem in the roots to reduce the transport of Na+ from the root to the shoot. However, the role of HKT1 in the leaves, if any, remains elusive. A primary role of HKT2 is the mediation of nutritional Na+ absorption and Na+ uptake from soil into the roots of K+-starved plants to compensate for the deficiency of K+ (Horie et al., 2009). The downregulation of TaHKT2 in salt-tolerant wheat genotypes has been reported to confer tolerance to salt stress (Singh et al., 2015). Although some reports challenge the assumption that Na+ exclusion leads to improved salinity tolerance, HKTs have emerged as crucial components of salt stress tolerance.
DNA methylation is one of the most studied epigenetic processes, because it results in a direct and heritable covalent modification triggered by external stimuli. Such modifications may be reversible and can be associated with the inactivation and activation of genes (Zemach et al., 2010). Demethylation of functionally inactive genes due to exposure to abiotic stresses may initiate their expression, and the stress may also cause heritable changes in cytosine methylation to form epialleles (Kou et al., 2011). The importance of epigenetic variations due to stressful condition arises from the fact that these epigenetic modulations can be inherited in the form of epigenetic memory (Boyko and Kovalchuk, 2010). Understanding the molecular mechanisms underlying stress-induced epigenetic regulation of gene expression may facilitate breeding programs in improving crop plants without excessive genetic modification.
Genome-wide high-resolution analysis of DNA methylation in rice revealed that 8% of the genes were methylated within their promoters, whereas 31% of the genes were methylated within their coding regions (Yan et al., 2010). Responses to environmental factors may vary among plant species; some of them can modulate the physiological and developmental machinery of plants to mitigate the effect of the stress. Mangroves growing in contrasting natural habitats (riversides or salt marshes) differed with respect to cytosine methylation despite the small genetic variation (Lira-Medeiros et al., 2010) between them. Thus, epigenetic variations need to be investigated so that epialleles can be identified and exploited in crop breeding programs to improve the adaptability of plants under changing climatic conditions (Kou et al., 2011).
Plants contain relatively high levels of 5-methylcytosine (5-mC), ranging from 6% to 25% of the total cytosine, depending on the species (Steward et al., 2002). Unlike DNA methylation in mammals, wherein it predominantly occurs in the CG context, DNA methylation in plants occurs in all three cytosine contexts: CG, CHG, and CHH (H = A, T, or C) (Wang et al., 2016). In Arabidopsis, symmetric CG and CHG methylation is maintained by DNA methyl transferase1 (MET1) and Chromomethylase3 (CMT3) during DNA replication, whereas asymmetric CHH methylation is established de novo by domains rearranged methylase2 (DRM2) via the RdDM pathway (Law and Jacobsen, 2010). The passive or active demethylation process may be used to remove 5-mCs. In plants, the active DNA demethylation pathway is initiated by a subfamily of typical HhH-GPD enzymes, including Repressor of Silencing-1 (ROS1), Demeter (DME), Demeter-like2 (DML2), and Demeter-like3 (DML3). Recently, Wang et al. (2016) reported MET18 to be a component of the active DNA demethylation pathway in plants and demonstrated that it plays an epigenetic role in the regulation of gene expression in Arabidopsis.
Wheat (Triticum aestivum L.) is one of the most widely cultivated cereals in the world. It is sensitive to soil salinity, which impedes its growth and development, resulting in reduced crop productivity or failure of the crop. Some wheat genotypes possess a unique ability to rapidly adapt to salt stress, whereas others are highly sensitive because of their genetic makeup and regulatory architecture. For instance, Kharchia-65 and KRL-210 are well-known salt-tolerant wheat genotypes (Sairam et al., 2005). On the basis of a multivariable (biochemical and physiological parameters) comprehensive analysis of wheat genotypes under salt stress, we identified Kharchia-65 and HD-2329 as the most contrasting pair of locally available wheat genotypes with regard to salt tolerance (Beena et al., unpublished data). However, differences in the methylation patterns and the epigenetic responses of these contrasting wheat genotypes under salt stress have been underexplored.
The present study examined the effects of salt stress on the extent and pattern of cytosine methylation and their effects on the expression of HKT genes in the two contrasting wheat genotypes, Kharchia-65 and HD-2329. We addressed the following two basic queries: (i) whether epigenetic changes, if any, are triggered by salt stress in bread wheat and (ii) whether epigenetic responses of the salt-tolerant (Kharchia-65) and salt-sensitive (HD-2329) genotypes are similar. Our investigation revealed that cytosine methylation was induced by salt stress in a genotype- and tissue-specific manner, which downregulated the expression of TaHKT2;1 and TaHKT2;3 in the shoots and roots of salt-tolerant and salt-sensitive genotypes. However, the root-specific downregulation of the TaHKT1;4 gene was not found to be controlled through the modulation in DNA methylation.
Materials and Methods
Plant materials and salt treatment
Two locally available, highly contrasting bread wheat genotypes (Kharchia-65, salt tolerant and HD-2329, salt sensitive) were used in the present investigation. The seeds of the contrasting wheat genotypes were surface sterilized by using 0.1% mercuric chloride for 2 min, followed by washing three times with sterilized distilled water. Six seeds were sown at equal intervals in 15-cm pots that were filled with agro-coir peat. Six pots for each genotype were grown under controlled conditions in a glasshouse at the National Phytotron Facility, IARI, New Delhi. On the basis of the results of our preliminary experiment (Beena et al., unpublished data), 14-day-old seedlings (in three pots) of each genotype were treated with 200 mM NaCl that was dissolved in half-strength Hoagland solution. The remaining three pots of each genotype were maintained untreated as controls. Salt stress treatment was continued for 14 days until the effects of salt stress were visible on the sensitive genotype. Fourteen days after the treatment (DAT), shoot and root samples were collected for molecular analyses.
Isolation of nucleic acids from plant tissues
Genomic DNA from plant tissues was isolated by using the DNeasy Plant Mini Kit (Qiagen). The shoot and root samples were first mechanically disrupted by using liquid nitrogen and then chemically lysed. RNAs were removed by using RNase A treatment during the lysis step by following the protocol prescribed by the manufacturer of the kit. The purified genomic DNA was eluted in low-salt buffer (AE buffer) and stored at −20°C for downstream use.
Total RNAs were isolated from 100 mg root and shoot tissue samples by using the RNeasy Plant Mini Kit (Qiagen) by following the manufacturer's instructions. The isolated RNAs were treated with RNase-Free DNase Set (Qiagen) for on-column digestion of DNA during RNA isolation. The quality of the isolated RNA samples was assessed through denaturing agarose (1%) gel electrophoresis. The quantification of isolated RNAs was performed by using the NanoDrop spectrophotometer (ND-1000), and the A260/280, A260/230 ratios were used to assess purity of the RNA.
PCR cloning of HKT1;4 gene
The genomic DNA (100 ng) isolated from the shoot tissues was used as a template for the amplification of HKT1;4 by using a primer pair (Table 1) that was designed for the last quarter of HKT1;4–2 (KF443079.1) CDS of Triticum durum. The following conditions were used for amplification of the gene: initial denaturation at 94°C for 5 min, followed by 36 cycles at 94°C for 1 min, 56°C for 1 min, 72°C for 1 min, and final extension at 72°C for 5 min. The PCR products were visualized on a 1.5% agarose gel, and the amplicons from Kharchia-65 and HD-2329 were cloned in pGEM-T Easy vector (Kumar and Saxena, 2016). The cloned fragments were outsourced for 5 × sequencing by Sanger's dideoxy method. The sequences were analyzed at NCBI and EMBL databases for homology search with the HKTs from other plant species. The partial HKT1;4 sequences were submitted to the EMBL database.
Table 1.
Primers Used for Cloning, RT-PCR, QPCR, and Bisulfite Sequencing Analyses of High-Affinity Potassium Transporter Genes in Bread Wheat
| S. No. | Sequence | Annealing temp. (°C) | Product size (bp) | Usage |
|---|---|---|---|---|
| HKT1;4 gene | ||||
| 1 | Forward primer: ATTCAGGCAACACCTAATCATGC | 56 | 473 | RT-PCR |
| Reverse primer: GCATCACAAGAATGAGGATGAGC | 581 | Cloning | ||
| 2 | Forward primer: TTTCTGTTCCAGGTACCTGCCTCCATACA | 49 | 384 | Bisulfite sequencing |
| Reverse primer: ARAARCCCCCATTTCCATCCRCACTRC | ||||
| 3 | Forward primer: ACCTCGCCATCTTCATCATC | 56 | 199 | qPCR |
| Reverse primer: GCTTCCATGAAGGAAACCAA | ||||
| Actin gene | ||||
| 4 | Forward primer: TGGGATGCCACCAAAGAC | 56 | 380 | RT-PCR and qPCR |
| Reverse primer: TGATACGCAAATGTTGAGC | ||||
| Ferredoxin-NADP(H) oxidoreductase (TaFNRII) gene | ||||
| 5 | Forward primer: CAGTGATCTTCACTTCTGAAC | 56 | 200 | RT-PCR and qPCR |
| Reverse primer: CGAGGACAAGAACGGGAAG | ||||
| HKT2;1 gene | ||||
| 6 | Forward primer: TATGTGATGAGTCGCAGCTTGAA | 56 | 316 | qPCR |
| Reverse primer: GCAACAAGAGGCCTGAATTCTTT | ||||
| 7 | Forward primer: TTYAATTYAGYYAAGAATGTAYAGAG | 49 | 254 | Bisulfite sequencing |
| Reverse primer: AARAACCATARTTTCATTTARARRCAC | ||||
| HKT2;3 gene | ||||
| 8 | Forward primer: TGAAGCCAAGCAACCCTAAC | 56 | 178 | qPCR |
| Reverse primer: CCAAGCAGGGAAACAAACAT | ||||
| 9 | Forward primer: GAATTATTTGGTGTTTTATTTTTYGGTTT | 51 | 369 | Bisulfite sequencing |
| Reverse primer: ACACRATAACCRATATAACTCTACTATC | ||||
HKT, high-affinity potassium transporter; RT-PCR, reverse transcription-polymerase chain reaction; qPCR, quantitative PCR.
Semi-quantitative expression analysis of TaHKT1;4 gene
RNA samples isolated from root and shoot tissues showing A260/280 between 1.8–2.0 and A260/230 > 2.0 were used for cDNA synthesis. First-strand cDNA was synthesized by using an equal amount (0.5 μg) of total RNA as the template and 2.0 μmol oligo-dT primer in a 20 μL reaction volume at 37°C for 1 h by using the Revert Aid Premium first-strand cDNA synthesis kit (Fermentas), as per the manufacturer's instructions, by using a Triple Master PCR system (Eppendorf). The first-strand cDNA (2.0 μL) was used for expression analysis of TaHKT1;4 gene by using the gene-specific primers. PCR conditions were as previously mentioned, and the number of PCR cycles for semi-quantitative analysis was optimized by assessing the amplification products after 20, 24, 28, 32, and 36 cycles on 1.5% agarose gel. Actin and Ferredoxin-NADP(H) oxidoreductase were used as reference genes. To ensure the reproducibility of the results, the experiment was repeated three times.
Estimation of genome-wide DNA methylation
The global DNA methylation status of shoot and root tissues of the contrasting wheat genotypes under salt stress and controlled conditions was estimated by using the MethylFlash Methylated DNA Quantification (Colorimetric) kit (Epigentek). Genomic DNA (100 mg) was used for the detection of 5-methylcytosine (5-mC) by using the kit that detects 5-mC by using a specific monoclonal antibody (along with a detection or secondary antibody). Negative and positive DNA controls provided in the kit were used in the assay for the preparation of the standard curve for the quantification of 5-mC with the help of a microplate reader by following the manufacturer's instructions. Genomic DNA was coated on the well of the assay plate in triplicate. After color development, absorbance was measured at 450 nm. Quantification of 5-mC (%) was performed by plotting a standard curve and with the help of the formula given next:
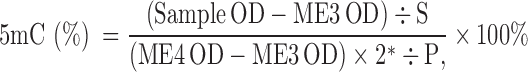 |
where S is the amount of input sample DNA in ng; P is the amount of input positive control (ME4) in ng; ME3 is the negative control; ME4 is the positive control; and 2* is a factor to normalize 5-mC in the positive control to 100%, as the positive control contains only 50% of 5-mC.
Bisulfite sequencing and data analysis
Approximately 100 ng of genomic DNA was modified by using the BisulFlash DNA Modification Kit (Epigentek) according to the manufacturer's instructions. Bisulfite modification converts unmethylated cytosine of DNA into uracil, leaving 5-mC unmodified. The bisulfite-modified genomic DNA was eluted with 20 μL elution solution and used for downstream processes. An aliquot (1 μL) of bisulfite-modified DNA was used for PCR in a reaction volume of 20 μL containing ExTaq DNA polymerase (Takara) and gene-specific primers (designed using MethPrimer software, www.urogene.org/methprimer). The PCR-amplified products were cloned by using the pGEM–T Easy vector, and 10 independent clones of each sample were outsourced for sequencing.
For a comparative analysis of the bisulfite sequence, the sequences for different genotypes, tissues, and treatments were aligned by using ClustalX software and visualized manually by using BioEdit graphical view (Srivastava et al., 2011). The methylation data were analyzed by using the Kismeth software, which allows the analysis of methylation at every particular site. The identification of the differentially methylated region (DMR) was carried out in every 100-bp window with a step size of 50 bp by comparing bisulfite sequences for different genotypes, tissues, and treatments by using the Fisher exact test with a p value cut-off of 0.05 (Wang et al., 2016). A window with ≥3 differentially 5-mCs and a ≥1.5-fold change in DNA methylation level was considered DMR.
Quantitative expression analysis of HKT genes
Quantitative analysis was performed by using reverse transcription-quantitative polymerase chain reaction (RT-qPCR) following the MIQE guidelines (Bustin et al., 2009) to correlate the probable effects of cytosine methylation with the expression levels of TaHKT1;4, TaHKT2;1, and TaHKT2;3. First-strand cDNA was synthesized from 0.5 μg of total RNA, diluted five-fold before using it in a 20 μL reaction volume containing 1 μL of cDNA and 10 pmol forward and reverse primers (Table 1; Singh et al., 2015). The RT-qPCR was performed by using the Mx3000PTM real-time PCR system with the SYBR Green qPCR Master Mix kit (BioRad). PCR amplification was performed with an initial denaturation at 95°C for 3 min, followed by 38 cycles each of 30 s denaturation at 94°C, 30 s annealing at 56°C, and 30 s extension at 72°C. Amplification data were collected at the end of each extension step.
To estimate the relative gene expression, Ct values (which are inversely related to the initial DNA concentration) for both target and reference genes were calculated on the basis of the mean value of three replications. Actin and Ferredoxin-NADP(H) oxidoreductase were used as reference or housekeeping genes. The Pfaffl formula [ΔΔCt = (ΔCtsample−ΔCtcontrol), where ΔCtsample = (ΔCttarget−ΔCtreference) for all samples against NaCl imposition; and ΔCtcontrol = (ΔCttarget−ΔCtreference)] (Pfaffl et al., 2001) was used to calculate the relative expression of HKTs under 200 mM NaCl salt stress after 14 days of treatment. Graphs were plotted for the treatments against the controls.
Statistical analysis
The data were analyzed by using one-way analysis of variance (ANOVA) by using statistical software (SPSS 19.0). Duncan's multiple-range tests were performed to determine the significant difference between means at a significance level of p < 0.05.
Results
Amplification, sequencing, and in silico analysis of HKT1;4 gene
Using homologous HKT1;4 gene-specific primers, approximately 0.6 kb of the last quarter of the HKT1;4 could be amplified from the shoot of salt-tolerant (Kharchia-65) and salt-sensitive (HD-2329) genotypes. Sequencing of the PCR products revealed that their size was 581 and 583 bp in the case of HD-2329 and Kharchia-65, respectively. Sequence search analysis exhibited 91% homology with the last quarter of TdHKT1;4–2 (KF443079) CDS of durum wheat (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/dna). Sequence alignment of the cloned partial sequences from Kharchia-65 and HD-2329 indicated that HD-2329 contains 2 deletions, 3 transitions, and 12 transversions (Supplementary Fig. S2A).
Conserved domain analysis indicated the presence of Ser–Gly–Gly–Gly and Gly–Arg motifs in TaHKT1;4 of Kharchia-65 but not in TaHKT1;4 of HD-2329 (Supplementary Fig. S2B). The gene from Kharchia-65 was named TaHKT1;4.2 and that from HD-2329 was named TaHKT1;4.3, as per the nomenclature system. The partial sequences of HKT1;4 genes were submitted to the EMBL database with the accession No. KR262818 (Kharchia-65) and KR262819 (HD-2329). Cloning, sequencing, and in silico analysis of TaHKT2;1 and TaHKT2;3 genes have been described earlier (Singh et al., 2015).
RT-PCR expression analysis of TaHKT1;4 gene
Semi-quantitative expression analysis of TaHKT1;4 (performed through RT-PCR) indicated that the gene is differentially expressed in the shoots and roots of the bread wheat genotypes. The gene was not expressed in the shoot of the wheat genotypes. Downregulation of TaHKT1;4.2 was observed in the root of the salt-tolerant (Kharchia-65) genotype, whereas the gene (TaHKT1;4.3) was observed to be upregulated in the salt-sensitive (HD-2329) genotype. Salt stress further downregulated the expression of the gene in the root of the salt-tolerant (Kharchia-65) genotype, whereas salt stress upregulated the expression of the gene in the root of the salt-sensitive genotype (Fig. 1). The expression level of the reference or housekeeping [Actin and Ferredoxin-NADP(H) oxidoreductase] genes was observed to be uniform in the shoots and roots of salt-sensitive and salt-tolerant genotypes under control and salt stress conditions. Semi-quantitative expression analysis of TaHKT2;1 and TaHKT2;3 genes in the shoots of the contrasting wheat genotypes has been reported earlier (Singh et al., 2015), wherein the differential expression of TaHKT2;1 and TaHKT2;3 in the contrasting wheat genotypes was presented. Salt stress was observed to upregulate the expression of TaHKT2;1 and TaHKT2;3 in the shoots of the salt-sensitive genotype, whereas these genes were downregulated in the salt-tolerant genotype.
FIG. 1.

Semi-quantitative expression analysis of TaHKT1;4 gene (performed through reverse transcription-polymerase chain reaction) in shoots and roots of the contrasting wheat genotypes Kharchia-65 (Kh, salt tolerant) and HD-2329 (HD, salt sensitive) under control (C) and 200 mM NaCl stress (T) conditions. Primers were designed from the last quarter of HKT1;4–2 (KF443079.1) CDS of Triticum durum. Actin and Ferredoxin-NADP(H) oxidoreductase (TaFNRII) were used as reference/housekeeping genes. M, Marker, 100 bp DNA ladder.
Variation in global DNA methylation due to salt stress
Estimation of the global 5-mC level in the shoot and root tissues of the contrasting wheat genotypes revealed that methylation level under control conditions in the shoot and root tissues of the salt-sensitive genotype was higher (4.05%–6.70%) compared with that in the salt-tolerant genotype (3.45%–5.40%). Moreover, the methylation level in the shoot tissues (of both the genotypes) was observed to be higher (5.40%–6.7%) under control conditions compared with that in the root tissues (3.45%–4.05%). Salt stress caused more than a 10% increase in global methylation level in the salt-tolerant genotype, whereas the increase was nonsignificant in the case of the salt-sensitive genotype (Fig. 2).
FIG. 2.

Global cytosine methylation (5-mC) level in shoot and root tissues of the contrasting wheat genotypes Kharchia-65 (Kh, salt tolerant) and HD-2329 (HD, salt sensitive) under control and 200 mM NaCl stress conditions. Bars represent standard deviation.
Variation in cytosine methylation in the coding region of HKTs
The coding region of TaHKT1;4 (264 bp, in the last quarter of the gene body) used for quantitative analysis of 5-mC was found to contain 25% of the cytosines in the CG context, 18% cytosine in the CHG context, and 57% cytosine in the CHH context. Context-specific variation in cytosine methylation was observed with respect to the genotypes, tissues, and salt stress (Fig. 3). An increase in the 5-mC level in the CG and CHH contexts was observed in the shoot of HD-2329 when salt stress was imposed. By contrast, no increase in 5-mC was observed in Kharchia-65. However, all the cytosines in the CHG context were observed to be methylated in Kharchia-65 (Fig. 3A, B). A decrease in the 5-mC level in the CHG and CHH contexts was observed in the roots of HD-2329 when salt stress was imposed. By contrast, an increase in the 5-mC level was observed in the CG context in the case of Kharchia-65 (Fig. 3C, D).
FIG. 3.

Cytosine methylation (5-mC) in TaHKT1;4 in different contexts (CG, CHG, and CHH) in the contrasting wheat genotypes Kharchia-65 and HD-2329 under control and 200 mM NaCl stress conditions. (A) Methylation pattern in shoots of salt-sensitive (HD-2329) genotype; (B) methylation pattern in shoots of salt-tolerant (Kharchia-65) genotype; (C) methylation pattern in roots of HD-2329; (D) methylation pattern in roots of Kharchia-65.
Alignment of multiple bisulfite sequences from the shoot and root tissues along with the unmodified/reference DNA sequences of the salt-sensitive and salt-tolerant genotypes indicated that salt stress caused the methylation of cytosine in only one CHH and one CG context in the shoot of HD-2329. However, demethylation of one cytosine in the CHG and CHH contexts in TaHKT1;4 was observed in the root of HD-2329 because of salt stress. By contrast, methylation of one cytosine in the CG context was observed in the root of Kharchia-65 because of salt stress (Supplementary Fig. S3). Thus, neither a considerable variation in cytosine methylation nor that in DMR was observed in TaHKT1;4.
The coding region of TaHKT2;1 (254 bp, in the second quarter of the gene body) used for quantitative analysis of 5-mC was found to contain 10% cytosine in the CG context, 23% cytosine in the CHG context, and 67% cytosine in the CHH context. Variations in 5-mC levels were observed with respect to the genotypes, tissues, and salt stress (Fig. 4). An increase in cytosine methylation in the CHG and CHH contexts (having all the cytosines methylated in the CG context) was observed in the shoot of HD-2329 when salt stress was imposed (Fig. 4A). An increase in the 5-mC level was observed in Kharchia-65 in all the three contexts when salt stress was imposed, but the maximum increase (14%, leading to all the cytosines methylated) was observed in the CG context (Fig. 4B). An increase in 5-mC was observed in all three contexts in the root of both the genotypes when salt stress was imposed, but the total methylation was higher (with all the cytosines in the CG context methylated) in the case of Kharchia-65 (Fig. 4C, D).
FIG. 4.

Cytosine methylation (5-mC) in TaHKT2;1 in different contexts (CG, CHG, and CHH) in the contrasting wheat genotypes Kharchia-65 and HD-2329 under control and 200 mM NaCl stress conditions. (A) Methylation pattern in shoots of salt-sensitive (HD-2329) genotype; (B) methylation pattern in shoots of salt-tolerant (Kharchia-65) genotype; (C) methylation pattern in roots of HD-2329; (D) methylation pattern in roots of Kharchia-65.
Alignment of multiple bisulfite sequences from the shoot and root tissues of the salt-sensitive and salt-tolerant genotypes with the reference sequence indicated that salt stress increased the methylation in the CG context to the maximum (100%) in both tissues and genotypes. The basic methylation level (under control conditions) in the shoot of the salt-tolerant genotype in the CHG and CHH contexts was found to be approximately 25% higher than that in the salt-sensitive genotype. Salt stress further increased the methylation level in these contexts. An 8% percent increase in 5-mC in the CHG context was observed in the root of the salt-tolerant genotype when salt stress was imposed compared with the methylation level in this context in the shoot of this genotype (Fig. 4B, D).
The coding region of the TaHKT2;3 (369 bp, in the first quarter of the gene body) used for the quantitative analysis of 5-mC contained 11%, 10%, and 79% of the cytosines in the CG, CHG, and CHH contexts, respectively. Variations in the quantity of 5-mC in different contexts was observed with respect to the genotypes, tissues, and salt treatments (Fig. 5). With all the cytosines methylated in the CG context, an increase in the methylation in the CHG and CHH contexts was observed in the shoots of HD-2329 when salt stress was imposed (Fig. 5A). An increase in methylation in the CG context only was observed in the shoot of Kharchia-65; however, totally, the methylation (94%) was considerably higher than that of HD-2329 (78%) (Fig. 5B). Only a minor increase in the methylation in the CHH context was observed in the root of HD-2329 in the presence of salt stress.
FIG. 5.

Cytosine methylation (5-mC) in TaHKT2;3 in different contexts (CG, CHG, and CHH) in the contrasting wheat genotypes Kharchia-65 and HD-2329 under control and 200 mM NaCl stress conditions. (A) Methylation pattern in shoots of salt-sensitive (HD-2329) genotype; (B) methylation pattern in shoots of salt-tolerant (Kharchia-65) genotype; (C) methylation pattern in roots of HD-2329; (D) methylation pattern in roots of Kharchia-65.
Although the basic methylation level was considerably high (94%), no further increase in 5-mC was observed in Kharchia-65 (Fig. 5C, D). The basic methylation level in the shoots of the salt-tolerant genotype was found to be 23% higher than that of the salt-sensitive genotype. The alignment of multiple bisulfite sequences from the shoots and roots of HD-2329 and Kharchia-65 genotypes indicated that salt stress caused the methylation of cytosines in the CHH and CHG contexts in HD-2329. Many of the cytosines were methylated in the roots of the salt-sensitive (HD-2329) genotype; however, they were found to be unmethylated in the shoots (Supplementary Fig. S4). Five cytosine residues (in the CHH context) were found to remain unmethylated in the salt-sensitive genotype, even under salt stress. Seven cytosine residues (in the CHG and CHH contexts) were found to show variations in methylation between the shoot and root tissues of the salt-sensitive genotype due to salt stress. Two of the cytosine residues (in the CHG and CHH contexts) showed variations in methylation in the shoot of the salt-sensitive genotype under control and salt stress conditions, whereas they were methylated in the root under both the conditions (Supplementary Fig. S4). Salt stress caused the methylation of 94% of the cytosines in the salt-tolerant genotype compared with only 78%–88% in the salt-sensitive genotype (Fig. 5). Thus, a significant variation in 5-mC and DMR was observed in TaHKT2;1 and TaHKT2;3 (Supplementary Fig. S4).
Expression profile of HKTs
The quantitative expression analyses of HKT genes revealed that under salt stress, the expression of TaHKT1;4 was 0.6-fold downregulated in the root of the salt-tolerant genotype, whereas it was 0.8-fold upregulated in the salt-sensitive genotype (Fig. 6A). By contrast, RT-qPCR analysis of TaHKT2;1 showed that under salt stress, the expression of the gene was four-fold downregulated in the shoot of the salt-tolerant genotype, but it was six-fold upregulated in the salt-sensitive genotype (Fig. 6B). In the root tissues, the gene was observed to be two-fold downregulated in the salt-tolerant genotype, and it was 1.1-fold downregulated in the salt-sensitive genotype under the stress (Fig. 6C). The quantitative analysis of the TaHKT2;3 expression revealed that it was eight-fold downregulated in the shoot of the salt-tolerant genotype but 10-fold upregulated in the salt-sensitive genotype (Fig. 6D). In the root tissues, the gene was observed to be 2.5-fold downregulated in the salt-tolerant genotype, but it was 1.4-fold downregulated in the salt-sensitive genotype under salt stress (Fig. 6E).
FIG. 6.

Quantitative expression analysis of HKT genes in the contrasting wheat genotypes. (A) Expression of TaHKT1;4 in roots of Kharchia-65 (Kh, salt tolerant) and HD-2329 (HD, salt sensitive); expression of TaHKT2;1 gene in (B) shoots and (C) roots of Kharchia-65 (Kh) and HD-2329 (HD); expression of TaHKT2;3 gene in (D) shoots and (E) roots of Kharchia-65 (Kh) and HD-2329 (HD). The results are presented as a mean fold change in relative expression over the control with three biological and three technical replicates, normalized with respect to the actin (reference) gene expression. Bars represent standard deviation.
Discussion
Plants cope with sodium toxicity by managing the accumulation of Na+ in the shoot (Munns and Tester, 2008). At the cellular level, Na+ accumulation in the cytoplasm is regulated by limiting Na+ entry into the (root) cells, transporting Na+ out of the (shoot) cells, and compartmentalizing Na+ into vacuoles (Shi et al., 2003). Davenport et al. (2007) reported that AtHKT1 directs the retrieval of Na+ from the xylem and the loading of Na+ into the root vacuoles. Thus, the apparent primary role of AtHKT1 is to retrieve Na+ from the xylem in roots to reduce the transport of Na+ from the roots to the shoots. Therefore, we selected HKTs to study the epigenetic effects of salt stress on the regulation of gene expression.
We observed the expression of TaHKT1;4 in the root but not in the leaf of the wheat genotypes. Our observation of the root-specific expression of TaHKT1;4 might be either because of the complete silencing of the gene in the shoot or because of its very low level of expression, which could not be detected during expression analysis. Moreover, the downregulation of TaHKT1;4 was observed in the roots of the salt-tolerant (Kharchia-65) genotype under salt stress. This gene was observed to be upregulated because of salt stress in the HD-2329 genotype, which might be responsible for making this genotype salt sensitive. Loss-of-function analysis of Arabidopsis and wheat HKT1 genes established that HTK1 transports Na+ into the cells and controls Na+ uptake in the roots (Rus et al., 2001; Laurie et al., 2002). The role of AtHKT1 in the leaves remains elusive (Baek et al., 2011). Møller et al. (2009) demonstrated that the overexpression of AtHKT1 in the root stele increased salt tolerance in transgenic plants, whereas the overexpression of AtHKT1 in the entire plant resulted in salt hypersensitivity. Thus, understanding tissue-specific expression of HKTs and their control mechanisms appears to be vital in elucidating the function of HKTs in the entire plant.
Several point mutations (nucleotide deletions, transversions, and transitions) were observed in TaHKT1;4.3 (Supplementary Fig. S2A), which appear to significantly affect its function as the conserved domains, for instance, “selectivity filter motif” (Ser–Gly–Gly–Gly) and Gly–Arg motifs were found to be absent in TaHKT1;4.2 but present in the TaHKT1;4.3 (Supplementary Fig. S2B). Platten et al. (2006) reported the structural analysis of plant HKTs, wherein they observed a conserved “selectivity filter” motif of Ser–Gly–Gly–Gly in case of HKT1 and Gly–Gly–Gly–Gly in case of HKT2. Ion channels are generally bound to the cellular interior by a highly conserved Gly–Arg motif, which has been reported to be a unique feature of the TrkH transporter of almost all bacterial superfamilies of K+ transporters (Cao et al., 2011).
All these results indicate that TaHKT1;4.2, but not TaHKT1;4.3, performed its activity efficiently and contributed toward providing salt tolerance to Kharchia-65, whereas TaHKT1;4.3 probably failed to do so in HD-2329. As per the suggested role of HKTs (Shi et al., 2003), TaHKT1;4.2 appeared to limit the entry of Na+ into root cells of Kharchia-65 by its downregulation under salt stress, but because of the upregulated expression of TaHKT1;4.3 under salt stress it could not limit Na+ entry into the root of HD-2329, which resulted in the salt sensitivity of the genotype.
We observed a significant increase in the genome-wide DNA methylation in the salt-tolerant (Kharchia-65) genotype when salt stress was imposed. The increase was more prominent (19%) in the root compared with that (11%) in the shoot. Such hypermethylation responses in tobacco cell culture (Kovarik et al., 1997) and Mesembryanthemum crystallinumin (Dyachenko et al., 2006) have been reported earlier under salt stress. By contrast, Wang et al. (2014) reported that salt stress reduced the global methylation level in wheat. An increase in genome-wide DNA methylation due to abiotic stress has been reported earlier in drought-tolerant rice genotypes; however, partial reversal in the methylation level was also reported after withdrawal of the stress (Kumar and Singh, 2016).
We observed the basic (under control conditions) methylation level of the shoot to be higher (5.40%–6.7%) than that (3.45%–4.05%) of the root, particularly in the salt-sensitive genotype (Fig. 2), which is consistent with the observation of Karan et al. (2012) in rice. Global DNA analysis and bisulfite DNA sequencing of Arabidopsis genome by Cokus et al. (2008) revealed that approximately 20% of cytosines in the genome were methylated, and methylation occurred either in the promoter region or in the gene body.
The coding region of TaHKT1;4 showed only a minor variation in 5-mC with respect to genotype, tissue, and salt treatment. Although total 5-mC content increased (3%) in the shoot of HD-2329 because of salt stress, it remained constant (with all the cytosines methylated in the CHG context) in Kharchia-65. By contrast, the total 5-mC content decreased (3%) in the root of HD-2329 because of salt stress, but it increased (2%) in the root of Kharchia-65 (Fig. 3). Overall, the 5-mC content of TaHKT1;4 was observed to be approximately 90% in both the tissues and genotypes under control and salt stress conditions. Such a variation in the 5-mC content in TaHKT1;4 due to salt stress could not be correlated with either gene expression levels or salt tolerance levels of the genotypes. Karan et al. (2012) also reported that no specific methylation pattern was observed in the roots and shoots of salt-tolerant or salt-susceptible genotypes of rice. However, they observed an association between salt treatment and methylation level in the shoots of the four rice genotypes, but in the roots of only two rice genotypes. In this regard, they suggested that many methylation changes are not directed by environmental perturbations. Although a tissue-specific variation in cytosine methylation was reported in sorghum, only a few of the tissue-specific DMRs could be correlated with the tissue-specific expression of the genes (Zhang et al., 2011).
Salt stress caused methylation of 88%–94% of the cytosines in the TaHKT2;1 genes and TaHKT2;3 genes of the salt-tolerant genotype compared with only 78%–88% in the salt-sensitive genotype (Fig. 4, 5). The expression profile of TaHKT2;1 genes revealed that with 88% methylation it was four-fold downregulated in the shoot of the salt-tolerant genotype, but with 84% (comparatively 4% lower) methylation it was six-fold upregulated (comparative difference of 10-fold) in the salt-sensitive genotype.
Similarly, with a methylation level of 88% in roots of the salt-sensitive genotype, the gene was 1.1-fold downregulated; however, with 90% (comparatively 2% higher) methylation in roots of the salt-tolerant genotype, the gene was two-fold downregulated (comparative difference of 0.9-fold). Correlation of methylation level and expression level of TaHKT2;3 also revealed that with a methylation level of 88% in roots of the salt-sensitive genotype, the gene was two-fold downregulated; however, with an increased methylation level of 94% in roots of the salt-tolerant genotype, the gene was 2.5-fold downregulated. In shoots of HD-2329, a 7% increase (from 77% to 84%) in methylation level resulted in a four-fold reduction (from 10-fold to 6-fold) in expression across the genes.
Context-specific analysis of 5-mC in TaHKT2;1 and TaHKT2;3 of salt-sensitive and salt-tolerant genotypes revealed that salt stress increased methylation in the CG context by as much as 97%–100% in both the tissues and genotypes. With all the cytosines in the CG context (in both the genes) being methylated in shoots of HD-2329 even under the control condition, the increase in total methylation downregulated the expression of the genes. Similarly, with an increase in methylation to approximately 100% in the CG context in TaHKT2;1 in roots (of both the genotypes) due to salt stress imposition, the increase in total methylation was confirmed to be responsible for the downregulated expression of the gene. The methylation level in CHG and CHH contexts in TaHKT2;1 in shoots of the salt-tolerant genotype under control conditions was found to be higher (25%) than that of the salt-sensitive genotype. Salt stress further increased the methylation level in all the cytosine contexts, resulting in the appearance of DMRs.
Despite the variation in cytosine methylation in different contexts, the total methylation increased significantly, and the increase could be correlated with downregulation of the genes. Unlike in mammals, cytosine methylation has been reported to occur in all three contexts in plants. Although CHG and CHH methylation is common in transposable elements (TEs) and repeat-enriched heterochromatin regions, gene bodies are mainly associated with CG methylation. Methylation in the coding region has been reported to inhibit gene expression (Hohn et al., 1996). Baek et al. (2011) reported higher CG methylation in the leaves than in the roots, leading to a higher expression of AtHKT1 in the roots than in the leaves. These changes in gene expression engender the unique biological functions that are performed by these tissues in response to salt stress.
Because roots experience the stress first, hypomethylation in root tissues suggests the preparedness of the genes to quickly respond to salt stress. Furthermore, certain genes with an increased CG methylation were found to be more highly expressed, but increased non-CG (CHG and CHH) methylation reduced their expression in Arabidopsis (Schmitz et al., 2013). Although the role of non-CG methylation in regulating gene expression in TEs through pre- and post-transcriptional silencing is well established, its effect on gene expression in plants remains underexplored. The salt-tolerant wheat genotype appears to modulate its TaHKT2;1 and TaHKT2;3 expression by changing cytosine methylation in the CG and non-CG contexts, thereby regulating the uptake of Na+ by the plant and its performance under the stress.
An analysis of different parts of the coding region of HKT genes confirmed that a variation in the 5′ proximal coding region of the gene has considerable effects on the expression of the gene. Methylation of the promoter and 5′ proximal parts of the coding region has been reported to affect the expression of genes (Hohn et al., 1996; Baek et al., 2011; Wang et al., 2014). However, the interaction between DNA methylation in different contexts in the gene body and gene expression has not been understood. In plants, the methylation level of some genes dynamically changes throughout the growth and development of the plant in response to environmental perturbations.
Conclusion
Genetic variability is a prerequisite for plant breeding for the improvement of crop plants. Cytosine methylations occurring consistently (even in absence of the triggering environment) at specific loci produce epialleles (Kou et al., 2011), which might be exploited in crop breeding programs to improve the tolerance of the plant to abiotic stress. We observed that salt stress induces hypermethylation, resulting in the downregulation of TaHKT2;1 and TaHKT2;3 in the shoots and roots of the salt-tolerant genotype. By contrast, TaHKT1;4 was found to be expressed in the root only and was downregulated by the stress in the salt-tolerant genotype.
The expression of this gene was not regulated through the modulation of 5-mC; however, it might be controlled through other molecular or epigenetic mechanisms. Thus, the present study on the differential genotype- and tissue-specific expression of HKT genes in bread wheat under salt stress regulated by cytosine methylation established the role of the epigenetic mechanism in stress adaptation. This finding on the epigenetic regulation of HKT genes will improve our understanding about salt tolerance in plants. Nevertheless, molecular control of DNA methylation and inheritance of epialleles are not currently practicable using the available knowledge, tools, and techniques of molecular biology. Hence, a better understanding of the role of DNA methylation, histone modifications, and small-RNAs in the regulation of gene expression (Irier and Jin, 2012) is crucial for elucidating the epigenetic mechanisms not only in mammals but also in plants.
Supplementary Material
Acknowledgments
The authors acknowledge the facilities provided by the Director, ICAR-Indian Agricultural Research Institute, New Delhi, India, to undertake the present study.
Disclosure Statement
The authors declare that there are no conflicts of interest.
References
- Baek D., Jiang J., Chung J.-S., Wang B., Chen J., Xin Z., and Shi H. (2011). Regulated AtHKT1 gene expression by a distal enhancer element and DNA methylation in the promoter plays an important role in salt tolerance. Plant Cell Physiol 52, 149–161 [DOI] [PubMed] [Google Scholar]
- Boyko A., and Kovalchuk I. (2010). Genome instability and epigenetic modification-heritable responses to environmental stress? Curr Opin Plant Biol 14, 260–266 [DOI] [PubMed] [Google Scholar]
- Brini F., Imen A., Kaouther F., Hanin M., Khoudi H., and Masmoud K. (2009). Physiological and molecular analyses of seedlings of two Tunisian durum wheat (Triticum turgidum L. subsp. Durum [Desf.]) varieties showing contrasting tolerance to salt stress. Acta Physiol Plant 31, 145–154 [Google Scholar]
- Bustin S.A., Benes V., Garson J., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L., Vandesompele, and Wittwer C.T. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622 [DOI] [PubMed] [Google Scholar]
- Cao Y., Jin X., Huang H., Derebe M.G., Levin E.J., Kabaleeswaran V., Pan Y., Punta M., Love J., and Weng J. (2011). Crystal structure of a potassium ion transporter, TrkH. Nature 471, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus S.J., Feng S., Zhang X., Chen Z., Merriman B., Haudenschild C.D., Pradhan S., Nelson S.F., Pellegrini M., and Jacobsen S.E. (2008). Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport R.J., Munoz-Mayor A., Jha D., Essah P.A., Rus A., and Tester M. (2007). The Na+ transporter AtHKT1;1 controls retrieval of Na+ from the xylem in Arabidopsis. Plant Cell Environ 30, 497–507 [DOI] [PubMed] [Google Scholar]
- Dyachenko O.V., Zakharchenko N.S., Shevchuk T.V., Bohnert H.J., Cushman J.C., and Buryanov Y.I. (2006). Effect of hypermethylation of CCWGG sequences in DNA of Mesembryanthemum crystallinum plants on their adaptation to salt stress. Biochemistry 71, 461–465 [DOI] [PubMed] [Google Scholar]
- Hamamoto S., Horie T., Hauser F., Deinlein U., Schroeder J., and Uozumi N. (2015). HKT transporters mediate salt stress resistance in plants: from structure and function to the field. Curr Opin Biotechnol 32, 113–120 [DOI] [PubMed] [Google Scholar]
- Hohn T., Corsten S., Rieke S., Muller M., and Rothnie H. (1996). Methylation of coding region alone inhibits gene expression in plant protoplasts. Proc Natl Acad Sci USA 93, 8334–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T., Hauser F., and Schroeder J.I. (2009). HKT transporter mediated salinity resistance mechanisms in Arabidopsis and monocot crop plants. Trends Plant Sci 14, 660–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irier H.A., and Jin P. (2012) Dynamics of DNA methylation in aging and Alzheimer's disease. DNA Cell Biol 31, S42–S48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan R., DeLeon T., Biradar H., and Subudhi P.K. (2012). Salt stress induced variation in DNA methylation pattern and its influence on gene expression in contrasting rice genotypes. PLoS ONE 7, e40203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kou H.P., Li Y., Song X.X., Ou X.F., and Xing S.C. (2011). Heritable alteration in DNA methylation induced by nitrogen-deficiency stress accompanies enhanced tolerance by progenies to the stress in rice (Oryza sativa L.). J Plant Physiol 168, 1685–1693 [DOI] [PubMed] [Google Scholar]
- Kovarik A., Koukalova B., Bezdek M., and Opatrny Z. (1997). Hypermethylation of tobacco heterochromatic loci in response to osmotic stress. Theor Appl Genet 95, 301–306 [Google Scholar]
- Kumar S., and Saxena S. (2016). Sequence characterized amplified regions linked with apomictic mode of reproduction in four different apomictic Cenchrus species. Mol Plant Breeding 7, 1–14 [Google Scholar]
- Kumar S., and Singh A. (2016). Epigenetic regulation of abiotic stress tolerance in plants. Adv Plants Agric Res 5, 00179 [Google Scholar]
- Laurie S., Feeny K.A., Maathuis F.J.M., Heard P.J., Brown S.J., and Leigh R.A. (2002). A role of HKT1 in sodium uptake by wheat roots. Plant J 32, 139–149 [DOI] [PubMed] [Google Scholar]
- Law J.A., and Jacobsen S.E. (2010). Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11, 04–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira-Medeiros C.F., Parisod C., Fernandes R.A., Mata C.S., and Cardoso M.A. (2010). Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 5, e10326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller I.S., Gilliham M., Jha D., Mayo G.M., Roy S.J., and Coates J.C. (2009). Shoot Na+ exclusion and increased salinity tolerance engineered by cell type-specific alteration of Na+ transport in Arabidopsis. Plant Cell 21, 2163–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munns R., and Tester M. (2008). Mechanisms of salinity tolerance. Annu Rev Plant Biol 59, 651–681 [DOI] [PubMed] [Google Scholar]
- Pfaffl M.W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platten J.D., Cotsaftis O., Berthomieu P., Bohnert H., Davenport R.J., Fairbairn D.J., Horie T., Leigh R.A., Lin H.X., and Luan S. (2006). Nomenclature for HKT transporters, key determinants of plant salinity tolerance. Trends Plant Sci 11, 372–374 [DOI] [PubMed] [Google Scholar]
- Rus A., Yokoi S., Sharkhuu A., Reddy M., Lee B.-H., and Matsumoto T.K. (2001). AtHKT1 is a salt tolerance determinant that controls Na+ entry into plant roots. Proc Natl Acad Sci USA 98, 14150–14155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sairam R.K., Srivastava G.C., Agarwal S., and Meena R.C. (2005) Differences in antioxidant activity in response to salinity stress in tolerant and susceptible wheat genotypes. Biol Plant 49, 85–91 [Google Scholar]
- Schmitz R.J., Schultz M.D., Urich M.A., Nery J.R., Pelizzola M., Libiger O., (2013). Patterns of population epigenomic diversity. Nature 495, 193–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H., Lee B.H., Wu S.J., and Zhu J.K. (2003). Overexpression of a plasma membrane Na+/H+ antiporter gene improves salt tolerance in Arabidopsis thaliana. Nat Biotechnol 21, 81–85 [DOI] [PubMed] [Google Scholar]
- Singh A., Bhushan B., Gaikwad K., Yadav O.P., Kumar S., and Rai R.D. (2015). Induced defence responses of contrasting bread wheat genotypes under differential salt stress imposition. Ind J Biochem Biophys 52, 75–85 [PubMed] [Google Scholar]
- Srivastava M.K., Yadav C.B., Bhat V., and Kumar S. (2011). Cloning and characterization of a partial cDNA encoding Xyloglucan endotransglucosylase in Pennisetum glaucum L. Afr J Biotechnol 10, 9242–9252 [Google Scholar]
- Steward N., Ito M., Yamaguchi Y., Koizumi N., and Sano H. (2002). Periodic DNA methylation in maize nucleosomes and demethylation by environmental stress. J Biol Chem 277, 37741–37746 [DOI] [PubMed] [Google Scholar]
- Wang M., Qin L., Xie C., Li W., Yuan J., Kong L., Yu W., Xia G., and Liu S. (2014). Induced and constitutive DNA methylation in a salinity-tolerant wheat introgression line. Plant Cell Physiol 55, 1354–1365 [DOI] [PubMed] [Google Scholar]
- Wang X., Li Q., Yuan W., Cao Z., Qi B., Kumar S., Li Y., and Qian W. (2016). The cytosolic Fe-S cluster assembly component MET18 is required for the full enzymatic activity of ROS1 in active DNA demethylation. Sci Rep 6, 26443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi T., Hamamoto S., and Uozumi N. (2013). Sodium transport system in plant cells. Front Plant Sci 4, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H.H., Kikuchi S., Neumann P., Zhang W.L., and Wu Y.F. (2010). Genome wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice. Plant J 63, 353–365 [DOI] [PubMed] [Google Scholar]
- Zemach A., McDaniel I.E., Silva P., and Zilberman D. (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916–919 [DOI] [PubMed] [Google Scholar]
- Zhang M., Xu C., Wettstein D.V., and Liu B. (2011). Tissue-specific differences in cytosine methylation and their association with differential gene expression in sorghum. Plant Physiol 156, 1955–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.