

Abstract
A continuous stream of activating and repressing signals is processed by the transcription complex paused at the promoter of the c-myc proto-oncogene. The general transcription factor IIH (TFIIH) is held at promoters prior to promoter escape and so is well situated to channel the input of activators and repressors to modulate c-myc expression. We have compared cells expressing only a mutated p89 (xeroderma pigmentosum complementation group B [XPB]), the largest TFIIH subunit, with the same cells functionally complemented with the wild-type protein (XPB/wt-p89). Here, we show structural, compositional, and functional differences in transcription complexes between XPB and XPB/wt-89 cells at the native c-myc promoter. Remarkably, although the mean levels of c-Myc are only modestly elevated in XPB compared to those in XPB/wt-p89 cells, the range of expression and the cell-to-cell variation of c-Myc are markedly increased. Our modeling indicates that the data can be explained if TFIIH integrates inputs from multiple signals, regulating transcription at multiple kinetically equivalent steps between initiation and promoter escape. This helps to suppress the intrinsic noise of transcription and to ensure the steady transcriptional output of c-myc necessary for cellular homeostasis.
The c-Myc transcription factor targets approximately 10% of genes, coordinating many essential cellular processes, including proliferation, growth, differentiation, metabolism, and apoptosis (37, 38). Small changes in c-Myc protein levels, either up or down, modify these processes, and so c-myc expression must be held to close tolerances (69). Although many proteins regulate c-myc expression (Fig. 1), none impose dominating influence over all the rest; the mechanisms integrating these multiple inputs to provide a controlled output have not been elucidated. Due to the fast turnover and low abundance of both c-myc mRNA and protein in most normal tissues, rapid feedback mechanisms must operate to constrain c-Myc levels (8, 22, 54, 61, 69). Otherwise, c-Myc levels would fluctuate because transcription from a single promoter is an intrinsically noisy process; promoter firing is a low-probability event on a molecular time scale (13, 55).
FIG. 1.

Schematic representation of transcription factors regulating c-myc promoter activity. Selected transcription activators and repressors that directly bind to the c-myc promoter are shown at their approximate binding sites. Transcriptionally engaged RNA polymerase II is paused immediately downstream of the major transcription start sites P1 and P2. DNase I hypersensitive sites (HS) indicative of altered chromatin structures are also shown. To maintain appropriate c-Myc levels, some mechanism should exist to integrate this plethora of signals.
The c-myc promoter is regulated by a transcriptionally engaged RNA polymerase, paused at the start site which must escape the promoter to commence elongation (44, 56, 63). Paused polymerases may be poised for rapid response but may also protect promoters from spurious activation, because activators facilitating preinitiation complex formation are thwarted until the start site is again available. To regulate promoters harboring paused polymerases, signals must be delivered to those components controlling early nascent transcript extension (Fig. 1). Although the regulatory mechanisms between initiation and promoter escape have been incompletely described, the general transcription factor IIH (TFIIH) operates throughout this interval. It is known that TFIIH is a multifunctional, multisubunit protein complex that plays central roles in transcription and DNA repair (nucleotide excision repair [NER]) (5, 64, 71). In addition, the three-subunit cyclin-dependent kinase (Cdk)-activating kinase (CAK)-kinase subcomplex of TFIIH, Cdk7/cyclin H/ménage à trois 1 (MAT1), separately contributes to cell cycle control. The two largest subunits of TFIIH, p89 (xeroderma pigmentosum complementation group B [XPB]) and p80 (XPD), have 3′-to-5′ and 5′-to-3′ helicase activities, respectively. TFIIH interacts with a variety of cellular and viral transcription activators and repressors that deliver signals to and receive signals from the transcription machinery. For example, transcription activation by steroid hormone receptors requires phosphorylation by Cdk7, and p53 both regulates and is phosphorylated by Cdk7 (3, 30, 43, 59). The activating events subsequent to Cdk7 action have been incompletely illuminated. Though some activators and repressors target p89/XPB helicase activity in vitro, the role of p89 action on native gene expression is largely unexplored (21, 23, 67, 76).
Joining preinitiation complexes after promoter selection, TFIIH contributes to basal transcription at several early stages of the transcription cycle (4, 11, 25, 26, 35, 50, 62). Generalizing from a limited number of promoters, p89/XPB first facilitates promoter melting and open complex formation; then ATP hydrolysis by the XPB helicase is required throughout the transitions, leading to promoter escape (16). In vitro, this requirement may be bypassed by premelting the start site (using mismatched bases to prop open the double helix) or, for some promoters, by providing supercoiled templates (19, 53). p80/XPD also contributes to nascent transcript growth, although XPD helicase activity is not required to support basal transcription, and so it has been proposed that p80 plays a structural role anchoring the CAK (9). The mutations of XPD that cause trichothiodystrophy impair TFIIH assembly and/or stability and so reduce basal transcription. CAK phosphorylates the carboxyl-terminal domain (CTD) of the largest subunit of RNA polymerase II. Phosphorylating serine-5 in the heptad repeat comprising the CTD, Cdk7 action enables the recruitment of factors required for mRNA capping; other Cdk7 targets likely contribute to the transcription and processing of RNAs in a gene-specific manner (32, 68).
The sequence-specific single-stranded binding transcription factor, FUSE-binding protein (FBP), and its antagonist, the FBP-interacting repressor (FIR), have been proposed to help impose tight regulation on c-myc transcription (10, 24, 40, 41, 46). FBP and FIR are recruited to single-stranded DNA at the FUSE sequence, far upstream of the major P2 promoter of the c-myc gene (10, 24, 40, 41, 46). Additionally, the conformation of FUSE DNA is especially sensitive to the torsional strain that is a transient by-product of transcription (24). The action of both FBP and FIR is channeled through TFIIH (40, 41). From several lines of experimental evidence, in vitro as well as in vivo, FBP and FIR modulate transcription; FBP hastens the passage from initiation through postinitiation steps until promoter escape, whereas FIR delays these transits. Recently, in vitro systems have shown that transcription activation via FBP and repression via FIR are lost in XPB and are impaired in XPD disease (41). Transfected FBP increases endogenous c-Myc levels in XPB cells only when coexpressed with wild-type p89, suggesting that FBP acts through TFIIH to help maintain proper c-myc regulation. It is expected that, lacking proper input from TFIIH-interacting factors such as FBP, FIR, and E2F, c-myc regulation in XPB cells would be disturbed.
This study shows that the tight control, characteristic of the c-myc gene, is compromised in XPB-mutant cells in a surprising manner. XPB cells exhibit striking cell-to-cell heterogeneity in c-Myc levels. Upon restitution of wild-type TFIIH, cellular c-Myc levels become much more uniform. Changes at the c-myc promoter indicate that the zone of TFIIH influence is contracted with mutation of p89/XPB and promoter escape occurs closer to the start site. As shown previously for FBP, FIR is found to depend on functional TFIIH to modulate endogenous c-Myc levels. Reasoning from these results, it is shown that by regulating transcription at multiple points between initiation and promoter escape, stochastic fluctuation of gene expression may be suppressed.
MATERIALS AND METHODS
Plasmid constructs.
pGS5 M/XPB (kindly provided by Kenneth Kraemer) was used as a source for human p89 cDNA. For generation of the inducible p89 expression system, a hemagglutinin (HA) tag was added to XPB cDNA by PCR, using Pfu polymerase. Clones were checked with direct sequencing. HA-tagged p89 was first cloned into pSP72 and then into pMEP4 (Invitrogen).
Cell culture, cell cycle manipulation, and transfection.
XPB-deficient lymphoblasts (GM02252C/XPB11BE from the Coriell Cell Repository, Camden, N.J.) were cultured in RPMI 1640 with 15% serum (Cellgro). Stable cell lines were generated by transfection with 1 μg of plasmid with FuGENE 6 (Roche), followed by selection under hygromycin (0.1 mg/ml). Protein expression was induced with cadmium and added directly into the cell culture medium at the indicated concentrations. BJAB cells were grown in RPMI 1640 with 10% serum (Cellgro). Cell number was determined by counting cells excluding trypan blue in a Neubauer chamber. Unless otherwise indicated, all experiments were performed at a cell density of 5 × 105 cells per ml to avoid biases due to differential growth kinetics. A previously described simian virus 40-transformed fibroblast system that was optimized for the purification of tagged TFIIH was not used to avoid the influences of chromosomal integration and repeated cycles of UV treatment used during clonal selection (74).
Cell survival assay.
Survival of UV-treated cells was assayed as previously described, with slight modifications (20). Briefly, cells were washed twice with 1× phosphate-buffered saline (PBS); 2 × 105 cells per 35-mm dish were plated, PBS was removed, and cells were treated with different doses of UV (0 to 50 J/m2) with a calibrated Stratalinker UV Cross-Linker (model 2400). Medium was added back to cells, and the survival rate was calculated after counting cells 72 h after UV treatment. For the proliferation assay, cells were plated (in triplicate) at the same density and cultured under the same conditions. Cell growth was determined by counting cell numbers daily.
Immunofluorescence microscopy.
Suspended cells were washed and spun onto coverslips prior to fixation. Cells were washed twice in 1× PBS (5 min) and fixed. For most staining procedures, cells were fixed for 12 min in 2% paraformaldehyde, washed twice in PBS, permeabilized for 5 min in permeabilization buffer (80 mM HEPES [pH 6.8], 5 mM EGTA, 1 mM MgCl2, 0.5% Triton X-100), and refixed for 6 min again in 2% paraformaldehyde, followed by four additional PBS washes. After fixation, cells were blocked for 12 min (0.5% Triton X-100 and 2% bovine serum albumin in 1× PBS) and incubated with primary antibody or species-identical nonspecific immunoglobulin G at the same concentration for 1 h.
Primary antibodies were as follows: α-HA (rabbit polyclonal; Roche), 1:50 dilution; α-cyclin H (mouse monoclonal; Austral), 1:50 dilution; and α-c-Myc (rabbit polyclonal; Upstate), 1:100 dilution. For staining c-Myc, incubation was performed overnight.
After incubation with primary antibody, cells were washed three times in PBS and incubated with fluorescein isothiocyanate-labeled secondary antibody (1:50 dilution; Jackson Immunochemicals) or tetramethyl rhodamine isocyanate-labeled secondary antibody (1:200 dilution; Sigma) for 30 min, followed by three additional washes. After DNA was counterstained with propidium iodide or 4′,6′-diamidino-2-phenylindole (DAPI), slides were mounted with Vectashield mounting solution (Vector Laboratories).
Flow cytometry for c-Myc quantification.
Cells were fixed and permeabilized with Intra stain (DAKO). After this, staining for c-Myc was performed as for immunofluorescence. Phycoerythrin-conjugated α-rabbit antibody (Molecular Probes) was used as a secondary antibody, and fluorescence was detected in the Fl-2 channel.
Western blotting.
Cells were washed twice in 1× PBS, resuspended in ice-cold radioimmunoprecipitation assay buffer (1% NP-40; 0.5% Na-deoxycholate, 0.1% sodium dodecyl sulfate [SDS]), swollen on ice for 20 min, homogenized, and disrupted by being passaged five times through a 25-gauge (5/8-in.) needle, incubated on ice for an additional 20 min, and centrifuged in a microcentrifuge at full speed for 30 min to recover total cell lysates. Protein lysates were separated on a 4 to 20% gradient gel (Novex) and blotted onto a nitrocellulose membrane. Primary antibodies were used as follows: α-HA (mouse monoclonal; Roche), 1:2,000 dilution; α-p89 (SC-293, rabbit polyclonal; Santa Cruz) that recognizes wild-type, but not C-terminal truncated p89), 1:500 dilution; α-p89 (Austral Biotech) that recognizes an N-terminal epitope of p89, in both wild-type and truncated p89/XPB; α-p62 (rabbit polyclonal; Santa Cruz), 1:500 dilution; α-c-Myc (N-262, rabbit polyclonal; Santa Cruz), 1:500 dilution; and α-actin (mouse monoclonal; Oncogene), 1:10,000 dilution.
RNase protection assay and nuclear run-on assay.
RNase protection assays (RPAs) and nuclear run-on experiments for determining c-myc mRNA levels were performed essentially as previously described (6).
KMnO4 footprinting.
Cells were harvested, washed once with room temperature PBS, resuspended in buffer A (15 mM Tris-HCl [pH 7.5], 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.5 mM EGTA, 300 mM sucrose), and incubated for 2 min at room temperature. Fresh KMnO4 was added to a final concentration of 25 mM and incubated at room temperature for 45 s. The reaction was stopped with β-mercaptoethanol (343 mM) and SDS (0.5%). Ligation-mediated PCR was performed as previously described and as modified (18, 45, 49). For heat shock response, cells were incubated at 42°C for 30 min and then incubated at 37°C for 30 min for recovery. Genomic DNA was treated with KMnO4 in vitro at 25°C for 1 min and treated as above (46).
Chromatin immunoprecipitation (ChIP).
The ChIP method was a variant of that previously described (73). Formaldehyde was added to cells in medium (final concentration of 1.0%). Cells were fixed for 15 min at 37°C, and the reaction was stopped by the addition of glycine to a final concentration of 125 mM. Cells were collected, washed twice with cold PBS, and then resuspended in Tris-EDTA (TE) with protease inhibitor cocktail (Calbiochem). The cell suspension was sonicated on ice with an ultrasonic sonicator for 6 pulses of 30 s each to an average DNA length of ∼1,000 bp. After centrifugation, SDS, sodium deoxycholate, Triton X-100, and NaCl were added to the chromatin solution to final concentrations of 0.1, 0.1, and 1.0% and 300 mM, respectively. The chromatin solutions were cleared with protein A-agarose beads (Roche) for 1 h at 4°C. For immunoprecipitations, chromatin solutions from 2 × 106 cells were mixed with the indicated antibodies, protein A-agarose beads preblocked with 0.1 mg of herring sperm DNA/ml, and 2 mg of bovine serum albumin/ml. After incubation at 4°C overnight, beads were washed twice with buffer 1 (1× TE, 0.1% SDS, 0.1% sodium deoxycholate, 1.0% Triton X-100, and 300 mM NaCl), once with buffer 2 (1× TE, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100, and 150 mM NaCl), twice with buffer 3 (1× TE, 0.25% sodium deoxycholate, 0.5% NP-40, and 250 mM LiCl) and once with TE. Immunoprecipitates were eluted from beads by incubation with elution buffer (50 mM Tris-HCl [pH 8.0], 1% SDS) at 65°C for 15 min. The beads were extracted again with TE, and proteinase K (final concentration, 0.5 mg/ml) was added to the combined elutes and incubated at 65°C overnight to reverse cross-linking. The samples were extracted with phenol and then phenol-chloroform, and DNA was precipitated with ethanol. A total of 10% of precipitated DNA was used for PCR. Antibodies used were p89 (recognizes an N-terminal epitope of p89, in both wild-type and truncated p89/XPB; Austral Biotech), cycH (Austral Biotech), hHSF1 (a kind gift of Carl Wu), and polymerase II (catalogue number 8WG16; Babco). Primers for PCR were as follows: for the c-myc promoter, the forward primer was 5′GGA TCG CGC TGA GTA TAA AAG CCG3′ and the reverse primer was 5′CTA TTC GCT CCG GAT CTC CCT TC3′; for the hsp70 promoter, the forward primer was 5′GCG AAA CCC CTG GAA TAT TCC CGA3′ and the reverse primer was 5′GAA GCC TTG GGA CAA CGG GAG TC3′. Titration of input DNA into PCR confirmed that amplifications occurred in the linear range.
RESULTS
A cell system to study the influence of p89/XPB on c-myc expression.
A model system was created to study the FBP/FIR/TFIIH regulatory system in cells with normal or defective TFIIH function. The extensively characterized XPB lymphoblast cell line from patient XP11BE served as the foundation for further investigation (27). Due to a splice mutation in the p89/xpb gene leading to a frameshift, the last 41 amino acids of the protein are out of frame. The mutant cells show a severe defect in NER with UV hypersensitivity and an impaired helicase activity, as well as reduced in vitro basal transcription activity. Wild-type p89 was expressed in XPB cells to rescue TFIIH function with an Epstein-Barr virus-derived episomal vector system with a metal-inducible promoter. The use of the episome ensured p89 expression in pools of cells without the confounding complications of clonal selection. Transfecting the empty vector and performing the same selection created a control cell line in addition to the parental mutant cells. The resulting cell lines were designated XPBo (the original XPB cells from which the others were derived), XPB (the XPBo cells harboring the empty vector), and XPB/wt-p89 (XPBo with an episome expressing wild-type p89). Microarray hybridization experiments revealed that the ratio of episomally expressed, wild-type p89 mRNA to endogenous, frameshifted p89 mRNA was about 4:1 under steady-state conditions without induction and was increased to more than 25:1 following treatment with cadmium (H.-J. Chung and D. Levens, unpublished data). p89 protein levels increased in a dose-dependent fashion along with mRNA expression upon the addition of cadmium. p89 protein was even more profoundly induced than the RNA, most likely due to accretion. Wild-type p89 was already detectable in XPB/wt-p89 cells even under uninduced steady-state conditions (Fig. 2A, lane 2), and was at levels comparable to those in the lymphoblast cell line BJAB (Fig. 2B, bottom panel, lanes 1 and 3). Although complemented cells showed modestly increased levels of p89 protein when an antibody recognizing both the truncated and wild-type p89 was used (Fig. 2C), restitution of wild-type p89 with uninduced expression did not obviously disturb the expression of other TFIIH components, since immunoblot analysis showed that levels of the p62 subunit were strikingly similar when levels of XPB, XPB/wt-p89, and the BJAB cells were compared (Fig. 2B, top panel). Thus, any difference between XPB cells and XPB/wt-p89 cells could be attributed to a difference in TFIIH quality rather that TFIIH quantity. XPB/wt-p89 B cells were stained with fluorescent antibodies to ascertain TFIIH assembly incorporating episomally encoded wild-type p89 in vivo and to visualize proper TFIIH subcellular localization. Wild-type p89 was found in a finely dotted pattern throughout the nucleus with relative sparing of the nucleoli, a pattern previously seen for TFIIH (28). Costaining for cyclin H, another component of TFIIH, revealed extensive colocalization of these two subunits, indicating proper assembly of the holoenzyme complex (Fig. 2D).
FIG. 2.

Inducible expression of wild-type p89 in XPB cells. (A) Expression of wild-type p89 can be induced by cadmium. HA-tagged wild-type p89 is under the control of a metal-inducible promoter. Wild-type p89 is detectable in XPB/wt-p89 cells even under steady-state conditions (lane 2) and can be induced by cadmium (lanes 3 to 8). No wild-type p89 is expressed even under high doses of cadmium in the XPB cell line (lane 1). (B) Quantification of p89 expression. A polyclonal antibody against the C terminus of p89 (SC-293) recognizes wild-type p89 but not truncated p89. Under steady-state conditions, XPB/wt-p89 cells express p89 to a level similar to that with the BJAB reference cell line. Probing for p62, another subunit of core TFIIH, shows similar levels of expression in all three cell lines, indicating that expressing wild-type p89 alters TFIIH quality but not protein turnover. (C) Quantification of p89 protein levels. XPB and XPB/wt-89 whole-cell lysates were subjected to Western blotting with α-p89 (Austral), which recognizes both truncated and wild-type p89. (D) Subcellular localization of wild-type p89 in XPB/wt-p89 cells. Wild-typep89 (detected by α-HA) is distributed in a finely dotted pattern throughout the nucleoplasm with relative sparing of nucleoli. Staining for cyclin H reveals colocalization with p89, indicating proper TFIIH assembly. Nuclei were counterstained with DAPI. (E) XPB/wt-p89 cells are DNA repair proficient. XPB cells, XPB/wt-p89 cells, XPB/wt-p89 cells under cadmium induction, and BJAB cells were UV irradiated with increasing doses, and survival rates (the ratio of treated cells/untreated cells) were assessed 4 days later. Low-dose UV irradiation dramatically reduced XPB cell survival, whereas XPB/wt-p89 cells were less sensitive. Comparison to a reference cell line (BJAB) indicates that the repair capacity is within normal range. Induction of wild-type p89 expression with 500 nM cadmium does not change survival compared to steady-state expression levels.
To assess the functional significance of the stable expression of wild-type p89, cells were subjected to a standard cell survival assay. Cells were UV irradiated with increasing doses (2 to 50 J/m2), and numbers of viable cells were counted 4 days after UV irradiation. Empty vector-transfected XPB lymphoblasts (XPB cells) showed reduced cell survival compared to the reference lymphoblast cell line, BJAB. In contrast, XPB lymphoblasts with wild-type p89 displayed UV sensitivity comparable that of BJAB cells. Thus, by stable transfection of wild-type p89, XPB cells were functionally corrected for survival after UV irradiation (Fig. 2E). Since uninduced levels of p89 expression obviously yielded sufficient TFIIH to sustain proper NER, the all of the following experiments were performed under basal levels of episomal wild-type p89 expression. Full rescue of TFIIH function in NER would be paralleled by the reversal of transcriptional deficits attributable to defective TFIIH. In addition, since XPB and XBP/wt-p89 cells differ only with respect to their p89 status but otherwise share the same genetic background, this model system seems suitable to explore the consequences of the XPB mutation on transcription in vivo.
c-Myc expression is coarse and heterogeneous in XPB versus XPB/wt-p89 cells.
In vitro, FBP accelerates and FIR delays promoter escape via TFIIH. The cell system described above was employed to assess the net consequences on c-myc expression of the TFIIH mutation in XPB disease. Reasoning that by simultaneously disabling opposing activators and repressors (41), c-myc regulation might become more erratic, the variation of endogenous c-Myc levels was assessed in individual cells by staining with anti-c-Myc, followed by flow cytometry. Comparison of the c-Myc distributions between uncorrected and corrected cells revealed two quantitative distinctions. First, the entire population of XPB cells showed ∼1.5-fold-higher mean levels of c-Myc than did the XPB/wt-p89 cells (375 versus 248 on an arbitrary scale corrected for the immunoglobulin G control) (Fig. 3A). Concomitant analysis of DNA content indicated that the difference in c-Myc was not due to a differential distribution in the cell cycle (data not shown). Second, the mutant cells indeed displayed a much broader distribution of c-Myc levels. Following restitution of wild-type p89, the variance of c-Myc distribution in the corrected cells was reduced to 31% of that of the mutant cells. The XPB cells had both higher levels of c-Myc and a higher coefficient of variation than the wild-type cells (0.53 versus 0.45, respectively); a higher level of expression would be expected to reduce the coefficient of variation.
FIG. 3.

TFIIH suppresses cell-to-cell variation in c-Myc. (A) To determine expression levels with single-cell resolution, cells were stained for c-Myc and analyzed by flow cytometry. (Top) Background from secondary antibody; (bottom) anti-c-Myc-specific fluorescence. Average c-Myc levels were 1.51-fold higher in XPB than in XPB/wt-p89 cells. Whereas expression levels of c-Myc resembled a Gaussian distribution in XPB/wt-p89 cells, c-Myc levels were more broadly distributed in XPB cells with some cells showing more than four times the mean of XPB/wt-p89 cells. The CV (coefficient of variation) of XPB and XPB/wt-p89 was 0.53 and 0.45, respectively. K-S statistics reveal these distributions to be different with P ≪ 0.001. (B) BJAB (normal TFIIH) cells show a distribution more similar to that of XBP/wt-p89 cells but distinct from the broad profile observed with XPB cells. Note that the absolute, but not relative, levels of staining would be expected to vary between experiments. The CVs for XPB, XPB/wt-p89, and BJAB were 0.52, 0.46, and 0.46, respectively (note that at high levels of c-Myc, the BJAB curve drops below the XPB/wt-p89 curve, allowing both cells to have similar CVs).
Analyzing BJAB cells revealed a c-Myc distribution similar to that seen with XBP/wt-p89 cells but distinct from the abnormally broad profile observed with XPB cells (Fig. 3B). Thus, even in lymphoma cells, c-myc expression is less variable than is seen upon mutation of TFIIH.
Concordant levels of c-myc mRNA and protein in XPB cells and XPB/wt-p89 cells.
The most simple explanation for the 1.5-fold increase in levels of c-Myc protein in cells with mutant p89/XPB would be a similar increase in c-myc mRNA, since TFIIH is a transcription factor. Therefore, RPAs were performed to ascertain whether the 50% increase in mean c-Myc protein measured by flow cytometry of XPB relative to that of XPB/wt-p89 lymphoblasts was paralleled at the c-myc mRNA level. In multiple independent RPA experiments, the uncorrected cells consistently contained at least 50% more c-myc mRNA than the corrected cells (Fig. 4A, lane 1 versus lane 2). Since c-myc mRNA and protein levels are tightly linked in most circumstances, c-Myc protein levels were compared to cross-check the differences between the corrected and uncorrected cells. Indeed, immunoblot blot analysis of XPB whole-cell extracts under steady-state conditions repeatedly revealed an increase of c-Myc protein, consistent with the levels determined by flow cytometry (Fig. 3) and commensurate with the increase in mRNA (Fig. 4A, lane 3 versus lane 4). Thus, at the RNA and protein levels, c-Myc expression was increased.
FIG. 4.

Steady-state endogenous c-myc levels are disturbed in XPB cells. (A) (Left) c-myc mRNA is expressed at higher steady-state levels in XPB than in XPB/wt-p89 cells (lanes 1 and 2). RNase protection with c-myc P1-P2 and exon 2 riboprobes is shown. Quantification of c-myc mRNA (normalized to gapdh mRNA) reveals 1.4- to 1.6-fold higher levels of c-myc mRNA in XPB than in XPB/wt-p89 cells. (Right) Comparison of c-Myc protein levels in XPB versus XPB/wt-p89 cells. Concordant with RNA levels, XPB cells have more c-Myc than cells expressing wild-type p89 (lanes 3 and 4). (B) (Left) Analysis of c-myc expression in response to UV irradiation by RNase protection 3 h after UV exposure (8 J/m2). Quantification of c-myc mRNA (normalized to gapdh mRNA) demonstrates 2.5- to 3.2-fold-higher levels of c-myc mRNA in XPB than XPB/wt-p89 cells. (Right) c-Myc protein levels parallel mRNA levels (lanes 3 and 4). (C) Analysis of the response of c-myc mRNA to TSA treatment. Cells were treated with TSA (500 ng/ml) or dimethyl sulfoxide (DMSO) (as the vehicle) for 4 h. The RPA reveals that c-myc mRNA is down-regulated more efficiently in XPB/wt-p89 than in XPB cells (lanes 5 and 6 versus 11 and 12). Undigested probes are shown in the left panel. Bands marked with an asterisk were from a shorter exposure to verify intensity.
To confirm the concordance between c-myc mRNA and protein levels in both the XPB and XPB/wt-p89 cells, maneuvers were attempted to alter transcriptional output of the gene. Reasoning that redeployment of TFIIH to sites of DNA damage might exacerbate the difference between them, XPB and XPB/wt-p89 cells were irradiated with UV light, a treatment known to down-regulate c-myc (1, 17, 47). RNA was collected 3 h postirradiation, and c-myc mRNA levels were measured by RPA. Because c-myc mRNA was more effectively down-regulated in XPB/wt-p89 than in XPB cells, the excess of c-myc transcripts in the mutant cells relative to the corrected increased to 2.7 fold, from 1.5 fold at steady state (Fig. 4B, lane 1 versus lane 2). c-myc shutoff was also delayed in the XPB cells; 3 h post-UV treatment, c-myc levels in the mutant cells persisted at 80 to 100% of steady state, whereas the c-myc levels in the corrected cells had declined by 50%. Eventually, 6 h after irradiation, c-myc mRNA bottomed at similarly low levels in both cell lines (data not shown). These changes in c-myc mRNA were reproducibly paralleled by the expected decline in c-Myc protein; following UV irradiation (Fig. 4B, lane 3 versus lane 4), the excess of c-Myc in XPB relative to XPB/wt-p89 cells became more pronounced.
Is the sluggish down-regulation of c-myc transcription in XPB cells secondary to the XP-NER deficit and so peculiar to UV irradiation, or does the XPB mutation generally impair c-myc repression due to a transcription defect at the promoter? Histone deacetylase inhibitors such as trichostatin A (TSA) and butyrate reduce c-myc promoter activity and induce differentiation in susceptible cells (2, 70). Indeed, after 4 h of TSA treatment, c-myc was down-regulated less effectively in XPB cells than in XPB/wt-p89; XPB/wt-p89 cells as well as BJAB cells (data not shown) exhibited the expected decline in c-myc RNA (Fig. 4C, lanes 5 and 6 versus lanes 11 and 12) (6). Similar results were seen upon histone deacetylase inhibition with butyrate (data not shown). Thus, under all conditions examined, the uncorrected cells expressed more c-Myc than the corrected cells. Though modest, the differences in c-myc expression due to the p89 mutation in XPB might be significant, since even subtle changes in c-Myc levels have been associated with alteration of cell size, proliferation, apoptosis, and organismal phenotypes. Although a defective basal transcription factor might be expected to impair transcription, failure to receive repressor input or to holdback at the c-myc promoter might yield a net increase in expression. The changes in c-myc expression seen in XPB cells might be due to defective TFIIH operating directly at the c-myc promoter or could be an indirect effect of the TFIIH mutation. If direct, the increased levels of c-myc expression could be due to sporadic hyperactivity from just a few c-myc promoters, while the remaining c-myc genes maintain normal promoter structure and function; in this case, the structure, function, and composition of complexes at the c-myc promoter would be similar between these cells. Alternatively, TFIIH may act uniformly at each c-myc promoter to hold expression to close tolerances; in this case, the mutant TFIIH should cause conformational, compositional, and functional differences when the c-myc promoters in XPB and XPB/wt-p89 cells are compared. Therefore, the c-myc promoters of XPB and XPB/wt-p89 cells were examined.
TFIIH mutations impose major conformational, compositional, and functional changes at the c-myc promoter.
Either by helping to set the trajectory of promoter DNA within transcription complexes or via its intrinsic helicase activity, TFIIH is equipped to manipulate DNA structure at transcription start sites. To assess the c-myc P2 promoter configuration in vivo, footprinting was performed using potassium permanganate as a DNA conformation-sensitive chemical probe; nucleotide +58 (using nucleotide +53 as a reference) was dramatically hyperreactive in cells with wild-type p89 but was much less reactive in XPB cells with mutant p89 (Fig. 5A). The start site region, in contrast, was somewhat less reactive in the corrected cells than in the mutant cells; thus, the overall reactivity was more closely localized to the initiation site in the absence of fully functional TFIIH. To be detectable, these changes must occur at a significant portion of the total population of c-myc promoters.
FIG. 5.

Holdback of c-myc transcription occurs in a broader zone in XPB/wt-p89 than in XPB cells. (A) Conformation-sensitive in vivo footprinting of the c-myc P2 promoter with KMnO4 shows that the region around +58 is hyperreactive (black arrow) in cells with wild-type p89; modest start-site hyporeactivity (grey arrows) is seen in XPB/wt-p89 versus XPB cells, consistent with a downstream shift of transcription complexes in XPB/wt-p89 cells. Blank regions are hyporeactive, due to a paucity of thymidines. Lane C, genomic DNA treated with KMnO4 in vitro for 1 min at 25°C. (B) ChIP analysis reveals diminished TFIIH binding at the c-myc P2 promoter in XPB cells, unless complemented with wild-type p89 (top panels, lanes 3); the same occurs at the hsp70 promoter (bottom panels, lanes 3). Binding of heat shock factor following heat shock is equivalent in both cells (lanes 8). Anti-p89 used in ChIP experiments recognizes both wild-type and truncated forms of p89. (C) ChIP analysis of the c-myc promoter in XPB and XPB/wt-p89 cells with α-p89 and α-CycH (Austral). (D) Nuclear run-on shows differential holdback at the major c-myc P2 promoter under steady-state conditions (left), as well as after UV irradiation (right). Hybridization with the second consecutive (downstream) P2 oligonucleotide is weaker for XPB cells, indicative of premature release of polymerase in cells with impaired TFIIH (lanes 1 and 3 versus 2 and 4). Differential release is exaggerated following UV irradiation (lanes 5 and 7 versus 6 and 8). Scans demonstrate relative intensities of the steady-state panel.
The structural perturbation at +58 seen in p89 wild-type but not mutant cells suggested that some component opening the promoter was defective or missing from this site. ChIP revealed RNA polymerase bound to the c-myc promoters of both cell lines (Fig. 5B, lane 4). Following heat shock treatment as a control (see below), RNA polymerase was reduced at the c-myc promoter consistent with the expected down-modulation of most transcription units (Fig. 5B, lane 10). In contrast, promoter-engaged TFIIH was efficiently recovered only from the XPB/wt-p89 cells, and the c-myc promoters in the XPB cells harbored only very low levels of TFIIH with antibodies against p89 and cyclin H (Fig. 5B, lane 3, and Fig. 5C). Apparently, mutant TFIIH is inefficiently recruited, prematurely discharged, or ineffectively retained at the c-myc promoter; nevertheless, these TFIIH-underloaded promoters support the same or higher levels of c-Myc.
To find out if the mutant TFIIH provoked changes in the activity, composition, or DNA conformation at promoters besides c-myc, the hsp70 promoter was also examined by RPA, in vivo footprinting with potassium permanganate, and ChIP. Starting from similar low basal levels, a pulse of heat shock effectively induced hsp70 expression in both XPB and XPB/wt-p89 cells; levels subsequently declined in both cell lines, although subtle details of the temporal profile may distinguish the two (hsp70 mRNA may linger in the mutant cells), as assayed by RPA (data not shown). Belying the overall similarity of the heat shock response of hsp70 in XPB (or XPBo) and XPB/wt-p89 cells, ChIP revealed the same striking difference found with the c-myc promoter; whether basal or heat shocked, the hsp70 promoter from the XPB mutant was dramatically deficient in TFIIH (Fig. 5B, lanes 3 and 9). In contrast, the hsp70 promoter from both the mutant and wild-type cells was effectively charged with RNA polymerase in both cell lines independent of heat shock, albeit with minor differences (Fig. 5B, lanes 4 and 10). Though substantially devoid of TFIIH, the XPB hsp70 promoter showed no unequivocal structural or functional changes attributable to the lack of TFIIH when probed with potassium permanganate in vivo. Apparently, the p89 mutation results in either less-efficient loading or more-efficiently discharge of TFIIH from the hsp70 promoter without dramatically perturbing transcription, even during heat shock.
The zone of transcription holdback is diminished in XPB.
If FBP, FIR, or other transacting factors use TFIIH to influence transcription between initiation and promoter escape at the c-myc promoter, then the distribution of engaged RNA polymerases might distinguish XPB and XPB/wt-p89 cells. To monitor the distribution of active transcription complexes at the c-myc promoter, nascent RNAs were labeled in nuclear run-on assays and hybridized to a panel of immobilized promoter segments (6). This method maps the zones of holdback or release of paused RNA polymerases, although the kinetics of transcription, the length of the labeled products, the duration of labeling reactions, and the size of the immobilized DNA sequences limit resolution.
In XPB cells and XPB/wt-p89 cells, as in most cell types, c-myc P1 promoter usage was very weak compared to P2. The run-on RNAs from the mutant and wild-type cells hybridized differentially to the sequential DNA segments downstream of P2 (Fig. 5D, lane 1 versus 2 and lane 3 versus 4). Prior to complementation, the maximum run-on hybridization signal mapped to the segment most proximal to P2 (nucleotides +1 to +50 (Fig. 5D, lanes 1 and 3). A dramatic fall-off of hybridization to the second segment (+51 to +100) indicated that without the TFIIH promoter escape occurs close to the promoter. Upon restitution of TFIIH, the zone of hybridization prior to the drop-off expanded to include the second segment oligonucleotides (Fig. 5D, lanes 2 and 4), suggesting repositioning of some paused transcription complexes to sites further downstream or an extended zone of holdback in the presence of functional TFIIH. Following UV irradiation, there was a greater reduction in the signal at P2 nucleotides +51 to +100 than in the signal at P2 nucleotides +1 to +50, in the XPB cells than in the XPB/wt-p89 cells (Fig. 5D, lanes 5 and 7, versus lanes 6 and 8). These results are consistent with the transition to elongation occurring within a narrower zone, closer to the start site in the mutant cells. Premature release of the paused polymerase may explain the delayed shutoff of c-myc mRNA seen in XPB but not in XPB/wt-p89 cells following UV irradiation.
FIR requires wild-type TFIIH to repress endogenous c-myc.
Why are the constitutional and conformational differences between the c-myc promoters of XPB and XPB/wt-p89 cells associated only with modest differences in c-myc mRNA levels? FBP and FIR operate through TFIIH to activate and repress transcription (41). Moreover, FBP modulation of endogenous c-myc is impaired by mutation of p89 in XPB cells (41). If FIR repression of c-myc were equivalently compromised in XPB disease, then one mutation would disable offsetting activating and repressing systems. To assess the influence of XPB/wt-p89 and FIR on endogenous c-myc expression, green fluorescent protein-tagged FIR was cotransfected into XPB fibroblasts with and without wild-type p89 or the appropriate empty vector. Immunostaining experiments showed that in the background of mutated p89, FIR failed to repress c-Myc levels (Fig. 6), but FIR sharply depressed endogenous c-Myc levels when cotransfected with wild-type p89. Thus, for FIR to represses endogenous c-Myc, TFIIH must be wild type. The simultaneous loss of FBP activation and FIR repression marginally shifts mean c-Myc levels within the total population but impairs fine tuning of c-Myc, leading to coarse regulation at the single-cell level. The operation of other activators and repressors operating through TFIIH may be similarly impaired.
FIG. 6.

FIR requires wild-type TFIIH to repress endogenous c-myc. Transfected green fluorescent protein-tagged FIR was coexpressed in the presence or absence of wild-type p89 and immunostained for c-Myc. In XPB cells, FIR expression alone does not alter levels of c-Myc, but coexpression of wild-type p89 enables FIR repression of c-Myc via wild-type TFIIH. Arrows indicate transfected cells.
DISCUSSION
The six-subunit core of TFIIH serves double duty, participating in NER as well as transcription (5, 12, 64, 71). Though XP has been considered a disease of defective DNA repair, mutations in XPB and XPD have the potential to compound the pathology via transcriptional deregulation of critical target genes (9, 30, 72). Mutations in XPB and XPD that disturb NER helicase subunits differentially affect DNA repair, basal transcription, or the response of TFIIH to activators and repressors (30, 71).
What does TFIIH do in transcription?
The role of TFIIH in the early stages of the transcription cycle has been studied in detail with only a small number of promoters (often artificial), mainly during basal transcription. As the last basal factor recruited to the preinitiation complex, TFIIH does not participate in promoter recognition. The most obvious contribution of TFIIH to transcription initiation is opening the transcription bubble via helicase activity. Yet most RNA polymerases (prokaryotic and eukaryotic, other than RNA polymerase II) have the intrinsic ability to open the template and so initiate transcription without recruiting an active, extrinsic helicase. Alone, purified RNA polymerase II also transcribes duplex DNA perfectly well, albeit nonspecifically in vitro. Moreover, preinitiation complexes assembled on some supercoiled templates support transcription without TFIIH. Possibly, TFIIH and XPB helicase activity operates at several stages between initiation and promoter escape (11, 25, 26, 31, 48). Even in the simplest phage systems, this transition involves a complicated structural reorganization of the template-bound transcription machinery (65, 77). On some RNA polymerase II promoters in vitro, promoter escape, and not preinitiation complex formation, is the slow step in transcript synthesis (14, 33, 34). The data in this work indicate that in vivo, functional TFIIH expands the zone at the endogenous c-myc promoter in which nascent transcript extension is regulated.
TFIIH oversees the transition from initiation to promoter escape.
These data and several previous key observations support the notion that TFIIH helps to supervise postinitiation, preescape c-myc transcription. (i) Most dramatically, by ChIP, TFIIH is almost absent from the c-myc promoter in the mutant cells, indicating that it is either inefficiently recruited or rapidly discharged. Surprisingly, this near-absence does not diminish overall levels of c-Myc. Alternatively transcriptionally generated supercoils might render TFIIH optional. (ii) By nuclear run-on, the paused polymerase at the c-myc promoter in XPB cells is released from holdback closer to the start site (within the first 50 nucleotides) than in normal cells that release downstream of +51. (iii) In cells with wild-type TFIIH, permanganate footprinting highlights an altered DNA conformation in the region spanning +53 to +58, consistent with an expanded zone of holdback compared with the XPB cells. (iv) FBP and FIR require wild-type TFIIH to modulate endogenous c-myc levels; these factors act at several postinitiation (but prepromoter) escape steps (41). (v) Mean c-Myc levels are modestly elevated but markedly variable in XPB mutant cells versus wild-type cells, suggesting an inability to finely adjust c-myc expression. (vi) Though footprinting and nuclear run-on studies reveal that in most situations the c-myc promoter is charged with polymerase (and so recruitment must not be rate limiting), no discrete pause site, pause sequence, or pause signals have yet been identified for the c-myc promoter (75). One way to account for the lack of a defined pause would be the asynchronous, slow progression of the c-myc transcription complex through several pauses prior to promoter escape.
A model for c-myc transcription.
Together, the above facts support the following model. After initiation, the transcription apparatus, without proper TFIIH, supports incremental transcript growth with pauses occurring at several, possibly variable, positions until promoter escape occurs early, well within the first 50 nucleotides. On some promoters in vitro, multiple sequential postinitiation, prepromoter escape transcription pauses have been observed; the exact number and sites of pausing are sequence dependent (14, 51, 52). If preinitiation complex formation is rapid on the c-myc promoter, as all evidence suggests, then promoter escape may be imagined as a queue of promoter-specific pauses. The duration of each individual pause and the interval from initiation until promoter escape are assumed to be Markovian, Poisson processes. Figure 7 shows two similar schemes compatible with the data. The total time of promoter escapes is then the sum of the intervals between the individual pauses, and the variance of this time is the summation of the variances at each pause. For simplicity, if each pause is imagined to be kinetically equivalent (although the general result is conceptually similar even relaxing this constraint), then the distribution of the time for promoter escape is described by an Erlang distribution, as follows.
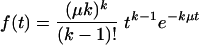 |
(1) |
The distribution is characterized by the number of pauses (k) and the average rate of promoter escape (μ), which is the reciprocal of the mean interval between sequential escapes (1/μ). The variance (1/kμ2) of the Erlang distribution decreases inversely with k; thus, the output becomes more regular as the number of steps goes up. The variance of c-Myc in XPB cells is threefold higher than in XPB/wt-p89. A combination of fewer and/or longer pauses could account for the increased variance.
FIG. 7.

Model describing the initiation to promoter escape transition as a series of steps, some accelerated and some slowed by properly functioning TFIIH. Assume that each transcript begins with the temporally random arrival of a transcription complex (M, a Markovian process). Subsequent transcript synthesis is controlled by a series of k steps leading to promoter escape. The rate of promoter escape is μ. Each individual step is a Markovian and Poisson process. Assume that the steps are kinetically equivalent (they may or may not be mechanistically equivalent). A series of k Poisson steps defines an Erlang k (Ek) distribution. Moreover, if each step is a Poisson step, then the frequency of the duration of each step is a γ-distribution (and so each is exponentially distributed). For an Ek distribution with a mean interval until promoter escape of 1/μ, the variance on this interval is 1/kμ2 and the coefficient of variation is k−1/2. Let transcripts be made on average at μ transcripts per unit of time, and steps occur on an average of λ steps per unit of time; so kμ = λ. Then the time for 1 step is 1/λ; the time for 1 transcript is 1/μ = K (1/λ) = k/λ. Because the steps occur through a Poisson process, the mean number of steps in a given time = λ.t = kμ.t with variance λ.t = kμ.t, and with a coefficient of variation c = (λ.t)−1/2. The mean number of transcripts in this time is (λ/k)t = μ.t and c = (kμ.t)−1/2. At t = 1/μ, c = k−1/2. The fluctuation of the number of promoter escapes per cell decreases as the number of steps required for each promoter escape increases. From the c values for the wild-type and mutant cells (observed by flow cytometry) (Fig. 3), the respective k values are 5 and 3.5; but since there is 1.5-fold more c-Myc in the mutant cells, there are 1.5-fold more promoter escapes in the same interval, so k/c-Myc is 2.4 for the mutant cells. So the mutant cells have approximately two to three fewer p89/XPB-dependent steps than the wild-type cells. The model is consistent with an early role for XPB in initiation and as a regulator of several subsequent steps leading to promoter escape as described for panels A and B. (A) Predicted distribution of c-Myc based on five steps leading to promoter escape in the wild-type cells versus two steps in the mutant, but in two-thirds the time in the mutant, so that on average the mutant cells have 1.5-fold more c-Myc than the wild-type cells (hence the area under the wild-type curve in panel C is two-thirds of that under the mutant curve). In panel B, promoter escape with wild-type TFIIH uses one initiation step and then two pauses, where as the pauses are lost in XPB, although the initiation step is slower (the TFIIH mutation influences the response to both activators and repressors). Again, five steps occur in the wild type in the same time that three steps occur in XPB. This scheme supports 1.7-fold more c-Myc in XPB than in wild-type cells. In both panels A and B, salmon-colored steps are wild-type XPB-facilitated initiation steps (and so occur faster with wild-type TFIIH), and green-colored steps represent TFIIH-dependent pauses. (C) An Erlang process composed of multiple steps confers temporal regularity. E1 is a γ-distribution value (related to Poisson n = 1), and events occur randomly. Promoter escape becomes more regular as k increases, even as the mean is held constant. Note that M/Ek/1 indicates Markovian (random) entry (in this case, PIC formation) into a queue; Ek indicates a queue composes of k Markovian (γ-distribution) steps where the probability of a backward step is minimal, and 1 indicates that only one product (nascent transcript) is manufactured at a time before promoter preescape. Adapted from Ivo Adan and Jacques Resing, Queueing theory (http://www.cs.duke.edu/∼fishhai/misc/queue.pdf).
In this scheme, wild-type TFIIH expands the zone of holdback, increases the total number of pauses (by two or three) (Fig. 7B versus 7A), and steadies promoter output. Yet this same stability might oppose the changes in c-myc expression demanded in vital situations. Unlike a pathway regulated at a single rate-limiting step, accelerating or decelerating any single link in this chain of kinetically equivalent steps has limited influence on the net reaction rate. If each transfactor were dedicated to the control of only a single step, then whole sets would be needed to drive up expression. In this situation, the step-specific transfactors would exhibit strong synergy, and incremental regulation would be difficult to achieve. To account for c-myc induction, activators modulating TFIIH at several stages, such as FBP, could influence expression alone and would cooperate additively with mechanistically similar activators to advance the transcription complex incrementally toward promoter escape; repressors such as FIR would behave oppositely. TFIIH would then govern the transcription machinery as it ratchets its way along the template before the transition to elongation. A mutation that simultaneously disables activation by FBP and repression by FIR would then not shift mean c-Myc levels, but k would be reduced by 2, coarsening expression. Current structures of multisubunit RNA polymerases have not revealed a power stroke coupling nucleoside triphosphate hydrolysis with translocation (7, 36), and so nascent strand growth may be accomplished with a diffusion-driven sliding clamp, a Brownian ratchet (15). Activated XPB helicase may help to thread premelted template into the transcription apparatus and so facilitate forward diffusion, whereas repressors driving TFIIH to its ground state would impose additional impediments to translocation. Thus, TFIIH would serve as a signal integrator incrementally hastening or delaying progression to promoter escape. The involvement of TFIIH throughout all the stages of early transcript synthesis makes it an attractive target for regulation at multiple steps of the transcription cycle. Besides modulation of helicase activity, sequential action of TFIIH's Cdk7 to multiply phosphorylate the CTD could serve an entirely analogous role buffering against stochastic bursts of promoter firing; in fact, any prepromoter escape step occurring on the same time scale would serve similarly. Recent evidence supports the notion that TFIIH may travel with the RNA polymerase through an expanded zone (60). The extent of TFIIH participation in the fine regulation of promoter escape may depend on the nature of the nearby DNA and chromatin-bound factors.
The XPB mutation removes the influence of at least some of the regulators operating through TFIIH, and so c-myc gene expression becomes more coarse. The influence of FIR in the presence of wild-type p89 exceeds that of FBP, and then the XPB mutation additionally nudges c-myc to higher levels, as observed. At other genes, the level of expression and the particular set of transfactors needed for proper regulation would determine the susceptibility to TFIIH mutations. Promoters regulated primarily at earlier stages of the transcription cycle (chromatin remodeling, preinitiation complex formation, or initiation) operating through transcription factors and coactivators targeting early steps in the transcription cycle such as occurs at the beta interferon gene would be less dependent on fully functional TFIIH for proper regulation (42, 66). Another mechanism to escape the TFIIH dependence may be to bypass particular TFIIH functions. Recently, it has been shown that the yeast heat shock factor can mediate phosphorylation of the CTD of the largest subunit of RNA polymerase II, thus making transcription independent of proper TFIIH function (58). This observation is in line with the lack of effect of the XPB mutation on the hsp70 promoter observed here. Thus, deficits in the general transcription factor TFIIH would be revealed as gene-specific defects in the expression of targets most dependent on TFIIH for proper regulation.
The XPB11BE mutation modestly increases mean c-Myc-levels, but dramatically expands the cell-to-cell variation of this key regulator of proliferation, growth, differentiation, and apoptosis. Do fluctuating levels of c-myc matter? Although the answer to this question is not yet known, it is apparent that increased absolute levels of expression are insufficient to account for the pathology elicited by deregulated c-myc genes; average c-Myc levels in tumors such as Burkitt's lymphoma may be only modestly elevated or even normal, belying the critical role for c-myc deregulation in the pathogenesis of cancer (29, 57). Many mRNAs are present at very low copy numbers, so low that the notion of “the average cell” has been questioned, due to the almost endless combinations that may be generated by variable regulation and stochastic fluctuations (39). For many, if not most, genes the organism is robust enough to withstand the vagaries of hit-or-miss expression. Yet it would be surprising if mechanisms had not evolved to ensure uniform, low-level expression of some important genes.
Acknowledgments
We thank Lance Liotta, Lyle Ungar, Louis Staudt, Deborah Boles, and Susan Mackem for helpful discussions and critical review of the manuscript. XP11BE lymphoblasts were from the Coriell Institute of Medical Research, Camden, N.J.
This study was supported by grants from the Deutsche Forschungsgemeinschaft (DFG) to A.W. (AW2397/1-1 and AW2397/2-1).
REFERENCES
- 1.Amundson, S. A., Q. Zhan, L. Z. Penn, and A. J. Fornace. 1998. Myc suppresses induction of the growth arrest genes gadd34, gadd45, and gadd153 by DNA-damaging agents. Oncogene 17:2149-2154. [DOI] [PubMed] [Google Scholar]
- 2.Chambers, A. E., S. Banerjee, T. Chaplin, J. Dunne, S. Debernardi, S. P. Joel, and B. D. Young. 2003. Histone acetylation-mediated regulation of genes in leukaemic cells. Eur. J. Cancer 39:1165-1175. [DOI] [PubMed] [Google Scholar]
- 3.Chen, D., T. Riedl, E. Washbrook, P. E. Pace, R. C. Coombes, J. M. Egly, and S. Ali. 2000. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell 6:127-137. [PubMed] [Google Scholar]
- 4.Coin, F., E. Bergmann, A. Tremeau-Bravard, and J. M. Egly. 1999. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 18:1357-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coin, F., and J. M. Egly. 1998. Ten years of TFIIH. Cold Spring Harb. Symp. Quant. Biol. 63:105-110. [DOI] [PubMed] [Google Scholar]
- 6.Collins, I., A. Weber, and D. Levens. 2001. Transcriptional consequences of topoisomerase inhibition. Mol. Cell. Biol. 21:8437-8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cramer, P. 2002. Multisubunit RNA polymerases. Curr. Opin. Struct. Biol. 12:89-97. [DOI] [PubMed] [Google Scholar]
- 8.Dani, C., N. Mechti, M. Piechaczyk, B. Lebleu, P. Jeanteur, and J. M. Blanchard. 1985. Increased rate of degradation of c-myc messenger-RNA in interferon-treated Daudi cells. Proc. Natl. Acad. Sci. USA 82:4896-4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubaele, S., L. Proietti De Santis, R. J. Bienstock, A. Keriel, M. Stefanini, B. Van Houten, and J. M. Egly. 2003. Basal transcription defect discriminates between xeroderma pigmentosum and trichothiodystrophy in XPD patients. Mol. Cell 11:1635-1646. [DOI] [PubMed] [Google Scholar]
- 10.Duncan, R., L. Bazar, G. Michelotti, T. Tomonaga, H. Krutzsch, M. Avigan, and D. Levens. 1994. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 8:465-480. [DOI] [PubMed] [Google Scholar]
- 11.Dvir, A., S. Tan, J. W. Conaway, and R. C. Conaway. 1997. Promoter escape by RNA polymerase II. Formation of an escape-competent transcriptional intermediate is a prerequisite for exit of polymerase from the promoter. J. Biol. Chem. 272:28175-28178. [DOI] [PubMed] [Google Scholar]
- 12.Egly, J. M. 2001. The 14th Datta lecture. TFIIH: from transcription to clinic. FEBS Lett. 498:124-128. [DOI] [PubMed] [Google Scholar]
- 13.Elowitz, M. B., A. J. Levine, E. D. Siggia, and P. S. Swain. 2002. Stochastic gene expression in a single cell. Science 297:1183-1186. [DOI] [PubMed] [Google Scholar]
- 14.Ferguson, H. A., J. F. Kugel, and J. A. Goodrich. 2001. Kinetic and mechanistic analysis of the RNA polymerase II transcription reaction at the human interleukin-2 promoter. J. Mol. Biol. 314:993-1006. [DOI] [PubMed] [Google Scholar]
- 15.Feynman, R., R. Leighton, and M. Sands. 1989. The Feynman lectures on physics: commemorative issue, vol. I. Addison-Wesley, Reading, Pa.
- 16.Fukuda, A., Y. Nogi, and K. Hisatake. 2002. The regulatory role for the ERCC3 helicase of general transcription factor TFIIH during promoter escape in transcriptional activation. Proc. Natl. Acad. Sci. USA 99:1206-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garmyn, M., M. Yaar, N. Holbrook, and B. A. Gilchrest. 1991. Immediate and delayed molecular response of human keratinocytes to solar-simulated irradiation. Lab. Investig. 65:471-478. [PubMed] [Google Scholar]
- 18.Giardina, C., M. Perez-Riba, and J. T. Lis. 1992. Promoter melting and TFIID complexes on Drosophila genes in vivo. Genes Dev. 6:2190-2200. [DOI] [PubMed] [Google Scholar]
- 19.Goodrich, J. A., and R. Tjian. 1994. Transcription factors IIE and IIH and ATP hydrolysis direct promoter clearance by RNA polymerase II. Cell. 77:145-156. [DOI] [PubMed] [Google Scholar]
- 20.Gozukara, E. M., C. N. Parris, C. A. Weber, E. P. Salazar, M. M. Seidman, J. F. Watkins, L. Prakash, and K. H. Kraemer. 1994. The human DNA repair gene, ERCC2 (XPD), corrects ultraviolet hypersensitivity and ultraviolet hypermutability of a shuttle vector replicated in xeroderma pigmentosum group D cells. Cancer Res. 54:3837-3844. [PubMed] [Google Scholar]
- 21.Greenblatt, J., and C. J. Ingles. 1996. Interaction between acidic transcriptional activation domains of herpes simplex virus activator protein VP16 and transcriptional initiation factor IID. Methods Enzymol. 274:120-133. [DOI] [PubMed] [Google Scholar]
- 22.Hann, S. R., and R. N. Eisenman. 1984. Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol. Cell. Biol. 4:2486-2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haviv, I., D. Vaizel, and Y. Shaul. 1996. pX, the HBV-encoded coactivator, interacts with components of the transcription machinery and stimulates transcription in a TAF-independent manner. EMBO J. 15:3413-3420. [PMC free article] [PubMed] [Google Scholar]
- 24.He, L., J. Liu, I. Collins, S. Sanford, B. O'Connell, C. J. Benham, and D. Levens. 2000. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 19:1034-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holstege, F. C., U. Fiedler, and H. T. Timmers. 1997. Three transitions in the RNA polymerase II transcription complex during initiation. EMBO J. 16:7468-7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holstege, F. C., P. C. van der Vliet, and H. T. Timmers. 1996. Opening of an RNA polymerase II promoter occurs in two distinct steps and requires the basal transcription factors IIE and IIH. EMBO J. 15:1666-1677. [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang, J. R., V. Moncollin, W. Vermeulen, T. Seroz, H. van Vuuren, J. H. J. Hoeijmakers, and J. M. Egly. 1996. A 3′→5′ XPB helicase defect in repair/transcription factor TFIIH of xeroderma pigmentosum group B affects both DNA repair and transcription. J. Biol. Chem. 271:15898-15904. [DOI] [PubMed] [Google Scholar]
- 28.Iben, S., H. Tschochner, M. Bier, D. Hoogstraten, P. Hozak, J. M. Egly, and I. Grummt. 2002. TFIIH plays an essential role in RNA polymerase I transcription. Cell 109:297-306. [DOI] [PubMed] [Google Scholar]
- 29.Keath, E. J., A. Kelekar, and M. D. Cole. 1984. Transcriptional activation of the translocated c-myc oncogene in mouse plasmacytomas: similar RNA levels in tumor and proliferating normal cells. Cell 37:521-528. [DOI] [PubMed] [Google Scholar]
- 30.Keriel, A., A. Stary, A. Sarasin, C. Rochette-Egly, and J. M. Egly. 2002. XPD mutations prevent TFIIH-dependent transactivation by nuclear receptors and phosphorylation of RARα. Cell 109:125-135. [DOI] [PubMed] [Google Scholar]
- 31.Kim, T. K., R. H. Ebright, and D. Reinberg. 2000. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science 288:1418-1422. [DOI] [PubMed] [Google Scholar]
- 32.Komarnitsky, P., E. J. Cho, and S. Buratowski. 2000. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14:2452-2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kugel, J. F., and J. A. Goodrich. 2000. A kinetic model for the early steps of RNA synthesis by human RNA polymerase II. J. Biol. Chem. 275:40483-40491. [DOI] [PubMed] [Google Scholar]
- 34.Kugel, J. F., and J. A. Goodrich. 1998. Promoter escape limits the rate of RNA polymerase II transcription and is enhanced by TFIIE, TFIIH, and ATP on negatively supercoiled DNA. Proc. Natl. Acad. Sci. USA 95:9232-9237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar, K. P., S. Akoulitchev, and D. Reinberg. 1998. Promoter-proximal stalling results from the inability to recruit transcription factor IIH to the transcription complex and is a regulated event. Proc. Natl. Acad. Sci. USA 95:9767-9772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Landick, R. 2004. Active-site dynamics in RNA polymerases. Cell 116:351-353. [DOI] [PubMed] [Google Scholar]
- 37.Levens, D. 2002. Disentangling the MYC web. Proc. Natl. Acad. Sci. USA 99:5757-5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levens, D. L. 2003. Reconstructing MYC. Genes. Dev. 17:1071-1077. [DOI] [PubMed] [Google Scholar]
- 39.Levsky, J. M., and R. H. Singer. 2003. Gene expression and the myth of the average cell. Trends Cell Biol. 13:4-6. [DOI] [PubMed] [Google Scholar]
- 40.Liu, J., L. He, I. Collins, H. Ge, D. Libutti, J. Li, J. M. Egly, and D. Levens. 2000. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol. Cell 5:331-341. [DOI] [PubMed] [Google Scholar]
- 41.Liu, J. H., S. Akoulitchev, A. Weber, H. Ge, S. Chuikov, D. Libutti, X. W. Wang, J. W. Conaway, C. C. Harris, R. C. Conaway, D. Reinberg, and D. Levens. 2001. Defective interplay of activators and repressors with TFIIH in xeroderma pigmentosum. Cell 104:353-363. [DOI] [PubMed] [Google Scholar]
- 42.Lomvardas, S., and D. Thanos. 2002. Modifying gene expression programs by altering core promoter chromatin architecture. Cell 110:261-271. [DOI] [PubMed] [Google Scholar]
- 43.Lu, H., R. P. Fisher, P. Bailey, and A. J. Levine. 1997. The CDK7-cycH-p36 complex of transcription factor IIH phosphorylates p53, enhancing its sequence-specific DNA binding activity in vitro. Mol. Cell. Biol. 17:5923-5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marcu, K. B., S. A. Bossone, and A. J. Patel. 1992. myc function and regulation. Annu. Rev. Biochem. 61:809-860. [DOI] [PubMed] [Google Scholar]
- 45.Michelotti, E. F., S. Sanford, and D. Levens. 1997. Marking of active genes on mitotic chromosomes. Nature 388:895-899. [DOI] [PubMed] [Google Scholar]
- 46.Michelotti, G. A., E. F. Michelotti, A. Pullner, R. C. Duncan, D. Eick, and D. Levens. 1996. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol. Cell. Biol. 16:2656-2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moberg, K. H., W. A. Tyndall, and D. J. Hall. 1992. Wild-type murine p53 represses transcription from the murine c-myc promoter in a human glial cell line. J. Cell Biochem. 49:208-215. [DOI] [PubMed] [Google Scholar]
- 48.Moreland, R. J., F. Tirode, Q. Yan, J. W. Conaway, J. M. Egly, and R. C. Conaway. 1999. A role for the TFIIH XPB DNA helicase in promoter escape by RNA polymerase II. J. Biol. Chem. 274:22127-22130. [DOI] [PubMed] [Google Scholar]
- 49.Mueller, P. R., and B. Wold. 1989. In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 246:780-786. [DOI] [PubMed] [Google Scholar]
- 50.Ostapenko, D., and O. Gileadi. 2000. Rad25p, a DNA helicase subunit of yeast transcription factor TFIIH, is required for promoter escape in vivo. Gene 245:109-117. [DOI] [PubMed] [Google Scholar]
- 51.Pal, M., and D. S. Luse. 2002. Strong natural pausing by RNA polymerase II within 10 bases of transcription start may result in repeated slippage and reextension of the nascent RNA. Mol. Cell. Biol. 22:30-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pal, M., D. McKean, and D. S. Luse. 2001. Promoter clearance by RNA polymerase II is an extended, multistep process strongly affected by sequence. Mol. Cell. Biol. 21:5815-5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parvin, J. D., and P. A. Sharp. 1993. DNA topology and a minimal set of basal factors for transcription by RNA polymerase II. Cell 73:533-540. [DOI] [PubMed] [Google Scholar]
- 54.Rabbitts, P. H., J. V. Watson, A. Lamond, A. Forster, M. A. Stinson, G. Evan, W. Fischer, E. Atherton, R. Sheppard, and T. H. Rabbitts. 1985. Metabolism of c-myc gene products: c-myc mRNA and protein expression in the cell cycle. EMBO J. 4:2009-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Raser, J. M., and E. K. O'Shea. 2004. Control of stochasticity in eukaryotic gene expression. Science 304:1811-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rougvie, A. E., and J. T. Lis. 1988. The RNA polymerase II molecule at the 5′ end of the uninduced hsp70 gene of D. melanogaster is transcriptionally engaged. Cell 54:795-804. [DOI] [PubMed] [Google Scholar]
- 57.Saez, A. I., M. J. Artiga, C. Romero, S. Rodriguez, J. C. Cigudosa, A. Perez-Rosado, I. Fernandez, M. Sanchez-Beato, E. Sanchez, M. Mollejo, and M. A. Piris. 2003. Development of a real-time reverse transcription polymerase chain reaction assay for c-myc expression that allows the identification of a subset of c-myc plus diffuse large B-cell lymphoma. Lab. Investig. 83:143-152. [DOI] [PubMed] [Google Scholar]
- 58.Sakurai, H., N. Hashikawa, H. Imazu, and T. Fukasawa. 2003. Carboxy-terminal region of the yeast heat shock factor contains two domains that make transcription independent of the TFIIH protein kinase. Genes Cells 8:951-961. [DOI] [PubMed] [Google Scholar]
- 59.Schneider, E., M. Montenarh, and P. Wagner. 1998. Regulation of CAK kinase activity by p53. Oncogene 17:2733-2741. [DOI] [PubMed] [Google Scholar]
- 60.Schwartz, B. E., S. Larochelle, B. Suter, and J. T. Lis. 2003. Cdk7 is required for full activation of Drosophila heat shock genes and RNA polymerase II phosphorylation in vivo. Mol. Cell. Biol. 23:6876-6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sears, R., F. Nuckolls, E. Haura, Y. Taya, K. Tamai, and J. R. Nevins. 2000. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 14:2501-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spangler, L., X. Wang, J. W. Conaway, R. C. Conaway, and A. Dvir. 2001. TFIIH action in transcription initiation and promoter escape requires distinct regions of downstream promoter DNA. Proc. Natl. Acad. Sci. USAProc 98:5544-5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spencer, C. A., and M. Groudine. 1991. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 56:1-48. [DOI] [PubMed] [Google Scholar]
- 64.Svejstrup, J. Q., P. Vichi, and J. M. Egly. 1996. The multiple roles of transcription/repair factor TFIIH. Trends Biochem. Sci. 21:346-350. [PubMed] [Google Scholar]
- 65.Tahirov, T. H., D. Temiakov, M. Anikin, V. Patlan, W. T. McAllister, D. G. Vassylyev, and S. Yokoyama. 2002. Structure of a T7 RNA polymerase elongation complex at 2.9 A resolution. Nature 420:43-50. [DOI] [PubMed] [Google Scholar]
- 66.Thanos, D., and T. Maniatis. 1995. Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83:1091-1100. [DOI] [PubMed] [Google Scholar]
- 67.Tong, X., R. Drapkin, D. Reinberg, and E. Kieff. 1995. The 62- and 80-kDa subunits of transcription factor IIH mediate the interaction with Epstein-Barr virus nuclear protein 2. Proc. Natl. Acad. Sci. USA 92:3259-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trigon, S., H. Serizawa, J. W. Conaway, R. C. Conaway, S. P. Jackson, and M. Morange. 1998. Characterization of the residues phosphorylated in vitro by different C-terminal domain kinases. J. Biol. Chem. 273:6769-6775. [DOI] [PubMed] [Google Scholar]
- 69.Trumpp, A., Y. Refaeli, T. Oskarsson, S. Gasser, M. Murphy, G. R. Martin, and J. M. Bishop. 2001. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414:768-773. [DOI] [PubMed] [Google Scholar]
- 70.Van Lint, C., S. Emiliani, and E. Verdin. 1996. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 5:245-253. [PMC free article] [PubMed] [Google Scholar]
- 71.van Steeg, H., and K. H. Kraemer. 1999. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol. Med. Today 5:86-94. [DOI] [PubMed] [Google Scholar]
- 72.Viprakasit, V., R. J. Gibbons, B. C. Broughton, J. L. Tolmie, D. Brown, P. Lunt, R. M. Winter, S. Marinoni, M. Stefanini, L. Brueton, A. R. Lehmann, and D. R. Higgs. 2001. Mutations in the general transcription factor TFIIH result in beta-thalassaemia in individuals with trichothiodystrophy. Hum. Mol. Genet. 10:2797-2802. [DOI] [PubMed] [Google Scholar]
- 73.Weinmann, A. S., and P. J. Farnham. 2002. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 26:37-47. [DOI] [PubMed] [Google Scholar]
- 74.Winkler, G. S., W. Vermeulen, F. Coin, J. M. Egly, J. H. Hoeijmakers, and G. Weeda. 1998. Affinity purification of human DNA repair/transcription factor TFIIH using epitope-tagged xeroderma pigmentosum B protein. J. Biol. Chem. 273:1092-1098. [DOI] [PubMed] [Google Scholar]
- 75.Wolf, D. A., L. J. Strobl, A. Pullner, and D. Eick. 1995. Variable pause positions of RNA polymerase II lie proximal to the c-myc promoter irrespective of transcriptional activity. Nucleic Acids Res. 23:3373-3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiao, H., A. Pearson, B. Coulombe, R. Truant, S. Zhang, J. L. Regier, S. J. Triezenberg, D. Reinberg, O. Flores, and C. J. Ingles. 1994. Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol. Cell. Biol. 14:7013-7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yin, Y. W., and T. A. Steitz. 2002. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science 298:1387-1395. [DOI] [PubMed] [Google Scholar]