

ABSTRACT
A previous bioinformatics analysis identified the Mycobacterium tuberculosis proteins Rv2125 and Rv2714 as orthologs of the eukaryotic proteasome assembly chaperone 2 (PAC2). We set out to investigate whether Rv2125 or Rv2714 can function in proteasome assembly. We solved the crystal structure of Rv2125 at a resolution of 3.0 Å, which showed an overall fold similar to that of the PAC2 family proteins that include the archaeal PbaB and the yeast Pba1. However, Rv2125 and Rv2714 formed trimers, whereas PbaB forms tetramers and Pba1 dimerizes with Pba2. We also found that purified Rv2125 and Rv2714 could not bind to M. tuberculosis 20S core particles. Finally, proteomic analysis showed that the levels of known proteasome components and substrate proteins were not affected by disruption of Rv2125 in M. tuberculosis. Our work suggests that Rv2125 does not participate in bacterial proteasome assembly or function.
IMPORTANCE Although many bacteria do not encode proteasomes, M. tuberculosis not only uses proteasomes but also has evolved a posttranslational modification system called pupylation to deliver proteins to the proteasome. Proteasomes are essential for M. tuberculosis to cause lethal infections in animals; thus, determining how proteasomes are assembled may help identify new ways to combat tuberculosis. We solved the structure of a predicted proteasome assembly factor, Rv2125, and isolated a genetic Rv2125 mutant of M. tuberculosis. Our structural, biochemical, and genetic studies indicate that Rv2125 and Rv2714 do not function as proteasome assembly chaperones and are unlikely to have roles in proteasome biology in mycobacteria.
KEYWORDS: Mycobacterium tuberculosis, proteasome, protein chaperone, structural biology
INTRODUCTION
Mycobacterium tuberculosis is the etiologic agent of tuberculosis, which kills approximately 1.4 million people each year and infects about one-third of the world's population. The microbe's growing resistance to antibiotics has made the search for new potential targets for therapy a priority. M. tuberculosis is the only known bacterial pathogen that has proteasomes (1). Previous studies have established that the proteasome system is essential for lethal tuberculosis infections in mice, making it an attractive target for therapeutics (2–7).
In bacteria, proteasomes are found in several species, including M. tuberculosis, Rhodococcus erythropolis, and Streptomyces coelicolor (8–10). We do not know if these bacteria acquired the proteasome system via horizontal gene transfer from eukaryotes or if the eukaryotic proteasome evolved from a bacterial ancestor (11). Bacterial proteasomes have similarities and differences compared with their eukaryotic and archaeal counterparts (12). Major similarities include the 20S proteasome core particle (CP) and the hexameric proteasomal ATPases (Mpa in M. tuberculosis, PAN in archaea, and Rpt1 to Rpt6 in eukaryotes) that unfold and translocate protein substrates for degradation by the 20S CP (13). Like eukaryotes, M. tuberculosis has an ATP-independent proteasome activator, PafE (14), but PafE forms a dodecamer, unlike the eukaryotic PA26 and PA28, which are heptameric (15). Finally, a major difference between bacteria and eukaryotes is in the substrate tagging system used for ATP-dependent degradation: eukaryotes use a β-grasp fold protein, ubiquitin, whereas bacteria use an intrinsically disordered protein, Pup (16). Ubiquitin is recognized by a non-ATPase proteasomal component, Rpn10 or Rpn13; Pup is recognized by the proteasomal ATPase Mpa (10, 17).
The assembly of the eukaryotic 20S proteasome is assisted by several chaperones, such as the proteasome biogenesis-associated complexes Pba1-Pba2 (called PAC1-PAC2 in mammals) and Pba3-Pba4 (PAC3-PAC4), as well as UMP1 (18). The heterodimeric Pba1-Pba2 and Pba3-Pba4 promote the assembly of the seven-membered α-ring (19–21), and UMP1 facilitates the incorporation of the β subunits for assembly of the half-proteasomes and their subsequent dimerization to form mature 20S CPs (22, 23). PbaA-PbaB, the archaeal homologue of the eukaryotic Pba1-Pba2 complex, associates with 20S CP assembly intermediates as well as with mature 20S CPs (24, 25). A recent bioinformatic analysis discovered that the M. tuberculosis genome encodes two orthologs of the eukaryotic PAC2 protein, Rv2125 and Rv2714 (26). Considering that the M. tuberculosis proteasome system has several parallels to the eukaryotic system, we considered whether Rv2125 and Rv2715 function as bacterial proteasome assembly chaperones. We conducted biochemical and structural studies of Rv2125 and found that it has an overall fold similar to that of members of the PAC2 family, including the archaeal proteasome activator PbaB and the yeast proteasome assembly chaperone Pba1 (25, 27). However, we found that neither Rv2125 nor Rv2714 (whose crystal structure was determined previously) (28) interacted with proteasome core particles. We also performed a proteomic analysis of an Rv2125 M. tuberculosis mutant and found that no known proteasome components or proteasome substrates were significantly altered, suggesting that Rv2125 is not involved in proteasome function in M. tuberculosis.
RESULTS
Overall crystal structure of Rv2125.
Full-length Rv2125 has 292 amino acids and a molecular mass of 31.8 kDa. We made several Rv2125 truncation constructs, but only one construct, consisting of amino acids Thr-16 through Ala-260 (Rv212516–260), could be crystallized. The initial crystals were thin sheets that diffracted X rays highly anisotropically. We optimized the initial crystal growth conditions by adding 1% Anapoe-20 and 2% benzamidine hydrochloride, and we obtained thicker hexagonal crystals that diffracted X rays with reduced anisotropy to a 3-Å resolution. The crystal belongs to the space group C2, with unit cell constants of a = 138.25 Å, b = 79.76 Å, c = 89.03 Å, and β = 103.57° (Table 1). We solved the structure by molecular replacement with a Streptomyces avermitilis homologue structure (Protein Data Bank [PDB] accession number 3MNF) (see Fig. S1 in the supplemental material) (29). The final structure of Rv2125 is superimposable on the S. avermitilis structure with a root mean square deviation (RMSD) of 1.9 Å. We found three monomers in the asymmetric unit that are related by a noncrystallographic 3-fold axis.
TABLE 1.
Data collection and refinement statistics
| Parameter | Value(s) for Rv212516–260a |
|---|---|
| Data collection statistics | |
| Wavelength (Å) | 1.100 |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 138.25, 79.76, 89.03 |
| α, β, γ (°) | 90.0, 103.57, 90.0 |
| Resolution range (Å) | 30.65–3.00 (3.16–3.00) |
| Rmerge (%) | 19.9 (50.2) |
| I/σI | 5.7 (2.5) |
| Completeness (%) | 98.9 (99.4) |
| Total no. of reflections | 67,525 (9,907) |
| Multiplicity | 3.6 (3.6) |
| Refinement statistics | |
| Resolution range (Å) | 30.65–3.00 |
| No. of unique reflections | 18,826 (2,748) |
| Rwork/Rfree | 0.236/0.289 |
| No. of non-H atoms | |
| Protein | 5,516 |
| Water | 86 |
| Average B-factor (Å2) | |
| Protein | 41.06 |
| Water | 23.62 |
| RMSD | |
| Bond length (Å) | 0.0098 |
| Bond angle (°) | 1.382 |
| Ramachandran statistics (%) | |
| Favored | 93.87 |
| Allowed | 6.13 |
| Outliers | 0 |
| PDB accession number | 5UN0 |
Data for the highest-resolution shell are shown in parentheses.
Rv2125 has a central β-sheet of seven β-strands flanked by four α-helices on one side and two α-helices on the other side (Fig. 1A). A β-hairpin formed by strands β3 and β4, inserted between the first (β2) and second (β5) strands of the central β-sheet, interacts with strand β9 of a neighboring monomer and appears to be responsible for trimerization (Fig. 1B and C). The buried surface area of one monomer in a trimer is 2,224 Å2, corresponding to about 20% of the total surface of a monomer. Rv212516–260 eluted in gel filtration chromatography at a volume corresponding to a molecular mass of 80 kDa (Fig. 2), indicating that the protein was already trimeric in solution; therefore, the observed Rv2125 trimer structure is not a crystallization artifact.
FIG 1.

Crystal structure of Rv2125. (A) The Rv2125 monomer. The secondary structural elements (β-strands, α-helices, and loops) are shown in magenta, cyan, and salmon, respectively. The N and C termini are labeled as NT and CT, respectively. The β3-β4 hairpin of the monomer interacts with the β9 strand of a neighboring monomer (β9′) within a trimer. (B) Secondary-structure topology of the Rv2125 monomer. The β3-β4 hairpin of the monomer interacts with the β9 strand of a neighboring monomer within a trimer. (C) The Rv2125 trimer in side (left) and bottom (right) views. Chains A, B, and C are colored cyan, magenta, and green, respectively. Surface representation of the trimer is in transparent light gray.
FIG 2.
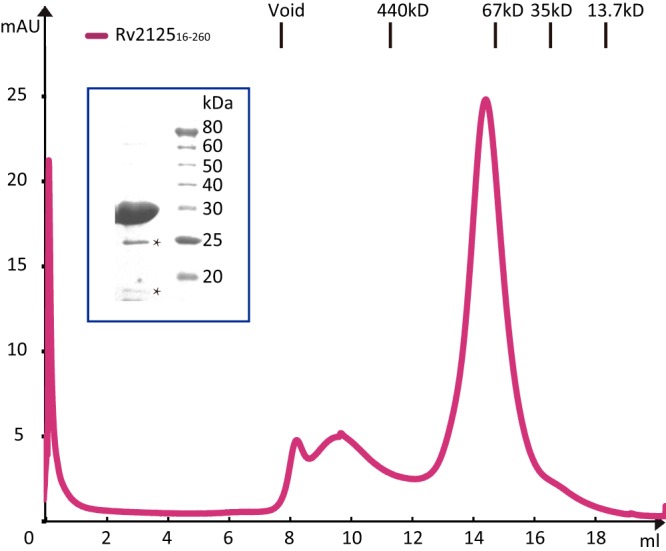
Size exclusion chromatography of Rv212516–260. Rv212516–260 was eluted as the major peak at about 14.2 ml, consistent with the expected elution volume of a globular protein of about 80 kDa, indicating that Rv2125 forms trimers. Size standards for calibration of the S200 size exclusion column are marked above the chromatographic panel. The inset is an SDS-PAGE gel of the elution peak. The major band at 27 kDa is the Rv2125 protein. The two lower bands marked by asterisks may be contaminants or breakdown products.
Similarities and differences between Rv2125 and other PAC2 homologues.
We first used the Dali server to search for structures closely related to that of Rv2125 and identified three unique homologues (30). The archaeal proteasome activator PbaB from Pyrococcus furiosus, a PAC2 family protein (25), shares 14% sequence identity with Rv2125. Except for several homologues of unknown function, PbaB had the highest structural similarity, with an RMSD of 3.0 Å over 244 Cα atoms (Fig. 3A and C) (PDB accession number 3VR0). Another PAC2 family protein, the yeast proteasome assembly chaperone Pba1 (27), is 10% identical with Rv2125 at the amino acid sequence level. They are structurally similar, with an RMSD of 3.8 Å over 200 Cα atoms (Fig. 3A and C) (PDB accession number 4G4S). In addition, Rv2125 shares 25% sequence identity with the other M. tuberculosis PAC2 homologue, Rv2714. Their structures are superimposable with an RMSD of 2.5 Å over 275 Cα atoms (Fig. 3B and C) (PDB accession number 2WAM) (28).
FIG 3.

Structure comparison and sequence alignment of Rv2125, Rv2714 (PDB accession number 2WAM), archaeal PbaB (PDB accession number 3VR0), and yeast Pba1 (PDB accession number 4G4S). (A) Superimposition of the crystal structures of Rv2125, PbaB, and Pba1. The C-terminal HbYX motif of Pba1 is shown as red spheres. The black oval marks a hook-like C-terminal segment of Rv2125 that is absent in the other two proteins. (B) Superimposition of the crystal structures of Rv2125 and Rv2714. The hook-like C-terminal segments of both M. tuberculosis proteins are marked by the black oval. (C) Sequence alignment of four different PAC2 homologues. Alignment was done using MultAlin multiple sequence alignments (46) and ESPript 2.2 (47). The secondary structural elements of Rv2125 are drawn above the primary sequences. Amino acid sequences used in the alignment are those of M. tuberculosis Rv2125 (GI 15609262), M. tuberculosis Rv2714 (GI 15609851), P. furiosus PbaB (P.f_PbaB; GI 474452817), and Saccharomyces cerevisiae Pba1 (S.c_Pba1; GI 1023942167).
We then analyzed the structural differences of Rv2125 and the three identified homologues. The archaeal PbaB assembles into homotetramers and functions as an ATP-independent proteasome activator as well as a molecular chaperone via its tentacle-like C-terminal segments (25). The subunit-subunit interfaces of the PbaB tetramer are 1,072 to 1,184 Å2, which is similar to that of Rv2125 (1,112 Å2). However, the structural elements at the interface, i.e., between β3 and β4 of one monomer and β9 of a neighbor, are quite different between PbaB and Rv2125 (Fig. 3A). PbaB binds to mature archaeal 20S CP with its C-terminal hydrophobic Tyr-X (HbYX) motif. The HbYX motif is conserved in archaeal and eukaryotic proteasome activators, whether they are ATP dependent (such as PAN and Rpt1 to Rpt6) or ATP independent (such as PA26 and PA28) (13). Both yeast Pba1 and Pba2 have the HbYX motif, and they form a heterodimer that binds mature 20S CP with those motifs (24, 27). Known M. tuberculosis proteasome activators, such as the ATP-dependent Mpa and the ATP-independent PafE, have a C-terminal GQYL motif that is functionally equivalent to the archaeal and eukaryotic HbYX motif (14, 31). A GQYL motif is not present in Rv2125 or Rv2714 (Fig. 3C). Furthermore, Rv2125 and Rv2714 both have an extra C-terminal α-helical segment that is not present in the yeast Pba1-Pba2 and archaeal PbaB (Fig. 3B and C). Superimposition of the Rv2125 monomer structure on the Pba1 structure in complex with the 20S CP revealed that the Rv2125 C-terminal α-helix is too long to be accommodated in the activator-binding pocket of the 20S CP (Fig. 4). For the same reason, Rv2714 was also unable to dock into the activator-binding pocket of the 20S CP.
FIG 4.
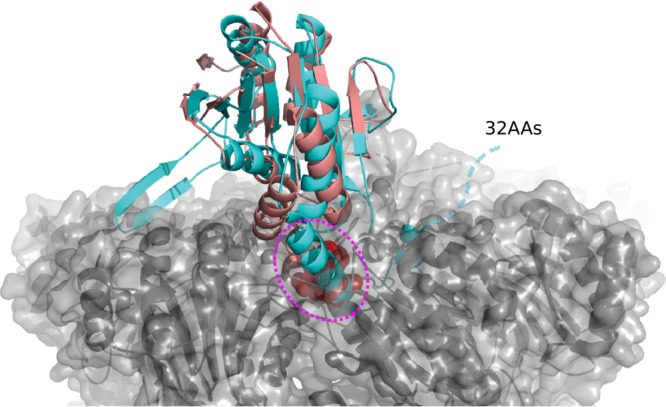
Superposition of the Rv212516–260 crystal structure with that of Pba1 in complex with a 20S CP (PDB accession number 4G4S). Rv212516–260 and Pba1 are shown in cyan and salmon, respectively. The C-terminal HbYX motif of Pba1 inserted in the binding pocket of the proteasome α-ring is shown as spheres and highlighted by a dashed magenta oval. The 20S CP is shown in a gray, semitransparent surface view. The dashed cyan line indicates the C-terminal 32 amino acids (32AAs) that were truncated in our Rv2125 construct. The C-terminal hook-like α-helix plus the additional 32-residue sequence are too long to be accommodated in the proteasome binding pocket.
Neither Rv2125 nor Rv2714 interacts with the M. tuberculosis 20S CP.
We next examined whether or not Rv2125 or Rv2714 physically interacts with the M. tuberculosis 20S CP. We used an open-gate M. tuberculosis 20S CP (20SOG) in which the N-terminal eight residues of the α subunits of the CP (which otherwise block the substrate entry port) are removed, making this complex more proteolytically active (6, 32) and better able to bind the ATP-dependent proteasomal activator Mpa and the ATP-independent activator PafE (15, 33). We purified the intact Rv2125 or Rv2714 and the 20SOG with 6×His tags on the C termini of the β subunits for interaction assays. In a pulldown assay with a Ni2+-nitrilotriacetic acid (NTA) column, both Rv2125 and Rv2714 washed off the column because they did not bind to the His-tagged 20SOG (Fig. 5A). As a positive control, the known M. tuberculosis proteasomal activator PafE was pulled down in the affinity column (14, 15, 34). We further carried out an isothermal titration calorimetry (ITC) experiment and found that there was no detectable binding between Rv2125 and M. tuberculosis 20SOG (Fig. 5B) or between Rv2714 and M. tuberculosis 20SOG (Fig. 5C). Rv2125 also did not bind the wild-type (WT) M. tuberculosis 20S CP (see Fig. S2 in the supplemental material). As a positive control, we produced a hybrid Rv2125 construct, Rv2125-TailPafE, in which the extra C-terminal α-helical segment of Rv2125 was deleted and then replaced with the C-terminal proteasome-binding motif of PafE. As expected, Rv2125-TailPafE bound tightly to the M. tuberculosis 20SOG with an estimated dissociation constant (Kd) of 0.65 μM (Fig. 5D).
FIG 5.

Rv2125 and Rv2714 do not interact with the M. tuberculosis 20S CP. (A) SDS-PAGE of purified input proteins (lanes 5 to 8) and samples eluted off of the Ni2+ affinity column by 500 mM imidazole (lanes 2 to 4). Full-length Rv2125 or Rv2714 with the 6×His tag removed was mixed with purified and 6×His-tagged M. tuberculosis 20SOG before being loaded into the affinity column. Lane 1 shows the molecular mass markers. Lanes 2 and 3 contain only the 20S α subunit (PrcA) and β subunit (PrcB) because Rv2125 (lane 2) and Rv2714 (lane 3) did not bind the His-tagged 20S CP on the Ni2+ beads and were therefore washed off the column. Lane 4 is a positive control with PafE. We used an inactive 20SOG variant, which carries a T1A substitution at the catalytic N terminus of the β subunit, to prevent potential degradation of the input proteins by the CPs. The two light bands in lane 5 marked by asterisks are either breakdown products or contaminants. (B to D) ITC of Rv2125 (B), Rv2714 (C), and Rv2125-TailPafE (D) titrated against the M. tuberculosis 20SOG did not detect any heat signal.
Disruption of Rv2125 in M. tuberculosis does not affect levels of proteasome components or substrates.
We isolated a mutant with a transposon insertion in Rv2125 from a library of ordered mutants (35). This mutant grew like the parental, wild-type strain H37Rv (not shown). We compared the proteomes of the wild-type and mutant strains using quantitative tandem mass tag labeling and mass spectrometry (TMT-MS) (36–38). Among approximately 3,100 proteins profiled, 10 proteins were reduced by about 2-fold and seven proteins were increased by more than 2-fold in the mutant (Table 2; see also Data Set S1 in the supplemental material). However, none of these proteins was a known substrate or component of the Pup-proteasome system (Table 2). Thus, Rv2125 is highly unlikely to be involved in proteasome assembly in M. tuberculosis.
TABLE 2.
TMT-MS analysis results for wild-type versus Rv2125 mutant M. tuberculosisa
| Protein | Protein description | No. of peptides | WT avg | Mutant avg | Fold change, mutant/WT |
|---|---|---|---|---|---|
| Rv2125.1 | 5 | 32.04 | 1.29 | 0.04 | |
| Proteins with >2-fold decrease | |||||
| Rv0011c.1 | CrgA | 1 | 28.75 | 4.58 | 0.16 |
| Rv1131.1 | Methylcitrate synthase PrpC | 2 | 28.18 | 5.16 | 0.18 |
| Rv1130.1 | Methylcitrate dehydratase PrpD | 13 | 27.25 | 6.08 | 0.22 |
| Rv1606.1 | Phosphoribosyl-AMP cyclohydrolase | 3 | 26.31 | 7.02 | 0.27 |
| Rv0863.1 | Hypothetical | 1 | 25.23 | 8.10 | 0.32 |
| Rv1624c.1 | Hypothetical | 1 | 24.43 | 8.90 | 0.36 |
| Rv1660.1 | Chalcone synthase Pks10 | 5 | 24.38 | 8.96 | 0.37 |
| Rv0367c.1 | Hypothetical | 1 | 24.19 | 9.14 | 0.38 |
| Rv2930.1 | Fatty acid coenzyme A ligase FadD | 9 | 22.89 | 10.44 | 0.46 |
| Rv3364c.1 | Hypothetical | 2 | 22.80 | 10.54 | 0.46 |
| Proteins with >2-fold increase | |||||
| Rv3875.1 | Early secretory antigenic target EsxA | 2 | 11.11 | 22.23 | 2.00 |
| Rv2628.1 | Hypothetical | 1 | 11.05 | 22.29 | 2.02 |
| Rv2055c.1 | 30S ribosomal protein S18 RpsR2 | 6 | 10.96 | 22.37 | 2.04 |
| Rv0106.1 | Hypothetical | 1 | 10.79 | 22.54 | 2.09 |
| Rv0572c.1 | Hypothetical | 4 | 10.55 | 22.79 | 2.16 |
| Rv2558.1 | Monooxygenase | 7 | 10.37 | 22.96 | 2.21 |
| Rv2056c.1 | Ribosomal protein | 2 | 8.08 | 25.26 | 3.13 |
| Proteins of the Pup-20S system | |||||
| Rv2097c.1 | Pup ligase PafA | 36 | 16.16 | 17.17 | 1.06 |
| Rv2109c.1 | 20S α subunit PrcA | 18 | 16.74 | 16.59 | 0.99 |
| Rv2110c.1 | 20S β subunit PrcB | 20 | 17.62 | 15.71 | 0.89 |
| Rv2111c.1 | Pup | 4 | 16.73 | 16.61 | 0.99 |
| Rv2112c.1 | Depupylase Dop | 26 | 16.65 | 16.69 | 1.00 |
| Rv2115c.1 | Mpa | 39 | 16.71 | 16.63 | 1.00 |
| Rv3780.1 | PafE | 24 | 16.12 | 17.22 | 1.07 |
Listed are proteins whose relative abundance is more than 2-fold higher or lower in the Rv2125 mutant (Rv2125::ΦMycoMarT7) strain.
DISCUSSION
In this work, we sought to test whether or not homologues of eukaryotic and archaeal proteasome assembly factors were required for proteasome assembly in M. tuberculosis. A previous bioinformatics analysis identified two proteins, Rv2125 and Rv2714, as possible PAC2-like proteasome chaperones (26). We determined the crystal structure of M. tuberculosis Rv2125 at a 3.0-Å resolution. Our crystallographic data strongly indicate that Rv2125 is unlikely to interact with proteasome core particles (unlike its homologues archaeal PbaB and yeast Pba1). Furthermore, neither pulldown nor ITC studies detected an interaction of Rv2125 trimers with the M. tuberculosis 20S CP. These data are consistent with our previous observation that neither Rv2125 nor Rv2714 coimmunoprecipitates with 20S CPs purified from M. tuberculosis (14). Finally, genetic disruption of Rv2125 did not affect the levels of any of the proteasome core or accessory factor proteins. It does not appear that Rv2125 participates in proteasome assembly or function despite its structural similarity to PbaB and Pba1.
Given that the M. tuberculosis 20S CP can be assembled when produced in Escherichia coli (32), it is possible that the bacterial proteasome assembly is autonomous. However, an archaeal 20S CP also assembles in vitro or when expressed in E. coli in the absence of an assembly chaperone (39), yet archaeal proteasome chaperones have been identified. Although Rv2125 is unlikely to function as an assembly chaperone, it remains to be determined if the M. tuberculosis 20S CP nonetheless requires assembly chaperones under certain circumstances. Finally, what are the functions of these Pba homologues in M. tuberculosis? Because several enzymes involved in methylcitrate metabolism appeared to be differentially regulated in the Rv2125 mutant, future work will be needed to understand if Rv2125 has direct or indirect effects on this pathway.
MATERIALS AND METHODS
Purification of the M. tuberculosis proteins Rv2125, Rv2714, 20SOG, and PafE.
For crystallization, a gene fragment encoding residues 16 to 260 of M. tuberculosis Rv2125 was amplified and cloned into a pET24b(+) vector (Novagen) at the NdeI and XhoI restriction sites. For binding studies, full-length Rv2125 and Rv2714 were cloned into pET28b(+) with an N-terminal 6×His tag followed by a thrombin cleavage site. Plasmids were then transformed into E. coli BL21(DE3). Bacteria were grown in Luria-Bertani (LB) broth with 25 μg/ml kanamycin at 37°C. When the optical density at 600 nm (OD600) reached approximately 0.6, isopropyl-β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.3 mM, and incubation was continued at 16°C for an additional 20 h. We harvested cells by centrifugation and resuspended the pellets in a lysis buffer containing 50 mM Tris (pH 8.0), 500 mM NaCl, 5% glycerol (vol/vol), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), and 20 mmol/liter imidazole. The cells were lysed by sonication. The lysates were loaded into a 5-ml Ni2+-nitrilotriacetic acid (Ni-NTA)–agarose column, and target proteins were eluted with a linear imidazole concentration gradient. Fractions containing proteins of interest were pooled, concentrated, and further purified in a Superdex 200 10/300 GL gel filtration column in a buffer containing 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl. Purified Rv212516–260 was concentrated to 20 mg/ml for crystallization trials. The N-terminal 6×His tags of the full-length Rv2125 and Rv2714 were cleaved using thrombin before they were used for binding studies with the His-tagged M. tuberculosis 20SOG. The open-gate 20S proteasome genes prcBA, where the first 24 nucleotides of the prcA coding sequence were deleted, were cloned into pET28b(+). PrcB was produced with a thrombin cleavage site followed by a C-terminal 6×His tag (32). The M. tuberculosis 20SOG was produced in E. coli and purified as described previously (40). PafE was purified as described previously (15). The purity of protein was assessed by SDS-PAGE. Protein concentration was determined by using a NanoDrop ND-1000 spectrophotometer (A280) or by the Bradford assay.
Crystallization, data collection, and structure determination.
A crystallization screen was done using the sitting-drop vapor diffusion method at 20°C. Crystals appeared after 2 days from drops consisting of 1 μl of protein solution and 1 μl of reservoir solution containing 0.1 M sodium chloride, 0.1 M HEPES (pH 7.5), 1.6 M ammonium sulfate, 1% (wt/vol) Anapoe-20, and 2% (wt/vol) benzamidine hydrochloride. Crystal diffraction data sets were collected at the X25 and X29 beamlines at the National Synchrotron Light Source, Brookhaven National Laboratory, and were processed with HKL2000 (41). The initial phases were obtained using the molecular replacement method (PDB accession number 3MNF) with the Phenix program (42). Coot was used to manually adjust and rebuild the models (43). Further model refinement was carried out using Refmac5 (44) in the CCP4 suite and Phenix (42). The refinement and manual rebuilding were iterated multiple times until acceptable Rwork (23.6%) and Rfree (28.9%) values were obtained. A selected region of the electron density map contoured at 1.2σ is presented to demonstrate the quality of the fitting between the map and the atomic model (see Fig. S1 in the supplemental material).
Pulldown experiment.
The purified, nontagged, full-length Rv2125, Rv2714, or PafE, plus the 6×His-tagged open-gate 20S proteasome, was used in the pulldown assay. We performed all pulldown experiments at room temperature. For immobilization, 0.1 mg of 6×His-tagged M. tuberculosis 20SOG was applied to Ni2+-NTA beads, which were subsequently incubated with 1 mg of intact Rv2125, Rv2714, or PafE for 30 min in 20 mM Tris-HCl (pH 8.0) and 150 mM NaCl. The beads were then washed three times with a buffer containing 20 mM imidazole. Finally, proteins bound to the beads were eluted by a buffer containing 500 mM imidazole and were analyzed by SDS-PAGE.
Isothermal titration calorimetry.
We used purified full-length Rv2125 and Rv2714 proteins and purified M. tuberculosis 20SOG and WT 20S CP for ITC analysis. For a positive control, the C-terminal proteasome binding motif of PafE was fused to the C terminus of Rv212516–242, in which the extra C-terminal α-helical segment was deleted, resulting in a fusion protein named Rv2125-TailPafE. It was expressed and purified the same way as Rv212516–260. All proteins were put in a solution containing 20 mM Tris (pH 8.0) and 150 mM NaCl. The experiment was run at 25°C using a VP-ITC microcalorimeter (MicroCal). We titrated 200 μM Rv2125 or 150 μM Rv2714 into 10 μM M. tuberculosis 20SOG or WT 20S CP. We titrated 30 μM Rv2125-TailPafE into 2 μM 20S CP and fitted the titration curve in program Origin 7.0.
TMT-MS analysis.
M. tuberculosis strains H37Rv (parental wild type) and MHD1050 (H37Rv Rv2125::MycoMarT7) were grown under routine culture conditions (Middlebrook 7H9 broth with albumin, glycerol, and glucose) to an optical density at 580 nm of 1.0. Bacteria were harvested by centrifugation (2,800 × g, 5 min) and washed with an equal volume of phosphate-buffered saline with 0.05% Tween 80. Insoluble proteins and cellular debris were removed by centrifugation and filtration of the lysate through a 0.45-μm syringe filter. For all samples, protein concentration was assessed by protein assay (Bio-Rad). Equal amounts of protein were precipitated by adding six volumes of cold acetone, incubated at −20°C for 20 min, and collected by 15 min of centrifugation at 16,000 × g at 4°C. Protein pellets were air dried.
Protein was resuspended in 6 M guanidine-HCl and quantified by bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, IL). Samples were reduced with 5 mM dithiothreitol (DTT), alkylated with 12 mM iodoacetamide, and digested for 2 h with endoproteinase Lys-C (Wako, Japan) at a ratio of 1:200 Lys-C to protein and then digested overnight with trypsin (Promega, Madison, WI) at a ratio of 1:100 trypsin to protein. The digest was acidified with formic acid to a pH of <3, and peptides were desalted using 50 mg of solid-phase C18 extraction cartridge (Waters, Milford, MA) and then lyophilized. Samples were resuspended in 100 μl of 200 mM HEPES (pH 8.5) with 30% acetonitrile and 10 μl of 20 μg/ml 6-plex TMT reagent in anhydrous acetonitrile added to each sample. The reaction proceeded for 1 h and then was quenched with hydroxylamine to a final concentration of 0.5%. Samples were then combined equally, desalted using homemade stage tips as previously described (45), and lyophilized.
After stage tip desalting, samples were resuspended in 0.1% formic acid and analyzed on an Orbitrap Elite (Thermo Fisher Scientific, San Jose, CA) using the Orbitrap LC-MS3 method as described previously (45). Spectra were matched against an M. tuberculosis H37Rv database (downloaded on 6 February 2013), and the protein false discovery rate was controlled to less than 1% using the reverse-database strategy. Reporter ion signal-to-noise (S/N) ratios for all peptides matching each protein were summed, and protein relative expression values were represented as a fraction of the total intensity for all TMT reporter ions for the protein.
Accession number(s).
The structure data for Rv2125 have been deposited in the Protein Data Bank (PDB) under accession number 5UN0.
Supplementary Material
ACKNOWLEDGMENTS
We thank David Nadziejka for helpful comments on a draft version of the manuscript. X-ray diffraction data were collected at X25 and X29A of the National Synchrotron Light Source of Brookhaven National Laboratory.
This work was supported by NIH grants AI070285 (to H.L.), AI088075 (to K.H.D.), GM067945 (to S.P.G.), and T32 AI007180 and F30 AI110067 (to J.B.J.) and by the Van Andel Research Institute (H.L.). K.H.D. holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00846-16.
REFERENCES
- 1.Nguyen L, Pieters J. 2009. Mycobacterial subversion of chemotherapeutic reagents and host defense tactics: challenges in tuberculosis drug development. Annu Rev Pharmacol Toxicol 49:427–453. doi: 10.1146/annurev-pharmtox-061008-103123. [DOI] [PubMed] [Google Scholar]
- 2.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 3.Gandotra S, Lebron MB, Ehrt S. 2010. The Mycobacterium tuberculosis proteasome active site threonine is essential for persistence yet dispensable for replication and resistance to nitric oxide. PLoS Pathog 6:e1001040. doi: 10.1371/journal.ppat.1001040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gandotra S, Schnappinger D, Monteleone M, Hillen W, Ehrt S. 2007. In vivo gene silencing identifies the Mycobacterium tuberculosis proteasome as essential for the bacteria to persist in mice. Nat Med 13:1515–1520. doi: 10.1038/nm1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Samanovic MI, Darwin KH. 2016. Game of 'somes: protein destruction for Mycobacterium tuberculosis pathogenesis. Trends Microbiol 24:26–34. doi: 10.1016/j.tim.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. 2009. Inhibitors selective for mycobacterial versus human proteasomes. Nature 461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin G, Chidawanyika T, Tsu C, Warrier T, Vaubourgeix J, Blackburn C, Gigstad K, Sintchak M, Dick L, Nathan C. 2013. N,C-capped dipeptides with selectivity for mycobacterial proteasome over human proteasomes: role of S3 and S1 binding pockets. J Am Chem Soc 135:9968–9971. doi: 10.1021/ja400021x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voges D, Zwickl P, Baumeister W. 1999. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 9.De Mot R, Nagy I, Baumeister W. 1998. A self-compartmentalizing protease in Rhodococcus: the 20S proteasome. Antonie Van Leeuwenhoek 74:83–87. doi: 10.1023/A:1001708012708. [DOI] [PubMed] [Google Scholar]
- 10.Jastrab JB, Darwin KH. 2015. Bacterial proteasomes. Annu Rev Microbiol 69:109–127. doi: 10.1146/annurev-micro-091014-104201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valas RE, Bourne PE. 2008. Rethinking proteasome evolution: two novel bacterial proteasomes. J Mol Evol 66:494–504. doi: 10.1007/s00239-008-9075-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darwin KH. 2009. Prokaryotic ubiquitin-like protein (Pup), proteasomes and pathogenesis. Nat Rev Microbiol 7:485–491. doi: 10.1038/nrmicro2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kish-Trier E, Hill CP. 2013. Structural biology of the proteasome. Annu Rev Biophys 42:29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jastrab JB, Wang T, Murphy JP, Bai L, Hu K, Merkx R, Huang J, Chatterjee C, Ovaa H, Gygi SP, Li H, Darwin KH. 2015. An adenosine triphosphate-independent proteasome activator contributes to the virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 112:E1763–E1772. doi: 10.1073/pnas.1423319112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai L, Hu K, Wang T, Jastrab JB, Darwin KH, Li H. 2016. Structural analysis of the dodecameric proteasome activator PafE in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 113:E1983–E1992. doi: 10.1073/pnas.1512094113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samanovic MI, Li H, Darwin KH. 2013. The pup-proteasome system of Mycobacterium tuberculosis. Subcell Biochem 66:267–295. doi: 10.1007/978-94-007-5940-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finley D. 2009. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu Rev Biochem 78:477–513. doi: 10.1146/annurev.biochem.78.081507.101607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matias AC, Ramos PC, Dohmen RJ. 2010. Chaperone-assisted assembly of the proteasome core particle. Biochem Soc Trans 38:29–33. doi: 10.1042/BST0380029. [DOI] [PubMed] [Google Scholar]
- 19.Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y, Natsume T, Tanaka K, Murata S. 2005. A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437:1381–1385. doi: 10.1038/nature04106. [DOI] [PubMed] [Google Scholar]
- 20.Le Tallec B, Barrault MB, Courbeyrette R, Guerois R, Marsolier-Kergoat MC, Peyroche A. 2007. 20S proteasome assembly is orchestrated by two distinct pairs of chaperones in yeast and in mammals. Mol Cell 27:660–674. doi: 10.1016/j.molcel.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 21.Kusmierczyk AR, Kunjappu MJ, Funakoshi M, Hochstrasser M. 2008. A multimeric assembly factor controls the formation of alternative 20S proteasomes. Nat Struct Mol Biol 15:237–244. doi: 10.1038/nsmb.1389. [DOI] [PubMed] [Google Scholar]
- 22.Hirano Y, Kaneko T, Okamoto K, Bai M, Yashiroda H, Furuyama K, Kato K, Tanaka K, Murata S. 2008. Dissecting beta-ring assembly pathway of the mammalian 20S proteasome. EMBO J 27:2204–2213. doi: 10.1038/emboj.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X, Kusmierczyk AR, Wong P, Emili A, Hochstrasser M. 2007. Beta-subunit appendages promote 20S proteasome assembly by overcoming an Ump1-dependent checkpoint. EMBO J 26:2339–2349. doi: 10.1038/sj.emboj.7601681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kusmierczyk AR, Kunjappu MJ, Kim RY, Hochstrasser M. 2011. A conserved 20S proteasome assembly factor requires a C-terminal HbYX motif for proteasomal precursor binding. Nat Struct Mol Biol 18:622–629. doi: 10.1038/nsmb.2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumoi K, Satoh T, Murata K, Hiromoto T, Mizushima T, Kamiya Y, Noda M, Uchiyama S, Yagi H, Kato K. 2013. An archaeal homolog of proteasome assembly factor functions as a proteasome activator. PLoS One 8:e60294. doi: 10.1371/journal.pone.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyer LM, Burroughs AM, Aravind L. 2008. Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biol Direct 3:45. doi: 10.1186/1745-6150-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stadtmueller BM, Kish-Trier E, Ferrell K, Petersen CN, Robinson H, Myszka DG, Eckert DM, Formosa T, Hill CP. 2012. Structure of a proteasome Pba1-Pba2 complex: implications for proteasome assembly, activation, and biological function. J Biol Chem 287:37371–37382. doi: 10.1074/jbc.M112.367003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grana M, Bellinzoni M, Miras I, Fiez-Vandal C, Haouz A, Shepard W, Buschiazzo A, Alzari PM. 2009. Structure of Mycobacterium tuberculosis Rv2714, a representative of a duplicated gene family in Actinobacteria. Acta Crystallogr Sect F Struct Biol Cryst Commun 65:972–977. doi: 10.1107/S1744309109035027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao B, Sugiman-Marangos S, Junop MS, Gupta RS. 2009. Structural and phylogenetic analysis of a conserved Actinobacteria-specific protein (ASP1; SCO1997) from Streptomyces coelicolor. BMC Struct Biol 9:40. doi: 10.1186/1472-6807-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holm L, Rosenstrom P. 2010. Dali server: conservation mapping in 3D. Nucleic Acids Res 38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Darwin KH, Lin G, Chen ZQ, Li HL, Nathan CF. 2005. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol 55:561–571. [DOI] [PubMed] [Google Scholar]
- 32.Lin G, Hu GQ, Tsu C, Kunes YZ, Li HL, Dick L, Parsons T, Li P, Chen ZQ, Zwickl P, Weich N, Nathan C. 2006. Mycobacterium tuberculosis prcBA genes encode a gated proteasome with broad oligopeptide specificity. Mol Microbiol 59:1405–1416. doi: 10.1111/j.1365-2958.2005.05035.x. [DOI] [PubMed] [Google Scholar]
- 33.Wang T, Li H, Lin G, Tang C, Li D, Nathan C, Darwin KH, Li H. 2009. Structural insights on the Mycobacterium tuberculosis proteasomal ATPase Mpa. Structure 17:1377–1385. doi: 10.1016/j.str.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Delley CL, Laederach J, Ziemski M, Bolten M, Boehringer D, Weber-Ban E. 2014. Bacterial proteasome activator bpa (rv3780) is a novel ring-shaped interactor of the mycobacterial proteasome. PLoS One 9:e114348. doi: 10.1371/journal.pone.0114348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Festa RA, Pearce MJ, Darwin KH. 2007. Characterization of the proteasome accessory factor (paf) operon in Mycobacterium tuberculosis. J Bacteriol 189:3044–3050. doi: 10.1128/JB.01597-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- 37.McAlister GC, Huttlin EL, Haas W, Ting L, Jedrychowski MP, Rogers JC, Kuhn K, Pike I, Grothe RA, Blethrow JD, Gygi SP.. 2012. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem 84:7469–7478. doi: 10.1021/ac301572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias JE, Gygi SP.. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods 4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 39.Maupin-Furlow JA, Humbard MA, Kirkland PA, Li W, Reuter CJ, Wright AJ, Zhou G. 2006. Proteasomes from structure to function: perspectives from Archaea. Curr Top Dev Biol 75:125–169. doi: 10.1016/S0070-2153(06)75005-0. [DOI] [PubMed] [Google Scholar]
- 40.Li D, Li H, Wang T, Pan H, Lin G, Li H. 2010. Structural basis for the assembly and gate closure mechanisms of the Mycobacterium tuberculosis 20S proteasome. EMBO J 29:2037–2047. doi: 10.1038/emboj.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Otwinowski Z, Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 42.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. 2002. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58:1948–1954. doi: 10.1107/S0907444902016657. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallographica Section D-Biological Crystallography 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Isasa M, Rose CM, Elsasser S, Navarrete-Perea J, Paulo JA, Finley DJ, Gygi SP.. 2015. Multiplexed, proteome-wide protein expression profiling: yeast deubiquitylating enzyme knockout strains. J Proteome Res 14:5306–5317. doi: 10.1021/acs.jproteome.5b00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res 16:10881–10890. doi: 10.1093/nar/16.22.10881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robert X, Gouet P. 2014. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res 42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.