

Abstract
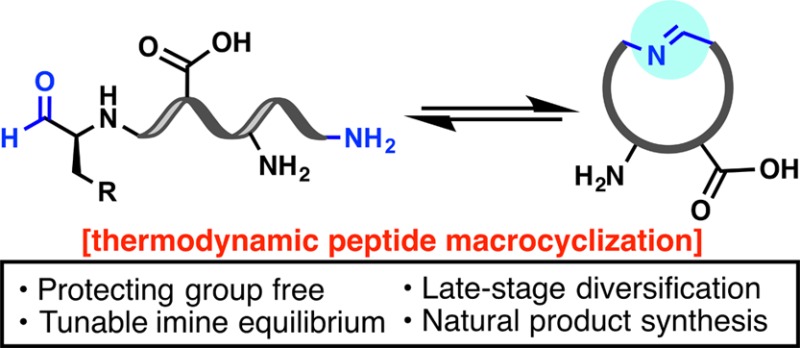
A thermodynamic approach to peptide macrocyclization inspired by the cyclization of non-ribosomal peptide aldehydes is presented. The method provides access to structurally diverse macrocycles by exploiting the reactivity of transient macrocyclic peptide imines toward inter- and intramolecular nucleophiles. Reactions are performed in aqueous media, in the absence of side chain protecting groups, and are tolerant of all proteinogenic functional groups. Macrocyclic products bearing non-native and rigidifying structural motifs, isotopic labels, and a variety of bioorthogonal handles are prepared, along with analogues of four distinct natural products. Structural interrogation of the linear and macrocyclic peptides using variable-temperature NMR and circular dichroism suggests that preorganization of linear substrates is not a prerequisite for macrocyclization.
Introduction
The quest to develop potent and selective new therapies has propelled pharmaceutical companies toward underexplored areas of chemical space, including the privileged intermediary between traditional small-molecule drugs (<500 Da) and high-value biologics (>5000 Da).1 The rise of protein–protein interactions (PPIs) as promising targets for therapeutic intervention has further contributed to the increasing molecular weight and complexity of drug leads, with medium-sized peptides and peptidomimetics emerging as able mimics of the protein surfaces and peptide epitopes involved in protein–protein binding.2 Nevertheless, most peptide leads suffer from low proteolytic stability and are historically poor drug candidates, rendering new synthetic strategies (e.g., macrocyclization, backbone and side chain modifications) aimed to enhance their druglike properties3 in exceedingly high demand in the pharmaceutical industry. As part of an ongoing collaboration with Bristol-Myers Squibb (BMS) on the development of PPI inhibitors, a synergistic academic–industrial collaboration4 was initiated to guide the development of a versatile peptide macrocyclization strategy. The goals were two-fold: (1) provide access to structurally diverse peptide products with immediate relevance to drug discovery, including access to non-natural and rigidifying motifs at the ring junction, and (2) facilitate macrocyclization through an unconventional, thermodynamic pathway. Such a method would complement other recent chemical strategies for peptide stapling and macrocyclization,5 including pioneering work by Yudin and co-workers on multicomponent peptide macrocyclizations.6
Inspiration for the design of a versatile macrocyclization could be found in the diversity of peptide macrocycles produced by the non-ribosomal peptide synthetase (NRPS) enzymatic machinery (Figure 1A).7 Following chain elongation on the non-ribosomal assembly line, release of the mature peptide from the enzyme complex can occur with concomitant macrocyclization catalyzed by a thioesterase domain to generate structurally complex macrolactones and macrolactams.8 Recently, head-to-tail macrocyclization through an intriguing imino linkage was shown to occur in certain bacteria following the reductive release of a peptide aldehyde mediated by a rare NAD(P)H-dependent reductase domain (Figure 1A).9 The nostocyclopeptide class of macrocyclic imines was the first group of natural products shown to be reductively cleaved from the NRPS9 and undergo spontaneous self-assembly.10 A number of additional imines—including scytonemide A,11 nostocyclopeptide M1,12 and koranimine13—have since been reported, with biosynthetic investigations into the latter suggesting the involvement of a reductive release mechanism. The biosynthesis of the promising antibiotic lugdunin,14 a thiazolidine macrocyclic peptide natural product isolated in 2016, also implicates a terminal reductase domain. Formation of the unique heterocyclic linkage is thought to occur via intramolecular trapping of a macrocyclic imine formed spontaneously upon reductive release from the NRPS complex.
Figure 1.
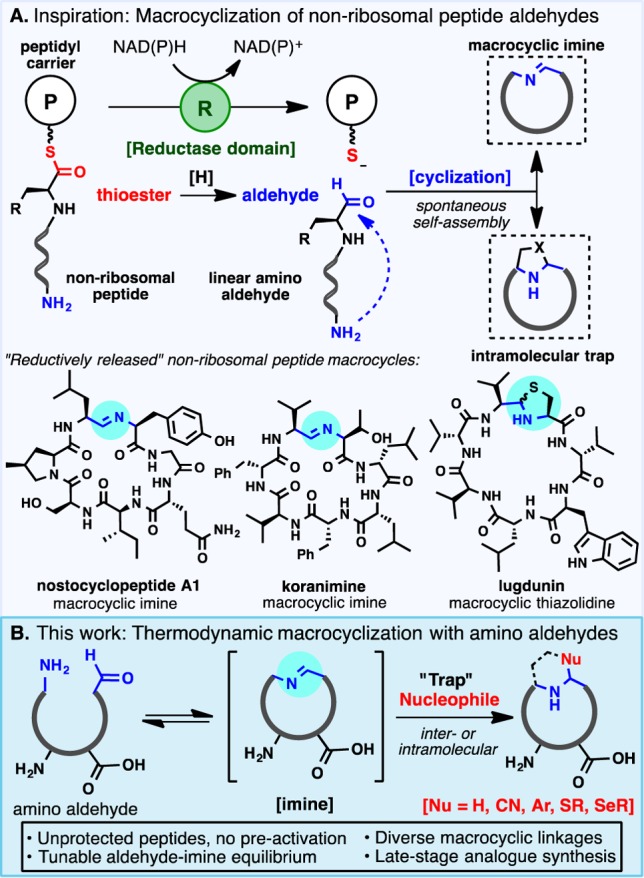
Peptide macrocyclization inspired by reductively released non-ribosomal peptide natural products.
These rare but structurally intriguing non-ribosomal peptide natural products prompted an exploration of imino macrocyclization as a unique and innately diversifiable mode of peptide cyclization. The formation of a reversible macrocyclic linkage sets the stage for a tunable, thermodynamic cyclization event, in contrast to reported modes of irreversible macrocyclization.15 Indeed, the reversible ring–chain equilibrium of imino macrocycles has previously been exploited by Marahiel and co-workers to gain a rare glimpse into the thermodynamics of macrocyclization.16 In the context of drug discovery, the trapping of a reactive imine intermediate with a variety of inter- or intramolecular nucleophiles would provide rapid access to structurally unique peptide macrocycles incorporating non-native motifs at the site of ring closure (Figure 1B). Due to the selective and reversible condensation of amines and aldehydes, diversification could proceed at a late stage with unprotected peptides, abrogating the need for preactivation and side-chain protection. Herein, this NRPS-inspired approach to macrocyclization is delineated along with applications to the preparation of diverse peptide macrocycles, including natural products and associated analogues. The solution structures of select linear and cyclic peptides are also probed using NMR techniques and circular dichroism, which demonstrate that macrocyclization efficiency is not predicated solely on substrate preorganization.
Synthesis of Linear Amino Aldehydes
Our investigations began with the synthesis of linear peptide amino aldehydes. A number of strategies exist for the preparation of peptide aldehydes,17 owing in part to their importance in bioconjugation18 and as potent protease inhibitors.19 Herein, two distinct solid-phase approaches20 were employed to assemble a variety of structurally diverse linear precursors (Scheme 1A,B; see the Supporting Information for additional details). The first method entailed incorporation of the key aldehyde unit by direct loading of commercially available aminoacetaldehyde dimethyl acetal onto the side chain of a resin-bound glutamic acid residue (Scheme 1A). Acidic cleavage from the resin facilitated global protecting group removal and release of a side-chain-appended glycinal unit following acetal hydrolysis. A second approach was employed to facilitate the synthesis of head-to-tail macrocycles and accommodate the incorporation of aldehydes bearing α-chirality. To this end, preformed Fmoc-amino aldehydes were prepared21 and loaded onto threonine–glycine (TG)-functionalized Rink amide resin (Scheme 1B). This resin linkage, first reported by Ede and Bray,22 relies on reversible oxazolidine formation between the aldehyde building block and the threonine 1,2-amino alcohol motif. The oxazolidine tether is stable to the reaction conditions employed in standard Fmoc-SPPS but is readily hydrolyzed upon treatment with aqueous acid to unveil a C-terminal peptide aldehyde. The assembled aldehyde sequences (28 examples in total) range from 5 to 10 amino acids in length and encompass all proteinogenic functional groups. To further increase the diversity of linear peptide amino aldehydes and limit the possibility of selecting substrates biased toward cyclization, a number of peptide sequences were designed using a random sequence generator (see the Supporting Information for details).
Scheme 1. Synthesis of Linear Peptide Amino Aldehydes: (A) Side Chain Incorporation of Aminoacetaldehyde Dimethyl Acetal and (B) Loading of Fmoc-Amino Aldehydes onto Rink TG Resin.

Intermolecular Imine Traps
Unlike the spontaneous cyclization implicated in the formation of the macrocyclic imine natural products, a variable propensity toward cyclization was observed for the synthetic amino aldehyde substrates. For some peptides, cyclization to the imine was observed directly upon LC–MS analysis of the cleaved peptide or upon incubation of the linear substrate in the presence of various additives (see the Supporting Information for details), while others persisted in linear form. Reasoning that a suitable imine trap might drive the equilibrium toward cyclization, the reactivity of the peptide substrates toward external nucleophiles was examined. Cyanide was quickly identified as a potent imine trap capable of facilitating efficient macrocyclization (Scheme 2A).
Scheme 2. Substrate Scope for Imine Macrocyclization Employing Intermolecular Imine Traps: (A) Strecker Macrocyclization, (B) Reductive Amination, and (C) Selective Cyclization of Substrates Bearing Internal Lysine Residues.

The resulting Strecker reaction proceeded in aqueous solution (1 mM in peptide) at rt with a slight excess of KCN (1.2 equiv) to afford macrocyclic α-aminonitriles23 (1–7) in good isolated yields. Interestingly, oligomerization products were not generally observed at 1 mM concentration—a possible consequence of reversible imine condensation, wherein linear oligomers may equilibrate to the target cyclic monomer prior to trapping. The reaction was tolerant of various functional groups—including carboxylic acids (3 and 5), primary amides, His imidazoles (5), phenols (6), and disulfides (7)—and proceeded in excellent yields with N-methylated derivatives (1 and 3), in contrast to conventional macrolactamization methods in which N-alkyl amines are often prohibitively sluggish. The enhanced reactivity for N-methylated peptides potentially lies in the formation of a highly reactive iminium intermediate upon reversible condensation of the N-methyl amine with the terminal aldehyde. Reactions typically afforded diastereomeric mixtures of α-aminonitriles that were inseparable by reverse-phase HPLC. In cases where quantification was possible, cyanide addition generally resulted in a 1:1 mixture of diastereomers, with the exception of disulfide-containing macrocycle 7 (3:1 dr). In this case, the steric bulk of the proximal StBu group may influence the approach of the nucleophile toward the cyclic imine. Substrates of various ring sizes (7–10 residues) and structural motifs were also amenable to cyclization, and a linear precursor bearing a randomly generated sequence of amino acids reacted smoothly to afford macrocycle 5 (86% yield). In some cases, slow macrocyclizations (e.g., 7) were accompanied by addition of cyanide to the open-chain aldehyde, affording a linear cyanohydrin. The inherent reversibility of this addition, however, allows gradual equilibration to the more stable cyclic α-aminonitrile. In contrast to conventional macrocyclizations, in which the yields of recalcitrant substrates are generally limited by the hydrolysis of an activated ester, this thermodynamic macrocyclization presents a viable approach to challenging cyclic targets. Importantly, the α-aminonitriles formed from the Strecker protocol were also amenable to further diversification, including nitrile hydrolysis to the corresponding primary amide (8).
Linear amino aldehydes were also efficiently trapped with NaBH3CN, establishing a robust reductive amination–macrocyclization protocol (Scheme 2B). Though previously explored for the purpose of peptide modification,24 reductive amination has not been widely probed as a means of peptide macrocyclization.25 We found that, in aqueous NaOAc buffer, reductive amination was efficient and highly tolerant of functional groups (9–18), leaving primary amides, disulfides (11 and 15), alcohols (13 and 14), acids (16), phenols (13 and 14), imidazoles (16), and sulfoxides (13) unscathed. Cyclic products ranging in size from 5 to 10 residues (15–32 atoms) were efficiently prepared. Although reactions were preferentially carried out at 1 mM concentration to minimize tendencies toward oligomerization, a thorough analysis of the effect of concentration on the cyclization yield of N-methylated peptide 10 revealed a tolerance for concentrations up to 10 mM (see the Supporting Information).
In another powerful example of the versatility of the reductive macrocyclization, we demonstrated the cyclization of the all-l-pentapeptide 17, which resisted conventional macrolactamization entirely at the same l-Leu/l-Pro junction.5a,26 On the basis of hydrogen-bonding networks identified using NMR techniques, the authors of the original study concluded that the linear substrate is not conformationally predisposed to cyclize at this junction,26 leading instead to exclusive hydrolysis of the activated C-terminal pentafluorophenyl ester. Although partial aldehyde reduction was observed herein as a consequence of reductive cyclization at the same conformationally disfavored junction, this competitive process did not preclude the desired cyclization pathway, with macrocycle 17 obtained in 36% isolated yield. In an effort to probe the influence of α-chirality on the cyclization of 17 and address the possibility of α-epimerization, a linear pentapeptide analogue bearing the corresponding C-terminal d-Leu aldehyde was also prepared. Cyclization of the d-variant occurred to form a distinct peptide product (18), indicating configurational stability in the macrocyclization event. Interestingly, the d-Leu variant cyclized rapidly and in higher yield than the l-Leu analogue, consistent with prior reports of improved cyclization efficiencies in linear peptides with configurationally mismatched termini.5a,27 These results highlight that while the reductive amination–macrocyclization is responsive to conformational bias, in this example, cyclic predisposition is not a strict prerequisite.
Probing the Impact of Internal Lysine Residues
The reversible aldehyde–imine equilibrium provides an intriguing platform for the evaluation of functional group specificity. In particular, the pKa difference between the peptide α-amine (pKa = 8.9) and the Lys ε-amine (pKa = 10.5) prompted an investigation into the possibility of α-amine selective cyclizations in the presence of unprotected Lys residues (Scheme 2C). Such selectivity is rare in base-mediated macrolactamization methods. Several linear amino aldehydes bearing unprotected Lys residues were prepared and cyclized using the Strecker (19 and 21) and reductive amination (22 and 24) macrocyclization methods. In each case, the major isolated cyclic products corresponded to α-amine-linked macrocycles (see the Supporting Information for NMR characterization). Importantly, when the side chain Lys group was masked with an Alloc protecting group (e.g., substrates 20 and 23), nearly identical yields of the cyclic products were obtained, suggesting that, in these cases, minor amounts of Lys-cyclized regioisomers were not removed during the course of HPLC purification. Although selectivity resulting from a conformational predisposition (rather than a difference in amine pKa) cannot be definitively excluded, the incorporation of two substrates with randomly generated amino acid sequences (21 and 24) and a detailed structural interrogation of the linear precursor to macrocycles 19 and 22 using NMR techniques and circular dichroism (vide infra) support the argument that a conformational predisposition of the linear precursor is not crucial to the observed selectivity.
Late-Stage Functionalization and Introduction of Bioorthogonal Handles
In addition to reliable functional group selectivity, the potential for late-stage substrate diversification is critical to the rapid development of new peptide-based drugs. From a single linear precursor, a variety of target macrocycles should ideally be accessible through targeted modification of the ring junction. Scheme 3 depicts a number of late-stage modifications facilitated by the imine macrocyclization protocol. Specifically, isotopic labeling for potential applications in metabolic studies was accomplished through Strecker macrocyclization with K13CN (Scheme 3A). Harnessing the synthetic power of native chemical ligation,28 mild and chemoselective acylation of 1,2-amino thiol-based macrocycles (e.g., 15) with peptide thioesters (e.g., S19) facilitates the rapid construction of complex peptide architectures (Scheme 3B). In addition, secondary amines formed through reductive amination can be N-acylated or N-alkylated with bioorthogonal handles, including biotin (27) or an alkyne moiety (28) (Scheme 3, parts C and D, respectively). Such tandem modifications enable rapid substrate diversification and have potential applications in bioconjugation studies.
Scheme 3. Late-Stage Peptide Diversification through the Introduction of Isotopic Labels and Bioorthogonal Handles.

Intramolecular Imine Traps
Analogous to the intramolecular trapping implicated in the biosynthesis of the macrocyclic natural product lugdunin (Figure 1), tethered nucleophiles were next evaluated as imine traps (Scheme 4). Aromatic rings, including indoles (29 and 31) and imidazoles (32), engaged in a Pictet–Spengler macrocyclization event29 to afford cyclic peptides with an embedded piperidine ring. Tethered thiol and selenol nucleophiles led to the corresponding thia- (33 and 35) and selenazolidines (36),30 the latter of which has garnered recent interest as a potential prodrug for selenocysteine.31 Following cyclization, these heterocyclic intermediates could be efficiently oxidized to introduce conformational rigidity as in the β-carboline (30) and thiazole variants (34). Incorporation of non-native, rigidifying motifs would typically require the synthesis of protected amino acid building blocks and laborious de novo peptide synthesis. The late-stage diversification herein is an attractive alternative to enable rapid analogue synthesis.
Scheme 4. Substrate Scope for Imine Macrocyclization Employing Intramolecular Imine Traps: (A) Pictet–Spengler Cyclization and (B) Thia-/Selenazolidine Macrocyclization.

Synthesis of Natural Products and Cyclic Analogues
In a final display of the versatility of the macrocyclization, late-stage diversification of non-ribosomal imine macrocycles to afford the natural products and associated analogues of scytonemide A (37),11 koranimine (38),13 and lugdunin (39 and 40)14 (Scheme 5) was pursued. Interestingly, the propensity of these cyclic imino natural products to spontaneously cyclize upon resin cleavage or LC–MS analysis was highly substrate dependent. The linear precursor to scytonemide A did not self-assemble, but a gradual titration of an amine base in organic solvent allowed for conversion to the cyclic imine (see the Supporting Information for NMR studies). In aqueous solution, the transient imino linkage could be effectively trapped with a hydride to afford macrocycle 37 in 66% isolated yield.
Scheme 5. Synthesis of Natural Products and Structural Analogues from Linear Amino Aldehyde Precursors.

Koranimine was observed as an equilibrium mixture of linear and cyclic forms following resin cleavage. Curiously, 1H NMR analysis of the peptide did not reveal the characteristic imine peak (∼6.8 ppm), an observation echoed in the original isolation paper.13 This behavior prompted a hypothesis that the N-terminal threonine residue may engage the imine intramolecularly to form the corresponding oxazolidine linkage at the ring junction. Such intramolecular trapping is supported by preliminary NMR data (see the Supporting Information). Nevertheless, the presence of macrocyclic imine as a contributor to the equilibrium mixture is evidenced by successful reduction to afford macrocycle 38 in excellent isolated yield.
The natural product lugdunin (39), isolated from the human-associated bacteria Staphylococcus lugdunensis in 2016 and the first example of a cyclic peptide thiazolidine natural product,14 exhibited a strong preference for spontaneous cyclization upon resin cleavage. An oxo-analogue of lugdunin (40) bearing an N-terminal threonine residue was isolated as an interconverting mixture of macrocyclic oxazolidines and the corresponding linear amino aldehyde. In this case, macrocyclization occurred at the analogous l-Val/l-Thr ring junction present in koranimine, lending further credence to the hypothesis that spontaneous oxazolidine formation may be a contributing mode of macrocyclization in non-ribosomal imine natural products.
In addition to the non-ribosomal natural products described above, sanguinamide A (43),32 a ribosomally synthesized and post-translationally modified peptide natural product, was also prepared. The key thiazole moiety was constructed using a thiazolidine cyclization–oxidation sequence. Interestingly, thermodynamic cyclization of the sanguinamide linear precursor as well as a seleno-analogue resulted in epimerization at the α-Ile aldehyde (41 and 42); such configurational instability at the α-position was not observed upon functionalization of the non-ribosomal imine natural products, a possible consequence of the thermodynamic self-assembly10 implicated in the biosynthesis of these molecules. Nevertheless, oxidation of the presanguinamide diastereomeric thiazolidine mixture (41) kinetically favored the native α-configuration, thus affording sanguinamide A (43) in 16% overall yield from the original resin loading.33
Structural Interrogation of Linear and Cyclic Peptides
With an aim to investigate the effect of peptide structure on the efficiency of macrocyclization, select amino aldehyde substrates and linear precursors to the non-ribosomal natural products were interrogated using variable-temperature NMR and circular dichroism (CD). Peptides were first dissolved in aqueous acetonitrile and analyzed using 1H and TOCSY NMR at 282, 291, 300, and 309 K (see the Supporting Information for details). The shift of each N–H bond as a function of temperature was evaluated and correlated to the extent of engagement in hydrogen-bonding interactions (Figure 2). On the basis of this analysis, linear peptide substrates S2 and S13 (which bears an internal Lys residue) did not exhibit any evidence of internal hydrogen bonding (Figure 2A). The N-terminal Trp peptide S20 exhibited shielding at Phe4, possibly resulting from an interaction with the Trp indole moiety. Importantly, the success of these substrates in imine macrocyclization chemistry was not strongly correlated to the presence of hydrogen-bonding interactions, with comparable yields for macrocyclization (54%, 57%, and 75% for the corresponding macrocyclic peptides 9, 22, and 29, respectively) obtained in all cases. Although this analysis does not rule out the influence of hydrophobic effects on cyclization efficiency, a qualitative link can be established between hydrogen-bonding interactions and the degree of structural organization, with macrocyclic products generally exhibiting smaller N–H shifts in response to temperature variation than their more flexible linear precursors. This observation was particularly evident for Lys-containing macrocycle 22, which exhibited a strong hydrogen-bonding interaction at Val5 as a result of cyclization.
Figure 2.

Structural interrogation of linear and cyclic peptides. Variable-temperature NMR studies on (A) model aldehydes and associated macrocycles and (B) natural products and linear precursors. (C) Circular dichroism studies.
In contrast to the generally disordered linear substrates examined in Figure 2A, the precursors to the imine natural products exhibited varying degrees of structural organization (Figure 2B). The variable-temperature NMR data agree with qualitative observations about the propensity of the linear natural product precursors to cyclize upon resin cleavage (vide supra). For example, linear scytonemide A (S26), experimentally characterized as the least prone toward cyclization, showed minimal hydrogen-bonding interactions. In contrast, linear koranimine (S27) engaged in a number of hydrogen-bonding interactions, and lugdunin, which cyclized spontaneously upon resin cleavage, had the most extensive hydrogen-bonding network. In the non-ribosomal peptides examined, a higher degree of sequence hydrophobicity (e.g., the presence of numerous Phe, Leu, and Val residues, as in peptides S27 and 39) tended to correlate to an increased number of observed hydrogen-bonding interactions, suggesting that both hydrophobic effects and hydrogen bonding may contribute to the spontaneous cyclization of imine natural products. Nevertheless, the macrocyclization efficiency of linear substrates S26 and S27 was independent of their disparate structural properties and amino acid sequences; similar yields were obtained for the reductive amination of both substrates (S26 to 37, 66% and S27 to 38, 71%), and the cyclization efficiency was comparable to that of the model peptides examined in Figure 2A. Notably, the natural-product-derived macrocycles 37 and 38 also displayed a higher degree of intramolecular hydrogen bonding than their linear precursors.
Variable-temperature NMR data was further supported by structural information obtained from CD analysis of the linear peptides (Figure 2C). Devoid of persistent hydrogen-bonding interactions, model aldehydes S2, S13, and scytonemide A aldehyde (S26) afforded spectra consistent with a random coil structure. In contrast, aldehyde S27 and lugdunin (39) exhibited more complex structural elements associated with ordered peptides. In the context of macrocyclization, these findings collectively highlight the versatility of imine macrocyclization, whereby the accessibility of a cyclic architecture is not inextricably wedded to a conformational preference in the linear substrate. Indeed, trapping of a minor component of a complex equilibrium mixture may be sufficient to drive a high-yielding macrocyclization reaction. Further studies probing the effect of structural and conformational elements on cyclization efficiency, including the role of sequence hydrophobicity, are currently underway.
Conclusions
This paper delineates a general strategy for macrocyclization inspired by the self-assembly of cyclic imines from linear, non-ribosomal peptide aldehydes. By exploiting the innate reactivity of imines toward inter- and intramolecular nucleophiles, a diverse array of macrocyclic peptides and natural products was obtained from unprotected amino aldehyde precursors. Characterized by operational simplicity, the reactions proceed in aqueous media and are tolerant of all proteinogenic functional groups. Importantly, the spontaneous, reversible condensation of amines and aldehydes also distances this thermodynamic macrocyclization approach from traditional methods reliant on activated C-terminal esters, thereby eliminating competitive hydrolysis as a limiting factor in the cyclization of difficult or non-predisposed peptide sequences. The generality of the approach is further supported by detailed NMR and CD experiments, which suggest that the efficiency of imino macrocyclization is largely independent of a distinct conformational predisposition in the linear substrate.
As with all synthetic methods, however, this macrocyclization protocol is not without limitations. Reactions require relatively high dilution to avoid oligomerization and care must be taken when handling amino aldehydes, as thermodynamic equilibration may result in erosion of α-chirality. Photographic guides, frequently encountered issues, and other useful information are described in the Supporting Information. Despite the aforementioned limitations, NRPS-inspired imine macrocyclization provides a unique and inherently diversifiable approach to the macrocyclization of unprotected linear precursors.34 Application of this methodology is already underway at BMS for the design of potent new PPI inhibitors. By expanding the scope of readily accessible macrocycles, it is anticipated that this approach will have a tangible impact on drug discovery.
Acknowledgments
Financial support for this work was provided by Bristol-Myers Squibb, NIH (F32GM117816 postdoctoral fellowship to L.R.M. and GM-118176), and NSF GRFP (J.N.D.). We thank Dr. David Langley and Dr. Claudio Mapelli (BMS) for helpful discussions and Dr. Brad Maxwell (BMS) for a sample of K13CN. We are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (The Scripps Research Institute) for assistance with nuclear magnetic resonance (NMR) spectroscopy.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01624.
Detailed experimental procedures and analytical data for linear and cyclic peptides (PDF)
Author Contributions
∥ L.R.M. and J.N.D. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. Chem. Biol. Drug Des. 2013, 81, 136. 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- a Wojcik P.; Berlicki L. Bioorg. Med. Chem. Lett. 2016, 26, 707. 10.1016/j.bmcl.2015.12.084. [DOI] [PubMed] [Google Scholar]; b Pelay-Gimeno M.; Glas A.; Koch O.; Grossmann T. N. Angew. Chem., Int. Ed. 2015, 54, 8896. 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Scott D. E.; Bayly A. R.; Abell C.; Skidmore J. Nat. Rev. Drug Discovery 2016, 15, 533. 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- a Di L. AAPS J. 2015, 17, 134. 10.1208/s12248-014-9687-3. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Adessi C.; Soto C. Curr. Med. Chem. 2002, 9, 963. 10.2174/0929867024606731. [DOI] [PubMed] [Google Scholar]
- Michaudel Q.; Ishihara Y.; Baran P. S. Acc. Chem. Res. 2015, 48, 712. 10.1021/ar500424a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a White C. J.; Yudin A. K. Nat. Chem. 2011, 3, 509. 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]; b Spokoyny A. M.; Zou Y.; Ling J. J.; Yu H.; Lin Y. S.; Pentelute B. L. J. Am. Chem. Soc. 2013, 135, 5946. 10.1021/ja400119t. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lautrette G.; Touti F.; Lee H. G.; Dai P.; Pentelute B. L. J. Am. Chem. Soc. 2016, 138, 8340. 10.1021/jacs.6b03757. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brown S. P.; Smith A. B. III J. Am. Chem. Soc. 2015, 137, 4034. 10.1021/ja512880g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Assem N.; Ferreira D. J.; Wolan D. W.; Dawson P. E. Angew. Chem., Int. Ed. 2015, 54, 8665. 10.1002/anie.201502607. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Mendive-Tapia L.; Preciado S.; Garcia J.; Ramon R.; Kielland N.; Albericio F.; Lavilla R. Nat. Commun. 2015, 6, 7160. 10.1038/ncomms8160. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Yudin A. K. Chem. Sci. 2015, 6, 30. 10.1039/C4SC03089C. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Rohrbacher F.; Deniau G.; Luther A.; Bode J. W. Chem. Sci. 2015, 6, 4889. 10.1039/C5SC01774B. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Lawson K. V.; Rose T. E.; Harran P. G. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, E3753. 10.1073/pnas.1311706110. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Lau Y. H.; de Andrade P.; Wu Y.; Spring D. R. Chem. Soc. Rev. 2015, 44, 91. 10.1039/C4CS00246F. [DOI] [PubMed] [Google Scholar]; k Noisier A. F.; Garcia J.; Ionut I. A.; Albericio F. Angew. Chem., Int. Ed. 2017, 56, 314. 10.1002/anie.201608648. [DOI] [PubMed] [Google Scholar]; l Zhang J.; Mulumba M.; Ong H.; Lubell W. D. Angew. Chem., Int. Ed. 2017, 10.1002/anie.201611685. [DOI] [PubMed] [Google Scholar]
- a Hili R.; Rai V.; Yudin A. K. J. Am. Chem. Soc. 2010, 132, 2889. 10.1021/ja910544p. [DOI] [PubMed] [Google Scholar]; b Frost J. R.; Scully C. C.; Yudin A. K. Nat. Chem. 2016, 8, 1105. 10.1038/nchem.2636. [DOI] [PubMed] [Google Scholar]
- Sieber S. A.; Marahiel M. A. J. Bacteriol. 2003, 185, 7036. 10.1128/JB.185.24.7036-7043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli R. M.; Walsh C. T. Chem. Commun. 2003, 297. 10.1039/b208333g. [DOI] [PubMed] [Google Scholar]
- Becker J. E.; Moore R. E.; Moore B. S. Gene 2004, 325, 35. 10.1016/j.gene.2003.09.034. [DOI] [PubMed] [Google Scholar]
- Kopp F.; Mahlert C.; Grunewald J.; Marahiel M. A. J. Am. Chem. Soc. 2006, 128, 16478. 10.1021/ja0667458. [DOI] [PubMed] [Google Scholar]
- Krunic A.; Vallat A.; Mo S.; Lantvit D. D.; Swanson S. M.; Orjala J. J. Nat. Prod. 2010, 73, 1927. 10.1021/np100600z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokela J.; Herfindal L.; Wahlsten M.; Permi P.; Selheim F.; Vasconcelos V.; Doskeland S. O.; Sivonen K. ChemBioChem 2010, 11, 1594. 10.1002/cbic.201000179. [DOI] [PubMed] [Google Scholar]
- Evans B. S.; Ntai I.; Chen Y.; Robinson S. J.; Kelleher N. L. J. Am. Chem. Soc. 2011, 133, 7316. 10.1021/ja2015795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipperer A.; Konnerth M. C.; Laux C.; Berscheid A.; Janek D.; Weidenmaier C.; Burian M.; Schilling N. A.; Slavetinsky C.; Marschal M.; Willmann M.; Kalbacher H.; Schittek B.; Brotz-Oesterhelt H.; Grond S.; Peschel A.; Krismer B. Nature 2016, 535, 511. 10.1038/nature18634. [DOI] [PubMed] [Google Scholar]
- In addition to refs (5) and (6), various methods for “Click” macrocyclization have been developed:; a van Maarseveen J. H.; Horne W. S.; Ghadiri M. R. Org. Lett. 2005, 7, 4503. 10.1021/ol0518028. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Turner R. A.; Oliver A. G.; Lokey R. S. Org. Lett. 2007, 9, 5011. 10.1021/ol702228u. [DOI] [PubMed] [Google Scholar]; c Aimetti A. A.; Shoemaker R. K.; Lin C. C.; Anseth K. S. Chem. Commun. 2010, 46, 4061. 10.1039/c001375g. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Chouhan G.; James K. Org. Lett. 2011, 13, 2754. 10.1021/ol200861f. [DOI] [PubMed] [Google Scholar]
- a Enck S.; Kopp F.; Marahiel M. A.; Geyer A. ChemBioChem 2008, 9, 2597. 10.1002/cbic.200800314. [DOI] [PubMed] [Google Scholar]; b Enck S.; Kopp F.; Marahiel M. A.; Geyer A. Org. Biomol. Chem. 2010, 8, 559. 10.1039/B917549K. [DOI] [PubMed] [Google Scholar]
- For a comprehensive overview of synthetic strategies toward peptide aldehydes, see the following:; a Moulin A.; Martinez J.; Fehrentz J. A. J. Pept. Sci. 2007, 13, 1. 10.1002/psc.787. [DOI] [PubMed] [Google Scholar]; b Melnyk O.; Fehrentz J. A.; Martinez J.; Gras-Masse H. Biopolymers 2000, 55, 165.. [DOI] [PubMed] [Google Scholar]
- El-Mahdi O.; Melnyk O. Bioconjugate Chem. 2013, 24, 735. 10.1021/bc300516f. [DOI] [PubMed] [Google Scholar]
- Lee D. H.; Goldberg A. L. Trends Cell Biol. 1998, 8, 397. 10.1016/S0962-8924(98)01346-4. [DOI] [PubMed] [Google Scholar]
- For a review on solid-phase approaches to C-terminally modified peptides, see the following:; a Alsina J.; Albericio F. Biopolymers 2003, 71, 454. 10.1002/bip.10492. [DOI] [PubMed] [Google Scholar]; For an innovative application of protected α-amino aldehydes on the solid-phase, see the following:; b Myers A. G.; Lanman B. A. J. Am. Chem. Soc. 2002, 124, 12969. 10.1021/ja027729n. [DOI] [PubMed] [Google Scholar]
- Wang G.; Mahesh U.; Chen G. Y.; Yao S. Q. Org. Lett. 2003, 5, 737. 10.1021/ol0275567. [DOI] [PubMed] [Google Scholar]
- Ede N. J.; Bray A. M. Tetrahedron Lett. 1997, 38, 7119. 10.1016/S0040-4039(97)01635-3. [DOI] [Google Scholar]
- For an example of an α-aminonitrile macrodiolide, see the following:Ma J.; Vannam R.; Terwilliger D. W.; Peczuh M. W. Tetrahedron Lett. 2014, 55, 4255. 10.1016/j.tetlet.2014.05.081. [DOI] [Google Scholar]
- a McFarland J. M.; Francis M. B. J. Am. Chem. Soc. 2005, 127, 13490. 10.1021/ja054686c. [DOI] [PubMed] [Google Scholar]; b Diaz D. B.; Scully C. C. G.; Liew S. K.; Adachi S.; Trinchera P.; St. Denis J. D.; Yudin A. K. Angew. Chem., Int. Ed. 2016, 55, 12659. 10.1002/anie.201605754. [DOI] [PubMed] [Google Scholar]
- For a macrocyclic reductive amination on peptidic ketones, see the following:; a Guéret S. M.; Meier P.; Roth H.-J. Org. Lett. 2014, 16, 1502. 10.1021/ol5003797. [DOI] [PubMed] [Google Scholar]; The reduction of cyclic iminoboronates has recently been applied to peptide cyclization:; b Bandyopadhyay A.; Gao J. J. Am. Chem. Soc. 2016, 138, 2098. 10.1021/jacs.5b12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U.; Langner J. J. Pept. Res. 1997, 49, 67. 10.1111/j.1399-3011.1997.tb01122.x. [DOI] [PubMed] [Google Scholar]
- a Brady S. F.; Varga S. L.; Freidinger R. M.; Schwenk D. A.; Mendlowski M.; Holly F. W.; Veber D. F. J. Org. Chem. 1979, 44, 3101. 10.1021/jo01332a003. [DOI] [Google Scholar]; b Ehrlich A.; Heyne H. U.; Winter R.; Beyermann M.; Haber H.; Carpino L. A.; Bienert M. J. Org. Chem. 1996, 61, 8831. 10.1021/jo951108d. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Muir T. W.; Clark-Lewis I.; Kent S. B. H. Science 1994, 266, 776. 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- Li X.; Zhang L.; Hall S. E.; Tam J. P. Tetrahedron Lett. 2000, 41, 4069. 10.1016/S0040-4039(00)00592-X. [DOI] [Google Scholar]
- Botti P.; Pallin T. D.; Tam J. P. J. Am. Chem. Soc. 1996, 118, 10018. 10.1021/ja954278g. [DOI] [Google Scholar]
- Short M. D.; Xie Y.; Li L.; Cassidy P. B.; Roberts J. C. J. Med. Chem. 2003, 46, 3308. 10.1021/jm020496q. [DOI] [PubMed] [Google Scholar]
- a Nielsen D. S.; Hoang H. N.; Lohman R. J.; Diness F.; Fairlie D. P. Org. Lett. 2012, 14, 5720. 10.1021/ol3027347. [DOI] [PubMed] [Google Scholar]; b Dalisay D. S.; Rogers E. W.; Edison A. S.; Molinski T. F. J. Nat. Prod. 2009, 72, 732. 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinamide A was previously prepared (see ref (32a)) in 10% overall yield. The thiazole was incorporated as a dipeptide surrogate.
- Young I. S.; Baran P. S. Nat. Chem. 2009, 1, 193. 10.1038/nchem.216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.