

Abstract
Nα-Acetyltransferase 60 (Naa60 or NatF) was recently identified as an unconventional N-terminal acetyltransferase (NAT) because it localizes to organelles, in particular the Golgi apparatus, and has a preference for acetylating N termini of the transmembrane proteins. This knowledge challenged the prevailing view of N-terminal acetylation as a co-translational ribosome-associated process and suggested a new mechanistic functioning for the enzymes responsible for this increasingly recognized protein modification. Crystallography studies on Naa60 were unable to resolve the C-terminal tail of Naa60, which is responsible for the organellar localization. Here, we combined modeling, in vitro assays, and cellular localization studies to investigate the secondary structure and membrane interacting capacity of Naa60. The results show that Naa60 is a peripheral membrane protein. Two amphipathic helices within the Naa60 C terminus bind the membrane directly in a parallel position relative to the lipid bilayer via hydrophobic and electrostatic interactions. A peptide corresponding to the C terminus was unstructured in solution and only folded into an α-helical conformation in the presence of liposomes. Computational modeling and cellular mutational analysis revealed the hydrophobic face of two α-helices to be critical for membranous localization. Furthermore, we found a strong and specific binding preference of Naa60 toward membranes containing the phosphatidylinositol PI(4)P, thus possibly explaining the primary residency of Naa60 at the PI(4)P-rich Golgi. In conclusion, we have defined the mode of cytosolic Naa60 anchoring to the Golgi apparatus, most likely occurring post-translationally and specifically facilitating post-translational N-terminal acetylation of many transmembrane proteins.
Keywords: acetylation, acetyltransferase, Golgi, lipid binding protein, membrane enzyme, N-terminal acetylation, NAT, Naa60, NatF, PI(4)P
Introduction
Nα-Acetyltransferase 60 (Naa60)3 belongs to a family of enzymes known as N-terminal acetyltransferases (NATs) (1). These modify protein N termini by catalyzing the transfer of an acetyl group onto the α-amino group of the protein backbone. Six human NATs are known to date and these are alphabetically named NatA to NatF according to acetyltransferase activity type (reviewed in Refs. 2 and 3). NAT activity is mediated by the catalytic Nα-acetyltransferase (Naa) subunits Naa10 to Naa60. Most of these join one or two auxiliary, often ribosome binding (4, 5), subunits to make up a NAT complex. In the case of Naa60, however, no auxiliary subunits seem necessary for NatF activity (1). Naa60 and the other catalytic Nα-acetyltransferases (Naas) share an evolutionarily conserved structural feature, a GNAT (GCN5 (general control nonderepressed 5-related N-acetyltransferase)-domain and belong to the GNAT superfamily (6, 7). Most Naas are strongly conserved in eukaryotes, however, fungi, including Saccharomyces cerevisiae, lack Naa60, which was secondarily lost during evolution (8).
N-terminal acetylation is mainly considered as a co-translational protein modification. It is highly abundant among eukaryotes, occurring on a majority of all protein species (9–12), and the acetylated N terminus has diverse effects for the substrate proteins (reviewed in Ref. 2). The N-terminal acetyl group may control protein interactions (13–15), localization/targeting (16–18), folding (19), or act as a degradation signal in a branch of the N-end rule pathway (20–22). The latter was recently linked to hypertension in humans (23, 24). Additional evidence for the essentiality of N-terminal acetylation in humans is its involvement in pathologies such as cancer, neurodegenerative disease, and genetic diseases like the lethal Ogden syndrome (3, 25–27). A physiological role of Naa60 has thus far not been established, however, depletion studies demonstrate its importance in maintaining the structural integrity of the Golgi apparatus in human cells (12), as well as for proper chromatid segregation during anaphase of Drosophila melanogaster cells (1).
Naa60 is the most recently identified enzyme among the human Nα-acetyltransferases (1), and interestingly it was characterized as the first organellar NAT as it primarily localizes to the Golgi (12). Another organellar NAT, Naa70/NatG, was very recently identified in plants as the first chloroplast NAT (28). These NATs challenge the prevailing view of N-terminal acetylation as a co-translational ribosome-associated process.
Although established as a membrane-localizing enzyme (12), the means by which Naa60 attaches to membranes is yet unresolved. Previous work placed Naa60 on the cytosolic side and also demonstrated it to N terminally acetylate transmembrane proteins with their N terminus facing the cytosol (12). Importantly, the C-terminal tail of Naa60 (amino acids 182–242), which constitutes an extra segment, not present in the other catalytic Naas, was defined as the part providing organellar localization. A recent crystal structure of Naa60 exposed the molecular determinants for substrate-specific acetylation and revealed that Naa60 employs a catalytic mechanism close to that of Naa50 (29). However, this structure does not contain the localization-determining C-terminal tail because it was necessary to omit this part to obtain purified Naa60 amenable to structural analysis (29). Another crystallization study of Naa60 met similar challenges and the resulting structure only shows amino acids 1–211, although the full-length protein (242 amino acids) was used (30). This structure shows an amphipathic α-helix (aa 190–202) that could conceivably be involved in membrane binding.
Here, we studied the localization-determining C-terminal region of Naa60 and defined the molecular determinants for the interaction of Naa60 with the membrane by means of computational modeling, in vitro liposome assays including intrinsic tryptophan fluorescence, circular dichroism, and isothermal titration calorimetry (ITC), and mutational analysis combined with localization determination in cellulo.
Results
Secondary structure predictions and implicit membrane model simulations suggest two putative membrane-binding regions in the C terminus of Naa60
We previously reported Naa60 to be a Golgi residing enzyme, and showed by truncation mutants that the last 61 amino acids (aa 182–242) of Naa60 were both necessary and sufficient to provide its organellar localization (12). We here expanded this work by further investigating how this C-terminal segment might interact with membranes and initially identified two α-helices that were predicted by PSIPRED (31), herein referred to as Pred-α1 and Pred-α2 (Fig. 1A), of which the former is in agreement with the Naa60-(1–211) crystal structure (30). Plotting helical wheels for both these predicted helices revealed their amphipathic character (Fig. 1B). The hydrophobic face of Pred-α1 consists of residues Ile190, Leu191, Tyr193, Ile194, Leu197, and Leu201 (Fig. 1B, left). The hydrophobic face of Pred-α2 consists of Val213, Tyr214, Leu220, Leu221, and Phe224 (Fig. 1B, I). Pred-α2 contains fewer aliphatic side chains (Ile, Leu) than Pred-α1.
Figure 1.

Secondary structure predictions, simulations, and initial experiments suggest two C-terminal α-helices to mediate Naa60 membrane interactions. A, sequence overview of Naa60 full-length (upper) with a detailed view of the C-terminal region of amino acids 182–242 (lower). The PSIPRED predicted α-helices, herein named “Pred-α1” (aa 190–202) and “Pred-α2” (aa 211–224) are indicated in blue. B, helical wheels for each of Pred-α1 and Pred-α2. The arrow and number inside each wheel correspond to the direction and magnitude of the helices hydrophobic moment, respectively. C and D, structures resulting from the implicit membrane simulations of the Naa60-(185–227) and Naa60-(185–242), respectively, seen from inside the membrane. The plane delimiting the lipid tails from the polar head region of the membrane is light gray, transparent and parallel to the page. The amino acids appearing clearly are those anchored in the hydrophobic region of the membrane model, whereas the other ones appearing with fading colors are either in the head group region or further away from the membrane. The color code for amino acids matches the one in B, but with fewer shades. E, contribution of each amino acid of the Naa60 C terminus (185–242) to the binding energy calculated with the IMM1 membrane model (ΔWAliph, ΔWarom and ΔWpolar, see ”Experimental procedures“). Yellow asterisks indicate those residues that constitute the hydrophobic face of Pred-α1 and Pred-α2 shown in B. F, localization of hNaa60-eGFP and hNaa60-(1–182)-eGFP ectopically expressed in S. cerevisiae. Scale bar, 2 μm.
Furthermore, we built a three-dimensional structure of the Naa60-(185–242) using I-TASSER and performed molecular dynamics (MD) simulations of two distinct systems: the whole C terminus (Naa60-(185–242)) and of only its predicted helical region (Naa60-(185–227)) in the presence of an implicit membrane model (IMM1). IMM1 provides an estimate of the solvation/desolvation energetic costs of inserting the peptide at the membrane interface (see ”Experimental procedures“). Resulting orientations of Naa60-(185–227) and Naa60-(185–242) are shown on Fig. 1, C and D, respectively. Simulations of Naa60-(185–242) result in both helices interacting with the membrane and inserting their aliphatic side chains into the hydrophobic region. This is in agreement with the amphipathic character of the predicted helices. Because of its higher number of aliphatic side chains Pred-α1 is more deeply anchored than Pred-α2, especially on its N-terminal end (Leu190, Ile191, and Ile194). The binding energies obtained for Naa60-(185–242) are clearly favorable (−19.08 to −12.74 kcal/mol depending on the starting orientation) showing that hydrophobic effect contributes significantly to membrane binding. Interestingly, our simulations indicate that the last 17 residues influence the binding of the helices as the truncated structure yields a shallower membrane binding of Pred-α2 (Fig. 1C) compared with the results of the full C terminus. This is also evident from the less favorable binding energies of the truncated structure (−13.59 to −5.59 kcal/mol). The contributions of amino acids 185 to 227 to the binding energy of the whole C terminus are plotted on Fig. 1E. All amino acids predicted to be on the hydrophobic faces of the helices yield favorable contribution to the binding energy, except Tyr193 and Tyr214. In addition, Trp227 has a small but favorable contribution in binding energy.
To provide a hint regarding the conservation of the membrane targeting mechanism of Naa60, we expressed the eGFP-tagged protein in S. cerevisiae. Human Naa60-eGFP displayed an organellar localization in yeast and this localization pattern was lost and replaced by a cytosolic signal upon truncation of the last 61 amino acids (Fig. 1F). This observation is in accordance with previous work in mammalian cells (12) and demonstrates that Naa60 uses a membrane targeting mechanism that is also functional in fungi, although Naa60 is not found in this kingdom (8).
Naa60-(189–242) peptide adopts helical secondary structure upon membrane interactions and interacts with Golgi-like liposomes through both hydrophobic and electrostatic interactions
The predicted amphipathic helices above, combined with the ability of human Naa60 to adopt membrane localization in yeast, suggested that the Naa60 C terminus might interact directly with intracellular membranes. To study the potential capability of Naa60 to interact with lipid bilayers, we used a Naa60 C-terminal peptide comprising amino acids 189–242 (Naa60-(189–242)) for in vitro liposome assays measuring intrinsic tryptophan fluorescence. The peptide contains only one tryptophan residue (Trp227) (Fig. 1A). After incubation of the Naa60-(189–242) peptide with Golgi-like liposomes composed of 52% phosphatidylcholine (PC), 20% phosphatidylethanolamine (PE), 15% cholesterol (CHOL), 8% phosphatidylinositol 4-phosphate (PI(4)P), and 5% phosphatidylserine (PS) (32), a clear blue shift from 355 to 348 nm was observed (Fig. 2A), hence indicating an interaction between Naa60-(189–242) and these liposomes, in agreement with simulations in Fig. 1D in which Trp227 dips into the membrane. Furthermore, this suggests that Naa60-(189–242) has intrinsic properties mediating interaction with liposomes, hence membrane binding appears to be independent of other factor(s), like protein interaction or lipid modification and thus these data support the presence of helical structure(s) shown in Fig. 1. To investigate the secondary structure of the Naa60 C terminus, we performed a far-UV circular dichroism (CD) study of Naa60-(189–242) in the presence or absence of Golgi-like liposomes. As seen in Fig. 2B, Naa60-(189–242) was unstructured in solution, but adopted a helical secondary structure in the presence of Golgi-like liposomes.
Figure 2.

Naa60-(189–242) incorporates in the bilayer through both electrostatic and hydrophobic interactions and adopts helical secondary structure upon membrane interaction. Tryptophan fluorescence spectra (A, C, and E) and circular dichroism (CD) spectra (B, D, and F) of Naa60-(189–242) peptide in the presence (filled line) or absence (dotted line) of various liposomes. A, combining 10 μm Naa60-(189–242) and 180 μm Golgi-like liposomes (52% PC, 20% PE, 15% cholesterol, 8% PI(4)P, and 5% PS) resulted in a blue shift in Trp fluorescence from 355 to 348 nm indicating interaction between Naa60-(189–242) and the Golgi-like liposomes. B, combining 20 μm Naa60-(189–242) and 360 μm Golgi-like liposomes resulted in a characteristic helical spectrum for the peptide. C, combining 10 μm Naa60-(189–242) with 180 μm 100% PC liposomes did not cause a Trp blue shift. D, combining 20 μm Naa60-(189–242) with 360 μm PC liposomes did not affect the CD spectrum of the Naa60 peptide. E, Naa60-(L197A/L201A/L204A) displayed a shift in the λmax from 355 to 348 nm in the presence of Golgi-like liposomes. F, CD spectra of 20 μm Naa60-(L197A/L201A/L204A) with and without 360 μm Golgi-like liposomes. RFI, relative fluorescence intensity.
One characteristic of the Golgi-like liposomes used above is their charge caused by PI(4)P and PS and we wondered whether this could be a contributing factor. We therefore performed the same experiments using neutral liposomes. When incubated with 100% PC liposomes, Naa60-(189–242) did not display a blue shift in Trp fluorescence (Fig. 2C) and also preserved its native unfolded state (Fig. 2D), thus indicating the necessity of a charged environment to initiate folding of the peptide and interaction with the membrane.
As shown in Fig. 1, Pred-α1 has a clear amphipathic nature with residues Ile190, Leu191, Ile194, Leu197, and Leu201 on the hydrophobic side. In a peptide in which selected hydrophobic residues were substituted with alanine (L197A, L201A, and L204A) the addition of Golgi-like liposomes still induced a blue shift from 355 to 348 nm (Fig. 2E), but for this peptide the α-helical content formed in the presence of Golgi-like liposomes was compromised (Fig. 2F). Overall, the Trp fluorescence demonstrates an intrinsic membrane-binding capacity of the Naa60 C-terminal tail and CD data supports the presence of helical secondary structure, which interestingly was dependent on the presence of liposomes.
Two different segments in the C terminus of Naa60 contribute to membrane binding in cellulo
The predictions and modeling indicated the presence of two α-helices (Fig. 1), of which the first one matched with previous crystallization data (30). Our CD also confirmed the presence of helical structure, although incapable of providing information on the number of helices. To investigate the importance of the two different C-terminal segments in membrane binding, we next studied the subcellular localizations of an extended panel of deletion and truncations mutants (Fig. 3A) in HeLa cells by microscopy. Based on previous data and experience, we found C-terminal eGFP tagging of Naa60 to be the best option for these studies (12). We first tested C-terminal truncations of Naa60-eGFP (Fig. 3B) as well as the targeting of eGFP by adding segments of the Naa60 C terminus (see Fig. 3C). As shown previously (12), truncating the last 61 amino acids (aa 182–242) caused a cytosolic localization, as opposed to the organellar localization of the full-length Naa60-eGFP (Fig. 3B). Hence the last 61 residues are essential for membrane localization. Attaching this 61-residue long segment onto the C terminus of eGFP (eGFP-Naa60-(182–242)) targeted eGFP to the same localization as Naa60 full-length (Fig. 3C), hence these 61 amino acids are not only necessary, but also sufficient for membrane targeting.
Figure 3.

Truncation and deletion constructs of eGFP-tagged Naa60 demonstrated regions in the C-terminal tail important for membrane association in cellulo. HeLa cells were transfected with the indicated eGFP-tagged Naa60 constructs (A) and imaged live (B–D) or subjected to subcellular fractionation followed by high pH/sodium carbonate-mediated protein extraction from membranes (E). A, the sequence of the Naa60 C-terminal tail as shown in Fig. 1, but with the truncated and deleted segments indicated. Red, green, yellow, and orange lines indicate positions of truncations and deletions shown in B–D. B, Naa60-eGFP C-terminal truncated constructs lacking the last 60 (1–182), 26 (1–216), or 17 (1–225) amino acids, compared with the full-length construct (1–242). The truncated segments are indicated with red lines in panel A. Scale bar is 10 μm and is representative for all microscopic images. C, eGFP targeting assay using segments of the Naa60 C-terminal tail attached to the C terminus of eGFP. Segments tested were the last 61 (182–242), 51 (192–242), 41 (202–242), and 26 (217–242) amino acids, each indicated with green lines in A. D, expressed Naa60-eGFP constructs with the indicated segments deleted. Deletions in Pred-α1 are indicated in yellow, whereas deletions in Pred-α2 are indicated in orange, with the corresponding colors used in A to indicate the positions of these segments. E, cells were transfected with the indicated constructs and subjected to high pH/sodium carbonate-mediated protein extraction from membranes following subcellular fractionation and immunoblotting. The peripheral membrane protein GM130 and transmembrane RCAS1 were used as controls for extractable and un-extractable modes of membrane binding, respectively.
Furthermore, we continued this eGFP targeting assay by adding shorter segments of Naa60 to eGFP, which caused the following: the last 51 aa provided organellar localization similar to the last 61 aa; the last 41 caused mixed organellar and cytosolic localization; and the last 26 aa caused cytosolic localization (Fig. 3C). Hence, aa 182–191 (N-terminal to Pred-α1) are dispensable in terms of membrane interaction contribution, whereas there is an essential (but not solo-contributing) element between aa 192 and 201 (the area of Pred-α1). Interestingly, truncation of the last 26 aa caused a mixed cytosolic and organellar localization (Fig. 3B), thus indicating an important contribution within these last 26 aa (part of Pred-α2). However, although the last 26 amino acids are clearly necessary, the eGFP-targeting assay (Fig. 3C) showed that this segment is not sufficient for membrane attachment, i.e. an important membrane-interacting region exist also N-terminal to Ala217. Truncating the last 17 aa also caused some (but less) cytosolic signal (Fig. 3B), although nothing was detected in the supernatant of fractionated cells (Fig. 3C). Nevertheless, the microscopy data suggest that residues also beyond Leu225 affect membrane binding capacity and thus support the IMM1 simulations that showed impaired binding for Pred-α2 for this truncated variant. Furthermore, truncating the last 12 aa gave similar results as truncating the last 17 aa, whereas truncating only the last 6 aa were similar to the full-length as observed by microscopy (Fig. 3B).
In addition to the truncation experiments, we also performed deletions of internal regions (Fig. 3D). Here, deletion of either Pred-α1 (Δ191–200) or Pred-α2 (Δ213–222) caused mixed cytosolic and organellar localizations, although the former had a higher degree of cytosolic over organellar signal. Combining the two deletions gave a complete cytosolic localization (Δ191–200 + Δ213–222). These data demonstrate important contributions from both regions, strong enough to partially obtain organellar association by itself. Interestingly, however, deletion of a prolonged Pred-α1-(191–209) caused a complete cytosolic localization, thus the presence of Pred-α2 is not sufficient for partial membrane association in this case. Similar enlarged deletions near Pred-α2 (Δ213–230 and Δ213–236) did successively worsen the mistargeting, however, not with the same severity as for the extended segment of Pred-α1. Deletion of the segment between the two helices (Δ203–210) peculiarly seemed to cause a shift in the organellar localization from Golgi to ER.
The microscopy data were supplemented with an extraction assay in which a sodium carbonate buffer of high pH is used to eliminate electrostatic interactions, whereas retaining the membrane and its hydrophobic protein interactions (refer to “Experimental procedures”). We applied the carbonate buffer to pellets resulting from subcellular fractionation to test the degree of extractability of various Naa60 mutants (Fig. 3E). In this way we were able to test the membrane binding capacity of partially mislocalizing mutants by measuring the extractability of the portion of molecules residing on the membranous fraction. Note that the protocol was slightly changed from that used in our previous study (12). With the improved protocol used in the current study, full-length Naa60 was completely pelleted in the organellar fraction (as described previously in Ref. 12) but now revealed partial extractability by high pH wash. The organelle pellet-associated molecules of the two Naa60 constructs containing deletions of either Pred-α1 (Δ191–200) or Pred-α2 (Δ213–222) were both well extracted (more than the WT/full-length). Furthermore, truncation of the last 17 aa gave a rather modest effect on the subcellular localization, but the membrane-bound portion washed out quite easily. All microscopy and sodium carbonate wash assay data obtained on the Naa60 mutants are summarized in supplemental Table S1.
Site-directed mutagenesis in Pred-α1 and Pred-α2 revealed residues important for Naa60 membrane binding in cellulo
Because the deletions above suggested membrane interaction by two regions matching the predicted helices, we further investigated the possible membrane interaction capacity of selected residues within and next to these helices by an extensive screen of point mutations (supplemental Table S1). We started by mutating hydrophobic amino acids in Pred-α1 (Fig. 4, A–C). The I190A/L191A/Y193A/I194A mutation, which involves two of the buried amino acids from Pred-α1, caused a mixed organellar and cytosolic localization (Fig. 4B). Hence, this mutation affected some amino acids that make a crucial contribution to Naa60 membrane association. Furthermore, a carbonate wash assay demonstrated that the partial membrane localization of this mutant was of a looser character, as it was extractable from the organellar fraction to a higher degree than the Naa60WT (Fig. 4C). Hence, this mutation either caused only partially impaired membrane integration of Pred-α1 or additional segments could be able to maintain some membrane association without Pred-α1. Support for the latter was indicated by a more drastic mutation strategy, in which the same amino acids were mutated to Glu rather than Ala, hence making Pred-α2 membrane interactions very unlikely. This corresponding Glu-mutant also had a mixed organellar and cytosolic signal, with a similar washing profile (supplemental Fig. S1). We further alanine mutated a slightly more C-terminal skewed segment, Leu197/Leu201/Leu204/Ile209, which involves two amino acids from Pred-α1, both buried in the membrane according to the IMM1 model. This mutation also gave a mixed localization (Fig. 4B), with a partial and almost completely extractable organellar fraction (Fig. 4C), but with a more severe degree of mislocalization compared with I190A/L191A/Y193A/I194A. The corresponding Glu-mutant, also had a more severe mislocalization with no observable organellar signal (supplemental Fig. S1).
Figure 4.

Mutated constructs of Naa60-eGFP revealed amino acids within Pred-α1 and Pred-α2 important for membrane association in cellulo. HeLa cells were transfected with the indicated Naa60-eGFP constructs (A) and imaged live (B and D) or subjected to subcellular fractionation followed by high pH/sodium carbonate-mediated protein extraction from membranes (C and E). A, the sequence of the Naa60 C-terminal tail as described in the legend to Fig. 1. Arrows indicate the positions of mutations shown in B–E, and are color coded accordingly. B, subcellular localization of Naa60-eGFP mutated constructs in the area of Pred-α1 and as indicated with corresponding colors in A. Scale bar is 10 μm and is representative for all microscopic images. C, cells were transfected with the indicated constructs and subjected to high pH/sodium carbonate-mediated protein extraction from membranes following subcellular fractionation and immunoblotting. The peripheral membrane protein GM130 and transmembrane RCAS1 were used as controls for extractable and un-extractable modes of membrane binding. D, as B, but for the area of Pred-α2. E, as C, but for the area of Pred-α2.
We next scaled down the mutations to three at a time and tested L197A/L201A/L204A (including two amino acids from Pred-α1 and matching the peptide tested in Fig. 2, E and F) and L201A/L204A/I209A (including one amino acid at the end of Pred-α1). Again, the more C-terminal skewed segment was slightly more severe in the degree of mislocalization (Fig. 4B). The corresponding Glu-mutants again made this difference clearer because L201E/L204E/I209E was completely cytosolic compared with L197E/L201E/L204E, which had some organellar localization (supplemental Fig. S1). This indicated that residues between the two α-helices are also of importance for Naa60 membrane binding. In an effort to identify single crucial residues, we further scaled down to mutating two residues at a time, however, here we could not observe any changes for I190A/L191A, Y193A/I194A, I194A/L197A, L197A/L201A, L201A/L204A, or L204A/I209A (supplemental Fig. S1).
The partially lost membrane eGFP localization of the L197A/L201A/L204A mutant cohered with the in vitro data above on the corresponding peptide (Fig. 2E) that indicated a capacity for interaction with liposomes. We supplemented these data with a sodium carbonate wash assay and found that eGFP-hNaa60-(L197A/L201A/L204A) was detected in both the pellet and supernatant fraction before sodium carbonate treatment (P1 and S1) (Fig. 4C), thus reflecting the same partial mislocalization as the microscopy data. Interestingly, this mutant was far more extractable by sodium carbonate as compared with Naa60WT (Fig. 3C), indicating a suboptimal interaction of Naa60-(L197A/L201A/L204A) with the intracellular membranes perhaps due to the reduced ability to adopt helical structure as suggested by the data in Fig. 2.
Additionally, we tested for the possible contribution of electrostatic interactions in the Naa60-membrane association. By mutating charged amino acids in Pred-α1, we found D192A/H196A to be unchanged in the localization pattern observed by microscopy and subcellular fractionation and was also extracted to the same extent as the wild type (supplemental Fig. S1). Furthermore, H185A/H196A was unchanged as observed by microscopy (supplemental Fig. S1).
Next, we applied the same site-directed mutagenesis strategy to the area of Pred-α2 (Fig. 4, A, D, and E). An I209A/V213A/Y214A mutant slightly mislocalized to the cytosol (Fig. 4D), whereas V213A/Y214A was undistinguishable from the wild type by microscopy (supplemental Fig. S2B). The corresponding Glu-mutants were partially mislocalized to the cytosol to a higher degree compared with Ala-mutants (supplemental Fig. S2B). L220A/L221A/F224A/L225A (covering all of the membrane-buried residues according to the IMM model) partially mislocalized, as did L220E/L221E (Fig. 4D and supplemental Fig. S2B). The L220A/L221A/F224A/L225A mutation also increased extractability in the carbonate buffer assay (Fig. 4E). A more C-terminal skewed mutant, F224A/L225A/W227A/I230A, as well as its corresponding Glu-mutant, also had a mixed cytosolic and organellar localization (Fig. 4D and supplemental Fig. S3) and the Glu-mutant also seemed slightly more susceptible to high pH extraction than the wild type (supplemental Fig. S2C). Mutating Phe224/Phe225 alone had undetectable effect in the case of the Ala-mutant and marginal effect for the Glu-mutant (supplemental Fig. S2B). Interestingly, a mutant combining F224A/L225A/W227A/I230A with L197A/L201A/L204A (hydrophobic residues of Pred-α1 area) completely abolished membraneous localization, rendering only cytosolic signal (Fig. 4D). These data again indicate that the two segments corresponding to Pred-α1 and -2 cooperate to provide Naa60 membrane binding.
Investigating if charge could contribute to binding for the second helix we made a H211A/R212A/R215A/H218A mutant, but could not observe altered localization compared by microscopy (Fig. 4D). Subcellular fractionation, however, indicated some degree of cytosolic localization, thus suggesting that these basic residues make contacts with the negatively charged lipid head groups contributing to the overall binding of Naa60 to the membrane (Fig. 4E).
Overall, our in cellulo experiments (listed in supplemental Table S1 and summarized in Figs. 3 and 4) revealed Naa60 membrane interaction to be rather rigorous and quite resistant to mutations. Several of the tested mutations partially abolished Naa60 membraneous localization, but only the more severe mutations and those involving several segments of the C-terminal tail could completely remove Naa60 from its organellar localization. Combined, these data support Naa60 membrane interaction to be mediated by hydrophobic residues within Pred-α1 and Pred-α2, but also by residues next to these helices.
Atomistic simulations show a membrane composition-dependent amphipathic helix interaction
The interaction of Naa60-(185–242) with membrane lipids was further investigated using atomistic simulations of the protein bound to POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) lipid bilayers. Three membrane bilayer compositions were used: POPC:PI(4)P, 92:8; POPC:POPS, 75:25; and POPC:PSM:CHOL, 70:20:10 (abbreviated PC:PI(4)P, PC:PS, and PC:SM, respectively). The last membrane bilayer has an overall neutral charge, whereas the two others are negatively charged (see “Experimental procedures” for more details). We performed MD simulations of Naa60-(185–242) docked on each of the three membrane bilayers using as starting orientation the one resulting from the implicit membrane simulations (see “Experimental procedures”). The helical structures remained stable on all membrane bilayers with root mean square deviations of the coordinates below 1 Å (except for Pred-α2 in PC:SM at 1.4 Å). The electron density profiles of the PC lipids and C-terminal helices (Naa60-(185–227)) are shown in Fig. 5A using plain and dashed lines, respectively. They illustrate how the membrane composition affects the position of the helices relative to the center of the lipid bilayers (Z = 0). With the anionic lipids (PC:PS and PC:PI(4)P), the density profiles of the helices (dashed lines) are centered around lower Z values than with the neutral bilayer (PC:SM). This reflects the fact that the two helices are anchored deeper in the membrane bilayer when it contains negatively charged lipids, PS or PI(4)P, hence validating the different liposome-interacting capacities observed for the C-terminal peptide with PI(4)P versus PC lipids (Fig. 2). The average number of contacts per frame between the helices (Naa60-(185–227)) and the lipid tails (Fig. 5B) are also higher for the bilayers containing anionic lipids. The same trend is observed for the average number of hydrogen bonds per frame between lipid head groups and amino acids of the helices (Naa60-(185–227)). These are reported in Table 1. Fig. 5, C and D, shows a snapshot of the simulation with the PC:PI(4)P bilayer illustrating that the helices are deeply anchored, remain parallel to the membrane plane, and engage in hydrogen bonds with PI(4)P lipids mainly through Arg212 and Arg215 of Pred-α2, and through Lys233 and Arg240 of the C-terminal tail.
Figure 5.

Effect of the membrane composition on the position of the Naa60 C terminus (185–242) and its interaction with lipids. Three lipid bilayers were considered POPC:PI(4)P, 92:8; POPC:POPS, 75:25; POPC:PSM:CHOL, 70:20:10, in atomistic simulations with the Naa60 C terminus (185–227). A, electron density along the normal to the membrane for the PC lipids (solid lines) and the Naa60 C terminus helices (185–227, dashed line) show that the presence of PI or PS lipids results in a deeper position of the protein. Zero corresponds to the plane between the two lipid layers. B, average number of hydrophobic contacts per trajectory frame between the lipid tails and the two helices (185–227). C and D, snapshot from the atomistic simulation of Naa60-(185–242) on a POPC:PI(4)P bilayer. Helices are shown in the same color scheme as in Fig. 1. C, side view showing that the helices are inserted beyond the phosphate atoms (yellow/tan spheres) of the lipids (PC in gray and PI(4)P in cyan). D, top view showing only the PI(4)P lipids of the upper leaflet. Four of the eight PI(4)P lipids are interacting with the side chains of charged amino acids (blue, stick).
Table 1.
Hydrogen bonds between phospholipids and amino acids of Naa60-(185–227) calculated from atomistic simulations
Average number of hydrogen bond per frame is reported for each bilayer. Only hydrogen bonds with average numbers above 0.5 are reported.
| Membrane composition | Amino acids - phospholipid | Hydrogen bonds |
|---|---|---|
| POPC:PI(4)P (PC:PI(4)P); | Arg212-PI(4)P106 | 1.66 |
| 92:8 | Tyr214-PI(4)P76 | 0.52 |
| Arg215-PI(4)P76 | 1.79 | |
| Lys233-PI(4)P116 | 0.71 | |
| Arg240-PI(4)P76 | 1.81 | |
| Arg240-PI(4)P72 | 1.86 | |
| POPC:POPS (PC:PS); | His185-PC23 | 0.52 |
| 75:25 | Arg212-PS94 | 0.99 |
| Lys233-PS87 | 0.52 | |
| POPC:PSM:CHOL (PC:SM); | Arg212-PC66 | 1.10 |
| 70:20:10 | Arg212-PC118 | 0.85 |
| Arg212-PC57 | 0.97 | |
| His185-PC31 | 0.61 |
Naa60-(189–242) peptide peripherally binds Golgi-like liposomes possibly with a horizontal orientation of helices relative to the membrane bilayer
Several experiments above indicated the membrane-interaction mode of Naa60 to involve the amphipathic helices modeled in Figs. 1 and 5. In the IMM1 and atomistic model these are predicted to bind the membrane with most of the residues of the hydrophobic face embedded completely inside the membrane plane. To further pursue this hypothesis experimentally, we performed a brominated PC (Br-PC) quenching assay (Fig. 6). Bromine is a tryptophan fluorescence quencher, and using lipids brominated at specific positions of the acyl chain, makes it possible to calculate the distance from the quenched tryptophan residue to the bilayer center (33, 34).
Figure 6.

Br-PC quenching assay with Naa60 peptides indicate peripheral binding to Golgi-like liposomes. A, WT Naa60-(189–242) peptide (W227) and mutated Naa60-(189–242) peptides (Y214W/W227F, I209W/W227F, or Y193W/W227F) were incubated with Br-Golgi-like liposomes (52% Br-PC, 20% PE, 15% cholesterol, 8% PI(4)P, and 5% PS). Vesicles contained PC molecules carrying a quenching Br-group at either position (11,12), (9,10), (6,7), or (4,5) in the acyl chain. The fluorescence measured for vesicles binding to the brominated vesicles (F) was divided by the fluorescence for vesicles binding to the non-brominated vesicles (F0). n = 2 per peptide. B, based on data in A, the position of Trp residues relative to the center of the bilayer was calculated using the parallax method (34).
We made Golgi-like liposomes as above, but using PC molecules carrying a quenching Bromo group at either positions (11,12), (9,10), (6,7), or (4,5) in the acyl chain, in addition to control liposomes with non-brominated PC molecules. These were used in Br-PC quenching assays combined with Naa60-(189–242) peptides that were tryptophan modified to test for the bilayer position of three different residues located at various positions of Naa60-(189–242). These were Tyr193 (Naa60-(Y193W/W227F)) at the hydrophobic side of Pred-α1; Ile209 (Naa60-(I209W/W227F)) between Pred-α1 and -α2; and Tyr214 (Naa60-(Y214W/W227F)) at the hydrophobic side of Pred-α2. In addition, the wild type peptide Naa60-(189–242), was used to study the depth of liposome interaction for Trp227, which is positioned C-terminal to Pred-α2.
Our data indicated that tryptophans at positions 193, 209, 214, and 227 were all quenched in a similar manner, hence showing that they all lie in the same plane of the bilayer (Fig. 6A). All tested tryptophan positions were differentially quenched in the order Br-(4,5) > Br-(6,7) > Br-(9,10) > Br-(11,12), indicating that the residues are positioned closer to the (4,5)- than to (11,12)-positions of the acyl chain, meaning they are all in closer proximity of the head groups than the bilayer center. In more detail, all the tested residues were calculated to be positioned at ∼11 Å from the bilayer center (Fig. 6B). These data therefore support the data in Fig. 5 in that the two helical segments in the Naa60 C terminus interact with the membrane bilayer in a horizontal peripheral rather than transmembrane fashion. Furthermore, these data suggest that residues next to the two modeled helices also take part in interfacial membrane interaction.
Naa60 has a preference for PI(4)P which may recruit it to the Golgi membrane
Naa60 specifically localizes to the Golgi apparatus within cells (12) and the Naa60-(189–242) peptide was shown here to interact with Golgi-like liposomes without the need of a protein partner in vitro. Thus, Naa60 is probably not inserted by the translocon machinery as it most likely is not transmembrane (Figs. 2 and 7). Moreover, our atomistic simulations above indicated a deeper anchoring of the amphipathic helices of Naa60 in membrane bilayers containing negatively charged lipids, PS or PI(4)P, of which the latter is a hallmark lipid of the Golgi membrane (35). We therefore wanted to investigate if Naa60 may be specifically recruited to the Golgi membrane through lipid preference.
Figure 7.

Pulldown ITC and CD experiments demonstrate the Naa60-(189–242) peptide to have a binding preference for PI(4)P. A, pulldown of Naa60-(189–242) using liposomes with 92 mol % PC and 8 mol % of either PI. Peptide and liposomes were incubated at room temperature before pelleting at 200,000 × g into supernatant (S) and pellet (P) fractions. Naa60-(189–242) was detected by means of a dot blot. n = 3–8 per vesicle type. Antibody was validated for detection of the peptide (supplemental Fig. S3). B, dot blot intensities of the pellet fractions in A were quantified (3 replications averaged) using ImageJ and expressed as percentage of bound Naa60 peptide based on P and S intensities. C, ITC experiments measuring dissociation constant for Naa60-(189–242) peptide with either PC-PI(3)P or PC-PI(4)P liposomes. Naa60-(189–242) peptide (50 μm) was injected in 20 successive injections of 2 μl with 180-s intervals into a total lipid concentration of 100 μm and heat rate was measured (upper). Dissociation constants were calculated based on peak area/molar ratio plots (lower). D, far-UV CD spectra of Naa60-(189–242) (20 μm) in the presence of 360 μm liposomes containing 92 mol % PC and 8 mol % of either PI(3)P or PI(4)P. Naa60-(189–242) with no liposomes (yellow), PI(3)P (pink), and PI(4)P (green). Spectra recorded between 180 and 260 nm.
The Golgi-like liposomes used above contained 8% PI(4)P, in addition to 5% PS, another anionic lipid. To test for PI preference we made liposomes composed of 92% PC and 8% of the various PIs found in the mammalian cell, in addition to two control vesicles composed of either 8% PS and 92% PC or 100% PC. Through pulldown experiments we found a strong preference for PI(4)P in comparison to all the other PIs (Fig. 7, A and B). The percentage of bound Naa60-(189–242) peptide was twice as high for PI(4)P as for the second most preferred PI, PI(3)P. Based on the control vesicles, these experiments also confirmed our previous results that the interaction between the Naa60-(189–242) peptide and vesicles requires a charged membrane as we were not able to pull down the Naa60 peptide after incubation with PC liposomes. Furthermore, we found that a mere charge itself was not the cause of interaction because PS vesicles were only weakly able to pull down Naa60-(189–242), hence demonstrating PI(4)P as the key anionic lipid in our Golgi-like vesicle (Fig. 7, A and B).
To further verify and quantify the interaction between Naa60-(189–242) peptide and PIs we conducted ITC experiments using liposomes composed of PC and either PI(3)P or PI(4)P (Fig. 7C). The dissociation constant for PI(4)P liposomes was 64 nm, thus demonstrating interaction between the Naa60-(189–242) peptide and PI(4)P. Comparatively the dissociation constant for PI(3)P liposome was 15-fold higher (957 nm), hence revealing a clear preference for PI(4)P over PI(3)P, and suggesting Naa60 to specifically bind to PI(4)P-containing membranes.
Because the above CD data indicated the presence of charged vesicles as a necessity for the Naa60-(189–242) peptide to display a spectrum characteristic of helical structure (Fig. 2B), we further tested the folding properties of Naa60-(189–242) in the presence of the different PIs. Since we identified a preference for PI(4)P in terms of binding capacity (Fig. 7, A–C), we hypothesized that folding characteristics might be affected by different PIs, which all bear their charge at a very specific position of their inositol ring. Indeed, as seen in Fig. 7D, this CD experiment showed that the PI(4)P containing liposomes induced a more typical helical spectrum of Naa60-(189–242), with distinctive negative bands at 208 and 222 nm and a positive band at 193 nm. Conversely, the CD spectra of Naa60-(189–242) incubated with other phosphoinositides (PIs) (supplemental Fig. S4) showed decreased ellipticity between 200 and 210 nm, reflecting suboptimal partial helical folding. Hence, stabilization of helical secondary structure is a likely feature through which PI(4)P facilitates interaction of the Naa60-(189–242) peptide with the vesicles.
Discussion
We here identified the membrane-binding ability of Naa60 to be mediated by two α-helices in the C terminus that attach horizontally and partly embed into the outer membrane layer. These α-helices partially overlap with two previously predicted transmembrane helices that were suggested to possibly form a transmembrane helical hairpin (12). However, we here found several pieces of evidence supporting the horizontal, peripheral binding mode. An improved protocol for the carbonate extraction assay employed previously (12) revealed Naa60 to be partly extractable from cellular membrane fractions, in contrast to transmembrane controls. Additionally, our Br-PC quenching data showed four residues that were spread throughout the C-terminal region to be positioned at approximately the same depth in the membrane, hence strongly disfavoring a two-helical hairpin transmembrane mode of interaction for Naa60, which was suggested by previous data.
Although peripheral rather than transmembrane, we found the membrane interaction of Naa60 to be strong, because carbonate wash only partially extracted the protein and further a completely abolished organellar localization was only observed for rather severe deletions and mutations. Combining the cellular localization assays with carbonate washes revealed further interesting characteristics, especially in those cases where the mutation partially abolished organellar localization. For example, a particular hydrophobic mutation in the first of the two helices (L197A/L201A/L204A) partially mislocalized Naa60 to the cytosol and interestingly, the pool of membrane-residing Naa60 molecules was almost completely extracted in the sodium carbonate assay, as opposed to the only partially extractable wild type Naa60. Moreover, the Naa60-(L197A/L201A/L204A) hydrophobic mutant peptide interacted with the Golgi-like liposomes, although under the same conditions underwent incomplete helical folding. This indicates a suboptimal insertion of Naa60-(L197A/L201A/L204A) in the cellular membrane, causing a looser attachment more susceptible to the high pH extraction. Sodium carbonate affects only electrostatic interactions and leaves the membranes and hydrophobic lipid-protein interactions intact. Hence, the resistance to extraction illustrate that Naa60 is anchored to the Golgi membrane predominately by hydrophobic interactions, whereas the small amount that is extracted indicates that electrostatic interactions also contribute. The atomistic model further supported importance of both charged and hydrophobic residues. Thus based on the various methods used here, it appears that the amphipathic helices are nicely stabilized at the interface between head groups and lipid chains where each side of the helices make either hydrophobic or ionic contacts.
Our previous study established that the main depot of Naa60 in the cell is at the Golgi membrane (12). In the atomistic model of the present study, we found deeper embedding of the two C-terminal helices in membranes containing PI(4)P or PS. Furthermore, in vitro experiments showed a Naa60 preference for PI(4)P. As PI(4)P is a characteristic feature of the Golgi membrane, we here propose that Naa60 could be targeted to the Golgi apparatus via specific interactions with PI(4)P. However, Naa60 does not harbor any of the known structured phospholipid-binding domains (36, 37), such as the PH domain. Nor was a polybasic motif identifiable from the sequence, however, the sequence contains basic amino acids that could in three-dimension arrange to form a site specific for PI(4)P lipids. And indeed, the simulations show hydrogen bond interactions of PI(4)P lipids with Arg212, Arg215 (of the second helix), and Arg240 (of the disordered segment C-terminal to the second helix and close to the C terminus end) (Table 1). Because atomistic modeling started from a computational model of the C-terminal structure that is partly disordered, and even though long simulations were performed, it is not sure that we have sampled the conformational space of the disordered region enough to make any firm conclusion on its actual three-dimensional structure. Based on the mutational cell data, the charge to alanine mutant Naa60-(H211A/R212A/R215A/H218A), was in part found in the supernatant fraction prior to sodium carbonate treatment, suggesting that at least some of these residues could form essential electrostatic interactions with the membrane head groups. Furthermore, the cell data support that the disordered regions next to the helices are involved in membrane binding.
The present study filled a knowledge gap that was caused by the difficulty to prepare full-length Naa60 for crystallographic studies, hence necessitating C-terminal truncation. Combining the C-terminal data obtained here with the published Naa60 crystal structures on C-terminal truncated variants (29, 30) makes it possible to view the whole protein and determine the position of the enzyme site relative to the membrane. Fig. 8 shows such a model and reveals the Ac-CoA binding site and catalytic domain to be on the side (i.e. not facing directly toward or away from the membrane) and hence available in the cytosol for Nα-acetylation of transmembrane proteins. This model also shows that the extended β3-β4 loop (around aa 78–87, green) is positioned near the membrane. Given that this is the segment that, in addition to the C terminus, is specific for Naa60 among the Naas, it is tempting to speculate that this loop has some function there, either in membrane binding or protein interaction.
Figure 8.
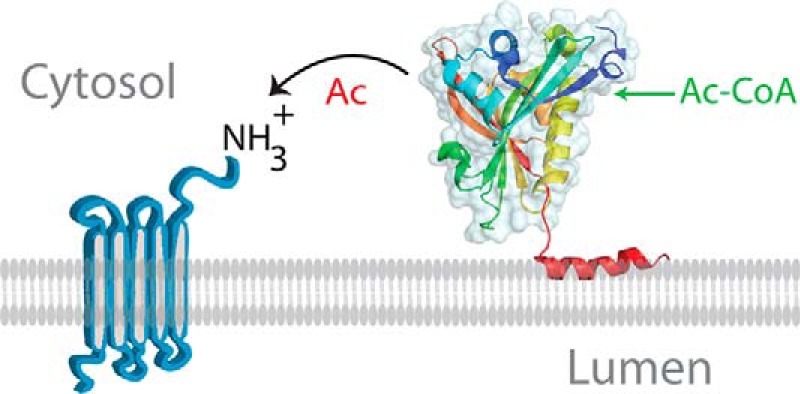
Model of Naa60 membrane association and N-terminal acetylation at the membrane. Shown is the structure of Naa60 (PDB code 5ICV) attached to the C-terminal α-helix predictied by PSIPRED. Only the first of the two amphipathic α-helices is shown. The helical anchor dipping horizontally into the membrane positions the enzymatic site of Naa60 available to acetylate N termini of transmembrane proteins. Whether Naa60 makes additional contact points with the membrane besides its two C-terminal helices remains to be revealed.
Naa60 was established as distinct from the rest of the catalytic NATs in that it prefers transmembrane substrates (12). The model in Fig. 8 suggests that Naa60 acts post-translationally at the Golgi membrane. If true, this would represent a completely new mode of NAT operation and new features of the NAT machinery could be considered, such as regulation of the NAT enzyme and reversibility of the reaction through a counteracting Golgi-residing N-terminal deacetylase.
In conclusion, we have defined Naa60 anchoring to the cytosolic side of the Golgi apparatus to involve two C-terminal amphipathic helices that, via hydrophobic and electrostatic interactions, bind the membrane directly in a parallel position relative to the lipid bilayer. These findings have implications for our understanding of Naa60 functioning, as they are strongly suggestive of Naa60 acting post-translationally to N terminally acetylate its many transmembrane substrates.
Experimental procedures
Helical wheel analysis
The helical wheel analysis was performed using the online Helical Wheel Projections tool (created by Don Armstrong and Raphael Zidovetzki (version ID: wheel.pl, v.1.4 2009–10-20).
IMM1 simulations
Molecular dynamics (MD) simulations were performed using an IMM1 (38), where the lipid bilayer is mimicked by a hydrophobic slab. The IMM1 model provides an estimation of the solvation/desolvation contribution of inserting the peptide at the membrane interface. The so-called effective energy (WIMM1) of a solute calculated with the IMM1 model is the sum of the intramolecular energy (Eintra) of the solute and of its solvation-free energy (ΔGsolv) (38). Simulations were conducted for 6 different starting orientations of the peptide with respect to the membrane plane. The six different orientations were obtained by rotating the peptide along the x and y axes as reported earlier (39). Three simulations were conducted for each of the six starting orientations and for up to 2 ns, with a time step of 0.002 ps. The hydrophobic region of the implicit membrane was 25 Å thick corresponding roughly to a 1,2-dideoyl-sn-glycero-3-phosphocholine bilayer. Binding energies for the peptide were calculated for each frame in the trajectory as the difference between effective energies of the membrane-bound and -solvated states (ΔWIMM1 = ΔEintra + ΔΔGsolv), and averaged over the last ns of the trajectory. The trajectory yielding the lowest ΔWIMM1 is reported. The solvation-free energy was further decomposed into contributions from each amino acid (38).
Preparation of lipid bilayers for atomistic simulations
We performed atomistic MD simulations of the Naa60 C terminus with three different bilayer compositions: one is neutral and the other two contain negatively charged lipids: POPC:PSM:CHOL, 70:20:10; POPC:POPS, 75:25; and POPC:PI(4)P, 92:8. Sphingomyelin is an important constituent of the Golgi membrane (35, 40, 41) and we chose a composition that has been investigated both by experimental and simulation studies (42, 43). The composition of the bilayers containing negatively charged lipids were chosen to be comparable with the accompanying experiments.
Each bilayer contained 256 lipids. For PI(4)P we used the SAPI14 lipid type in the CHARMM force field 18:0/20:4 (5, 7, 9, 11). The CHARMM force field (c36) (44, 45), including the CMAP corrections and update for lipids (46) was used for all simulations. The PC:SM:CHOL bilayer was prepared using the CHARMM-GUI (47), shortly equilibrated (500 ps) in the NVT ensemble and simulated further during 500 ns in the NPT ensemble. The PC:PS bilayer was taken from Chon et al. (48). The PC:PI(4)P bilayer was also built using the CHARMM-GUI and was simulated for 200 ns in the NPT ensemble after a short NVT equilibration of 500 ps. All simulations were performed using NAMD (v2.10) (49) with an integration time step of 2 fs. We used the Langevin piston method (50) (target pressure: 1 atm, oscillation period: 200 fs, damping time scale: 50 fs) and Langevin dynamics to control the temperature (temperature damping coefficient: 1.0). All simulations were performed at 310 K temperature. A multiple time step algorithm (51) was used to integrate the equations of motions; bonded interactions and short-range nonbonded forces were evaluated every step, long-range electrostatics every second step. Force-based switching was used for both electrostatic and van der Waals interactions, with a switch distance of 11 Å and a cutoff distance of 12 Å. Particle mesh Ewald (52) was used for long-range electrostatic interactions. SHAKE (53) was applied to constrain all bonds to hydrogen atoms. We calculated the area per lipid of the simulated bilayers along simulation time. The average value for POPC:PSM:CHOL, 70:20:10, was 56.8 ± 0.8 Å2, which is in agreement with a simulation study (43) of a close composition (POPC:PSM:CHOL, 69:23:8). For POPC:PI(4)P, 92:8, the average area per lipid was 65.7 ± 0.9 Å2 close to what we obtained earlier for a pure POPC bilayer, i.e. 65.5 ± 0.8 Å2 (53).
Atomistic simulations of the Naa60 C terminus (185–242) with lipid bilayers
The orientation was taken from the results of the IMM1 simulations. The domain was then placed so that the COM of its two helices (aa 185–227) was placed at 20 Å from the bilayer center. Overlapping water and lipid molecules were removed. 9, 12, and 8 lipid molecules were deleted from the upper leaflets of the PC:PI(4)P, PC:PS, and PC:SM:CHOL bilayers, respectively. Membrane and protein were then submitted to energy minimization using positional harmonic restraints (150 kcal/mol/Å2 for protein backbone and water molecules; 100 kcal/mol/Å2 for membrane atoms located further than 5 Å from the protein and protein side chain atoms located further than 5 Å from any lipid, 50 kcal/mol/Å2 for membrane atoms located within 5 Å of the protein, 15 kcal/mol/Å2 for protein side chain atoms located at 5 Å or less from any lipid). The minimization was performed using the CHARMM simulation package (v38) (54) and consisted of 20 consecutive cycles of 600 minimization steps (500 steepest descent, 100 conjugated gradients) with restraints being scaled by 0.65 after each cycle. Using VMD (55) the optimized structures were then solvated in a pre-equilibrated box of TIP3 water molecules and neutralized with Cl− atoms. The systems were then equilibrated for 500 ps in the NPT ensemble with constrained protein backbone and the constraints were removed for the production run. The characteristics for each simulated system are given in Table 2.
Table 2.
Atomistic simulations of Naa60 docked on lipid bilayers
| Composition | Dimensions | Atoms | Number of ions | Simulation time |
|---|---|---|---|---|
| Å | ns | |||
| PC:PI(4)P | 120.9 × 118.7 × 97.6 | 81,866 | 2 Cl− | 1000 |
| PC:PS | 110.1 × 122.1 × 97.9 | 79,026 | 4 Cl− | 700 |
| PC:SM:CHOL | 111.6 × 108.5 × 105.6 | 85,701 | 2 Cl− | 900 |
Analyses of the atomistic simulations
The simulations were performed for the last 300 ns of the simulations, where the depths of anchorage of the helices were found to be stable. Electron densities along the normal bilayer were calculated using the VMD MEMBPLUGIN (56). The densities were calculated for the PC lipids and the two helices (185–227) of the C-terminal domain. We also calculated the average number of hydrophobic contacts per trajectory frame. Hydrophobic contacts are considered to exist if two unbound candidate atoms are within 3 Å for at least 10 ps. Candidate atoms are atoms in aliphatic groups of amino acids side chains (CHARMM force field nomenclature: ca; cb; cg1; cg2; ha*; hb*; hg; hg2*; type cg except the one of hsd, hse, Asn, Asp; type hg1 except for Cys, Thr, Ser; type cd except for Arg, Gln, Glu; type cd1; type cd2 except for hsd, hse; type ce1, ce2, cz and associated hydrogens of Phe, Tyr; type cd1, cd2, ce2, ce3, cz2, cz3, and associated hydrogens of Trp; type cay and type hy*). The criteria for counting the hydrogen bonds are as follows: acceptor (A) to donor (D) distance ≤3.0 Å and angle A-D-H ≥36°. These two criteria must be met for at least 10 ps. The donor and acceptor definitions are from the CHARMM force field. Hydrogen bonds are reported in terms of number of the fraction of trajectory frames where the interaction is present. Due to lipid diffusion, a given residue can interact with different DMPC molecules along the simulation, but we consider the membrane as a whole and lipids as interchangeable. We also reported the average Z coordinate of the center of mass of each residue as an indication of the depth of anchorage of that amino acid. All coordinates statistics were done using VMD (55).
Plasmids and cloning
Plasmids for pNAA60-EGFP, pNAA60-(1–182)-EGFP, and pNAA60-(1–225)-EGFP were created by inserting the respective NAA60 ORF fragment between sites NheI and KpnI of pEGFP-N1 (Clontech). All mutations were performed by a QuikChange Lightning Multi Site-directed Mutagenesis Kit (Agilent) on either NAA60-EGFP full-length or derivatives of this plasmid. pEGFP-NAA60-(217–242) and pEGFP-NAA60-(182–242) were made by insertion of NAA60 fragments between SacI and KpnI sites in pEGFP-C1. Plasmids for expression in yeast were generated from the mammalian EGFP versions by insertion of NAA60 ORF or truncated NAA60 ORF with the EGFP tag into pBEVY-U vector between XbaI and SalI sites. Yeast procedures were performed as described previously (58).
Cell culture transfections
HeLa cells (ATCC, CCL-2) were cultured in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Lonza, 12-604) supplemented with 10% fetal bovine serum (FBS), 3% l-glutamine, and 1% penicillin/streptomycin. Sodium carbonate wash assay was performed as previously (12), except for some adaptations mentioned under “Results.”
Microscopy
HeLa cells were seeded in Lab-Tek Chambered Coverglass #1 (Nunc), transfected after 2–4 h using FuGENE 6 (Promega) according to protocols and imaged live after 18–24 h. Fluorescent images were obtained on a Leica DMI6000 B wide field microscope equipped with a Leica DC500 camera and a HCX PL APO 100 × 1.4 NA oil objective in addition to a ×1.5 or 2 magnification lens. The acquired images were processed using the Photoshop CS5 image software (Adobe Systems).
Sodium carbonate extraction
HeLa cells were transfected with various plasmids encoding C-terminal eGFP-tagged Naa60 constructs as described above. 48 h post-transfection, cells were collected in ice-cold PBS and centrifuged at 1,000 × g for 5 min at 4 °C. The supernatant was discarded and the cell pellet resuspended in homogenization buffer (0.25 m sucrose, 1 mm EDTA, 20 mm HEPES-NaOH, pH 7.4). Cells were homogenized using a ball-bearing cell cracker (EMBL, Heidelberg), with ball size of 8.002 mm giving 9 μm clearance, 20 strokes. The cell lysate was spun at 3,000 × g for 5 min at 4 °C to remove nuclei and cell debris before loading equal volumes in ultracentrifugation tubes. Organelles were pelleted by centrifugation at 45,000 rpm (ravg 95,100 × g) using a MLA-130 rotor (Beckman Coulter), for 1 h at 4 °C. The supernatant (S1) was collected and pellets were resuspended with 550 μl of homogenization buffer (P1, put a side), sucrose buffer (320 mm sucrose, 4 mm HEPES-KOH, pH 7.4), or 0.1 m sodium carbonate, pH 11.5. Pellets resuspended in sucrose or sodium carbonate buffer were incubated on ice for 30 min as previously described (57) before pelleting by ultracentrifugation as above. Supernatants (S2) were collected and mixed with sample buffer. Pellets (P2) were resuspended in 550 μl of homogenization buffer and added to 10× SDS sample buffer. All buffers included in the experiment were supplemented with fresh Protease Inhibitor Mixture EDTA free (Roche Applied Science). Note that the protocol was performed as described previously (12), except for one improvement: all pellets were dissolved in exactly the same volume from which they were pelleted. Supernatant and pellet samples were analyzed by immunoblotting. Primary antibodies used were mouse mAb to GFP (Roche Applied Science, 11814460001), rabbit mAb to GM130 (EP892Y, Abcam, ab52649), and rabbit mAb to RCAS1 (D2B6N, Cell Signaling, 12290). Secondary antibodies used were ECL anti-rabbit IgG, HRP-linked from donkey (GE Healthcare, NA934), and ECL anti-mouse IgG, HRP-linked from sheep (GE Healthcare, NA931).
Peptides
All peptides were purchased from Biogenes (Berlin, Germany; >90% purity), solubilized in ddH2O, aliquoted, and stored at −20 °C. The peptides used were: Naa60-(189–242), (TILDYIQHLGSALASLSPCSIPHRVYRQAHSLLCSFLPWSGISSKSGIEYSRTM), Naa60-(L197/L201/L204A), (TILDYIQHAGSAAASASPCSIPHRVYRQAHSLLCSFLPWSGISSKSGIEYSRTM), Naa60-(Y193W/W227F), (TILDWIQHLGSALASLSPCSIPHRVYRQAHSLLCSFLPFSGISSKSGIEYSRTM), Naa60-(I209W/W227F), (TILDYIQHLGSALASLSPCSWPHRVYRQAHSLLCSFLPFSGISSKSGIEYSRTM), and Naa60-(Y214W/W227F), (TILDYIQHLGSALASLSPCSIPHRVWRQAHSLLCSFLPFSGISSKSGIEYSRTM).
Lipids
The following lipids were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL): egg PC (840051P), brain PS (840032P), egg PE (840021P), egg PA (840101P), brain PI(4)P (840045P), cholesterol (700000P), 1-palmitoyl-2-(4,5-dibromo)stearoyl-sn-glycero-3-phosphocholine (850479P), 1-palmitoyl-2-(6,7-dibromo)stearoyl-sn-glycero-3-phosphocholine (850480P),1-palmitoyl-2-(9,10-dibromo)stearoyl-sn-glycero-3-phosphocholine (850481P), and 1-palmitoyl-2-(11,12-dibromo)stearoyl-sn-glycero-3-phosphocholine (850482P). The following lipids were purchased from Echelon Biosciences, Inc. (Salt Lake City, UT): phosphatidylinositol 3-phosphate diC16 (P-3016); phosphatidylinositol 4-phosphate diC16 (P-4016), phosphatidylinositol 5-phosphate diC16 (P-5016), phosphatidylinositol 3,4-bisphosphate diC16 (P-3416), phosphatidylinositol 3,5-bisphosphate (P-3516), phosphatidylinositol 4,5-bisphosphate diC16 (P-4516), and phosphatidylinositol 3,4,5-trisphosphate diC16 (P-3916). All lipids were reconstituted in chloroform, or a mixture of chloroform, methanol, and water (1:2:0.8) and kept at −20 °C.
Large unilamellar vesicle preparation
To prepare liposomes, lipids were mixed and dried under a flow of nitrogen gas to make a lipid film. The lipid film was hydrated with H2O to yield a final lipid concentration of 1 mm before incubation overnight at 37 °C, 250 rpm. The mixture was freeze-thawed five times using liquid nitrogen before passing through a 100-nm pore size polycarbonate filter, 21 times using a mini extruder (Avanti Polar Lipids, Inc.) to create large unilamellar liposomes. Liposomes were stored at room temperature and used within 3 days. The Golgi mixture liposomes contained (mol %): 52% egg PC or brominated PC, 20% egg PE, 5% brain PS, 15% cholesterol, and 8% brain PI(4)P (32). Other liposomes were made of (mol %) 92% PC and 8% of particular lipid of interest.
Circular dichroism
All CD experiments were executed using 20 μm peptide and 100–360 μm liposomes. Far-UV CD spectra were recorded using a Jasco-810 circular dichroism spectropolarimeter, scan range 180–260 nm, data pitch 0.2 nm, scan speed 20 nm min−1, response 2 s, and 3 accumulations. All experiments were performed at 20 °C using a circulating water bath, and with a N2 flow at 10 liters min−1.
Tryptophan fluorescence
Tryptophan fluorescence emission spectra were recorded from 310 to 400 nm after excitation at 295 nm at 20 °C using a LS 50 B (PerkinElmer Life Sciences) instrument with a temperature-controlled water bath. 5–10 μm peptide was incubated with 180–360 μm vesicles. The peptides were centrifuged at 17,000 × g for 10 min at 4 °C after thawing and their concentrations determined prior to use.
Brominated phosphatidylcholine quenching
5 μm peptide (Naa60-(189–242), Naa60-(Y193W/W227F), Naa60-(I209W/W227F), or Naa60-(Y214W/W227F)) was mixed with 300 μm Golgi mixture liposomes where egg PC was substituted by various brominated PC variants. Fluorescence emission spectra were recorded as described for tryptophan fluorescence.
Liposome pulldown
5 μm peptide (Naa60-(189–242)) was incubated with 300 μm liposomes in a total volume of 100 μl for 20 min at room temperature before sedimentation of liposomes at 65,000 rpm (200,000 × g) using a MLA130 rotor (Beckman Coulter) for 30 min at 20 °C. The supernatant was collected, and the pellet fraction was washed with water and resuspended in 100 μl before being subjected to a dot blot. The level of peptide in the pellet and supernatant fractions, respectively, was visualized using a 1:2000 diluted antibody rabbit anti-Naa60-(192–241) (Abcam, ab103800).
Isothermal titration calorimetry
ITC experiments were carried out on a Nano ITC low volume calorimeter (TA Instruments) and all experiments were performed at 25 °C. PI(3)P or PI(4)P liposomes with a total lipid concentration of 100 μm were placed in the instrument, and the Naa60-(189–242) peptide with a concentration of 50 μm was injected in 20 successive injections of 2 μl with 180-s intervals. Titrations in the absence of liposomes were performed under identical conditions to account for the heat signal that arise from titration of the peptide into water. The data were analyzed with NanoAnalyze (TA Instruments) using the Independent model, and the first data point was always omitted from the modeling.
Author contributions
H. A. and M. G. conducted most of the wet lab experiments, analyzed the results, and wrote the paper. Ø. S. and A. D. supported M. G. in the execution and analysis of in vitro experiments. Ø. S. conducted the ITC experiments. Q. W. and N. R. performed bioinformatics and modeling. T. A. conceived, initiated, and supervised the project with the support of H. A., and wrote the paper together with H. A. and M. G. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Øyvind Halskau, Aurélia E. Lewis, Aurora Martinez, Anne Baumann, and Diana Turcu for discussions, advice, and use of equipment.
This work was supported by Research Council of Norway Grants 230865 and 249843 (to T. A.), grants from the Norwegian Cancer Society (to T. A.), the Bergen Research Foundation BFS (to T. A.), and fellowship Grant DR 998/2-1 from the Deutsche Forschungsgemeinschaft (to A. D.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table S1 and Figs. S1–S4.
- Naa60
- Nα-acetyltransferase 60
- NAT
- N-terminal acetyltransferase
- CHOL
- cholesterol
- ITC
- isothermal titration calorimetry
- Naa
- Nα-acetyltransferase
- NatF
- N-terminal acetyltransferase F
- PC
- phosphatidylcholine
- PE
- phosphatidylethanolamine
- PI(4)P
- phosphatidylinositol 4-phosphate
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PS
- phosphatidylserine
- IMM
- implicit membrane model
- Br-PC
- brominated PC
- SM
- sphingomyelin
- VMD
- visual molecular dynamics
- aa
- amino acid
- eGFP
- enhanced green fluorescent protein
- POPS
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine.
References
- 1. Van Damme P., Hole K., Pimenta-Marques A., Helsens K., Vandekerckhove J., Martinho R. G., Gevaert K., and Arnesen T. (2011) NatF contributes to an evolutionary shift in protein N-terminal acetylation and is important for normal chromosome segregation. PLoS Genet. 7, e1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aksnes H., Drazic A., Marie M., and Arnesen T. (2016) First things first: vital protein marks by N-terminal acetyltransferases. Trends Biochem. Sci. 41, 746–760 [DOI] [PubMed] [Google Scholar]
- 3. Drazic A., Myklebust L. M., Ree R., and Arnesen T. (2016) The world of protein acetylation. Biochim. Biophys. Acta 1864, 1372–1401 [DOI] [PubMed] [Google Scholar]
- 4. Polevoda B., Brown S., Cardillo T. S., Rigby S., and Sherman F. (2008) Yeast N(α)-terminal acetyltransferases are associated with ribosomes. J. Cell Biochem. 103, 492–508 [DOI] [PubMed] [Google Scholar]
- 5. Gautschi M., Just S., Mun A., Ross S., Rücknagel P., Dubaquié Y., Ehrenhofer-Murray A., and Rospert S. (2003) The yeast Nα-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell Biol. 23, 7403–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Neuwald A. F., and Landsman D. (1997) GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154–155 [DOI] [PubMed] [Google Scholar]
- 7. Polevoda B., Norbeck J., Takakura H., Blomberg A., and Sherman F. (1999) Identification and specificities of N-terminal acetyltransferases from Saccharomyces cerevisiae. EMBO J. 18, 6155–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rathore O. S., Faustino A., Prudêncio P., Van Damme P., Cox C. J., and Martinho R. G. (2016) Absence of N-terminal acetyltransferase diversification during evolution of eukaryotic organisms. Sci. Rep. 6, 21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arnesen T., Van Damme P., Polevoda B., Helsens K., Evjenth R., Colaert N., Varhaug J. E., Vandekerckhove J., Lillehaug J. R., Sherman F., and Gevaert K. (2009) Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. U.S.A. 106, 8157–8162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goetze S., Qeli E., Mosimann C., Staes A., Gerrits B., Roschitzki B., Mohanty S., Niederer E. M., Laczko E., Timmerman E., Lange V., Hafen E., Aebersold R., Vandekerckhove J., Basler K., Ahrens C. H., Gevaert K., and Brunner E. (2009) Identification and functional characterization of N-terminally acetylated proteins in Drosophila melanogaster. PLos Biol. 7, e1000236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bienvenut W. V., Sumpton D., Martinez A., Lilla S., Espagne C., Meinnel T., and Giglione C. (2012) Comparative large scale characterization of plant versus mammal proteins reveals similar and idiosyncratic Nα-acetylation features. Mol. Cell. Proteomics 11, M111.015131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aksnes H., Van Damme P., Goris M., Starheim K. K., Marie M., Støve S. I., Hoel C., Kalvik T. V., Hole K., Glomnes N., Furnes C., Ljostveit S., Ziegler M., Niere M., Gevaert K., and Arnesen T. (2015) An organellar Nα-acetyltransferase, Naa60, acetylates cytosolic N termini of transmembrane proteins and maintains Golgi integrity. Cell Rep. 10, 1362–1374 [DOI] [PubMed] [Google Scholar]
- 13. Coulton A. T., East D. A., Galinska-Rakoczy A., Lehman W., and Mulvihill D. P. (2010) The recruitment of acetylated and unacetylated tropomyosin to distinct actin polymers permits the discrete regulation of specific myosins in fission yeast. J. Cell Sci. 123, 3235–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Monda J. K., Scott D. C., Miller D. J., Lydeard J., King D., Harper J. W., Bennett E. J., and Schulman B. A. (2013) Structural conservation of distinctive N-terminal acetylation-dependent interactions across a family of mammalian NEDD8 ligation enzymes. Structure 21, 42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scott D. C., Monda J. K., Bennett E. J., Harper J. W., and Schulman B. A. (2011) N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 334, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Behnia R., Panic B., Whyte J. R., and Munro S. (2004) Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 6, 405–413 [DOI] [PubMed] [Google Scholar]
- 17. Setty S. R., Strochlic T. I., Tong A. H., Boone C., and Burd C. G. (2004) Golgi targeting of ARF-like GTPase Arl3p requires its Nα-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 6, 414–419 [DOI] [PubMed] [Google Scholar]
- 18. Forte G. M., Pool M. R., and Stirling C. J. (2011) N-terminal acetylation inhibits protein targeting to the endoplasmic reticulum. PLos Biol. 9, e1001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holmes W. M., Mannakee B. K., Gutenkunst R. N., and Serio T. R. (2014) Loss of amino-terminal acetylation suppresses a prion phenotype by modulating global protein folding. Nat. Commun. 5, 4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hwang C. S., Shemorry A., and Varshavsky A. (2010) N-terminal acetylation of cellular proteins creates specific degradation signals. Science 327, 973–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shemorry A., Hwang C. S., and Varshavsky A. (2013) Control of protein quality and stoichiometries by N-terminal acetylation and the N-end rule pathway. Mol. Cell 50, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim H. K., Kim R. R., Oh J. H., Cho H., Varshavsky A., and Hwang C. S. (2014) The N-terminal methionine of cellular proteins as a degradation signal. Cell 156, 158–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Park S. E., Kim J. M., Seok O. H., Cho H., Wadas B., Kim S. Y., Varshavsky A., and Hwang C. S. (2015) Control of mammalian G protein signaling by N-terminal acetylation and the N-end rule pathway. Science 347, 1249–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aksnes H., Drazic A., and Arnesen T. (2015) (Hyper)tension release by N-terminal acetylation. Trends Biochem. Sci. 40, 422–424 [DOI] [PubMed] [Google Scholar]
- 25. Myklebust L. M., Van Damme P., Støve S. I., Dörfel M. J., Abboud A., Kalvik T. V., Grauffel C., Jonckheere V., Wu Y., Swensen J., Kaasa H., Liszczak G., Marmorstein R., Reuter N., Lyon G. J., Gevaert K., and Arnesen T. (2015) Biochemical and cellular analysis of Ogden syndrome reveals downstream Nt-acetylation defects. Hum. Mol. Genet. 24, 1956–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rope A. F., Wang K., Evjenth R., Xing J., Johnston J. J., Swensen J. J., Johnson W. E., Moore B., Huff C. D., Bird L. M., Carey J. C., Opitz J. M., Stevens C. A., Jiang T., Schank C., et al. (2011) Using VAAST to identify an X-linked disorder resulting in lethality in male infants due to N-terminal acetyltransferase deficiency. Am. J. Hum. Genet. 89, 28–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Saunier C., Støve S. I., Popp B., Gerard B., Blenski M., AhMew N., de Bie C., Goldenberg P., Isidor B., Keren B., Leheup B., Lampert L., Mignot C., Tezcan K., Mancini G. M., et al. (2016) Expanding the phenotype associated with NAA10-related N-terminal acetylation deficiency. Hum. Mutat. 37, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dinh T. V., Bienvenut W. V., Linster E., Feldman-Salit A., Jung V. A., Meinnel T., Hell R., Giglione C., and Wirtz M. (2015) Molecular identification and functional characterization of the first Nα-acetyltransferase in plastids by global acetylome profiling. Proteomics 15, 2426–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Støve S. I., Magin R. S., Foyn H., Haug B. E., Marmorstein R., and Arnesen T. (2016) Crystal structure of the Golgi-associated human Nα-acetyltransferase 60 reveals the molecular determinants for substrate-specific acetylation. Structure 24, 1044–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen J. Y., Liu L., Cao C. L., Li M. J., Tan K., Yang X., and Yun C. H. (2016) Structure and function of human Naa60 (NatF), a Golgi-localized bi-functional acetyltransferase. Sci. Rep. 6, 31425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miller M. B., Vishwanatha K. S., Mains R. E., and Eipper B. A. (2015) An N-terminal amphipathic helix binds phosphoinositides and enhances kalirin Sec14 domain-mediated membrane interactions. J. Biol. Chem. 290, 13541–13555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carney J., East J. M., Mall S., Marius P., Powl A. M., Wright J. N., and Lee A. G. (2006) Fluorescence quenching methods to study lipid-protein interactions. Curr. Protoc. Protein Sci. Chapter 19, Unit 19 12 [DOI] [PubMed] [Google Scholar]
- 34. Chattopadhyay A., and London E. (1987) Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry 26, 39–45 [DOI] [PubMed] [Google Scholar]
- 35. van Meer G., Voelker D. R., and Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell. Biol. 9, 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammond G. R., and Balla T. (2015) Polyphosphoinositide binding domains: key to inositol lipid biology. Biochim. Biophys. Acta 1851, 746–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lemmon M. A. (2008) Membrane recognition by phospholipid-binding domains. Nat. Rev. Mol. Cell Biol. 9, 99–111 [DOI] [PubMed] [Google Scholar]
- 38. Lazaridis T. (2003) Effective energy function for proteins in lipid membranes. Proteins 52, 176–192 [DOI] [PubMed] [Google Scholar]
- 39. Hajjar E., Mihajlovic M., Witko-Sarsat V., Lazaridis T., and Reuter N. (2008) Computational prediction of the binding site of proteinase 3 to the plasma membrane. Proteins 71, 1655–1669 [DOI] [PubMed] [Google Scholar]
- 40. Banfield D. K. (2011) Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 3, a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. van Meer G., and de Kroon A. I. (2011) Lipid map of the mammalian cell. J. Cell Sci. 124, 5–8 [DOI] [PubMed] [Google Scholar]
- 42. Fritzsching K. J., Kim J., and Holland G. P. (2013) Probing lipid-cholesterol interactions in DOPC/eSM/Chol and DOPC/DPPC/Chol model lipid rafts with DSC and 13C solid-state NMR. Biochim. Biophys. Acta 1828, 1889–1898 [DOI] [PubMed] [Google Scholar]
- 43. Sodt A. J., Pastor R. W., and Lyman E. (2015) Hexagonal substructure and hydrogen bonding in liquid-ordered phases containing palmitoyl sphingomyelin. Biophys. J. 109, 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 46. Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr, and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jo S., Kim T., Iyer V. G., and Im W. (2008) CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 [DOI] [PubMed] [Google Scholar]
- 48. Chon N. L., Osterberg J. R., Henderson J., Khan H. M., Reuter N., Knight J. D., and Lin H. (2015) Membrane docking of the synaptotagmin 7 C2A domain: computation reveals interplay between electrostatic and hydrophobic contributions. Biochemistry 54, 5696–5711 [DOI] [PubMed] [Google Scholar]
- 49. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Feller S. E., Zhang Y., Pastor R. W., and Brooks B. R. (1995) Constant-pressure molecular-dynamics simulation: the Langevin piston method. J. Chem. Phys. 103, 4613–4621 [Google Scholar]
- 51. Izaguirre J. A., Reich S., and Skeel R. D. (1999) Longer time steps for molecular dynamics. J. Chem. Phys. 110, 9853–9864 [Google Scholar]
- 52. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 53. Andersen H. C. (1983) Rattle: a velocity version of the shake algorithm for molecular-dynamics calculations. J. Comput. Phys. 52, 24–34 [Google Scholar]
- 54. Brooks B. R., Brooks C. L. 3rd, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph 14, 33-38, 27–38 [DOI] [PubMed] [Google Scholar]
- 56. Guixà-González R., Rodriguez-Espigares I., Ramírez-Anguita J. M., Carrió-Gaspar P., Martinez-Seara H., Giorgino T., and Selent J. (2014) MEMBPLUGIN: studying membrane complexity in VMD. Bioinformatics 30, 1478–1480 [DOI] [PubMed] [Google Scholar]
- 57. Fujiki Y., Hubbard A. L., Fowler S., and Lazarow P. B. (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J. Cell Biol. 93, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aksnes H., Osberg C., and Arnesen T. (2013) N-terminal acetylation by NatC is not a general determinant for substrate subcellular localization in Saccharomyces cerevisiae. PLoS One 8, e61012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.