

Abstract
Chemical or electrochemical one‐electron oxidation of 5‐N‐arylaminothiazoles was found to afford stable radical cations. For chemical oxidation, 1 equivalent of [(4‐BrC6H4)3N][SbCl6] (Magic Blue, MB) was added to CH2Cl2 solutions of the thiazoles, and the thus‐obtained radicals showed light absorption in the near‐infrared region. Electrochemical oxidation also led to bathochromic shifts in the absorption bands, and the obtained spectra were similar to those derived from the chemically oxidized species. These radicals afforded electron paramagnetic resonance (EPR) spectra that are consistent with the notion of stable nitrogen radicals (half‐life≤385 h). The EPR spectrum of a thiazole containing 4‐dimethylaminophenyl groups on the nitrogen atom at the 5‐position changed significantly upon adding >3 equivalents of MB. Details of the electronic structures of the experimentally obtained radical cations were generated from theoretical calculations.
Keywords: absorption spectroscopy, electron paramagnetic resonance, heterocycles, oxidation, radical ions
1. Introduction
Despite their usually low stability, organic radical cations1, 2 have raised substantial interest in a wide range of research areas owing to their critical importance in a multitude of organic and biological reactions, for which they act as reactive intermediates. Furthermore, radical cations that absorb light in the near‐infrared (NIR) region have attracted much attention in the materials science community, as this feature may potentially enhance the efficiency of solar cells.3 The core structure of radical cations is usually based on fused rings and/or metal complexes. Radical cations with monocyclic rings have received substantially less attention in comparison.4 The core units of these molecules usually contain heteroatoms such as nitrogen, oxygen, and sulfur.1b, 2i, 2j, 5, 6 The properties of triarylamine‐derived radical cations have especially been studied in great detail in this context.7 In general, their absorption bands at approximately λ=700 nm are subject to a bathochromic shift. Moreover, N,N‐dimethylaminophenyl‐substituted triphenylamines show absorption bands at approximately λ=1300 nm. However, most of these compounds are unstable and/or are present only at low temperature. For example, the radical cation of 1,3,6‐tris(di‐p‐anisylamino)pyrene, which contains fused rings, exhibits a half‐life of 3 h.8 In addition, several radical cations generated from sulfur‐containing monocyclic aromatics, such as thiophenes9 and thiazoles,10 have been reported. Some thiazole radical cations, such as 1‐butyl‐5‐methylthiazolium,9d have been reported, but their photophysical properties have not been disclosed. Furthermore, even the radical cations derived from large molecules with heterocyclic rings, such as azathia[7]helicene, exhibit half‐lives of only approximately 8 h.2h
In this context, we recently developed 5‐aminothiazoles with various substituents (Scheme 1) that could be readily prepared from thioformamides and secondary thioamides. These 5‐aminothiazoles represent the first examples of thiazoles that contain diarylamino groups at the 5‐position,11 and they show a wide range of absorption and fluorescence properties that change upon exposure to external stimuli, such as the addition of acids. Thiazoles contain electron‐donating (D) and electron‐accepting (A) moieties that deviate from coplanar alignment. These D‐A structures usually lead to fluorescence from intramolecular charge transfer (ICT) states.12 Therefore, the electrons of compounds having the D‐A structure are delocalized over the molecule in stages of absorption and emission. Thus, this structure is expected to lead to stabilized charge‐separated species.
Scheme 1.
5‐N‐Arylaminothiazoles 1–4.
Thiazole derivatives moreover exhibit a reversible one‐electron oxidation wave in their cyclic voltammograms (Figure S1, Supporting Information). For example, the half‐wave potential of 1 in CH3CN appears at +0.623 V, whereas that of 3, which contains a 4‐methoxyphenyl electron donor at the 5‐position, appears at +0.407 V. This result indicates that 3 oxidizes more easily than 1. In contrast, the cyclic voltammogram of 4,13 which bears a 4‐dimethylaminophenyl group at the 5‐position, exhibits two reversible oxidation waves. The oxidation potential and waveform of 5‐aminothiazoles can thus be easily controlled by judicious choice of the substituents at the 5‐position. On the basis of these results, we expected that chemical oxidation of 5‐aminothiazoles would generate stable radicals, for which we anticipated absorption in the UV/Vis–NIR region. Herein, we report the chemical and electrochemical oxidation of 5‐aminothiazoles to generate radical cations that show UV/Vis–NIR absorption bands. In one case, a half‐life time of more than 300 h was observed.
2. Results and Discussion
To identify appropriate reagents for the chemical oxidation of 5‐aminothiazole 1, BF2CF3 ⋅OEt,14 NO[SbF6],15 and [(4‐BrC6H4)3N][SbCl6] (Magic Blue, MB)16 were added to solutions of 1 in CH2Cl2 (Scheme 2). Upon the addition of BF2CF3 ⋅OEt, the color of the solution changed from yellow to orange. However, the thus‐obtained compounds were very unstable. The use of NO[SbF6] did not afford any new species, as evident from quantitative recovery of the starting materials. Notably, a single chemical species was only obtained with MB, and therefore, we subsequently used MB to generate single chemical species of 1–4.
Scheme 2.
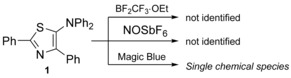
Reactions between thiazole 1 and BF2CF3 ⋅OEt, NO[SbF6], and [(4‐BrC6H4)3N][SbCl6] (Magic Blue).
We then measured the absorption spectra of the radical cations derived from 1–4 (Figures 1 and S2). Whereas 1–4 showed absorption maxima at approximately λ=358–410 nm (Figure 1 a), we expected bathochromic shifts in their absorption bands upon the addition of MB. The addition of 1 equivalent of MB to 1 afforded new absorption bands at λ=900 and 650 nm (Table 1). After the addition of 1 equivalent of MB, the absorption maxima of 2 and 3, both of which contain methoxy groups, were observed at λ=918 and 932 nm. Furthermore, the addition of 1 equivalent of MB to thiazole 4, which contains the strongest electron donor, resulted in a strong absorption in the NIR region (λ=1180 nm). These results suggest that the wavelengths and strength of the absorptions can be adjusted by the substituents introduced on the thiazole rings.
Figure 1.
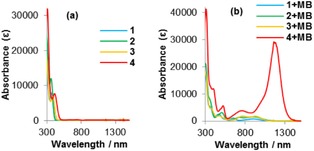
Absorption spectra of a) 1–4 and b) 1+MB to 4+MB in CH2Cl2, [solute]=1×10−4 M.
Table 1.
UV/Vis absorption spectra of thiazoles 1–4 with MB.[a]
| Thiazole | λabs [nm] | Species | λabs [nm] |
|---|---|---|---|
| 1 | 364 | 1+MB | 900, 650, 466 |
| 2 | 358 | 2+MB | 918, 726, 506 |
| 3 | 382 | 3+MB | 932, 768, 590, 480 |
| 4 | 410 | 4+MB (1 equiv) 4+MB (3 equiv) | 1180, 1006, 760, 522 2726, 1474, 796, 456 |
[a] In CH2Cl2, [solute]=1×10−4 m.
The addition of >3 equivalents of MB to 4 also induced a shift in the absorption band (Figure S3). The absorption of 4 gradually changed from λ=760, 1006, and 1180 nm to 796, 1474, and 2726 nm, whereas the color of the solution changed from red to green. These spectra remained unchanged after 1 day. These results corroborate that thiazole 4 generates different radical species, depending on the amount of MB added.
To compare the photophysical properties of the chemically oxidized species with those of the electrochemically oxidized species,17 we measured the cyclic voltammograms and absorption spectra of 1. In CH2Cl2, 1 showed a one‐electron oxidation wave with peaks at E=1.04 and 0.73 V (Figure S4). Thus, a potential of 1.04 V was applied to a CH2Cl2 solution of 1 for the electrochemical oxidation while the time‐dependent absorption spectra of this solution were measured (Figure 2). The oxidation potentials and the waveforms of the oxidation waves could be controlled by judicious choice of the substituents of the 5‐aminothiazoles. The observed spectra of electrochemically oxidized 1 were found to be similar to those of chemically oxidized 1 (Figure S5). Hence, the spectral changes should be attributed to the generation of radical species, and the radical species derived from 1 showed absorption bands at λ≈450 and 650 nm.
Figure 2.
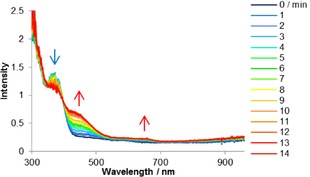
Time‐dependent absorption spectra of 1 after application of 1.04 V in CH2Cl2; [1]=1×10−4 m, 0.1 m [nBu4N][ClO4]; scan rate=100 mV s−1; reference electrode: Ag/AgCl, counter electrode: Pt, and working electrode: Pt gauze.
To elucidate the properties of the chemically oxidized species, the electron paramagnetic resonance (EPR) spectra were measured at room temperature for CH2Cl2 solutions of 1–4 that contained 1 equivalent of MB (1+MB to 4+MB, Figure 3). In all cases, the formation of radical species was observed, and the EPR parameters are summarized in Table 2. The g tensor and hyperfine coupling constant (hfcc, A) values were determined from spectral simulations, whereby A N and A H refer to interactions with nitrogen and hydrogen atoms, respectively. The g values of the spectra for 1+MB to 4+MB (g=2.0030–2.0034) differ substantially from the g value of a pure MB solution (g=2.0102). Thiazoles 3 and 4, with electron‐donating groups at the 5‐positions, showed higher g values than 1 and 2. Moreover, upon adding >3 equivalents of MB to 4, the g value of the 4+MB solution changed (g=2.0044) relative to that observed for the addition of 1–2 equivalents of MB. This result indicates that the properties of the radical species of 4 drastically change upon adding an excess amount of MB.
Figure 3.
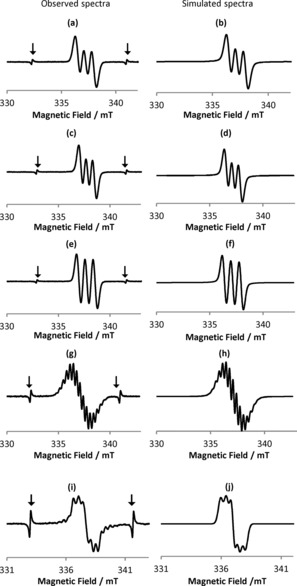
Observed EPR spectra of a) 1, c) 2, e) 3, and g) 4 with 1 equivalent of MB, as well as that of i) 4 with 3 equivalents of MB; the spectra in panels b, d, f, h, and j are the corresponding simulated spectra. Signals marked with arrows in panels a, c, e, g, and i represent the Mn2+ marker.
Table 2.
EPR data of solutions of thiazoles 1–4 with MB.[a]
| Thiazole | MB [equiv] | g value | A N [mT] | τ 1/2 [h][b] |
|---|---|---|---|---|
| 1 | 1 | 2.0030 | 0.72 | 91 |
| 2 | 1 | 2.0031 | 0.63 | 74 |
| 3 | 1 | 2.0034 | 0.78 | 385 |
| 4 | 1 | 2.0033 | 0.64 (one N atom) 0.34 (two N atom) 0.29 (eight H atom) | 3 |
| 3 | 2.0044 | 0.43 |
[a] In CH2Cl2 (distilled from P2O5), [solute]=1×10−4 m. The g and A N values were determined from spectral simulations. [b] Calculated by integrating the area of the EPR signals, normalized to the peak height of the signals from the Mn2+ marker as g standard.
To elucidate further details from the EPR spectra, we simulated the spectra for 1+MB to 4+MB with software for isotropic EPR spectra provided by JEOL (Figure 3 b, d, f, h, j). In the spectra of 1+MB to 3+MB, only the nitrogen atom at the 5‐postition affected the spectral line shape. Interestingly, the EPR spectrum of 4+MB, which exhibited two reversible oxidation waves in its cyclic voltammogram, included components of both the nitrogen atoms and hydrogen atoms, which implies that the unpaired electron is more delocalized in 4+MB than in 1+MB to 3+MB, despite the relatively short half‐life of 4+MB (3 h).
5‐Aminothiazoles with electron‐donating groups at the 5‐position and electron‐accepting groups at the 2‐position easily adopt ICT states derived from their D‐A structures. The smallest A N value among 1–3 was observed for 2 (0.63 mT), which contains an electron‐donating group at the 2‐position. A slightly higher A N value was observed for 3 (0.78 mT), which contains an electron‐donating group at the 5‐position, and this value is also marginally higher than that of 1 (0.72 mT).18 Nevertheless, these A N values are much smaller than that of 2,2,6,6‐tetramethylpiperidin‐1‐oxyl (TEMPO, 1.59 mT),19 in which an unpaired electron is localized on the nitrogen atom. This result implies that thiazole‐based radicals contain more delocalized electrons than, for example, TEMPO, and hence exhibit higher levels of stability, which is reflected in the relatively long half‐lives of 1+MB to 4+MB (e.g. 3+MB: τ 1/2=385 h, ≈16 days; Table 2). Interestingly, 4+MB exhibited, depending on the amount of MB added, two types of EPR spectra. Upon the addition of 1–2 equivalents MB, 4+MB showed multiplet signals, whereas upon the addition of >3 equivalents of MB, the signals associated with 4+MB disappeared.
The simulated spectrum of 4+MB (Figure 3 h) was very similar to the experimentally observed spectrum (Figure 3 g) and reflected interactions with one nitrogen atom (A N=0.64), two other nitrogen atoms (A N=0.34), and eight hydrogen atoms (A H=0.29). In contrast, the addition of >3 equivalents of MB to 4 furnished another radical species (Figure 3 i). The simulated spectrum (Figure 3 j) reflects interactions with two nitrogen atoms (A N=0.43) and is in good agreement with the experimentally observed spectrum.
Time‐dependent density functional theory (TD‐DFT) calculations for neutral 1 showed that the maximum absorption band (λ=416 nm) should be assigned predominantly to the HOMO→LUMO transition (97 % contribution). The compositions of the HOMO and LUMO are shown in Figure S6. The HOMO is mainly localized on the substituent at the 5‐position and the thiazole ring, whereas the LUMO is located predominantly on the phenyl group at the 2‐position and the thiazole ring. Accordingly, a correction is required for long‐range interactions. For that purpose, we used the long‐range‐corrected CAM‐B3LYP20 functional instead of the B3LYP functional to recalculate the UV/Vis absorption spectrum. The recalculated spectrum revealed an absorption maximum at λ=349 nm, which is in good agreement with the experimentally observed value (λ=364 nm).
We also calculated the UV/Vis absorption spectrum for the radical cation of 1. At the B3LYP level, an absorption band at λ=890 nm was obtained, which is in excellent agreement with the experimental value (λ=900 nm). This excitation was assigned predominantly to the cation‐HOMO→SOMO transition (97 % contribution) shown in Figure S6. The cation‐HOMO is very similar to the HOMO−1 of neutral 1, whereas the SOMO is essentially the singly occupied HOMO of neutral 1. A detailed comparison between the HOMO of neutral 1 and the SOMO showed a slightly higher contribution from the phenyl group at the 2‐position for the latter, which was probably caused by increased charge transfer from the substituent at the 5‐position. As a result, the calculated N−C(5‐position of thiazole) and C(2‐position of thiazole)−C(2‐position phenyl group) bond lengths contract upon removal of one electron from 1.399 (N−C neutral) to 1.367 Å (N−C radical cation) and from 1.472 (C−C neutral) to 1.455 Å (C−C radical cation).
The cation‐HOMO and SOMO essentially exhibit contributions from the same region, which suggests that the consideration of long‐range corrections for the hybrid B3LYP method is not necessary. The calculated cation‐HOMO–SOMO energy gap (2.10 eV) is much smaller than the HOMO–LUMO gap in neutral 1 (B3LYP: 3.56 eV; CAM‐B3LYP: 5.96 eV). This difference should be responsible for the dramatically redshifted UV/Vis absorption after the one‐electron oxidation.
For organic radicals, the B3LYP method can be used to predict the EPR parameters.21 The g tensor shifted from the free‐electron value, and the A N values were predicted by DFT calculations for our radical‐cation species. The calculated g tensor and the isotropic Fermi A N values for the N atom at the 5‐position (2.0029, 0.74 mT) agree very well with the experimental data (2.0030, 0.72 mT) shown in Table S1, which verifies the validity of the results obtained from calculations at the B3LYP/6‐31+G(d) level of theory in this particular case.22
The spin density map for the radical cation of 1 (Figure 4) shows positive (blue) and negative (green) spin densities at an isodensity level of 0.004 e bohr−1. The spin density is mostly delocalized over the N atom at the 5‐position and the neighboring π‐conjugated −C=C−N=C− fragment in the thiazole ring. The radical character of the cation is also reflected in the occupancy (0.88 e) of the orbital of the lone pair of electrons for the N atom at the 5‐position analyzed by NBO 6.0.23
Figure 4.
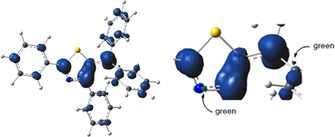
Mulliken spin‐density map for the radical cation of 1 calculated at the B3LYP/6‐31+G(d)/PCM(CH2Cl2) level of theory.
3. Conclusions
In conclusion, radical cations 1+MB to 4+MB were generated by adding 1 equivalent of Magic Blue (MB) to thiazoles 1–4. The electron paramagnetic resonance (EPR) spectra of 1+MB to 4+MB indicated that it was the nitrogen atom at the 5‐postition that mainly affected the spectral line shape of these thiazole‐based radical cations. Radical cations with electron‐donating groups at the 5‐position (i.e. 3+MB and 4+MB) showed larger g values than the other radical cations. Moreover, upon the addition of 3 equivalents of MB to 4 a different radical species was obtained. Radicals 1+MB to 4+MB showed smaller A N values than 2,2,6,6‐tetramethylpiperidin‐1‐oxyl (TEMPO), which suggested a correspondingly higher level of delocalization and, consequently, more stable radical cations. Among these radical cations, 3+MB was the most stable (half‐life of 385 h). Thus, compounds with strong intramolecular charge‐transfer character that contain one moiety that is able to react with oxidants may represent a new concept for the design of stable organic radicals. The absorption wavelengths of 1–4 showed a bathochromic shift upon the addition of 1 equivalent of MB, and the absorption bands of 1+MB to 4+MB were observed in the near‐infrared region. The absorption spectra of chemically oxidized 1 showed changes similar to those observed in the spectra of electrochemically oxidized 1. Therefore, chemical and electrochemical oxidation methods can be chosen for the generation of similar chemical species. Further studies on the applications of 5‐N‐arylaminothiazoles as stable dyes with a wide absorption and emission range are currently in progress in our laboratory.
Experimental Section
General Remarks
High‐resolution mass spectra were recorded with a double‐focusing mass spectrometer in the EI mode. EPR spectra and their simulations were obtained with a JES‐TE200. UV/Vis absorption spectra were measured with a Hitachi U‐4100 spectrometer. Electrochemical measurements were performed by using a platinum working electrode, a platinum gauze working electrode, a platinum wire counter electrode, and a Ag/Ag+ [0.01 m AgCl] reference electrode in CH2Cl2 with 0.1 m [nBu4N][ClO4]. Measurements were recorded with a BAS CS‐3A Cell Stand and SEC2000 Spectra System. Prior to use, CH2Cl2 was distilled from P2O5. All other chemicals were used as received without further purification. Photophysical properties were measured under atmospheric conditions at 23 °C. EPR measurements were performed under an atmosphere of argon at 23 °C. Thiazoles 1–4 were prepared according to literature procedures.11a
General Procedure for the Preparation of the Radical Cations
A CH2Cl2 solution of the corresponding thiazole (1×10−3 m, 0.1 mL) was diluted in CH2Cl2 (0.8 mL), and the mixture was stirred for 1 min. A CH2Cl2 solution of Magic Blue (1×10−3 m, 0.1 mL) was then added, which afforded a CH2Cl2 solution of the radical cation (1×10−4 m, 1 mL). Then, an aliquot (0.3 mL) of this solution was transferred to an EPR sample tube (diameter=1 mm), and the measurements were conducted. To measure the photophysical properties, 3 mL of the same solution was prepared.
Figure 5.
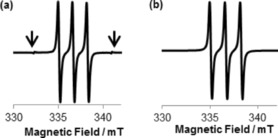
a) EPR spectrum of a solution of TEMPO and b) EPR simulated spectrum. The signals indicated with arrows in panel a are the Mn2+ marker.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the following grants‐in‐aid: Scientific Research on Innovative Areas (Ministry of Education, Culture, Sports, Science and Technology, MEXT), “Stimuli‐Responsive Chemical Species for the Creation of Functional Molecules” [2408], 15H00933 (T.M.), 16K05748 (S.K.) and 24109013(N.T.), as well as ACT‐C from the Japan Science and Technology Agency (JST).
K. Yamaguchi, T. Murai, S. Kutsumizu, Y. Miwa, M. Ebihara, J.-D. Guo, N. Tokitoh, ChemistryOpen 2017, 6, 282.
References
- 1.
- 1a. Geiser U., Schlueter J. A., Chem. Rev. 2004, 104, 5203–5241; [DOI] [PubMed] [Google Scholar]
- 1b. Shuvaev K. V., Passmore J., Coord. Chem. Rev. 2013, 257, 1067–1091; [Google Scholar]
- 1c. Engesser T. A., Lichtenthaler M. R., Schleep M., Krossing I., Chem. Soc. Rev. 2016, 45, 789–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Yasui S., Yamazaki S., Chem. Lett. 2015, 44, 422–424; [Google Scholar]
- 2b. Yamamoto K., Nakamura T., Higashibayashi S., Chem. Lett. 2015, 44, 1229–1231; [Google Scholar]
- 2c. Pandit P., Nakamura T., Higashibayashi S., Chem. Lett. 2015, 44, 1336–1338; [Google Scholar]
- 2d. Yoshida K., Suzuki S., Kozaki M., Okada K., Chem. Lett. 2016, 45, 203–205; [Google Scholar]
- 2e. Kayahara E., Kouyama T., Kato T., Yamago S., J. Am. Chem. Soc. 2016, 138, 338–344; [DOI] [PubMed] [Google Scholar]
- 2f. Sato C., Suzuki S., Kozaki M., Okada K., Org. Lett. 2016, 18, 1052–1055; [DOI] [PubMed] [Google Scholar]
- 2g. Morris S. A., Wang J., Zheng N., Acc. Chem. Res. 2016, 49, 1957–1968; [DOI] [PubMed] [Google Scholar]
- 2h. Wang Y., Zhang H., Pink M., Olankitwanit A., Rajca S., Rajca A., J. Am. Chem. Soc. 2016, 138, 7298–7304; [DOI] [PubMed] [Google Scholar]
- 2i. Ito A., Kurata R., Noma Y., Hirao Y., Tanaka K., J. Org. Chem. 2016, 81, 11416–11420; [DOI] [PubMed] [Google Scholar]
- 2j. Regnier V., Molton F., Philouze C., Martin D., Chem. Commun. 2016, 52, 11422–11425; [DOI] [PubMed] [Google Scholar]
- 2k. Wang X., Zhang F., Schellhammer K. S., Machata P., Ortmann F., Cuniberti G., Fu Y., Hunger J., Tang R., Popov A. A., Berger R., Müllen K., Feng X., J. Am. Chem. Soc. 2016, 138, 11606–11615; [DOI] [PubMed] [Google Scholar]
- 2l. Uebe M., Kawashima K., Takahashi K., Ito A., Chem. Eur. J. 2016, 23, 278–281. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hua C., Lee K., Huang J., Hsu C., Kuo T., Yang D., Ho K., Sol. Energy Mater. Sol. Cells 2009, 93, 2102–2107; [Google Scholar]
- 3b. Bar G., Larina N., Grinis L., Lokshin V., Gvishi R., Kiryuschev I., Zaban A., Khodorkovsky V., Sol. Energy Mater. Sol. Cells 2012, 99, 123–128; [Google Scholar]
- 3c. Chen W., Huang L., Qiao X., Yang J., Yua B., Yan D., Org. Electron. 2012, 13, 1086–1091; [Google Scholar]
- 3d. Van Nguyen T., Maeda T., Nakazumi H., Yagi S., Chem. Lett. 2016, 45, 291–293; [Google Scholar]
- 3e. Higashino T., Sugiura K., Tsuji Y., Nimura S., Ito S., Imahori H., Chem. Lett. 2016, 45, 1126–1128. [Google Scholar]
- 4.
- 4a. Sun D., Lindeman S. V., Rathore R., Kochi J. K., J. Chem. Soc. Perkin Trans. 1 2001, 2, 1585–1594; [Google Scholar]
- 4b. Rathore R., Burns C. L., Deselnicu M. I., Org. Lett. 2001, 3, 2887–2890; [DOI] [PubMed] [Google Scholar]
- 4c. Baciocchi E., Gerini M. F., Lanzalunga O., Mancinelli S., Tetrahedron 2002, 58, 8087–8093; [Google Scholar]
- 4d. Ratera I., Veciana J., Chem. Soc. Rev. 2012, 41, 303–349; [DOI] [PubMed] [Google Scholar]
- 4e. Tamura R., Suzuki K., Uchida Y., Noda Y., Electron Paramagn. Reson. 2013, 23, 1–21; [Google Scholar]
- 4f. Turek A. K., Hardee D. J., Ullman A. M., Nocera D. G., Jacobsen E. N., Angew. Chem. Int. Ed. 2016, 55, 539–544; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 549–554. [Google Scholar]
- 5.For tertiary amines, see:
- 5a. Ito A., Taniguchi A., Yamabe T., Tanaka K., Org. Lett. 1999, 1, 741–743; [DOI] [PubMed] [Google Scholar]
- 5b. Michinobu T., Tsuchida E., Nishide H., Bull. Chem. Soc. Jpn. 2000, 73, 1021–1027; [Google Scholar]
- 5c. Low P. J., Paterson M. A. J., Puschmann H., Goeta A. E., Howard J. A. K., Lambert C., Cherryman J. C., Tackley D. R., Leeming S., Brown B., Chem. Eur. J. 2004, 10, 83–91; for SOMO catalysis, see: [DOI] [PubMed] [Google Scholar]
- 5d. Beeson T. D., Mastracchio A., Hong J., Ashton K., MacMillan D. W. C., Science 2007, 316, 582–585; [PubMed] [Google Scholar]
- 5e. Bushby R. J., Kilner C. A., Taylor N., Vale M. E., Tetrahedron 2007, 63, 11458–11466; [Google Scholar]
- 5f. Zalibera M., Rapta P., Gescheidt G., Christensen J. B., Hammerich O., Dunsch L., J. Phys. Chem. C 2011, 115, 3942–3948; [Google Scholar]
- 5g. Zalibera M., Jalilov A. S., Stoll S., Guzei I. A., Gescheidt G., Nelsen S. F., J. Phys. Chem. A 2013, 117, 1439–1448; [DOI] [PubMed] [Google Scholar]
- 5h. Sakamaki D., Ito A., Furukawa K., Kato T., Tanaka K., J. Org. Chem. 2013, 78, 2947–2956; [DOI] [PubMed] [Google Scholar]
- 5i. Sakamaki D., Yano S., Kobashi T., Seki S., Kurahashi T., Matsubara S., Ito A., Tanaka K., Angew. Chem. Int. Ed. 2015, 54, 8267–8270; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8385–8388; [Google Scholar]
- 5j. Kobashi T., Sakamaki D., Seki S., Angew. Chem. Int. Ed. 2016, 55, 8634–8638; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 8776–8780. [Google Scholar]
- 6.For radical cations of sulfur- and nitrogen-containing heterocycles, see: Barlow S., Risko C., Odom S. A., Zheng S., Coropceanu V., Beverina L., Brédas J., Marder S. R., J. Am. Chem. Soc. 2012, 134, 10146–10155. [DOI] [PubMed] [Google Scholar]
- 7. Ito A., Kurata R., Sakamaki D., Yano S., Kono Y., Nakano Y., Furukawa K., Kato T., Tanaka K., J. Phys. Chem. A 2013, 117, 12858–12867. [DOI] [PubMed] [Google Scholar]
- 8. Nie H., Yao C., Shao J., Yao J., Zhong Y., Chem. Eur. J. 2014, 20, 17454–17465. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bourgeaux M., Skene W. G., J. Org. Chem. 2007, 72, 8882–8892; [DOI] [PubMed] [Google Scholar]
- 9b. Haubner K., Tarabek J., Ziegs F., Lukes V., Jaehne E., Dunsch L., J. Phys. Chem. A 2010, 114, 11545–11551; [DOI] [PubMed] [Google Scholar]
- 9c. Lukes V., Rapta P., Idzik K. R., Beckert R., Dunsch L., J. Phys. Chem. B 2011, 115, 3344–3353; [DOI] [PubMed] [Google Scholar]
- 9d. Beneduci A., Cospito S., La Deda M., Veltri L., Chidichimo G., Nat. Commun. 2014, 5, 3105; [DOI] [PubMed] [Google Scholar]
- 9e. Dicka J. E., Poirel A., Ziessel R., Bard A. J., Electrochim. Acta 2015, 178, 234–239; [Google Scholar]
- 9f. Wu X., Wang W., Li B., Hou Y., Niu H., Zhang Y., Wang S., Bai X., Spectrochim. Acta Part A 2015, 140, 398–406; [DOI] [PubMed] [Google Scholar]
- 9g. Watanabe M., Chang Y. J., Chou P.-T., Staykov A., Shibahara M., Sako K., Ishihara T., Chow T. J., Tetrahedron Lett. 2015, 56, 1548–1551; [Google Scholar]
- 9h. García-Rodríguez A., Rodríguez A. M., Prieto P., Andreu R., Merino S., Rodríguez-López J., Eur. J. Org. Chem. 2015, 2394–2404; [Google Scholar]
- 9i. Raju M. V. N., Mohanty M. E., Ranjan Bangal P., Vaidya J. R., J. Phys. Chem. C 2015, 119, 8563–8575; [Google Scholar]
- 9j. Smith C. E., Odoh S. O., Ghosh S., Gagliardi L., Cramer C. J., Frisbie C. D., J. Am. Chem. Soc. 2015, 137, 15732–15741; [DOI] [PubMed] [Google Scholar]
- 9k. Cospito S., Beneduci A., Veltri L., Salamonczykc M., Chidichimo G., Phys. Chem. Chem. Phys. 2015, 17, 17670–17678; [DOI] [PubMed] [Google Scholar]
- 9l. Gadgil B., Damlin P., Dmitrieva E., Ääritalo T., Kvarnström C., RSC Adv. 2015, 5, 42242–42249; [Google Scholar]
- 9m. Goraa M., Pluczyk S., Zassowski P., Krzywiec W., Zagorska M., Mieczkowski J., Lapkowski M., Pron A., Synth. Met. 2016, 216, 75–82; [Google Scholar]
- 9n. Hayashi N., Saito Y., Zhou X., Yoshino J., Higuchi H., Mutai T., Tetrahedron 2016, 72, 4159–4168; [Google Scholar]
- 9o. Zhang Y., Steyrleuthner R., Bredas J.-L., J. Phys. Chem. C 2016, 120, 9671–9677; [Google Scholar]
- 9p. Maltese V., Cospito S., Beneduci A., Simone B. C. D., Russo N., Chidichimo G., Janssen R. A. J., Chem. Eur. J. 2016, 22, 10179–10186; [DOI] [PubMed] [Google Scholar]
- 9q. Barbosa C. G., Bento D. C., Péres L. O., Louarn G., de Santana H., J. Mater. Sci. Mater. Electron. 2016, 27, 10259–10269. [Google Scholar]
- 10.
- 10a. Reddy M. V. V. S., Celalyan-Berthier A., Geoffrey M., Morgantini P. Y., Weber J., Bernardinellis G., J. Am. Chem. Soc. 1988, 110, 2748–2753; [Google Scholar]
- 10b. Tormos G. V., Bakker M. G., Wang P., Lakshmikantham M. V., Cava M. P., Metzger R. M., J. Am. Chem. Soc. 1995, 117, 8528–8535; [Google Scholar]
- 10c. Tsai A., Berka V., Chen P., Palmer G., J. Biol. Chem. 1996, 271, 32563–32571; [DOI] [PubMed] [Google Scholar]
- 10d. McGibbon G. A., Hrušák J., Lavorato D. J., Schwarz H., Terlouw J. K., Chem. Eur. J. 1997, 3, 232–236; [DOI] [PubMed] [Google Scholar]
- 10e. Belkheiri N., Bouguerne B., Bedos-Belval F., Duran H., Berni C., Salvayre R., Nègre-Salvayre A., Baltas M., Eur. J. Med. Chem. 2010, 45, 3019–3026; [DOI] [PubMed] [Google Scholar]
- 10f. Huang P., Fu X., Liang Y., Zhang R., Dong D., Aust. J. Chem. 2012, 65, 121–128; [Google Scholar]
- 10g. Shkrob I. A., Marin T. W., Luo H., Dai S., J. Phys. Chem. B 2013, 117, 14372–14384; [DOI] [PubMed] [Google Scholar]
- 10h. Ling Y., Mierloo S. V., Schnegg A., Fehr M., Adriaensens P., Lutsen L., Vanderzande D., Maes W., Goovaertsa E., Doorslaer S. V., Phys. Chem. Chem. Phys. 2014, 16, 10032–10040. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Yamaguchi K., Murai T., Hasegawa S., Miwa Y., Kutsumizu S., Maruyama T., Sasamori T., Tokitoh N., J. Org. Chem. 2015, 80, 10742–10756; [DOI] [PubMed] [Google Scholar]
- 11b. Yamaguchi K., Murai T., Guo J.-D., Sasamori T., Tokitoh N., ChemistryOpen 2016, 5, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Lakowicz J. R., Principles of Fluorescence Spectroscopy , 3rd. ed., Springer, New York, 2006; [Google Scholar]
- 12b. Banerjee M., Lindeman S. V., Rathore R., J. Am. Chem. Soc. 2007, 129, 8070–8071; [DOI] [PubMed] [Google Scholar]
- 12c. Ookubo Y., Wakamiya A., Yorimitsu H., Osuka A., Chem. Eur. J. 2012, 18, 12690–12697; [DOI] [PubMed] [Google Scholar]
- 12d. Nandi A., Ghosh R., Palit D. K., J. Photochem. Photobio. A Chem. 2016, 321, 171–179; [Google Scholar]
- 12e. Sasaki S., Drummen G. P. C., Konishi G., J. Mater. Chem. C 2016, 4, 2731–2743. [Google Scholar]
- 13.For aminothiophenes as analogues of 5-aminothiazole 4, see:
- 13a. Sasaki S., Iyoda M., Chem. Lett. 1995, 1011–1012; [Google Scholar]
- 13b. Rapta P., Haubner K., Machata P., Lukeš V., Rosenkranz M., Schiemenz S., Klod S., Kivelä H., Kvarnström C., Hartmann H., Dunsch L., Electrochim. Acta 2013, 110, 670–680; [Google Scholar]
- 13c. Dessì A., Consiglio G. B., Calamante M., Reginato G., Mordini A., Peruzzini M., Taddei M., Sinicropi A., Parisi M. L., de Biani F. F., Basosi R., Mori R., Spatola M., Bruzzi M., Zani L., Eur. J. Org. Chem. 2013, 1916–1928. [Google Scholar]
- 14. Nishida T., Fukazawa A., Yamaguchi E., Oshima H., Yamaguchi S., Kanai M., Kuninobu Y., Chem. Asian J. 2014, 9, 1026–1030. [DOI] [PubMed] [Google Scholar]
- 15. Kayahara F., Kouyama T., Kato T., Takaya H., Yasuda N., Yamago S., Angew. Chem. Int. Ed. 2013, 52, 13722–13726; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13967–13971. [Google Scholar]
- 16.
- 16a. Bell F. A., Ledwith A., Sherrington D. C., J. Chem. Soc. C 1969, 2719–2720; [Google Scholar]
- 16b. Connelly N. G., Geiger W. E., Chem. Rev. 1996, 96, 877–910; [DOI] [PubMed] [Google Scholar]
- 16c. Dou H., Ni H., Zhao C., Zhang Y., J. Fluorine Chem. 2005, 126, 1130–1133; [Google Scholar]
- 16d. Dou H., Ni H., Zhao C., Zhang Y., Magn. Reson. Chem. 2006, 44, 515–520; [DOI] [PubMed] [Google Scholar]
- 16e. Yoshifuji M., Arduengo A. J., Konovalova T. A., Kispert L. D., Kikuchi M., Ito S., Chem. Lett. 2006, 35, 1136–1137; [DOI] [PubMed] [Google Scholar]
- 16f. Suzuki T., Tamaoki H., Katoono R., Fujiwara K., Ichikawa J., Fukushima T., Chem. Lett. 2013, 42, 703–705; [Google Scholar]
- 16g. Christensen M. A., Parker C. R., Sørensen T. J., de Graaf S., Morsing T. J., Brock-Nannestad T., Bendix J., Haley M. M., Rapta P., Danilov A., Kubatkin S., Hammerich O., Nielsen M. B., J. Mater. Chem. C 2014, 2, 10428–10438; [Google Scholar]
- 16h. Kuramoto Y., Matsui Y., Ohta E., Sato H., Ikeda H., Tetrahedron Lett. 2014, 55, 4366–4369; [Google Scholar]
- 16i. Davis C. M., Ohkubo K., Ho I., Zhang Z., Ishida M., Fang Y., Lynch V. M., Kadish K. M., Sessler J. L., Fukuzumi S., Chem. Commun. 2015, 51, 6757–6760; [DOI] [PubMed] [Google Scholar]
- 16j. Pirovano P., Farquhar E. R., Swart M., Fitzpatrick A. J., Morgan G. G., McDonald A. R., Chem. Eur. J. 2015, 21, 3785–3790; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16k. Satoh T., Minoura M., Nakano H., Furukawa K., Matano Y., Angew. Chem. Int. Ed. 2016, 55, 2235–2238; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 2275–2278. [Google Scholar]
- 17.For details on electrochemical oxidations, see:
- 17a. Zak J. K., Miyasaka M., Rajca S., Lapkowski M., Rajca A., J. Am. Chem. Soc. 2010, 132, 3246–3247; [DOI] [PubMed] [Google Scholar]
- 17b. Kagan E. S., Yanilkin V. V., Morozov V. I., Nastapova N. V., Zhukova I. Y., Kashparov I. I., Kashparov V. P., Russ. J. Gen. Chem. 2009, 79, 1001–1003. [Google Scholar]
- 18.Nitrogen hyperfine coupling constants increase with the number of electron-donor substituents, see: Improta R., Barone V., Chem. Rev. 2004, 104, 1231–1254. [DOI] [PubMed] [Google Scholar]
- 19.The EPR spectrum of TEMPO was measured in CH2Cl2 at 23 °C, [solute]=10−4 m The g (2.0062) and A N (1.59 mT) values were determined from spectral simulations. See Figure 5.
- 20. Yanai T., Tew D., Handy N., Chem. Phys. Lett. 2004, 393, 51–55. [Google Scholar]
- 21.
- 21a. Malkina O. L., Vaara J., Schimmelpfenning B., Munzarova M., Malkin V. G., Kaupp M., J. Am. Chem. Soc. 2000, 122, 9206–9218; [Google Scholar]
- 21b. Neese F., J. Chem. Phys. 2001, 115, 11080–11096. [Google Scholar]
- 22.We also used the well-defined basis set IGLO-III. However, the hyperfine coupling constant value for the N atom at the 5-position thus obtained was only 0.54 Gauss. For IGLO-III, see: Kutzelnigg W., Fleischer U., Schindler M., “The IGLO-Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities” in NMR Basic Principles and Progress Vol. 23: Deuterium and Shift Calculation, Springer, Heidelberg, 1990, . [Google Scholar]
- 23.NBO 6.0. E. D. Glendening, J, K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 The spin density maps and UV/Vis spectra for the species derived from 4+MB were also calculated (Figure S7). The calculated spectra were in good agreement with those in Figure S3.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary