

Abstract
Kinases are amongst the largest families in the human proteome and serve as critical mediators of a myriad of cell signaling pathways. Since altered kinase activity is implicated in a variety of pathological diseases, kinases have become a prominent class of proteins for targeted inhibition. Although numerous small molecule and antibody-based inhibitors have already received clinical approval, several challenges may still exist with these strategies including resistance, target selection, inhibitor potency and in vivo activity profiles. Constrained peptide inhibitors have emerged as an alternative strategy for kinase inhibition. Distinct from small molecule inhibitors, peptides can provide a large binding surface area that allows them to bind shallow protein surfaces rather than defined pockets within the target protein structure. By including chemical constraints within the peptide sequence, additional benefits can be bestowed onto the peptide scaffold such as improved target affinity and target selectivity, cell permeability and proteolytic resistance. In this review, we highlight examples of diverse chemistries that are being employed to constrain kinase-targeting peptide scaffolds and highlight their application to modulate kinase signaling as well as their potential clinical implications.
Keywords: constrained peptide, kinase inhibitor, stapled peptide, peptide macrocycle
1. Introduction
Kinases play the fundamental role of phosphorylating various substrates, and this seemingly minor event causes profound effects on cell signaling. The kinase superfamily is encoded by one of the largest gene families in the human genome and is composed of approximately 539 kinases (Brognard & Hunter, 2011). Since kinases are key regulators of various cellular pathways, altered activity or regulation of kinases can result in dire consequences including diabetes, inflammation and cancer (Fabbro, Cowan-Jacob, & Moebitz, 2015). As such, kinases have garnered much attention by the pharmaceutical industry where numerous kinases are the focus of targeted inhibition.
The vast majority of kinase inhibitors are ATP-competitive small molecule inhibitors that target the ATP-binding pocket centered between the small and large lobes of the kinase domain (J. Zhang, Yang, & Gray, 2009). However, several challenges exist with small molecule targeting including target selectivity and inhibitor resistance (J. Zhang, et al., 2009). To date, small molecule kinase inhibitors that have been developed for clinical applications have effectively targeted only a relatively small proportion of the kinome since only a subset of kinases has been viewed as highly sought-after targets. However, a major remaining challenge is that many patients develop resistance to clinical kinase inhibitors (Smith, 2016; H. Zhang, 2016) and many disease-relevant kinases still lack clinically relevant therapeutic inhibitors. Another alternative strategy for kinase inhibition is the use of monoclonal antibodies. Since antibodies are not cell-permeant, they require an extracellular surface for binding and thus are limited to receptor kinases such as EGFR and Her2. While antibodies can achieve high target specificity, a major limitation is that the vast majority of kinases lack extracellular features that can be targeted using this approach. Thus, there is a significant need for alternative targeting approaches.
One strategy that has emerged is that of constrained peptide inhibitors. Unlike small molecules, peptides present a large binding surface area and therefore can bind relatively large, shallow surfaces on the target protein. Further, by applying constraints to the peptide scaffold, compound properties can be altered to gain features such as cell permeability, improved proteolytic resistance, improved binding affinity and improved target potency. Constrained peptide scaffolds have been used to target various features of kinases including the area surrounding the ATP-binding site, ligand-binding site, substrate-binding site, and protein-protein interactions that regulate kinase activity. In addition, peptide-based allosteric inhibitors were also developed as kinase inhibitors. By targeting alternative sites on the surfaces of kinases, areas can be targeted with greater evolutionary divergence as compared to the ATP-binding site, thereby lowering the bar for target specificity. Here, we highlight several examples of synthetic and target site strategies for modulation of kinase signaling.
2. Constrained peptide-small molecules targeting the kinase ATP-binding site
Small molecule inhibitors are routinely designed to target the ATP-binding site of kinases; however, selectivity remains a major challenge due to conservation of the binding pocket across the kinase superfamily. As a strategy to improve selectivity of the promiscuous kinase inhibitor staurosporine, bivalent peptide-small molecule ligands were developed to simultaneously target the active site as well as a nearby surface on the large lobe of the kinase domain of Protein Kinase A (PKA) (Figure 1) (Meyer, Shomin, Gaj, & Ghosh, 2007). To achieve this, carboxylated staurosporine was non-covalently attached to head-to-tail cyclized peptides by employing the heterodimer of transcription factors Jun and Fos, which interact to form a coiled coil. In this strategy, the Jun-conjugated staurosporine derivative and Fos-conjugated peptide self-assemble, thereby simultaneously presenting a molecule to target the ATP-binding site and another to interact with a neighboring site on PKA. Phage display was used to create the library of Fos-cyclic peptide variants that incorporated only natural amino acids for peptide residues 2–7 (Meyer, et al., 2007). Multiple rounds of phage display selection were used to identify a lead cyclic peptide that could bind PKA and later demonstrated micromolar inhibition of PKA activity. This cyclic peptide was then covalently attached to carboxylated staurosporine via a short PEG linker, which led to a significant improvement in inhibitory potency in vitro as compared to the staurosporine analog alone (IC50 values of 2.6 nM versus 159 nM). Moreover, when the bivalent inhibitor was compared against the staurosporine derivative in a panel of six different kinases (PKA, ASK1, CaMKIIβ, c-Src, EphA5, and Mnk2), the bivalent inhibitor demonstrated considerable in vitro selectivity towards PKA. Additional modifications to this bivalent inhibitor demonstrated that the individual components (small molecule, linker or peptide) could be altered in a modular fashion to improve inhibitory potency and target selectivity (Shomin, Meyer, & Ghosh, 2009). The generality of this approach therefore has the potential to be applied to diverse kinases during inhibitor development by bestowing improved selectivity and affinity on lead small molecule inhibitors. Although peptide potency can be enhanced using this synthetic strategy, the peptide alone remained a relatively weak binder to its intended target. Additionally, a major caveat of this approach is that the overall compound size is significant since the bifunctional peptide-small molecule conjugate is required for high affinity binding, and may therefore have limited therapeutic potential for targeting intracellular kinase domains.
Figure 1. Peptide-molecule conjugates targeting the ATP-binding site.

An ATP-competitive compound can be linked to constrained peptide macrocycles to create a bivalent inhibitor that blocks ATP with increased affinity while also bestowing improved selectivity on the small molecule where the peptide binds the adjacent surface on the kinase domain.
3. Constrained peptides targeting the kinase ligand-binding site
Peptides targeting ligands or the ligand-binding domain of enzyme-linked receptors can be designed to serve as modulators of kinase activity, thereby regulating signal transduction cascades that contribute to a variety of cellular processes. Of the enzyme-linked receptors, receptor tyrosine kinases have received much attention as targets for the development of anti-proliferative, anti-metastatic, and anti-angiogenic compounds in cancer due to their roles in cell growth and motility (Regad, 2015). A variety of constrained peptides have been developed to target ligand-induced activation of receptor tyrosine kinases by blocking the receptor-binding surface of the ligand or by occluding the ligand-binding site of the receptor (Blaskovich, et al., 2000; De Rosa, et al., 2014; Diana, et al., 2011; Guardiola, et al., 2016; Lamberto, et al., 2014; Lamberto, et al., 2012; Murai, et al., 2003; Nakamura, et al., 2005; Tam, et al., 2009; Vicari, Foy, Liotta, & Kaumaya, 2011). By blocking ligand binding, the peptides can prevent the conformational change and dimerization that promotes kinase activation and subsequent tyrosine phosphorylation events (Figure 2).
Figure 2. Inhibition of receptor kinases via ligand-binding site inhibition.
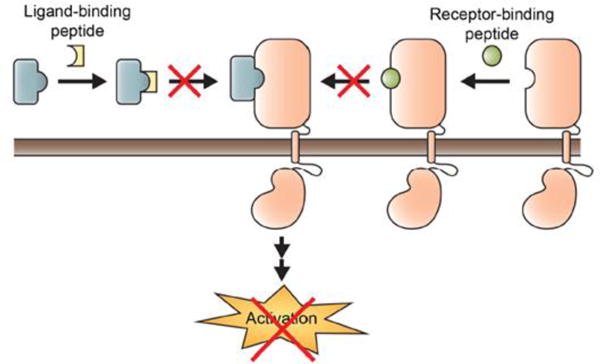
Receptor kinases can be activated by extracellular signals that bind the ectodomain of the kinase. These extracellular ligand-binding sites can be selectively targeted to ultimately inhibit kinase activation. Examples include the interaction between PDGF and PDGFR, EGF and EGFR, HGF and the Met receptor and VEGF and VEGFR.
The development of ligand-targeted compounds capable of blocking the ligand-receptor interaction has been a considerable challenge due to the large size of the receptor-binding surface on growth factors. However, multiple approaches, including functionalized scaffolds, miniproteins, and phage display, have been employed to develop peptide-based compounds capable of binding such surfaces (Blaskovich, et al., 2000; Guardiola, et al., 2016; Tam, et al., 2009). In one example of a ligand-targeted approach, a peptide-functionalized scaffold was developed to sequester platelet-derived growth factor (PDGF) and thereby block platelet-derived growth factor receptor (PDGFR) activation in cancer (Blaskovich, et al., 2000). PDGFR has roles in growth and motility that may contribute to proliferation and angiogenesis in cancer and has thus received attention as a potential target for therapeutic development (Andrae, Gallini, & Betsholtz, 2008). The PDGF hetero- or homodimer binds to PDGFR, inducing dimerization and activation of the receptor (Andrae, et al., 2008). Thus, compounds capable of blocking PDGF binding to the receptor may block PDGFR activation. A library of cyclic hexapeptides was designed using a 3-aminomethylbenzoate dipeptide mimic to cyclize the peptide via amide bonds (Blaskovich, et al., 2000). The cyclic peptides were attached to a calix[4]arene scaffold to generate a functionalized surface that could bind the receptor-binding surface on PDGF. The library was modified to test the effect of different charged and hydrophobic patches on binding to PDGF. A peptide complementary to the basic and hydrophobic surface of PDGF blocked the PDGF-B homodimer, PDGF-BB, from binding to PDGFR with an IC50 value of 250 nM as measured in cells. This compound was reported to inhibit PDGFR phosphorylation and downstream processes including DNA synthesis, tumor growth and angiogenesis. While the large size of these molecules may affect delivery of the peptide, cyclization may enhance peptide stability towards proteases. This approach may also offer an alternative to ligand- or receptor-targeted antibodies that block receptor tyrosine kinase activation.
In another ligand-targeted approach, peptides were developed to target hepatocyte growth factor (HGF) and block its binding to the Met receptor (Tam, et al., 2009). Like other receptor tyrosine kinases, the Met receptor dimerizes and is activated by binding of its ligand, HGF, promoting signaling pathways involved in cell cycle regulation (Gherardi, Birchmeier, Birchmeier, & Woude, 2012). As dysregulation of HGF and Met is observed in a variety of cancers, blocking HGF binding to Met may prevent ligand-induced activation of Met in cancer. Phage display was used to identify and optimize linear and disulfide cross-linked peptides with affinity towards HGF, resulting in a disulfide-bridged peptide capable of binding the β-chain of HGF. The 15-residue peptide was constrained to a hairpin structure containing a type-I β-turn and could inhibit Met in vitro with an IC50 of 450 nM. In the absence of the disulfide cross-link, activity of the peptide was diminished, and thereby demonstrates the importance of the constraint for the antagonistic effect of the peptide. Interestingly, this peptide did not disrupt binding of full length HGF to Met, yet it was capable of blocking Met phosphorylation, revealing the importance of the β-chain of HGF in Met activation.
In addition to PDGF and HGF, epidermal growth factor (EGF) has also received considerable attention as a target in the development of peptides blocking ligand-receptor interactions. Binding of EGF to its receptor, the epidermal growth factor receptor (EGFR), induces dimerization and activation of EGFR (Lemmon, Schlessinger, & Ferguson, 2014). Deregulated EGFR signaling promotes proliferative signaling in cancer. Thus the EGFR signaling pathway is an attractive target for therapeutic development. In a recent study, a panel of peptides was developed to bind to EGF and block activation of EGFR signaling pathways (Guardiola, et al., 2016). In one strategy, a miniprotein was designed to mimic the helix and loop structures of EGFR that interact with EGF. The peptide was stabilized by installing a disulfide bridge between the terminal cysteine residues, thereby fashioning a cyclized peptide. This peptide displayed micromolar affinity towards EGF, blocked the interaction between EGF and EGFR with an IC50 value of 60.6 μM using a cell-based EGF-EGFR inhibition assay. Further the peptide was shown to inhibit EGF-mediated cell proliferation. Since the mini-protein contains both loop and helical structures, further studies with additional mutations or truncations of the mini-protein may reveal the contribution of specific features, such as the helix, to binding and inhibition.
Another strategy to develop inhibitors of the ligand-binding site is through the use of monoclonal antibodies that recognize a specific conformation-epitope (Fukumoto, et al., 1998). Using an anti-EGF monoclonal antibody, a disulfide-constrained phage display library was screened to identify lead peptides with an enhanced affinity towards the EGF-targeted antibody (Nakamura, et al., 2005). A lead candidate peptide was subsequently modified to contain an intramolecular disulfide bridge. One disulfide-bridged lead was identified that could bind EGFR, block EGF binding with an IC50 value of 70 μM through an EGF competition assay in cells and inhibit EGFR phosphorylation and mitogenesis in cells. Although micromolar concentrations of the constrained peptide were required to inhibit ligand-induced proliferation, this may serve as a scaffold for further optimization to improve potency.
Another receptor tyrosine kinase, vascular endothelial growth factor receptor (VEGFR), has roles in angiogenesis (Shibuya, 2013), and has become an attractive target for the development of anti-angiogenic peptides (De Rosa, et al., 2014; Diana, et al., 2011; Vicari, et al., 2011). A peptide mimicking a receptor-binding loop of vascular endothelial growth factor (VEGF) was developed to bind VEGFR-2 and block its activation in cancer (Vicari, et al., 2011). The peptide was designed to mimic the antiparallel β-sheet conformation of the native structure, with a disulfide cross-link between strands to constrain the loop. This peptide was reported to bind VEGFR-2 with a KD of 11 nM in vitro and decrease VEGFR-2 phosphorylation, proliferation and wound healing. The anti-tumor activity of the constrained peptide was also evaluated in a double transgenic mouse model, VEGF(+/−) Neu2–5(+/−). Using this model, the constrained peptide appeared to reduce tumor volume highlighting its translational potential to serve as an anti-tumor agent.
In a similar study, peptides were constrained with a disulfide bond to mimic a β-hairpin feature of placental growth factor (PIGF) and block activation of VEGFR-1 (De Rosa, et al., 2014). In this approach, additional conformational stability was achieved via aromatic interactions between tryptophan residues from the opposing strands that helped to stabilize the type-II β-turn structure. Although no direct binding or inhibitory effect on VEGFR was reported, the peptides were capable of blocking downstream signaling events and decreasing VEGF-induced proliferation in cells. Interestingly, in the absence of the disulfide bond, the peptide was reported to have agonistic activity (Diana, et al., 2011), suggesting the importance of the disulfide cross-link for antagonistic activity.
Altered activity stemming from the ephrin type-A receptor 4 (EphA4) is linked to cancer, neurodegenerative diseases and neural repair (Faruqi, 2012), leading to efforts to disrupt EphA4 activity. Peptides with affinity towards EphA4 were identified using a random 12-mer phage display library and screened against EphA4, leading to the development of a disulfide bridged EphA4 binding peptide (Lamberto, et al., 2014; Lamberto, et al., 2012; Murai, et al., 2003). The lead peptide, termed APY, was subsequently crystalized with the ligand binding domain of EphA4, leading to rational design in efforts to improve efficacy (Lamberto, et al., 2014). The original APY sequence was optimized to stabilize the hairpin structure by introducing a C-terminal amide while releasing strain at the turn residues by substituting glycine with a β-alanine residue. These modifications improved in vitro IC50 values from 1.4 μM for the parent APY peptide to 30 nM for the optimized sequence. Further, the improved peptide could disrupt EphA4 activation without toxicity in cells. Since the original APY peptide showed decreased activity after incubation with serum, it would be interesting to determine whether the modifications, particularly the C-terminal amide, improve proteolytic stability because this would affect its translational potential.
A major advantage of targeting ligands or ligand-binding domains is that cell permeability of the targeting agent is not necessary since the target is extracellular. Furthermore, given these constrained peptides do not target the highly conserved kinase active site and there is greater diversity amongst the ligand binding sites, this may limit off-target effects that are often observed with ATP-competitive inhibitors. However, the peptide must outcompete the ligand, which may be overexpressed in some disease states such as cancer. Further, this targeting strategy is largely limited to the receptor tyrosine kinase subfamily since the vast majority of kinases lack extracellular ligand binding domains. From a synthetic standpoint, many of the constrained peptides discussed here have used disulfide bridging to stabilize the secondary structure of the targeting peptide. A major shortcoming of this chemistry is that disulfide bridging is susceptible to reduction of the bond and loss of the constraint under reducing conditions. Exploration of alternative bridging strategies may lead to identification of improved pharmacological compound properties including enhanced activity and stability.
4. Constrained peptides targeting the kinase substrate-binding site
While specificity remains a major challenge for targeting the ATP-binding site of kinases, the substrate-binding site represents an alternative strategy since this site is more divergent amongst the kinase superfamily. However, this binding site is relatively flat and elongated on the surface of the large lobe of the kinase domain, and therefore lacks features that make it amenable to small molecule targeting approaches. As increasing numbers of synthetic and natural peptide substrate sequences have been identified, significant efforts have been put forth to generate constrained peptides that target the substrate binding site and thus downregulate kinase activity (Figure 3).
Figure 3. Peptides targeting the substrate-binding site.
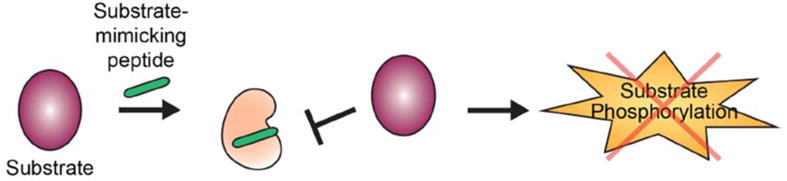
Constrained peptides mimicking peptide substrates can be designed to occlude the substrate-binding groove, thereby inhibiting substrate phosphorylation for a kinase of interest. Successfully targeted kinases using this strategy include c-Src, Abl, Akt and CDK2.
An early example of a substrate-binding peptide macrocycle was developed by building upon previous work that identified a c-Src-selective peptide substrate sequence from a “one-bead one-peptide” approach (Lam, Wu, & Lou, 1995). Using this sequence, amino acid substitutions and disulfide bridging was introduced into the Src peptide substrate to produce constrained β-turn peptides with IC50 values ranging from 26 μM to 130 nM in vitro against Src (Alfaro-Lopez, et al., 1998). Further, they demonstrated that their lead compounds were selective in vitro for Src over Lyn or Lck by up to 1000-fold in some cases. The cyclized peptides were slightly more potent than their linear counterparts, thus inferring that a secondary structural fold may impart improved binding on Src and may have more favorable entropic conformer for binding. An interesting observation was that inclusion of tyrosine mimetics that could not be phosphorylated resulted in the most potent inhibitors identified from this work, albeit this modification also resulted in reduced selectivity between Src and Lck.
In a similar study, a previously identified c-Src pseudosubstrate inhibitor peptide (Kamath, Liu, Enstrom, Lou, & Lam, 2003) was conformationally constrained and modified in a medicinal chemistry approach to improve the potency of the parent compound (Kumar, et al., 2006). Using the non-specific tyrosine kinase substrate peptide polyE4Y as a means to assess Src inhibition, they sought to improve inhibitor potency by modifying the functionality of the tyrosine residues while either leaving the peptide linear or constraining it via head-to-tail, side chain or terminus-to-side chain bridging. Interestingly, simply replacing tyrosine on position 5 of the linear peptide with a 4-nitrophenylalanine led to a significant increase in inhibitory potency as compared to the parent compound. However, the addition of constraints on the parent sequence also conferred improved inhibitory properties when head-to-tail cyclization or side chain bridging was introduced. Additional work also demonstrated that head-to-tail cyclized peptides with alternating basic and aromatic residues inhibit Src activity in vitro with IC50 values as low as 280 nM (Shirazi, et al., 2013). Moreover, inclusion of arginine instead of lysine, ring size and overall peptide length of cyclized peptides could further enhance potency of these pseudosubstrate Src inhibitors.
Bcr-Abl is an oncogenic fusion protein that arises as a result of a chromosomal translocation event (Nowell, 2007). While Abl kinase activity is normally autoinhibited, the Bcr-Abl fusion maintains constitutive activity and activates oncogenic signaling and transformation in chronic myeloid leukemia (CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL) (Hantschel, 2012). Although numerous ATP-competitive inhibitors have been developed to target Bcr-Abl, drug resistance remains a major challenge (Zabriskie, et al., 2014). As an alternative targeting strategy, substrate-based peptide inhibitors were developed to target the Abl kinase domain (Huang, et al., 2015). In this work, cyclotides were employed as a strategy to stably present an Abl substrate. Cyclotides are protease-resistant, cysteine-rich peptides that are head-to-tail cyclized and contain a cyclic cysteine knot (CCK) motif (Colgrave & Craik, 2004). One such cyclotide, termed MCoTI-II, was also shown to be cell-permeant (Greenwood, Daly, Brown, Stow, & Craik, 2007) and was therefore used to incorporate a known Abl substrate sequence (Huang, et al., 2015). A library was first generated where analogs of the Abl substrate peptide abltide were grafted into MCoTI-II. Using in vitro assays, several of the cyclotide inhibitors were found to inhibit wild-type Abl or a mutant form of Abl (T315I) with IC50 values as low as 1.3 or 12.2 μM, respectively. Several lead inhibitors were also found to gain intracellular access to varying extents, albeit to a lesser degree as compared to a TAT sequence. Nonetheless, the peptides were strikingly resistant to proteolytic degradation. Three peptides demonstrated no detectable loss of the parent compound after a 24-hour incubation period with human serum, thereby illustrating the potential for long anticipated half-lives in vivo. A separate strategy to inhibit proteolytic degradation of an Abl peptide substrate was also demonstrated by incorporating a small, constrained β-turn peptide structure on the N-terminus of a linear substrate sequence (S. Yang, et al., 2013).
Akt (Protein Kinase B) is a member of the AGC kinase subfamily and phosphorylates numerous substrates to regulate processes such as metabolism, proliferation and survival (Manning & Cantley, 2007). Overexpression of Akt and aberrant Akt-mediated signaling is found in early and late state cancers (Manning & Cantley, 2007) and therefore serves as a major oncogenic drug target (Mundi, Sachdev, McCourt, & Kalinsky, 2016). A well-known substrate of Akt is GSK3β, and phosphorylation of this substrate causes GSK3β inactivation. Structural studies of a GSK3β peptide demonstrated that the peptide binds the substrate-binding site of Akt in an extended conformation (J. Yang, et al., 2002). As a strategy to synthetically mimic this extended structure, peptidomimetics were designed to replace two residues with an azabicycloalkane dipeptide surrogate using a GSK3β peptide sequence (Ranatunga & Del Valle, 2011). In vitro characterization of the peptidomimetic library was performed using purified Akt, where low micromolar IC50 values (3 μM) were achieved. Subsequent studies replaced the azabicycloalkane dipeptide surrogate with an imidazopyridine dipeptide surrogate as an alternative to mimic the extended Akt substrate peptide (Kim, et al., 2014). This synthetic modification improved in vitro potency with an IC50 value in the high nanomolar range (640 nM) and also improved proteolytic stability as compared to the non-modified parent sequence. GSK3β peptides were also designed as macrocyclic Akt substrates via urea backbone cyclization or head-to-tail amide backbone cyclization, of which several cyclized peptides were found to have increased in vitro inhibitory activity against Akt as compared to their linear counterparts (Tal-Gan, et al., 2011). Since Akt has over 100 known substrates (Mundi, et al., 2016), these strategies outlined above may provide additional opportunities to design alternative constrained pseudosubstrate targeting sequences for Akt inhibition.
Cyclin-dependent kinases (CDKs) regulate cell cycle transitions, and cyclin dependent kinase 2 (CDK2) is a key player in multiple aspects of these transitions including DNA replication and cell cycle progression (Boulikas, 1994). Altered regulation of CDK2 was identified in multiple cancers and has become the focus of targeted cancer therapies (Asghar, Witkiewicz, Turner, & Knudsen, 2015). CDK2 can phosphorylate substrates either through interactions on the substrate binding site of the kinase domain or through interactions with cyclin A when it is in a CDK2-cyclin A protein complex (Boulikas, 1994). Since some CDK2 substrates require recognition by cyclin A prior to CDK2 phosphorylation, one approach to inhibit kinase activity is to prevent substrates from docking on the cyclin A binding groove (CBG) to thereby prevent substrate presentation to CDK2 (Adams, et al., 1996; Andrews, et al., 2004). Using the crystal structure of a protein fragment from a known CDK2 inhibitor, p27KIP1, bound to the CBG of cyclin A in the CDK2-cyclin A complex (Russo, Jeffrey, Patten, Massague, & Pavletich, 1996), a macrocyclic peptide was designed using side-chain-to-C-terminal tail bridging to conformationally constrain the inhibitor peptide (Andrews, et al., 2004). Increased in vitro potency was observed for some of the constrained peptides with the lowest IC50 values reaching 360 nM; however, activity was dependent on overall ring size. This compound was further characterized by x-ray crystallography in complex with cyclin A, and this data revealed the additional energetically favorable interactions between the compound and the CBG of cyclin A. Thus, this structure-guided approach yielded a constrained analog with increased potency by inhibiting substrate presentation to CDK2.
5. Constrained peptides as allosteric disruptors of kinases
In addition to ligand and substrate binding, kinase activity can be regulated through allosteric mechanisms. Dimerization of receptor tyrosine kinases, such as EGFR, contributes to activation of the kinase domain and subsequent tyrosine phosphorylation events. Thus, disruption of dimerization or allosteric activation using constrained peptides is a promising strategy for modulation of kinase activity (Figure 4). Constrained peptides have been developed to disrupt EGFR dimerization in order to block EGFR signaling pathways in cancer. One strategy focuses on the disruption of the receptor dimer by targeting the EGFR dimerization arm, a β-loop structure that stabilizes the EGFR dimer (Hanold, Oruganty, et al., 2015; Hanold, Watkins, et al., 2015; Mizuguchi, et al., 2012; Mizuguchi, et al., 2009; Toyama, Mizuguchi, Nomura, & Tamamura, 2016). In this approach, cyclic peptides mimicking the EGFR dimerization arm were developed to occlude the dimerization arm-binding site. Multiple constraints have been applied to these peptides to improve activity and stability, including introduction of disulfide, selenylsulfide and triazolyl bridges. In some cases, cell-based inhibition was demonstrated with 10 μM IC50 values (Hanold, Oruganty, et al., 2015; Toyama, et al., 2016).
Figure 4. Allosteric regulation of kinases using peptide-based compounds.
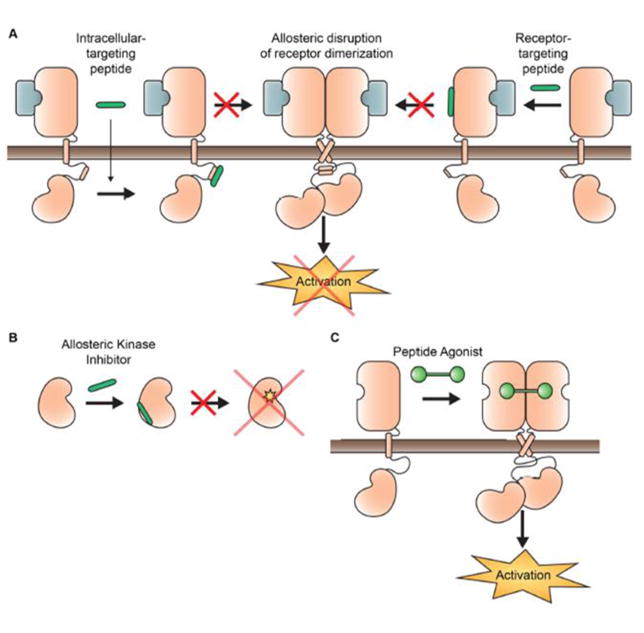
A) Dimerization of the kinase receptor, such as EGFR and Her2, can be blocked in a ligand-independent manner to block activation of the intracellular kinase domain. B) Peptides can be used to target an allosteric site of the kinase domain that is critical for activation, such as one on Aurora kinase A, thereby sequestering the kinase in an inactive state. C) Dimerization can also be induced by creating an activating peptide agonist that promotes receptor kinase dimerization and activation. An example of this targets the Met receptor.
Interactions modulating kinase activity also occur between intracellular regions of receptor tyrosine kinases. Upon receptor dimerization the EGFR kinase domains form an activated asymmetric dimer, and additional regulatory interactions occur between juxtamembrane regions of EGFR (Jura, et al., 2009; X. Zhang, Gureasko, Shen, Cole, & Kuriyan, 2006). To disrupt the juxtamembrane interactions and block kinase activation, all-hydrocarbon stapled peptides mimicking the helical structure of the juxtamembrane region were developed (Sinclair, Denton, & Schepartz, 2014; Sinclair & Schepartz, 2014). These peptides were similar to unconstrained peptides targeting the EGFR juxtamembrane (Boran, et al., 2012) and were able to block EGFR signaling and cell proliferation with IC50 values in the 10 μM range. Furthermore, these peptides demonstrated increased helicity compared to the unconstrained peptide. It is possible that higher concentrations of receptors at the membrane may lead to an increased presence of preformed dimers, and the efficacy of these strategies of dimer disruption may be dependent on expression levels of the receptor. Thus, determination of the effects of receptor expression levels on the ability of the peptides to disrupt dimerization could reveal the applicability of this strategy to different cell lines that express varying levels of EGFR.
Another member of the EGFR family is human epidermal growth factor receptor 2, Her2, which dimerizes with members of the EGFR family to promote kinase activity and proliferative signaling in cancer (Lemmon, et al., 2014). Thus, Her2 has received attention as a target for therapeutic development. Peptides mimicking interactions of trastuzumab, a Her2-targeted antibody that binds to domain IV of the ectodomain, were developed to disrupt Her2 signaling (Banappagari, Ronald, & Satyanarayanajois, 2011; Kanthala, Gauthier, & Satyanarayanajois, 2014; Satyanarayanajois, Villalba, Jianchao, & Lin, 2009). Through phosphorylation assays, a lead compound was identified that could inhibit proliferation at sub-micromolar levels (373 nM) and successfully inhibit Her2 dimerization. By targeting the dimerization interface of the ectodomain of the EGFR family proteins, one can strategically disrupt homo- and heterodimerization amongst the EGFR family members by targeting unique interaction surfaces. Although the IC50 values are relatively high, these strategies provide proof-of-principle that extracellular surfaces may be amenable for selective targeting.
In contrast to kinase inhibition by dimer disruption, constrained peptides may be developed to induce receptor dimerization and promote kinase activation. The Met receptor has received attention as a potential target for regenerative medicine due to its role in cell growth and motility (Ito, et al., 2015). A constrained peptide that acts as a Met agonist was reported to activate Met signaling pathways by inducing dimerization. In this study, peptides with affinity towards the Met receptor were generated using a random non-standard peptide integrated discovery (RaPID) approach. The peptides were cyclized by formation of a thioether bond between cysteine and an N-terminal chloroacetyl substituent, which helped to improve the in vitro binding affinity of the peptides to KD values as low as 2.3 nM. The resulting macrocyclic peptides were then cross-linked to form dimers that were capable of binding to an allosteric site, inducing receptor dimerization and exhibiting similar maximal activity as HGF in cell-based assays. Although, the peptide was designed for an extracellular target, determining whether or not similar peptides can penetrate cells could expand the application of this strategy to activate intracellular targets.
Bivalent approaches have also been explored as a means to increase the specificity of kinase inhibitors by covalently attaching a tyrosine kinase inhibitor to a peptide targeting an allosteric site of the kinase domain. This approach has been applied to Aurora kinase A (Shomin, Restituyo, Cox, & Ghosh, 2011), a serine/threonine kinase with roles in cell cycle regulation (Dar, Goff, Majid, Berlin, & El-Rifai, 2010). Peptides targeting Aurora kinase A were developed using phage display of disulfide bridged peptides in an approach similar to that of the bivalent, peptide-small molecule PKA inhibitors (Meyer, et al., 2007; Shomin, et al., 2009; Shomin, et al., 2011). Cyclic peptides binding Aurora A functioned in vitro as selective noncompetitive inhibitors of Aurora kinase A with IC50 values as low at 6 μM. While these peptides inhibited kinase activity in vitro, their activity in cells was not reported. Thus, the ability of these peptides to penetrate cells and access intracellular targets may warrant further exploration.
The use of constrained peptides targeting allosteric sites of kinases is a promising approach to modulation of kinase activity. This approach offers the advantage of regulating kinase activity without targeting highly conserved active sites, thereby potentially limiting off-target effects. Further, these peptides are not required to compete with ligand binding. However, the disruption of dimer interactions to modulate kinase activity may depend on the expression levels of the protein, especially in the presence of preformed dimers (Freed, Alvarado, & Lemmon, 2015).
6. Constrained peptides as disruptors of kinase-mediated protein-protein interfaces
Since kinases engage in multiple protein-protein interactions (PPIs) to regulate signaling cascades, numerous efforts have been put forth to block essential PPI interfaces as a strategy to downregulate kinase signaling (Figure 5). One example of this targeted approach aims to block an interface involved in regulating Protein Kinase A (PKA) activity. A-kinase Anchoring Proteins (AKAPs) serve as spatial and temporal regulators of the PKA holoenzyme complex. AKAPs are highly diverse in function and localization, but share the common defining feature of anchoring PKA in an isoform-specific manner via interactions with the regulatory subunits of PKA (PKA-R) (Kennedy & Scott, 2015). Although numerous peptide inhibitors were previously designed to target the AKAP-PKA interface as a means to regulate PKA in a spatial and temporal manner (reviewed in (Kennedy & Scott, 2015; Troger, Moutty, Skroblin, & Klussmann, 2012), cell-permeant “stapled” alpha-helical peptides were more recently designed to disrupt the same PPI interface (Wang, et al., 2014; Wang, et al., 2015). These stapled AKAP inhibitors have the added advantage of being both cell permeant and constrained in a pre-binding state. Therefore, these inhibitors do not require transfections or other methods such as TAT conjugation or aliphatic anchors for delivery. By targeting this PPI, these chemical probes allow one to perturb the role of AKAPs in various signaling processes within cells while simultaneously leaving the catalytic activity of PKA intact with in vitro KD values as low as 2 nM for the intended isoform-specific target and inhibitory effects in the low μM range in cells. However, a limitation of targeting this particular PPI is that current AKAP inhibitors act as universal disruptors by blocking PKA anchoring in an isoform-specific manner, i.e. all RI- or all RII-selective AKAPs (Kennedy & Scott, 2015; Troger, et al., 2012; Wang, et al., 2015), and therefore no cell-permeant inhibitors exist which target individual AKAPs. Since most cells express 10–15 AKAPs simultaneously (Lester, Coghlan, Nauert, & Scott, 1996), selective disruption of individual AKAPs will be ideal for investigating an AKAP of interest. This may be achieved by targeting unique, non-overlapping PPIs for individual AKAPs.
Figure 5. Disruption of protein-protein interactions involving kinases.
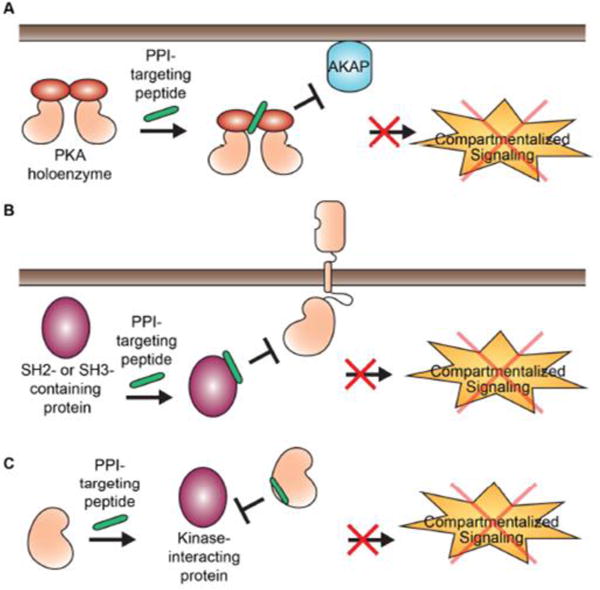
A) Spatiotemporal regulation of kinases can be disrupted by blocking PPI interfaces between kinases and adaptor proteins. Illustrated in this example is the inhibited interaction between PKA and AKAP proteins to disrupt compartmentalized signaling. B) SH2 or SH3 domains can also be selectively targeted using peptide-based compounds to block localized signaling interactions with kinases. C) Some kinases have defined protein interactions that are distinct from the substrate-binding site such as the RH domain of GRK5 and 6, the polo-box domain of Plk1 or the helical domain of PI3K. These unique interaction sites can be selectively targeted for peptide-based inhibition.
In addition to targeting PPIs regulating PKA, a PPI interface was also targeted to disrupt regulation of Casein Kinase II (CKII). CKII regulates a myriad of cellular processes including differentiation and proliferation. Like PKA, CKII forms a dynamic tetrameric holoenzyme complex consisting of two catalytic (CKIIα and CKIIα′) and two regulatory subunits (CKIIβ). The catalytic subunits of CKII are constitutively active, thus the regulatory subunits serve to regulate both activity and intracellular location (Bibby & Litchfield, 2005). In order to synthetically dissect the tetrameric complex, constrained peptides were designed to mimic a C-terminal sequence stemming from CKIIβ (Laudet, et al., 2007). Then, by introducing and characterizing alanine substitutions around a conserved sequence stretch on the C-terminal portion of CKIIβ, the binding hot spots of the interface between CKIIα and CKIIβ were identified. A 13-mer peptide was designed based on the conserved sequence from a beta-loop structure of CKIIβ that encompassed two of the identified hotspots. The loop structure was constrained by disulfide bridging between two Cys residues and was engineered to target the binding surface of CKIIα. Binding assays with purified protein demonstrated inhibition of the CKII tetrameric holoenzyme complex in the presence of increasing concentrations of the constrained inhibitory peptide, and in vitro phosphorylation assays with a purified CKII substrate showed inhibited phosphorylation by the inhibitor peptides with IC50 values ranging down to 6.5 μM (Laudet, et al., 2007). Although this CKII disruptor was not shown to cross the cell membrane, it validates a targeting site to disrupt the CKII holoenzyme complex and provides a basis for further optimization and design of cell-permeant peptide scaffolds.
Another strategy to modulate kinase activity is through inhibition of Src Homology 2 (SH2) domain interactions. SH2 domains recognize and bind phosphotyrosine-containing sequences, thereby compartmentalizing and regulating signal transduction in a phosphorylation-dependent manner. Initial efforts were focused on the development of head-to-tail cyclized hexameric peptides that were derived from PDGFRβ and designed to target the SH2 domain of PI3K (Burke, et al., 1994). Significant efforts have also focused on targeting the SH2 domain of growth factor receptor-binding protein (Grb2). Grb2 is an adaptor protein that links proteins with both receptor and non-receptor tyrosine kinases including Her2 (Belov & Mohammadi, 2012). Initial efforts identified a minimal pentameric phosphotyrosine-containing sequence, that once cyclized by disulfide or lactam bridging, selectively bound the SH2 domain of Grb2 (Ettmayer, et al., 1999; Gay, et al., 1997). Subsequent efforts include the design of β-turn loops using olefin metathesis (Gao, Voigt, Wu, Yang, & Burke, 2001). Through in vitro binding studies, several peptide candidates were identified with IC50 values as low as 20 nM. Further, these peptides appeared to permeate the cell membrane and phosphorylation of Her2 was found to be downregulated after treating whole cells with the Grb2 macrocycle antagonists. Multiple variations of cyclized Grb2 inhibitors were later developed with the installment of C-terminal β-functionalized allylglycines for ring-closing metathesis (RCM) (Oishi, et al., 2005), azide-alkyne cycloaddition macrocycles (Choi, et al., 2006), thioether-bridged macrocyclization (Jiang, et al., 2009) and peptide bicycles containing both head-to-tail and side chain cyclization (Quartararo, Wu, & Kritzer, 2012). Many of these peptides demonstrated improved properties including increased affinity, improved inhibition and enhanced proteolytic stability. A shortcoming of many of the SH2-targeting Grb2 inhibitor peptides discussed may include limited cell permeability. However, due to their high affinity, modifications may be introduced into the optimized scaffolds to increase access to the intracellular environment.
Since Grb2 is a desirable therapeutic target due to its key role in cancer cell signaling by mediating receptor tyrosine kinase signaling, constrained peptides were also developed to target other domains of this protein (Liu, et al., 2011). Grb2 contains a central SH2 domain bordered by SH3 domains on both sides. SH3 domains mediate protein-protein interactions with proline-rich sequences through a general Pro-X-X-Pro (PxxP) motif contained in either polyproline type I or polyproline type II helices. Type II helices adopt a distinct left-handed, extended structure (Adzhubei, Sternberg, & Makarov, 2013), and this structure was mimicked via macrocyclization to inhibit Grb2 (Liu, et al., 2011). Alkenyloxy-bearing proline analogs were introduced into the PxxP motif and cyclized using RCM chemistry in order to stabilize the binding motif. Interestingly, macrocyclization led to a loss of binding affinity in in vitro binding assays for the compounds tested, from a KD value of 11 μM for the parent compound to 12 to 173 μM for the derivatives. However, the macrocyclic peptides had increased helicity and were able to effectively block Grb2-Sos1 interactions at micromolar concentrations in cell lysates.
A constrained type I polyproline helix was also synthesized to target the SH3 domain of Src. Src is a non-receptor tyrosine kinase that regulates a multitude of signaling pathways and is implicated in a variety of cancers (Roskoski, 2015). Using a previously identified type I sequence that binds the SH3 domain of Src (Feng, Kasahara, Rickles, & Schreiber, 1995), constrained versions were designed with the aim of improving target affinity (Tiwari, Brown, Narramaneni, Sun, & Parang, 2010). Three different constrained peptides were generated: N-terminal-to-side chain, C-terminal-to-side chain and head-to-tail macrocycles. By in vitro binding assays, two versions of the constrained peptide demonstrated decreased affinity towards Src SH3; however, the C-terminal-to-side chain cyclized peptide had a comparable target affinity as compared to the linear control (KD values of 350 nM versus 340 nM for the control using in vitro binding studies with purified SH3). Thus,this work demonstrated that placement of the constraint may disrupt key critical interactions for SH3 binding.
The SH2 domain of Src was also explored as a target for constrained peptide scaffolds (Nam, Ye, Sun, & Parang, 2004). Tetrameric, phospho-tyrosine bearing sequences were designed with the intent to boost interactions with the P and P+3 sites on the SH2 domain. As compared to their linear analogs, cyclization of the peptides significantly boosted in vitro inhibitory activity towards purified SH2 with IC50 values of 6.5 μM for the linear peptide up to 1.1 μM for a cyclized analog. Molecular modeling further revealed that constraining the peptide scaffold enhanced interactions with the two key binding sites (P and P+3) on the surface of the SH2 domain. The key interaction distances between the SH2-binding sites that were defined by this work could serve to design optimized SH2-interacting ligands.
G-protein coupled receptors (GPCRs) undergo phosphorylation by a class of serine/threonine kinases termed G-protein coupled receptor kinases (GRKs). An allosteric site in the regulator G protein signaling homology (RH) domain of GRK5 and 6 was first identified and found to contain multiple conserved residues that are important for catalytic activity (Baameur, et al., 2010). Importantly, this region is located outside of the kinase domain yet directly influences substrate phosphorylation (Baameur, et al., 2010). As a strategy to disrupt GRK5/6 activity, a helix derived from the RH domain, helix 9, was synthesized using side chain-to-side chain cyclization so as to chemically reinforce the alpha-helical structure of the peptides. At high concentrations, the constrained peptides were shown to reduce GRK5/6-mediated phosphorylation of rhodopsin in vitro using purified protein, albeit to a similar level as compared to their non-constrained counterpart with an approximate IC50 value of 30 μM. These inhibition studies highlight the significance of the RH domain on activation of GRK5/6 kinase activity and represent an indirect strategy for targeted inhibition of GRK kinases. Effective targeting of the GRKs would serve to both downregulate intracellular GRK-mediated signaling but would also serve the benefit of downregulating GPCR signaling.
The catalytic subunit of phosphatidylinositol-4,5-bisphosphate 3-kinase (PIK3CA, p110α) is a lipid kinase that relays extracellular signals, leading to the activation of various intracellular signaling events. Acquired mutations are frequently found in PI3K in diverse cancers, leading to overactivation of PI3K signaling in cells (Thorpe, Yuzugullu, & Zhao, 2015). Most mutations were found at two sites within p110α: either within the helical domain or within the kinase domain of p110α and have different effects on oncogenic signaling. Mutations found in the helical domain frequently occur at several acidic residues including E542 and E545, and, unlike the wild-type, several of these helical domain p110α mutants were found to interact with insulin receptor substrate 1 (IRS1) in a phosphorylation-independent manner and stabilize the catalytic subunit of mutant p110α (Hao, et al., 2013). Since the interaction between IRS1 and the p110α mutant E545K was further found to be required for growth of xenograft tumors (Hao, et al., 2013), a stapled peptide was designed to mimic the helical domain of the p110α E545K mutant as a strategy to block interactions with IRS1. This stapled peptide inhibited binding interactions between IRS1 and p110α E545K at 50 μM in both cell lysates and live cells. They further demonstrated that the constrained peptide could inhibit tumor growth in mouse xenograft models bearing the E545K mutation. Since this mutant form of p110α causes a modified protein-protein interaction with IRS1 in cancer, this work demonstrates the feasibility of targeting this PPI in signaling as a strategy for selective targeting of altered cell-signaling pathways in cancer.
Polo-like kinase 1 (Plk1) regulates mitosis and cell proliferation, and targeted inhibition of Plk1 was shown to inhibit mitosis and promote apoptosis (Gutteridge, Ndiaye, Liu, & Ahmad, 2016). Since Plk1 is overexpressed in diverse cancers, it has become an appealing anticancer target. The C-terminus of Plk1 contains a conserved polo-box domain (PBD) that binds phosphorylated serine/threonine sequences to regulate interactions of the Plk1 kinase domain with its substrates (Park, et al., 2010). Using a previously identified phospho-threonine-containing pentameric sequence as a scaffold to target the PBD domain (Yun, et al., 2009), constrained cyclized peptides or peptomers were designed to inhibit Plk1 (Murugan, et al., 2013). Cyclization was performed using either head-to-tail or side chain-to-tail linkages while exploring different bridging chemistries. The peptide/peptomer library was screened for Plk1 inhibition using an ELISA-based assay to identify inhibitory scaffolds. Two cyclized peptomers were identified from in vitro binding assays and showed an improved binding affinity for the PBD of Plk1 as compared to the linear control (1.1 and 2.6 μM versus 32 μM for the control). They also demonstrated binding selectivity for the PBD of Plk1 over the PBD derived from the closely related proteins Plk2 and Plk3. Since the PBD is a key regulator of Plk1 substrate phosphorylation, this serves as an alternative strategy for inhibiting Plk1 without the need for targeting its kinase domain.
7. Clinical implications and perspectives
Due to the critical roles of kinases in cellular processes, dysregulation of kinases is often associated with a variety of disease states. As such, kinases are widely targeted in the clinic by numerous approved inhibitors that have recently been reviewed (Fabbro, 2015; Muller, Chaikuad, Gray, & Knapp, 2015; Wu, Nielsen, & Clausen, 2016). The primary strategies used to target kinases have focused on the development of small molecule kinase inhibitors or monoclonal antibodies so as to disrupt kinase signaling. Although these strategies have shown promising successes in the clinic, each possesses certain limitations or drawbacks that have prompted discovery of alternative targeting strategies. Small molecules designed to target the kinase active site often exhibit off-target effects due to the high conservation of this site between kinase families (De Smet, Christopoulos, & Carmeliet, 2014; Wu, Clausen, & Nielsen, 2015). Furthermore, as mutations at the inhibitor binding sites contribute to resistance, targeting alternative surfaces may provide opportunities to modulate activity of mutant kinase forms.
The discovery of compounds targeting allosteric sites or disrupting protein-protein interactions is a promising alternative for therapeutic development and has been extensively reviewed (Nero, Morton, Holien, Wielens, & Parker, 2014; Petta, Lievens, Libert, Tavernier, & De Bosscher, 2015; White, Westwell, & Brahemi, 2008; Wu, et al., 2015). However, disruption of protein interactions may also present a challenge for small molecule discovery as many interfaces are characterized by large relatively shallow surfaces that lack the hydrophobic pockets favorable for small molecule binding (Ivanov, Khuri, & Fu, 2013). While monoclonal antibodies provide a large surface area for binding and can be highly specific for their intended target, they are unable to penetrate cells and are therefore limited to extracellular targets such as receptors. Thus, alternative targeting strategies enabling binding to both intracellular targets and large, relatively featureless surfaces while remaining target-selective are desirable in the development of additional kinase targeting therapeutics.
Constrained peptides are a promising class of compounds that are attracting more interest in therapeutic development. The structural characteristics of peptides may be more favorable for binding to sites that are generally difficult to target with small molecules by providing a larger surface area for binding and bypassing the need for a binding pocket within the protein structure. In addition, since peptides provide a larger binding surface area as compared to small molecule scaffolds, target selectivity may be enhanced. Further, some peptides are capable of penetrating the cell or can be modified to increase cell penetration, expanding accessibility to include intracellular proteins (Derossi, Joliot, Chassaing, & Prochiantz, 1994; Fawell, et al., 1994; Vivès, Brodin, & Lebleu, 1997; Walensky, et al., 2004; Wang, et al., 2014).
Early clinical applications of peptides were limited by their unfavorable pharmacological properties. While non-constrained peptides have numerous setbacks for drug development including poor proteolytic stability and loss of secondary structure, advances in peptide chemistry have led to modifications that increase both proteolytic and conformational stability as discussed in recent reviews (Cromm, Spiegel, & Grossmann, 2015; Hill, Shepherd, Diness, & Fairlie, 2014; Lau, de Andrade, Wu, & Spring, 2015; Northfield, et al., 2014; Pelay-Gimeno, Glas, Koch, & Grossmann, 2015; Tsomaia, 2015). Using chemical constraints, peptide instability that was previously observed could be overcome, thereby enhancing the pharmacokinetic properties of various peptides and increasing their promise as potential therapeutics.
Yet, multiple challenges remain with peptide therapeutics including cell permeation, synthetic strategies for stabilization of diverse secondary structures and delivery methods. Some peptide constraints such as peptide stapling demonstrate improved cell permeability (Chu, et al., 2015), however, an overall net negative charge can still impede cell uptake in spite of this chemical modification (Chu, et al., 2015). Additionally, although peptides provide a large binding surface area, this also opens the possibility for peptides to interact with other off-target proteins within the cellular environment. Extensive chemistries have also been explored to design constrained alpha-helical structures, however, synthetic strategies to stabilize such structures as beta-sheets, -turns and -loops have not been as extensively investigated and thus it remains a challenge to effectively design peptide mimics and induce a cellular response. Moreover, systemic delivery of peptide therapeutics is challenging since peptides are inherently more susceptible to multiple factors including proteolytic degradation, poor absorption, immunogenicity and poor bioavailability and are thus often delivered via parenteral administration to enhance bioavailability (Antosova, Mackova, Kral, & Macek, 2009). Other alternative delivery routes are being explored including airway delivery (Andrade, Videira, Ferreira, & Sarmento, 2011), topical delivery (Witting, Obst, Friess, & Hedtrich, 2015) and delivery through the buccal mucosa (Caon, Jin, Simoes, Norton, & Nicolazzo, 2015). Although extensive research efforts are also focused on oral delivery of biologics, effective delivery of peptide-based therapeutics through this route remains a significant challenge with low bioavailability despite advances in delivery technologies (Aguirre, et al., 2016).
Of the body of work concerning kinases targets, the majority of constrained peptides are still in early development phases and have relatively low potency as compared to clinical kinase inhibitors. In addition, cellular activity, in vivo efficacy and off-target binding have not been thoroughly explored for many of these studies. At the molecular level, structural studies are still lacking for many constrained peptides bound to their kinase targets. This would provide tremendous insight to elucidate peptide inhibitory activity including specificity and mechanism of action and may provide direction for improvement of compound efficacy. Two studies in particular, a stapled alpha-helical VEGF inhibitor (Vicari, et al., 2011) and a disulfide-bridge beta-turn disruptor that blocks interactions between p110α E545K and IRS1 (Hao, et al., 2013), show considerable promise for therapeutic development. Both inhibitors were shown to have efficacy in cell-based assays and further demonstrated either tumor regression or delayed tumor development in mice. Although potency and bioavailability will likely require further optimization, these studies aid in target validation and demonstrate the translational potential of constrained peptides for therapeutic kinase targets.
Although the pharmacokinetic properties of peptides are often considered undesirable for therapeutic development, several constrained peptides have received approval or entered clinical trials for the treatment of various diseases or conditions (Fosgerau & Hoffmann, 2015; Kaspar & Reichert, 2013). One of the simplest cyclization techniques, the side chain-linked disulfide bridge, is incorporated into clinically approved peptides such as desmopressin and oxytocin (Mannucci, Ruggeri, Pareti, & Capitanio, 1977; Seitchik & Castillo, 1982). Cyclosporine, an N-methylated, backbone-cyclized peptide, is approved as an immunosuppressant to reduce the risk of transplant rejection (Calne, et al., 1979; Canadian Multicentre Transplant Study, 1983; European Multicentre Trial, 1983). In addition to cyclosporine, another cyclic peptide, pasireotide, was approved for the treatment of acromegaly (Colao, et al., 2014; Petersenn, et al., 2014; Petersenn, et al., 2010; Sheppard, et al., 2015). In recent years, two stapled peptides have entered FDA clinical trials: ALRN-5281 (ClinicalTrials.gov ID: NCT01775358) for the treatment of growth hormone deficiency and ALRN-6924 (ClinicalTrials.gov ID: NCT02264613), which is a dual inhibitor of MDM2 and MDMX. ALRN-6924 was developed from a precursor that was identified through phage display screening, ATSP-7041 (Chang, et al., 2013). Currently, no constrained peptide scaffolds that specifically target kinases are FDA approved. However, the approval and evaluation of other constrained peptides in the clinic demonstrates the therapeutic promise of this strategy and its application for the disruption of kinase signaling in various disease states.
Abbreviations
- AKAP
A-kinase anchoring protein
- Akt
protein kinase B
- ALL
acute lymphoblastic leukemia
- CBG
cyclin binding groove
- CDK2
cyclin dependent kinase 2
- CKII
casein kinase II
- CKIIα
casein kinase II subunit alpha
- CKIIβ
casein kinase II subunit beta
- CML
chronic myeloid leukemia
- EGF
epidermal growth factor
- EGFR
epidermal growth factor receptor
- Eph4A
ephrin type-A receptor 4
- Fos
proto-oncogene c-Fos
- GPCR
G-protein coupled receptors
- Grb2
growth factor receptor-binding protein 2
- GRK
G-protein coupled receptor kinase
- Her2/Neu
human epidermal growth factor receptor 2
- HGF
hepatocyte growth factor
- IRS1
insulin receptor substrate 1
- PBD
polo-box domain
- PDGF
platelet-derived growth factor
- PDGFR
platelet-derived growth factor receptor
- PIK3CA/p110α
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PKA
protein kinase A
- PKA-R
regulatory subunit of PKA
- PlGF
placental growth factor
- Plk1
Polo-like kinase 1
- PPI
protein-protein interaction
- RaPID
random non-standard peptide integrated discovery
- RCM
ring-closing metathesis
- RH
regulator G protein signaling homology domain
- SH2
Src Homology 2
- SH3
Src Homology 3
- Src/c-Src
proto-oncogene tyrosine-protein kinase
- VEGFR
vascular endothelial growth factor receptor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest Statement
The authors declare that there are no conflicts of interest.
References
- Adams PD, Sellers WR, Sharma SK, Wu AD, Nalin CM, Kaelin WG. Identification of a cyclin-cdk2 recognition motif present in substrates and p21-like cyclin-dependent kinase inhibitors. Molecular and Cellular Biology. 1996;16:6623–6633. doi: 10.1128/mcb.16.12.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei AA, Sternberg MJ, Makarov AA. Polyproline-II helix in proteins: structure and function. J Mol Biol. 2013;425:2100–2132. doi: 10.1016/j.jmb.2013.03.018. [DOI] [PubMed] [Google Scholar]
- Aguirre TA, Teijeiro-Osorio D, Rosa M, Coulter IS, Alonso MJ, Brayden DJ. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv Drug Deliv Rev. 2016;106:223–241. doi: 10.1016/j.addr.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Alfaro-Lopez J, Yuan W, Phan BC, Kamath J, Lou Q, Lam KS, Hruby VJ. Discovery of a novel series of potent and selective substrate-based inhibitors of p60c-src protein tyrosine kinase: conformational and topographical constraints in peptide design. J Med Chem. 1998;41:2252–2260. doi: 10.1021/jm9707885. [DOI] [PubMed] [Google Scholar]
- Andrade F, Videira M, Ferreira D, Sarmento B. Nanocarriers for pulmonary administration of peptides and therapeutic proteins. Nanomedicine (Lond) 2011;6:123–141. doi: 10.2217/nnm.10.143. [DOI] [PubMed] [Google Scholar]
- Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes & Development. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews MJ, McInnes C, Kontopidis G, Innes L, Cowan A, Plater A, Fischer PM. Design, synthesis, biological activity and structural analysis of cyclic peptide inhibitors targeting the substrate recruitment site of cyclin-dependent kinase complexes. Org Biomol Chem. 2004;2:2735–2741. doi: 10.1039/B409157D. [DOI] [PubMed] [Google Scholar]
- Antosova Z, Mackova M, Kral V, Macek T. Therapeutic application of peptides and proteins: parenteral forever? Trends Biotechnol. 2009;27:628–635. doi: 10.1016/j.tibtech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baameur F, Morgan DH, Yao H, Tran TM, Hammitt RA, Sabui S, McMurray JS, Lichtarge O, Clark RB. Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol. 2010;77:405–415. doi: 10.1124/mol.109.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banappagari S, Ronald S, Satyanarayanajois SD. Structure-activity relationship of conformationally constrained peptidomimetics for antiproliferative activity in HER2-overexpressing breast cancer cell lines. MedChemComm. 2011;2:752–759. doi: 10.1039/C1MD00126D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov AA, Mohammadi M. Grb2, a double-edged sword of receptor tyrosine kinase signaling. Sci Signal. 2012;5:pe49. doi: 10.1126/scisignal.2003576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2beta. Int J Biol Sci. 2005;1:67–79. doi: 10.7150/ijbs.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich MA, Lin Q, Delarue FL, Sun J, Park HS, Coppola D, Hamilton AD, Sebti SM. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat Biotech. 2000;18:1065–1070. doi: 10.1038/80257. [DOI] [PubMed] [Google Scholar]
- Boran ADW, Seco J, Jayaraman V, Jayaraman G, Zhao S, Reddy S, Chen Y, Iyengar R. A Potential Peptide Therapeutic Derived from the Juxtamembrane Domain of the Epidermal Growth Factor Receptor. PLoS One. 2012;7:e49702. doi: 10.1371/journal.pone.0049702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulikas T. Control of DNA replication by protein phosphorylation. Anticancer Res. 1994;14:2465–2472. [PubMed] [Google Scholar]
- Brognard J, Hunter T. Protein kinase signaling networks in cancer. Curr Opin Genet Dev. 2011;21:4–11. doi: 10.1016/j.gde.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TR, Jr, Nomizu M, Otaka A, Smyth MS, Roller PP, Case RD, Wolf G, Shoelson SE. Cyclic peptide inhibitors of phosphatidylinositol 3-kinase p85 SH2 domain binding. Biochem Biophys Res Commun. 1994;201:1148–1153. doi: 10.1006/bbrc.1994.1825. [DOI] [PubMed] [Google Scholar]
- Calne RY, Rolles K, Thiru S, McMaster P, Craddock GN, Aziz S, White DJG, Evans DB, Dunn DC, Henderson RG, Lewis P. Originally published as Volume 2, Issue 8151CYCLOSPORIN A INITIALLY AS THE ONLY IMMUNOSUPPRESSANT IN 34 RECIPIENTS OF CADAVERIC ORGANS: 32 KIDNEYS, 2 PANCREASES, AND 2 LIVERS. The Lancet. 1979;314:1033–1036. doi: 10.1016/s0140-6736(79)92440-1. [DOI] [PubMed] [Google Scholar]
- Canadian Multicentre Transplant Study, G. A Randomized Clinical Trial of Cyclosporine in Cadaveric Renal Transplantation. New England Journal of Medicine. 1983;309:809–815. doi: 10.1056/NEJM198310063091401. [DOI] [PubMed] [Google Scholar]
- Caon T, Jin L, Simoes CM, Norton RS, Nicolazzo JA. Enhancing the buccal mucosal delivery of peptide and protein therapeutics. Pharm Res. 2015;32:1–21. doi: 10.1007/s11095-014-1485-1. [DOI] [PubMed] [Google Scholar]
- Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013;110:E3445–3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi WJ, Shi ZD, Worthy KM, Bindu L, Karki RG, Nicklaus MC, Fisher RJ, Burke TR., Jr Application of azide-alkyne cycloaddition ‘click chemistry’ for the synthesis of Grb2 SH2 domain-binding macrocycles. Bioorg Med Chem Lett. 2006;16:5265–5269. doi: 10.1016/j.bmcl.2006.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Q, Moellering RE, Hilinski GJ, Kim YW, Grossmann TN, Yeh JTH, Verdine GL. Towards understanding cell penetration by stapled peptides. Med Chem Commun. 2015;6:111–119. [Google Scholar]
- Colao A, Bronstein MD, Freda P, Gu F, Shen CC, Gadelha M, Fleseriu M, van der Lely AJ, Farrall AJ, Hermosillo Reséndiz K, Ruffin M, Chen Y, Sheppard M. Pasireotide Versus Octreotide in Acromegaly: A Head-to-Head Superiority Study. The Journal of Clinical Endocrinology and Metabolism. 2014;99:791–799. doi: 10.1210/jc.2013-2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgrave ML, Craik DJ. Thermal, chemical, and enzymatic stability of the cyclotide kalata B1: the importance of the cyclic cystine knot. Biochemistry. 2004;43:5965–5975. doi: 10.1021/bi049711q. [DOI] [PubMed] [Google Scholar]
- Cromm PM, Spiegel J, Grossmann TN. Hydrocarbon Stapled Peptides as Modulators of Biological Function. ACS Chem Biol. 2015;10:1362–1375. doi: 10.1021/cb501020r. [DOI] [PubMed] [Google Scholar]
- Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora Kinase Inhibitors - Rising Stars in Cancer Therapeutics? American Association for Cancer Research. 2010;9:268–278. doi: 10.1158/1535-7163.MCT-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa L, Diana D, Basile A, Russomanno A, Isernia C, Turco MC, Fattorusso R, D’Andrea LD. Design, structural and biological characterization of a VEGF inhibitor β-hairpin-constrained peptide. European Journal of Medicinal Chemistry. 2014;73:210–216. doi: 10.1016/j.ejmech.2013.12.016. [DOI] [PubMed] [Google Scholar]
- De Smet F, Christopoulos A, Carmeliet P. Allosteric targeting of receptor tyrosine kinases. Nat Biotech. 2014;32:1113–1120. doi: 10.1038/nbt.3028. [DOI] [PubMed] [Google Scholar]
- Derossi D, Joliot AH, Chassaing G, Prochiantz A. The third helix of the Antennapedia homeodomain translocates through biological membranes. Journal of Biological Chemistry. 1994;269:10444–10450. [PubMed] [Google Scholar]
- Diana D, Basile A, De Rosa L, Di Stasi R, Auriemma S, Arra C, Pedone C, Turco MC, Fattorusso R, D’Andrea LD. β-Hairpin Peptide That Targets Vascular Endothelial Growth Factor (VEGF) Receptors: DESIGN, NMR CHARACTERIZATION, AND BIOLOGICAL ACTIVITY. Journal of Biological Chemistry. 2011;286:41680–41691. doi: 10.1074/jbc.M111.257402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettmayer P, France D, Gounarides J, Jarosinski M, Martin MS, Rondeau JM, Sabio M, Topiol S, Weidmann B, Zurini M, Bair KW. Structural and conformational requirements for high-affinity binding to the SH2 domain of Grb2(1) J Med Chem. 1999;42:971–980. doi: 10.1021/jm9811007. [DOI] [PubMed] [Google Scholar]
- European Multicentre Trial, G. Originally published as Volume 2, Issue 8357CYCLOSPORIN IN CADAVERIC RENAL TRANSPLANTATION: ONE-YEAR FOLLOW-UP OF A MULTICENTRE TRIAL. The Lancet. 1983;322:986–989. [PubMed] [Google Scholar]
- Fabbro D. 25 Years of Small Molecular Weight Kinase Inhibitors: Potentials and Limitations. Molecular Pharmacology. 2015;87:766–775. doi: 10.1124/mol.114.095489. [DOI] [PubMed] [Google Scholar]
- Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR Review 14. Br J Pharmacol. 2015;172:2675–2700. doi: 10.1111/bph.13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqi M. Neurodegenerative disease: EPHA4 inhibition rescues neurodegeneration in ALS. Nat Rev Drug Discov. 2012;11:747. doi: 10.1038/nrd3853. [DOI] [PubMed] [Google Scholar]
- Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:664–668. doi: 10.1073/pnas.91.2.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Kasahara C, Rickles RJ, Schreiber SL. Specific interactions outside the proline-rich core of two classes of Src homology 3 ligands. Proc Natl Acad Sci U S A. 1995;92:12408–12415. doi: 10.1073/pnas.92.26.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discovery Today. 2015;20:122–128. doi: 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Freed DM, Alvarado D, Lemmon MA. Ligand regulation of a constitutively dimeric EGF receptor. Nat Commun. 2015;6:7380. doi: 10.1038/ncomms8380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto T, Torigoe N, Kawabata S, Murakami M, Uede T, Nishi T, Ito Y, Sugimura K. Peptide mimics of the CTLA4-binding domain stimulate T-cell proliferation. Nat Biotechnol. 1998;16:267–270. doi: 10.1038/nbt0398-267. [DOI] [PubMed] [Google Scholar]
- Gao Y, Voigt J, Wu JX, Yang D, Burke TR., Jr Macrocyclization in the design of a conformationally constrained Grb2 SH2 domain inhibitor. Bioorg Med Chem Lett. 2001;11:1889–1892. doi: 10.1016/s0960-894x(01)00316-x. [DOI] [PubMed] [Google Scholar]
- Gay B, Furet P, Garcia-Echeverria C, Rahuel J, Chene P, Fretz H, Schoepfer J, Caravatti G. Dual specificity of Src homology 2 domains for phosphotyrosine peptide ligands. Biochemistry. 1997;36:5712–5718. doi: 10.1021/bi962642y. [DOI] [PubMed] [Google Scholar]
- Gherardi E, Birchmeier W, Birchmeier C, Woude GV. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12:89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- Greenwood KP, Daly NL, Brown DL, Stow JL, Craik DJ. The cyclic cystine knot miniprotein MCoTI-II is internalized into cells by macropinocytosis. Int J Biochem Cell Biol. 2007;39:2252–2264. doi: 10.1016/j.biocel.2007.06.016. [DOI] [PubMed] [Google Scholar]
- Guardiola S, Díaz-Lobo M, Seco J, García J, Nevola L, Giralt E. Peptides Targeting EGF Block the EGF–EGFR Interaction. Chembiochem. 2016;17:702–711. doi: 10.1002/cbic.201500525. [DOI] [PubMed] [Google Scholar]
- Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol Cancer Ther. 2016;15:1427–1435. doi: 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanold LE, Oruganty K, Ton NT, Beedle AM, Kannan N, Kennedy EJ. Inhibiting EGFR dimerization using triazolyl-bridged dimerization arm mimics. PLoS One. 2015 doi: 10.1371/journal.pone.0118796. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanold LE, Watkins CP, Ton NT, Liaw P, Beedle AM, Kennedy EJ. Design of a selenylsulfide-bridged EGFR dimerization arm mimic. Bioorganic & Medicinal Chemistry. 2015;23:2761–2766. doi: 10.1016/j.bmc.2015.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer. 2012;3:436–446. doi: 10.1177/1947601912458584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Wang C, Cao B, Hirsch BM, Song J, Markowitz SD, Ewing RM, Sedwick D, Liu L, Zheng W, Wang Z. Gain of interaction with IRS1 by p110alpha-helical domain mutants is crucial for their oncogenic functions. Cancer Cell. 2013;23:583–593. doi: 10.1016/j.ccr.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining Cyclic Peptides To Mimic Protein Structure Motifs. Angew Chem Int Ed. 2014;53:13020–13041. doi: 10.1002/anie.201401058. [DOI] [PubMed] [Google Scholar]
- Huang YH, Henriques ST, Wang CK, Thorstholm L, Daly NL, Kaas Q, Craik DJ. Design of substrate-based BCR-ABL kinase inhibitors using the cyclotide scaffold. Scientific Reports. 2015;5 doi: 10.1038/srep12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Sakai K, Suzuki Y, Ozawa N, Hatta T, Natsume T, Matsumoto K, Suga H. Artificial human Met agonists based on macrocycle scaffolds. Nat Commun. 2015;6 doi: 10.1038/ncomms7373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AA, Khuri FR, Fu H. Targeting protein–protein interactions as an anticancer strategy. Trends in Pharmacological Sciences. 2013;34:393–400. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang S, Liao C, Bindu L, Yin B, Worthy KW, Fisher RJ, Burke TR, Jr, Nicklaus MC, Roller PP. Discovery of thioether-bridged cyclic pentapeptides binding to Grb2-SH2 domain with high affinity. Bioorg Med Chem Lett. 2009;19:2693–2698. doi: 10.1016/j.bmcl.2009.03.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jura N, Endres NF, Engel K, Deindl S, Das R, Lamers MH, Wemmer DE, Zhang X, Kuriyan J. Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell. 2009;137:1293–1307. doi: 10.1016/j.cell.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath JR, Liu R, Enstrom AM, Lou Q, Lam KS. Development and characterization of potent and specific peptide inhibitors of p60(c-src) protein tyrosine kinase using pseudosubstrate-based inhibitor design approach. Journal of Peptide Research. 2003;62:260–268. doi: 10.1046/j.1399-3011.2003.00094.x. [DOI] [PubMed] [Google Scholar]
- Kanthala S, Gauthier T, Satyanarayanajois S. Structure-activity relationships of peptidomimetics that inhibit PPI of HER2-HER3. Biopolymers. 2014;101:693–702. doi: 10.1002/bip.22441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar AA, Reichert JM. Future directions for peptide therapeutics development. Drug Discovery Today. 2013;18:807–817. doi: 10.1016/j.drudis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Kennedy EJ, Scott JD. Selective disruption of the AKAP signaling complexes. Methods Mol Biol. 2015;1294:137–150. doi: 10.1007/978-1-4939-2537-7_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YB, Kang CW, Ranatunga S, Yang H, Sebti SM, Del Valle JR. Imidazo[1,2-a]pyridine-based peptidomimetics as inhibitors of Akt. Bioorganic & Medicinal Chemistry Letters. 2014;24:4650–4653. doi: 10.1016/j.bmcl.2014.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Ye GF, Wang YH, Lin XF, Sun GQ, Parang K. Synthesis and structure-activity relationships of linear and conformationally constrained peptide analogues of CIYKYY as src tyrosine kinase inhibitors. Journal of Medicinal Chemistry. 2006;49:3395–3401. doi: 10.1021/jm060334k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KS, Wu JZ, Lou Q. Identification and Characterization of a Novel Synthetic Peptide Substrate-Specific for Src-Family Protein-Tyrosine Kinases. International Journal of Peptide and Protein Research. 1995;45:587–592. doi: 10.1111/j.1399-3011.1995.tb01323.x. [DOI] [PubMed] [Google Scholar]
- Lamberto I, Lechtenberg BC, Olson EJ, Mace PD, Dawson PE, Riedl SJ, Pasquale EB. Development and Structural Analysis of a Nanomolar Cyclic Peptide Antagonist for the EphA4 Receptor. ACS Chemical Biology. 2014;9:2787–2795. doi: 10.1021/cb500677x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberto I, Qin H, Noberini R, Premkumar L, Bourgin C, Riedl Stefan J, Song J, Pasquale Elena B. Distinctive binding of three antagonistic peptides to the ephrin-binding pocket of the EphA4 receptor. Biochemical Journal. 2012;445:47–56. doi: 10.1042/BJ20120408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau YH, de Andrade P, Wu Y, Spring DR. Peptide stapling techniques based on different macrocyclisation chemistries. Chem Soc Rev. 2015;44:91–102. doi: 10.1039/c4cs00246f. [DOI] [PubMed] [Google Scholar]
- Laudet B, Barette C, Dulery V, Renaudet O, Dumy P, Metz A, Prudent R, Deshiere A, Dideberg O, Filhol O, Cochet C. Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem J. 2007;408:363–373. doi: 10.1042/BJ20070825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J, Ferguson KM. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb Perspect Biol. 2014;6 doi: 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester LB, Coghlan VM, Nauert B, Scott JD. Cloning and characterization of a novel A-kinase anchoring protein. AKAP 220, association with testicular peroxisomes. Journal of Biological Chemistry. 1996;271:9460–9465. doi: 10.1074/jbc.271.16.9460. [DOI] [PubMed] [Google Scholar]
- Liu F, Giubellino A, Simister PC, Qian W, Giano MC, Feller SM, Bottaro DP, Burke TR., Jr Application of ring-closing metathesis to Grb2 SH3 domain-binding peptides. Biopolymers. 2011;96:780–788. doi: 10.1002/bip.21692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannucci PM, Ruggeri ZM, Pareti FI, Capitanio A. 1-Deamino-8-d-arginine vasopressin: a new pharmacological approach to the management of haemophilia and von Willebrands’ diseases. Lancet (London, England) 1977;1:869–872. doi: 10.1016/s0140-6736(77)91197-7. [DOI] [PubMed] [Google Scholar]
- Meyer SC, Shomin CD, Gaj T, Ghosh I. Tethering small molecules to a phage display library: Discovery of a selective bivalent inhibitor of protein kinase A. Journal of the American Chemical Society. 2007;129:13812–13813. doi: 10.1021/ja076197d. [DOI] [PubMed] [Google Scholar]
- Mizuguchi T, Ohara N, Iida M, Ninomiya R, Wada S, Kiso Y, Saito K, Akaji K. Evaluation of dimerization-inhibitory activities of cyclic peptides containing a b-hairpin loop sequence of the EGF receptor. Bioorg Med Chem. 2012;20:5730–5737. doi: 10.1016/j.bmc.2012.08.013. [DOI] [PubMed] [Google Scholar]
- Mizuguchi T, Uchimura H, Kakizawa T, Kimura T, Yokoyama S, Kiso Y, Saito K. Inhibitory effect of a dimerization-arm-mimetic peptide on EGF receptor activation. Bioorg Med Chem Lett. 2009;19:3279–3282. doi: 10.1016/j.bmcl.2009.04.080. [DOI] [PubMed] [Google Scholar]
- Muller S, Chaikuad A, Gray NS, Knapp S. The ins and outs of selective kinase inhibitor development. Nat Chem Biol. 2015;11:818–821. doi: 10.1038/nchembio.1938. [DOI] [PubMed] [Google Scholar]
- Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharmacol. 2016 doi: 10.1111/bcp.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai KK, Nguyen LN, Koolpe M, McLennan R, Krull CE, Pasquale EB. Targeting the EphA4 receptor in the nervous system with biologically active peptides. Molecular and Cellular Neuroscience. 2003;24:1000–1011. doi: 10.1016/j.mcn.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Murugan RN, Park JE, Lim D, Ahn M, Cheong C, Kwon T, Nam KY, Choi SH, Kim BY, Yoon DY, Yaffe MB, Yu DY, Lee KS, Bang JK. Development of cyclic peptomer inhibitors targeting the polo-box domain of polo-like kinase 1. Bioorg Med Chem. 2013;21:2623–2634. doi: 10.1016/j.bmc.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Takasugi H, Aizawa T, Yoshida M, Mizuguchi M, Mori Y, Shinoda H, Hayakawa Y, Kawano K. Peptide mimics of epidermal growth factor (EGF) with antagonistic activity. Journal of Biotechnology. 2005;116:211–219. doi: 10.1016/j.jbiotec.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Nam NH, Ye G, Sun G, Parang K. Conformationally constrained peptide analogues of pTyr-Glu-Glu-Ile as inhibitors of the Src SH2 domain binding. J Med Chem. 2004;47:3131–3141. doi: 10.1021/jm040008+. [DOI] [PubMed] [Google Scholar]
- Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- Northfield SE, Wang CK, Schroeder CI, Durek T, Kan MW, Swedberg JE, Craik DJ. Disulfide-rich macrocyclic peptides as templates in drug design. Euro J Med Chem. 2014;77:248–257. doi: 10.1016/j.ejmech.2014.03.011. [DOI] [PubMed] [Google Scholar]
- Nowell PC. Discovery of the Philadelphia chromosome: a personal perspective. J Clin Invest. 2007;117:2033–2035. doi: 10.1172/JCI31771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi S, Shi ZD, Worthy KM, Bindu LK, Fisher RJ, Burke TR., Jr Ring-closing metathesis of C-terminal allylglycine residues with an N-terminal beta-vinyl-substituted phosphotyrosyl mimetic as an approach to novel Grb2 SH2 domain-binding macrocycles. Chembiochem. 2005;6:668–674. doi: 10.1002/cbic.200400298. [DOI] [PubMed] [Google Scholar]
- Park JE, Soung NK, Johmura Y, Kang YH, Liao C, Lee KH, Park CH, Nicklaus MC, Lee KS. Polo-box domain: a versatile mediator of polo-like kinase function. Cell Mol Life Sci. 2010;67:1957–1970. doi: 10.1007/s00018-010-0279-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew Chem Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersenn S, Farrall AJ, Block C, Melmed S, Schopohl J, Caron P, Cuneo R, Kleinberg D, Colao A, Ruffin M, Hermosillo Reséndiz K, Hughes G, Hu K, Barkan A. Long-term efficacy and safety of subcutaneous pasireotide in acromegaly: results from an open-ended, multicenter, Phase II extension study. Pituitary. 2014;17:132–140. doi: 10.1007/s11102-013-0478-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersenn S, Schopohl J, Barkan A, Mohideen P, Colao A, Abs R, Buchelt A, Ho YY, Hu K, Farrall AJ, Melmed S, Biller BMK. Pasireotide (SOM230) Demonstrates Efficacy and Safety in Patients with Acromegaly: A Randomized, Multicenter, Phase II Trial. The Journal of Clinical Endocrinology & Metabolism. 2010;95:2781–2789. doi: 10.1210/jc.2009-2272. [DOI] [PubMed] [Google Scholar]
- Petta I, Lievens S, Libert C, Tavernier J, De Bosscher K. Modulation of protein-protein interactions for the development of novel therapeutics. Mol Ther. 2015 doi: 10.1038/mt.2015.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartararo JS, Wu P, Kritzer JA. Peptide bicycles that inhibit the Grb2 SH2 domain. Chembiochem. 2012;13:1490–1496. doi: 10.1002/cbic.201200175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranatunga S, Del Valle JR. Synthesis of GSK3beta mimetic inhibitors of Akt featuring a novel extended dipeptide surrogate. Bioorg Med Chem Lett. 2011;21:7166–7169. doi: 10.1016/j.bmcl.2011.09.079. [DOI] [PubMed] [Google Scholar]
- Regad T. Targeting RTK Signaling Pathways in Cancer. Cancers (Basel) 2015;7:1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol Res. 2015;94:9–25. doi: 10.1016/j.phrs.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Russo AA, Jeffrey PD, Patten AK, Massague J, Pavletich NP. Crystal structure of the p27Kip1 cyclin-dependent-kinase inhibitor bound to the cyclin A-Cdk2 complex. Nature. 1996;382:325–331. doi: 10.1038/382325a0. [DOI] [PubMed] [Google Scholar]
- Satyanarayanajois S, Villalba S, Jianchao L, Lin GM. Design, Synthesis, and Docking Studies of Peptidomimetics Based on HER2–Herceptin Binding Site with Potential Antiproliferative Activity Against Breast Cancer Cell lines. Chemical Biology & Drug Design. 2009;74:246–257. doi: 10.1111/j.1747-0285.2009.00855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitchik J, Castillo M. Oxytocin augmentation of dysfunctional labor: I. Clinical data. American Journal of Obstetrics and Gynecology. 1982;144:899–905. doi: 10.1016/0002-9378(82)90181-8. [DOI] [PubMed] [Google Scholar]
- Sheppard M, Bronstein MD, Freda P, Serri O, De Marinis L, Naves L, Rozhinskaya L, Hermosillo Reséndiz K, Ruffin M, Chen Y, Colao A. Pasireotide LAR maintains inhibition of GH and IGF-1 in patients with acromegaly for up to 25 months: results from the blinded extension phase of a randomized, double-blind, multicenter, Phase III study. Pituitary. 2015;18:385–394. doi: 10.1007/s11102-014-0585-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya M. VEGFR and Type-V RTK Activation and Signaling. Cold Spring Harbor Perspectives in Biology. 2013;5 doi: 10.1101/cshperspect.a009092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirazi AN, Tiwari RK, Brown A, Mandal D, Sun GQ, Parang K. Cyclic peptides containing tryptophan and arginine as Src kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2013;23:3230–3234. doi: 10.1016/j.bmcl.2013.03.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomin CD, Meyer SC, Ghosh I. Staurosporine tethered peptide ligands that target cAMP-dependent protein kinase (PKA): Optimization and selectivity profiling. Bioorganic & Medicinal Chemistry. 2009;17:6196–6202. doi: 10.1016/j.bmc.2009.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shomin CD, Restituyo E, Cox KJ, Ghosh I. Selection of cyclic-peptide inhibitors targeting Aurora kinase A: Problems and solutions. Bioorganic & Medicinal Chemistry. 2011;19:6743–6749. doi: 10.1016/j.bmc.2011.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair JKL, Denton EV, Schepartz A. Inhibiting Epidermal Growth Factor Receptor at a Distance. Journal of the American Chemical Society. 2014;136:11232–11235. doi: 10.1021/ja504076t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair JKL, Schepartz A. Influence of Macrocyclization on Allosteric, Juxtamembrane-Derived, Stapled Peptide Inhibitors of the Epidermal Growth Factor Receptor (EGFR) Org Lett. 2014;16:4916–4919. doi: 10.1021/ol502426b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CI. Enigmas in tumor resistance to kinase inhibitors and calculation of the drug resistance index for cancer (DRIC) Semin Cancer Biol. 2016 doi: 10.1016/j.semcancer.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Tal-Gan Y, Hurevich M, Klein S, Ben-Shimon A, Rosenthal D, Hazan C, Shalev DE, Niv MY, Levitzki A, Gilon C. Backbone Cyclic Peptide Inhibitors of Protein Kinase B (PKB/Akt) Journal of Medicinal Chemistry. 2011;54:5154–5164. doi: 10.1021/jm2003969. [DOI] [PubMed] [Google Scholar]
- Tam EM, Runyon ST, Santell L, Quan C, Yao X, Kirchhofer D, Skelton NJ, Lazarus RA. Noncompetitive Inhibition of Hepatocyte Growth Factor-dependent Met Signaling by a Phage-derived Peptide. Journal of Molecular Biology. 2009;385:79–90. doi: 10.1016/j.jmb.2008.09.091. [DOI] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari R, Brown A, Narramaneni S, Sun G, Parang K. Synthesis and evaluation of conformationally constrained peptide analogues as the Src SH3 domain binding ligands. Biochimie. 2010;92:1153–1163. doi: 10.1016/j.biochi.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Toyama K, Mizuguchi T, Nomura W, Tamamura H. Functional evaluation of fluorescein-labeled derivatives of a peptide inhibitor of the EGF receptor dimerization. Bioorganic & Medicinal Chemistry. 2016;24:3406–3412. doi: 10.1016/j.bmc.2016.05.026. [DOI] [PubMed] [Google Scholar]
- Troger J, Moutty MC, Skroblin P, Klussmann E. A-kinase anchoring proteins as potential drug targets. Br J Pharmacol. 2012;29:1476–5381. doi: 10.1111/j.1476-5381.2011.01796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsomaia N. Peptide therapeutics: Targeting the undruggable space. Euro J Med Chem. 2015;94:459–470. doi: 10.1016/j.ejmech.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Vicari D, Foy KC, Liotta EM, Kaumaya PTP. Engineered conformation-dependent VEGF peptide mimics are effective in inhibiting VEGF signaling pathways. Journal of Biological Chemistry. 2011 doi: 10.1074/jbc.M110.216812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivès E, Brodin P, Lebleu B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. Journal of Biological Chemistry. 1997;272:16010–16017. doi: 10.1074/jbc.272.25.16010. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Kormeyer SJ. Activation of Apoptosis in Vivo by a Hydrocarbon-Stapled BH3 Helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ho TG, Bertinetti D, Neddermann M, Franz E, Mo GC, Schendowich LP, Sukhu A, Spelts RC, Zhang J, Herberg FW, Kennedy EJ. Isoform-selective disruption of AKAP-localized PKA using hydrocarbon stapled peptides. ACS Chem Biol. 2014;9:635–642. doi: 10.1021/cb400900r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ho TG, Franz E, Hermann JS, Smith FD, Hehnly H, Esseltine JL, Hanold LE, Murph MM, Bertinetti D, Scott JD, Herberg FW, Kennedy EJ. PKA-type I selective constrained peptide disruptors of AKAP complexes. ACS Chem Biol. 2015;10:1502–1510. doi: 10.1021/acschembio.5b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AW, Westwell AD, Brahemi G. Protein–protein interactions as targets for small-molecule therapeutics in cancer. Expert Reviews in Molecular Medicine. 2008;10:e8. doi: 10.1017/S1462399408000641. [DOI] [PubMed] [Google Scholar]
- Witting M, Obst K, Friess W, Hedtrich S. Recent advances in topical delivery of proteins and peptides mediated by soft matter nanocarriers. Biotechnol Adv. 2015;33:1355–1369. doi: 10.1016/j.biotechadv.2015.01.010. [DOI] [PubMed] [Google Scholar]
- Wu P, Clausen MH, Nielsen TE. Allosteric small-molecule kinase inhibitors. Pharmacology & Therapeutics. 2015;156:59–68. doi: 10.1016/j.pharmthera.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: an analysis of FDA-approved drugs. Drug Discovery Today. 2016;21:5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nature Structural Biology. 2002;9:940–944. doi: 10.1038/nsb870. [DOI] [PubMed] [Google Scholar]
- Yang S, Proctor A, Cline LL, Houston KM, Waters ML, Allbritton NL. beta-Turn sequences promote stability of peptide substrates for kinases within the cytosolic environment. Analyst. 2013;138:4305–4311. doi: 10.1039/c3an00874f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SM, Moulaei T, Lim D, Bang JK, Park JE, Shenoy SR, Liu F, Kang YH, Liao C, Soung NK, Lee S, Yoon DY, Lim Y, Lee DH, Otaka A, Appella E, McMahon JB, Nicklaus MC, Burke TR, Jr, Yaffe MB, Wlodawer A, Lee KS. Structural and functional analyses of minimal phosphopeptides targeting the polo-box domain of polo-like kinase 1. Nat Struct Mol Biol. 2009;16:876–882. doi: 10.1038/nsmb.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabriskie MS, Eide CA, Tantravahi SK, Vellore NA, Estrada J, Nicolini FE, Khoury HJ, Larson RA, Konopleva M, Cortes JE, Kantarjian H, Jabbour EJ, Kornblau SM, Lipton JH, Rea D, Stenke L, Barbany G, Lange T, Hernandez-Boluda JC, Ossenkoppele GJ, Press RD, Chuah C, Goldberg SL, Wetzler M, Mahon FX, Etienne G, Baccarani M, Soverini S, Rosti G, Rousselot P, Friedman R, Deininger M, Reynolds KR, Heaton WL, Eiring AM, Pomicter AD, Khorashad JS, Kelley TW, Baron R, Druker BJ, Deininger MW, O’Hare T. BCR-ABL1 compound mutations combining key kinase domain positions confer clinical resistance to ponatinib in Ph chromosome-positive leukemia. Cancer Cell. 2014;26:428–442. doi: 10.1016/j.ccr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer. Drug Des Devel Ther. 2016;10:3867–3872. doi: 10.2147/DDDT.S119162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]