

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a disease where detection preceding clinical symptoms significantly increases the life expectancy of patients. In this study, a recombinant antibody microarray platform was used to analyze 213 Chinese plasma samples from PDAC patients and normal control (NC) individuals. The cohort was stratified according to disease stage, i.e. resectable disease (stage I/II), locally advanced (stage III) and metastatic disease (stage IV). Support vector machine analysis showed that all PDAC stages could be discriminated from controls and that the accuracy increased with disease progression, from stage I to IV. Patients with stage I/II PDAC could be discriminated from NC with high accuracy based on a plasma protein signature, indicating a possibility for early diagnosis and increased detection rate of surgically resectable tumors.
Keywords: Pancreatic cancer, Biomarker signatures, Early detection, Antibody microarrays, Recombinant antibodies
Highlights
Blood samples contain sufficient information to discriminate PDAC stages I–IV using multiparametric analysis.
PDAC was classified with increased accuracy from stage I to IV. Candidate markers for early stage PDAC were identified.
The technical transferability of a recombinant antibody microarray platform was demonstrated.
Plasma signatures identified in Chinese PDAC patients correlated to serum signatures in late stage Caucasian patients.
A blood test could be utilized for earlier diagnosis and increased detection rate of surgically resectable PDAC tumors.
Abbreviations
- ANOVA
analysis of variance
- AUC
area under curve
- BE
backward elimination
- CV
coefficient of variance
- HDL
high-density lipoprotein
- K–L
Kullback–Leibler
- NC
normal controls
- PBSMT
phosphate buffered saline with 1% milk, 1% Tween-20
- PBST
phosphate buffered saline with 1% Tween-20
- PCA
principal component analysis
- PDAC
pancreatic ductal adenocarcinoma
- ROC
receive operating characteristics
- RT
room temperature
- scFv
single-chain fragment variable
- SVM
support vector machine
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers with a 5‐year survival rate of 3–4%. A key driver behind this poor prognosis is the current inability to diagnose patients at an early stage. Data supports that it takes more than five years from tumor initiation until the acquisition of metastatic ability (Yachida et al., 2010), which clearly demonstrates a window of opportunity for early detection if accurate markers were available. At the time of diagnosis, patients have often developed late‐stage disease, and only approximately 15–20% of the patients have resectable tumors (Conlon et al., 1996; Sohn et al., 2000; Zhang et al., 2016). The 5‐year survival of these patients, displaying large resected tumors, is only 10–20% (Conlon et al., 1996; Sohn et al., 2000). However, the 5‐year survival increases to 30–60% if tumors ≤20 mm (stage I–II) can be resected (Furukawa et al., 1996; Shimizu et al., 2005). The late diagnosis (stage III–IV) is due to unspecific clinical symptoms in combination with the lack of markers for early diagnosis. Interestingly, studies suggest that pancreatic tumors could be resectable as early as six months prior to clinical diagnosis at an asymptomatic stage (Gangi et al., 2004; Pelaez‐Luna et al., 2007).
The so far most evaluated marker for PDAC, CA19‐9, suffers from poor specificity, with elevated levels in several other indications, as well as a complete absence in patients that are genotypically Lewis a−b− (5% of the population). Consequently, the use of CA19‐9 for pancreatic cancer screening is not recommended (Locker et al., 2006). Today, no other single biomarker has been shown to accurately diagnose PDAC, although recent discovery studies have demonstrated that both exosomes and nucleosomes contain information associated with pancreatic cancer (Bauden et al., 2015; Melo et al., 2015). However, the field of cancer diagnostics is today moving towards panels of markers, since this yields increased sensitivity and specificity (Brand et al., 2011; Bunger et al., 2011).
Inflammation is a critical component of tumor progression (Coussens and Werb, 2002) and the immunoregulatory plasma proteome may be a source of potential cancer biomarkers. Considering the systemic effect, as well as the pluripotency of many proteins of the immune system, small panels of markers will not be sufficiently specific for pancreatic cancer, particularly when e.g. trying to discriminate pancreatic cancer from pancreatitis. Previous studies have also demonstrated that an increased number of immunoregulatory proteins in combination with cancer‐associated markers (n = 20–25) will yield disease‐specific signatures, reflecting the systemic response to disease (Carlsson et al., 2011b; Ingvarsson et al., 2008; Wingren et al., 2012). However, analysis of the immunoregulatory and tumor secretion proteome is associated with several challenges, such as, (i) plasma proteins display a vast dynamic concentration range; (ii) cancer markers are more likely to be found among the most low‐abundant proteins (Haab et al., 2005; Surinova et al., 2011); (iii) disease‐associated changes in plasma levels of low‐abundant markers are expected to be small, requiring a significant number of samples for adequate statistics (Alonzo et al., 2002).
To meet these challenges, we have in the present pilot study analyzed 213 plasma samples from Chinese patients with pancreatic cancer stage I–IV and normal controls, using a sensitive antibody microarray platform. The aim was to identify stage‐associated PDAC markers by comparing control samples to stage I–IV and the results support the concept that the information content in a blood sample is sufficient to discriminate even the earlier disease stages. Consequently, this could pave the way for early diagnosis of PDAC, particularly for the benefit of patients at high risk, such as chronic pancreatitis, hereditary PDAC, and Peutz‐Jeghers syndrome patients.
2. Material and methods
2.1. Plasma samples
This retrospective study was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital (TMUCIH). After informed consent, blood was collected at TMUCIH, plasma was isolated and stored at −80 °C. A total of 213 plasma samples, collected from Jan‐01 2012 to Dec‐13 2013, was used (Table 1). The enrolled PDAC patients (n = 118) were all Chinese Han ethnicity and treated at TMUCIH. None of the patients had received chemotherapy or radiotherapy at the time of blood draw. All PDAC samples were cytology confirmed by experienced pathologists. Patients were diagnosed with PDAC with the following exceptions: Malignant serous cystadenoma (n = 1), pancreatic sarcoma (n = 2), tubular papillary pancreatic adenocarcinoma (n = 1). Five patients were diagnosed with PDAC with liver metastasis. Data on tumor stage and size at diagnosis, and tumor location within the pancreas were based on clinical pathology. Staging was performed according to the American Cancer Society's guidelines (Table 1) and the extent of resection was classified as R0. Normal control (NC) samples (n = 95) were collected from healthy inhabitants of Tianjin at their routine physical examination at TMUCIH, and were genetically unrelated to the PDAC patients (Table 1). Sample IDs were recoded and randomized at labeling, and sample annotation and clinical data was blinded to the operator at all downstream experimental procedures. All samples were labeled at one single occasion, using a previously optimized protocol (Wingren et al., 2012) (Supplementary Methods).
Table 1.
Clinical samples.
| Group | No of samples | Gender (M/F) | Median age (range) | Tumor location | Stage groupinga |
|---|---|---|---|---|---|
| PDAC | 118 | 76/42 | 59 (21–83) | Head = 69, Body/Tail = 43, Neck = 4, Neck + Body = 1, Head + Tail = 1 | |
| Stage I | 11 | 6/5 | 59 (48–71) | Head = 6, Body/Tail = 5 | T1/2, N0, M0 |
| Stage II | 33 | 16/17 | 59 (46–83) | Head = 27, Body/Tail = 6 | T1‐3, N0/1, M0 |
| Stage III | 38 | 28/10 | 59 (21–75) | Head = 22, Body/Tail = 12, Neck = 2, Neck + Body = 1 | T4, any N, M0 |
| Stage IV | 36 | 26/10 | 59 (38–75) | Head = 14, Body/Tail = 20, Neck = 1, Head + Tail = 1 | Any T & N, M1 |
| NC | 95 | 20/75 | 63 (52–74) | N/A | |
| Total | 213 | 96/117 | 62 (21–83) |
Staging according to the guidelines of the American Cancer Society.
2.2. Generation of antibody microarrays
The antibody microarrays contained 350 human recombinant scFv antibodies (Supplementary Table 1), selected and generated from in‐house designed phage display antibody libraries, produced in E. coli as previously been described (Pauly et al., 2014) and printed onto slides in 14 arrays/slide and 3 replicate spots/array (see Supplementary Methods for details). The VH/VL framework is the same for all scFvs, based on a molecular design of high on‐chip stability, adapted for microarray applications (Borrebaeck and Wingren, 2011). The antibodies were selected against plasma proteins involved in immune regulation but also against proteins previously associated with different cancer indications. The specificity, affinity, and on‐chip functionality have been assured using stringent phage‐display screening and selection protocols using different sample formats ranging from pure proteins, mixtures of pure proteins to crude samples (Soderlind et al., 2000). In addition, the specificity of selected antibodies has been validated using pure proteins, mixtures of pure proteins, as well as well‐characterized, standardized serum samples i) with known levels of the targeted analyte(s), ii) spiked with known level of specific protein(s), and/or iii) depleted of the targeted protein(s), and/or orthogonal methods such as mass spectrometry (affinity pull‐down experiments), ELISA, Meso Scale Discovery assay and cytometric bead assay, as well as spiking and blocking experiments (Supplementary Table 1) (Carlsson et al., 2011, 2010, 2011, 2007, 2008, 2012, 2013, 2007). Despite stringent selection and validation protocols, one limitation is the lack of information on fine specificity concerning proteoforms, translational modifications and potential complex formations. Eighty‐six antibodies raised against cancer‐related proteins as part of the AFFINOMICS project (Stoevesandt and Taussig, 2012) were novel to this study, however the on‐chip functionality of the scFv framework used has been demonstrated in an independent study (Säll et al., in press). All slides used for this study were printed at a single occasion, shipped to TMUCIH, and used for analysis within four weeks after printing.
2.3. Antibody microarray analysis
Ten slides (140 individual subarrays) were processed per day with randomized sample order as described in Supplementary Methods. Briefly, arrays were blocked with PBSMT, washed with PBST, and incubated with biotinylated plasma samples for 2 h at RT. Unbound proteins were washed off, and bound proteins were detected using 1 μg/mL Alexa Fluor647‐Streptavidin (1 h at RT). Excess reagent was washed off, and slides were dried and immediately scanned in a LuxScan 10K Microarray scanner (CapitalBio Corp., Beijing, China) at 10 μm resolution using the 635 nm and the 532 nm excitation lasers.
2.4. Data acquisition, quality control and pre‐processing
Signal intensities were quantified by two trained analysts (ASG and MN), blinded to patient ID and clinical data, using the ScanArray Express software version 4.0 (PerkinElmer Life and Analytical Sciences), with the fixed‐circle option. For each microarray, a grid was positioned using the Alexa Fluor555 signals from microarray printing, and used to quantify the Alexa Fluor647 signal corresponding to the relative level of bound protein. Eleven samples (10 PDAC, 1 NC) were not quantified due to poor quality images resulting from of high background and/or low overall signals. The excluded samples were three stage II (two head, one body/tail tumors), four stage III (head tumors), and three stage IV (body/tail tumors). For quantified arrays, the spot saturation, mean intensity and signal‐to‐noise ratio of each spot were evaluated. Fourteen antibodies were excluded because (i) the median signal intensity was below the cut‐off limit, defined as the background (average PBS signal) +2 standard deviations (n = 8), (ii) saturated signal in the lowest scanner intensity setting in more than 50% of samples (n = 1), or (iii) inadequate antibody printing (n = 5). Based on the remaining 202 samples and 336 antibodies, a dataset was assembled using the mean spot intensity after local background subtraction. Each data point represented an average of the three replicate spots, unless the replicate CV exceeded 15% from the mean value, in which case it was discarded and the average of the two remaining replicates was used instead. The average replicate CV was 7.9% (±4.1%). Applying a cut‐off CV of 15%, 79% of data values were calculated from all three replicates and the remaining 21% from two replicates. The logged data was normalized, using the empirical Bayes algorithm ComBat (Johnson et al., 2007) by applying the ComBat function in the SVA package for R, to adjust for technical variation, followed by a linear scaling of data from each array to adjust for variations in sample background level. The scaling factor was based on the 20% of antibodies with the lowest standard deviation across all samples and was calculated by dividing the intensity sum of these antibodies on each array with the average sum across all arrays (Carlsson et al., 2008; Ingvarsson et al., 2008). The primary data is available from the corresponding author upon request.
2.5. Data analysis
The sample and variable distribution was analyzed and visualized using a principal component analysis (PCA) based program (Qlucore, Lund, Sweden). ANOVA was applied for initial filtering within the PCA analysis for display of data distribution in the PDAC and NC subgroups. The performance of individual markers was evaluated using Wilcoxon t‐test, Benjamini Hochberg procedure for false discovery rate control, and fold changes. Separation of different subgroups was assessed using support vector machine (SVM), applying a linear kernel with the cost of constraints set to 1. Initial SVM analyses were performed using a leave‐one‐out cross validation approach. To minimize over‐interpretation and to demonstrate robustness of the data, sample data was then randomly divided into training and test sets, and an SVM‐based backward elimination algorithm previously described (Carlsson et al., 2011a) was applied in the training sets to filter data. The Kullback–Leibler (K–L) error in the classification was plotted against the number of eliminated antibodies, and the optimal antibody signature was defined as the one applied when the minimum K–L error was obtained. Consequently, biomarker signatures were generated in training sets consisting of 2/3 of the total samples from each subgroup, and models based on the resulting antibody signatures were evaluated in test sets containing the remaining 1/3 of samples. Ten different pairs of training and test sets were created for this purpose, resulting in a consensus list in which each antibody was given an elimination score corresponding to its median order of elimination in the ten training sets. The performance was assessed using receiver operating characteristics (ROC) curves and reported as area under the curve (AUC) values. The sensitivity and specificity, positive and negative predictive values of each signature in its respective test set were noted for the SVM decision value threshold corresponding to the maximum sum of sensitivity and specificity.
To investigate whether bilirubin levels or age were confounding factors in the antibody microarray analysis, patients with jaundice (n = 27) were compared to patients without jaundice (n = 81) and the oldest 50% of the patient was compared to the youngest 50% of the patients. Similarly, a gender‐adjusted dataset, as well as a gender‐matched sub‐dataset were generated to assess whether gender was a confounding factor.
3. Results
3.1. Discrimination between cases and controls
Initially, we assessed whether PDAC cases could be discriminated from normal controls, using PCA, q‐value filtration, and SVM analysis with leave‐one‐out cross validation. Principal component analysis revealed a moderate separation of PDAC and NC samples (Figure 1A), and differential analysis with a q‐value cut‐off of 0.1 resulted in 11 antibodies displaying significantly different expression levels in PDAC vs. NC (Figure 1B and Supplementary Figure 1). Three of these were targeting Apolipoprotein A1 (Apo‐A1) and showed decreased proteins levels in PDAC vs. NC. Properdin, C1q, C3, IgM and IL‐8 also showed reduced levels in PDAC, while VEGF, MAPK‐8 and CHP‐1 were elevated in the cancer group. Initial SVM analysis with leave‐one‐out cross validation using data from all 336 antibodies, demonstrated that PDAC and NC were separated with an AUC‐value of 0.88 (p‐value = 6.4 × 10−21, Figure 1C).
Figure 1.
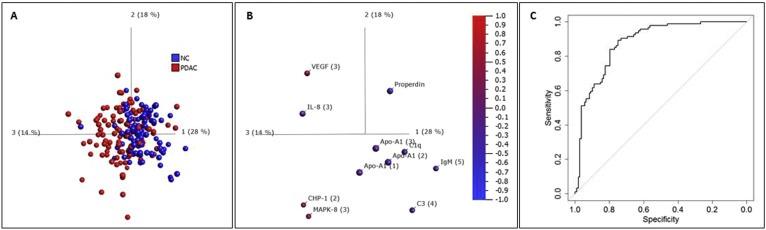
Discrimination of PDAC vs. NC. (A) Principal component analysis (PCA) of PDAC (red) and NC (blue). The data was filtered to q < 0.1 using ANOVA; (B) Relative protein levels demonstrated by the 11 antibodies that remained after filtering in a PCA plot synchronized to the one in (A). Red = up‐regulated levels, blue = down‐regulated levels in PDAC vs. NC; (C) ROC‐curve with AUC of 0.88 from SVM analysis with leave‐one‐out cross‐validation of PDAC vs. NC based on unfiltered data (using data from all antibodies).
Of note, the 25% of patients with jaundice could not be significantly discriminated from patients without jaundice (AUC 0.64), although this analysis was hampered by the more early stage (I/II, 36%) than late stage (III/IV, 16%) patients presented with jaundice. Similarly, the oldest 50% of the cancer patients could not be separated from the youngest 50% (AUC 0.48). These results indicated that neither age nor hyperbilirubinemia were confounding factors in the microarray analysis. The gender distribution was biased in the cohort, thus several measures were performed to investigate the effect of gender on the analysis. A gender‐normalized dataset was generated, showing that PDAC vs. NC was equally well (AUC 0.89 vs. 0.88) discriminated when the gender factor had been eliminated. The lists of significant (p < 0.05) antibodies identified in the original and the gender‐normalized data were also highly similar (Supplementary Table 2). Moreover, a smaller, completely gender matched dataset of 19 male, 19 female in both the PDAC and the NC group was generated in which male and female could not be separated (AUC 0.59), while PDAC and NC were still discriminated with high accuracy (AUC 0.84). As a third measure, the PDAC vs. NC comparison was performed in the male and female sample groups separately, showing that PDAC vs. NC could be discriminated in both groups (AUC 0.87 in female, AUC 0.83 in male) despite that these comparisons were affected by skewed PDAC and NC distribution. Finally, male and female could not be well separated (AUC 0.62) in early stage (I/II) samples only, the only subset of samples for which the number of male (n = 20) and female (n = 21) samples were even. Taken together, these results pointed to the fact that gender could not be considered a confounding factor.
3.2. Identification of plasma protein signatures associated with pancreatic cancer
The high level of differentiation between PDAC and NC based on unfiltered data (AUC 0.88) motivated in‐depth data filtering for identifying a condensed PDAC‐associated protein signature. To avoid over‐fitting the model to the data, samples were first separated into training sets for generating antibody signatures models, which then were evaluated using separate test sets. In the training sets, antibodies were filtered using SVM‐based backward elimination and the Kullback–Leibler (K–L) error in the classification was plotted against the number of eliminated antibodies. Figure 2A illustrates the elimination process in the first training set, in which a distinct minimum of the error was observed after 313 iterations, corresponding to a 23 antibody signature. Based on this signature, an SVM model was constructed in the training set and evaluated in a separate test set, where it generated an AUC‐value of 0.87 (Figure 2B). To test the robustness of the data set this elimination procedure was repeated in a total of ten different, randomly generated pairs of training and test sets, which in term generated ten signatures identified for optimal separation of PDAC vs. NC. The length of signatures ranged from 17 to 29 antibodies (median 23.5). The AUC‐values in the test sets ranged from 0.77 to 0.87, with an average of 0.83 (Figure 2C). The sensitivity and specificity had average values of 0.77 (ranging 0.56–0.94) and 0.86 (ranging 0.55–0.97), respectively, with the corresponding average positive predictive value of 0.86 (ranging 0.71–0.97) and average negative predictive value of 0.77 (ranging 0.64–0.89).
Figure 2.
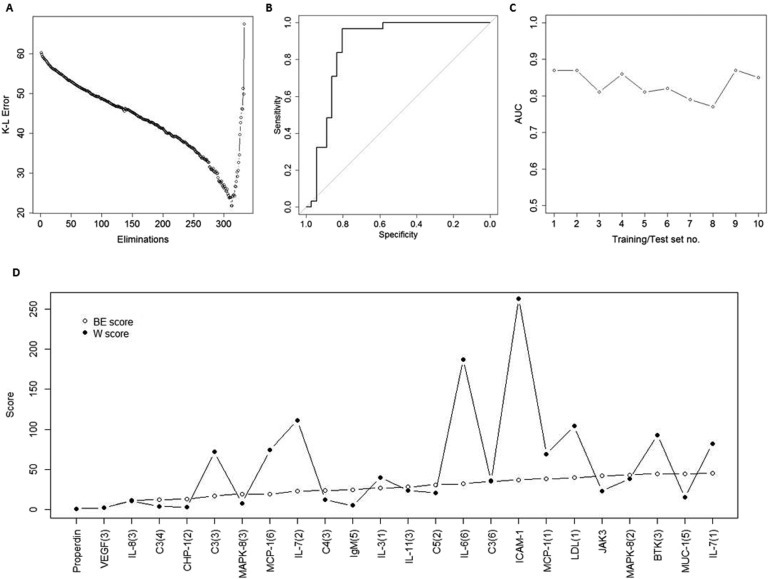
Identification of plasma protein signatures for PDAC. A training set and a test set was generated by randomized selection of 2/3 of samples from each group (PDAC and NC) to the training set, and the remaining 1/3 of samples to the test set. The training set was used to define a condensed signature for discriminating PDAC from NC. (A) Filtering of variables was conducted by a SVM‐based stepwise backward elimination of the antibodies in the training set. In each iterative step, the Kullback–Liebler (K–L) error of the classification was determined and plotted. The antibodies that remained in the elimination process when the classification error reached its minimum value were used as a unique signature for constructing a new model in the training set; (B) ROC‐curve resulting from the signature model from the training set, “frozen” and directly applied onto the previously unseen test set samples; (C) The procedure was repeated to a total of ten times, in ten different sets of randomly created training and test sets. The area under the ROC‐curve (AUC values) generated by the frozen biomarker signature models in each corresponding test set were plotted; (D) The antibody score derived from the overall ranking in the backward elimination (BE) process (open circles) was compared to the score based on the Wilcoxon (W) test ranking (filled circles).
Each antibody was scored based on the reverse order of elimination, with number one being the last antibody to be eliminated, and ranked in order of their median elimination score from the ten sequential elimination rounds. Table 2 lists the 25 highest ranked antibodies, with their p‐ and q‐values, and the p‐value ranking for PDAC vs. NC. The top two antibodies, with median elimination scores of 1 and 2, respectively, were the last eliminated antibodies in 9/10 (Properdin), and the second last eliminated in 8/10 (VEGF) training sets. The top 25 ranked antibodies together represented 20 different specificities.
Table 2.
Top 25 signature candidate analytes.
| Elimination rank | Name | Median elimination score | Wilcoxon p‐value | Benjamini–Hochberg q‐value | Wilcoxon rank | Previous elimination rank (Wingren et al., 2012) |
|---|---|---|---|---|---|---|
| 1 | Properdin | 1 | 6.18E‐15 | 2.08E‐12 | 1 | 3 |
| 2 | VEGF (3) | 2 | 1.84E‐08 | 3.10E‐06 | 2 | 17 |
| 3 | IL‐8 (3) | 11.5 | 1.99E‐03 | 6.07E‐02 | 11 | 114 |
| 4 | C3 (4) | 12.5 | 3.74E‐05 | 3.15E‐03 | 4 | N/A |
| 5 | CHP‐1 (2) | 13 | 4.47E‐06 | 5.00E‐04 | 3 | N/A |
| 6 | C3 (3) | 17 | 1.31E‐01 | 6.01E‐01 | 72 | N/A |
| 7 | MAPK‐8 (3) | 19.5 | 1.85E‐04 | 7.76E‐03 | 8 | N/A |
| 8 | MCP‐1 (6) | 19.5 | 1.32E‐01 | 6.01E‐01 | 74 | N/A |
| 9 | IL‐7 (2) | 23 | 2.53E‐01 | 7.60E‐01 | 111 | 21 |
| 10 | C4 (3) | 23.5 | 2.96E‐03 | 8.29E‐02 | 12 | N/A |
| 11 | IgM (5) | 24.5 | 5.61E‐05 | 3.77E‐03 | 5 | N/A |
| 12 | IL‐3 (1) | 27 | 7.36E‐02 | 5.72E‐01 | 40 | 18 |
| 13 | IL‐11 (3) | 28 | 2.17E‐02 | 3.03E‐01 | 24 | 35 |
| 14 | C5 (2) | 31 | 1.81E‐02 | 2.84E‐01 | 21 | 1 |
| 15 | IL‐6 (6) | 32 | 4.95E‐01 | 8.88E‐01 | 187 | N/A |
| 16 | C3 (6) | 35 | 6.06E‐02 | 5.66E‐01 | 36 | N/A |
| 17 | ICAM‐1 | 37 | 7.36E‐01 | 9.37E‐01 | 263 | N/A |
| 18 | MCP‐1 (1) | 38.5 | 1.22E‐01 | 5.89E‐01 | 69 | 15 |
| 19 | LDL (1) | 39.5 | 2.33E‐01 | 7.52E‐01 | 104 | N/A |
| 20 | JAK3 | 42 | 1.96E‐02 | 2.86E‐01 | 23 | 34 |
| 21 | MAPK‐8 (2) | 43 | 6.79E‐02 | 5.72E‐01 | 38 | N/A |
| 22 | MUC‐1 (5) | 44.5 | 1.02E‐02 | 2.29E‐01 | 15 | 53 |
| 23 | BTK (3) | 44.5 | 2.06E‐01 | 7.26E‐01 | 93 | N/A |
| 24 | IL‐7 (1) | 45 | 1.61E‐01 | 6.61E‐01 | 82 | 88 |
| 25 | LUM | 46.5 | 1.04E‐01 | 5.89E‐01 | 55 | N/A |
The backward elimination procedure was designed to identify the optimal combination of antibodies, not taking into consideration one‐dimensional separation of data based on individual antibodies, and the consensus signature presented in Table 2 is based solely on the backward elimination ranking. However, the top two antibodies were also the two highest ranked on basis of p‐ and q‐values. In fact, the five highest ranked antibodies all displayed highly significant p‐ and q‐values for PDAC vs. NC (p < 4.47E‐06 and q < 5.00E‐04). The backward elimination rank (BE‐score) and the t‐test rank (W‐score) for the consensus signature antibodies were plotted together in Figure 2D. The W‐score starts to deviate from the BE‐score after the top five antibodies, and then lost any correlation. Thus, the five highest ranked antibodies (Properdin, VEGF, IL‐8, C3, and CHP‐1) make out a stable core of the consensus signature, as indicated by both the backward elimination procedure and the univariate differential expression analysis. However, the current data, consistent with previous datasets analyzed with similar approaches, shows that the signature core needs to be supplemented by orthogonal markers to reach a clinically relevant level of accuracy in terms of sensitivity and specificity, for discriminating PDAC vs. NC.
3.3. Discrimination between stages of pancreatic cancer
Discrimination between different stages of PDAC is pertinent for being able to diagnose PDAC in its non‐invasive stages and the PDAC samples were therefore stratified according to disease stage. SVM with leave‐one‐out cross validation showed that all PDAC stages could be separated from NC and that classification accuracy increased with disease stage, with AUC‐values of 0.71, 0.86, 0.90 and 0.93 for discriminating NC from stage I, II, III, and IV, respectively (Figure 3A). The subgrouping into stages resulted in smaller sample numbers, ranging from n = 11 for stage I to n = 34 for stage III patients. For increased statistical power, a grouping into early confined disease (stage I/II) and late invasive disease (stage III/IV) was also performed. These groups were discriminated from NC with AUC values of 0.80 (early stage) and 0.96 (late stage). Figure 3B shows all antibodies displaying significant (Wilcoxon p < 0.05) differential protein levels in at least one of the stage groups when compared to NC (see Supplementary Figure 2 for stage‐separated dot plots for all patients). Among the five described core candidate markers (Properdin, VEGF, IL‐8, C3, and CHP‐1), Properdin was down‐regulated in all stages, CHP‐1 was up‐regulated in all stages, and IL‐8 was down‐regulated in stage III/IV disease only.
Figure 3.
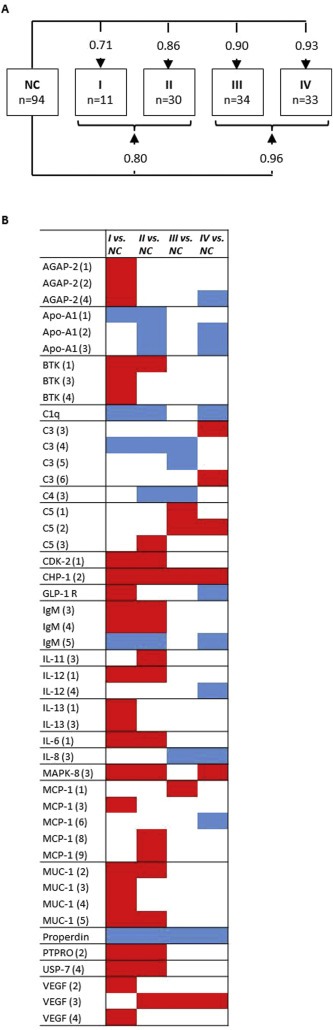
Discrimination of PDAC stages vs. controls. (A) AUC‐values from SVM analysis with leave‐one‐out cross validation using unfiltered data (all antibodies), comparing NC to patients grouped according to their PDAC stage. (B) Antibodies with Wilcoxon p < 0.05 in one or more PDAC stages vs NC. Red = up‐regulated, blue = down‐regulated in PDAC vs NC, white = no significant difference.
While the vast majority of antibodies of the same specificity showed a consistent binding pattern, it was observed that VEGF (clone 3) showed elevated VEGF levels in stages II–IV, while VEGF (clone 2) and (clone 4) indicated elevated levels in stage I patients only. Similarly, C3 (clone 4) and (clone 5) showed down‐regulated C3 levels in stage I–III disease, while clone 3 and clone 6 showed increased levels in stage IV. The specificity for some of these clones, including VEGF (clone 3) which was part of the PDAC vs. NC core signature, have been validated using mass spectrometry but the precise epitope specificities are not presently known. Hence, the differential stage‐associated signals could possibly be explained by different epitope accessibility due to splicing events, during disease progression from stage I–IV.
It is noteworthy that in our previous study on serum markers in stage III/IV Caucasian PDAC patients, C5 was ranked as the most prominent marker in a backward elimination filtering analysis, while the current study ranked the same antibody as number 14 (Table 2). However, when stage‐specific analysis was performed it was evident that C5 was elevated only in late stage disease, which is coherent with the former study (Wingren et al., 2012). Of note, the stage‐specific analysis pointed out several early marker candidates, e.g. elevated levels of BKT, CDK2, MAPK‐8, AGAP‐2, IL‐13, IL‐6, PTPRO, USP‐7, MUC‐1, and reduced levels of Apo‐A1 and C1q, measurable already at stage I/II disease.
3.4. Correlation between markers derived from Caucasian and Chinese populations
We next assessed the concordance of the plasma protein signature in the Chinese PDAC subjects with the serum protein signature previously identified in a late stage PDAC Caucasian cohort, analyzed in relation to normal controls as well as to patients with pancreatitis (Wingren et al., 2012). Of note, the antibody microarray content had expanded significantly since the former study, limiting measures of correlation of the two studies. The former study contained only 36% of the antibodies currently used and did for example not include the core marker CHP‐1. However, the remaining four core signature proteins (Properdin, VEGF, IL‐8 and C3) were all part of the previously published serum PDAC signature. In addition to the core panel, there was a clear overlap between the two ethnic cohorts when comparing the 25 highest ranked antibodies in the two studies (Table 2), indicating that blood‐based proteomics analysis is less affected by the genetic make‐up of sample donors (Caucasian/Asian), or sample format (serum/plasma).
In the previous Caucasian cohort it was also shown that PDAC could be differentiated both from normal controls and pancreatitis (Wingren et al., 2012). Since our Asian cohort did not include chronic pancreatitis samples, we instead compared significant (Wilcoxon p < 0.05) markers present in this and previous studies (Sandstrom et al., 2012; Wingren et al., 2012). Five markers (C3, C5, IL‐12, IL‐8, Properdin) were commonly expressed in both serum and plasma PDAC samples, as well as in pancreatitis samples (Supplementary Figure 3). Despite this overlap, the PDAC‐associated profile was notably different compared to chronic pancreatitis. Moreover, the current and previous studies have demonstrated that it is the combination of markers in a multiplexed signature that will deliver the most precise accuracy, regardless of whether a subset of the markers are overlapping with inflammatory or non‐related indications.
3.5. Markers associated with tumor location
PDAC tumors located in different parts of the pancreas appear to have different biology and aggressiveness; hence markers for discriminating tumor location could be of clinical relevance. Therefore, the samples were also grouped by the primary tumor location in the pancreas, and plasma from tumors located in the head of pancreas was compared to those located in the body and/or tail of pancreas. Samples with tumors at other locations (neck = 4, neck + body = 1, head + tail = 1) were excluded from this analysis. Applying a cut‐off of Wilcoxon p < 0.05, 37 antibodies showed significantly different intensity levels in Head vs. Body/Tail (Figure 4). The AUC for Head (n = 63) vs. Body/Tail (n = 39) localized tumors was 0.64 (p = 5.4e‐3). Although this modest AUC value showed that the groups could not be distinctly separated by SVM analysis, the 37 significant antibodies identified by univariate analysis overlapped remarkably well with the signals derived from the same antibodies identified by the same approach in a previous study (Figure 4), despite differences in regards to sample format (serum/plasma), ethnicity (Caucasian/Asian), technical processes (assay protocol and instrumentation) and data processing (normalization procedures) (Gerdtsson et al., 2015). The only protein not correlating with the former study was C3, which was found to be elevated in plasma but reduced in serum in Head vs. Body/Tail localized tumors. Complement proteins are, however, sensitive to serum preparation and their concentrations are known to differ between serum and plasma, which may explain the observed discrepancy of C3 in the two studies. Of note, the proteins that discriminated between Head and Body/Tail localized tumors (Figure 4) were also distinctly different from the protein signatures for PDAC vs. NC (Table 2).
Figure 4.
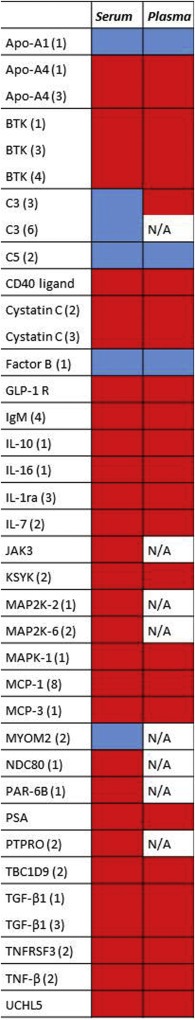
Differentiation of primary tumor location and comparison to a previous study in serum (Gerdtsson et al., 2015). Red = up‐regulated, blue = down‐regulated in Head vs Body/Tail tumors, N/A = antibody not included in study.
4. Discussion
This study describes the use of a recombinant (scFv) antibody microarray for plasma profiling of immunoregulatory and cancer‐associated proteins in PDAC. The technology has previously been applied for accurate (AUC > 0.95) prediction of late (stage III/IV) PDAC vs. NC in serum (Gerdtsson et al., 2015; Ingvarsson et al., 2008; Wingren et al., 2012). Here, an updated version of the platform was applied to a cohort of Chinese PDAC and NC plasma samples. Due to regulatory reasons, the analyses were for the first time conducted in an external (Chinese) laboratory. Despite the technology transfer and vast differences in study design, including sample format (plasma vs. serum), instrumentation (incubators, scanners), microarray content, layout, and production, as well as data analyses approaches, several “core” proteins identified here as potential markers for PDAC vs. NC correlated to findings in previously analyzed cohorts. Apart from the technology transfer, this is also the first time this platform has been used for stage‐specific (stage I–IV) analysis of PDAC.
For the identification of a plasma marker signature, two complementary strategies were used, (i) univariate differential expression analysis generating a multiple‐testing corrected q‐value for each antibody in the assay, and (ii) a backward elimination approach designed to identify the optimal combination of markers, contributing with orthogonal information for discriminating cases and controls (Carlsson et al., 2011a). The two strategies resulted in the identification of a robust core signature of the five proteins Properdin, VEGF, IL‐8, C3, and CHP‐1. The specificity of the Properdin antibody has been validated in crude serum samples of known concentrations (Ingvarsson et al., 2007), and it has been shown in two previous independent studies that Properdin is strongly down‐regulated in PDAC serum (Wingren et al., 2012) and plasma (Gerdtsson et al., unpublished observations). VEGF, associated with the angiogenesis‐dependence of tumor growth, is known to be upregulated in many cancers including PDAC (Itakura et al., 1997), and the present VEGF antibody, for which the specificity has been validated by mass spectrometry, has also in earlier studies demonstrated a significantly elevated VEGF level in PDAC (Gerdtsson et al., 2015; Wingren et al., 2012). IL‐8 and Complement Factor C3 have also previously been shown by us and others to be associated with late stage PDAC (Chen et al., 2013; Shaw et al., 2014; Wingren et al., 2012). In contrast, CHP‐1, Calcineurin Homologous Protein‐1, has not previously been measured by us and we believe its association with PDAC is novel to this study. CHP‐1 is part of the Ca2+‐binding family, and is a widely expressed protein localized in multiple subcellular compartments. Apart from being involved in trafficking across the plasma membrane, the functions of this presumably pluripotent protein is largely unknown (Jimenez‐Vidal et al., 2010), although its isoform CHP‐2 has been shown to promote tumor growth, invasion and metastasis in ovarian cancer (Jin et al., 2007).
The results showed that the core signature needed to be supplemented with additional proteins in order to achieve the highest possible sensitivity and specificity. Here, approximately 23 markers were shown to deliver an optimal discrimination of PDAC vs. NC. In addition to the five protein core, several potential markers of interest were identified. Apo‐A1, the major component of high density lipoprotein (HDL), was one of the strongest differentially expressed proteins, as shown by all three Apo‐A1‐specific antibodies (q‐values 0.003–0.02), consistent with previous observations in PDAC plasma (Honda et al., 2012) (Gerdtsson et al., unpublished observations). Decreased level of HDL is associated with poor cardiovascular health, and thus may reflect the association of PDAC with smoking and obesity. C1q was another down‐regulated protein in accordance with our previous study (Wingren et al., 2012). It is noteworthy that although both Apo‐A1 and C1q were among the top markers based on univariate analysis, they were not included in the consensus signature derived from the backward elimination analysis, which only takes into account the performance of the combined signature and not the individual markers therein. In contrast, MAPK‐8, a serine/threonine protein kinase involved in several cellular processes and signaling pathways, has not previously been analyzed using this platform, and was identified as a high scoring marker by both approaches. The signature of the five core proteins with the supplemental tentative markers identified in the current study will form the basis in follow‐up analyses, focusing in particular on analyzing high‐risk groups, such as patients with hereditary pancreatic cancer, chronic pancreatitis and late onset diabetes (Pannala et al., 2009).
Since early diagnosis significantly increases the life expectancy of PDAC patients (Furukawa et al., 1996; Shimizu et al., 2005), the defined markers associated with stage I/II are of particular importance when designing a clinically relevant test. Although the discrimination of PDAC vs. NC increased with PDAC stage, also stage I/II patients could be differentiated from NC despite limited sample numbers. The stage‐specific signatures presented here were based on univariate analysis using all data from the microarray analysis, and could probably be refined using larger cohorts. Several proteins, including AGAP‐2, BTK, CDK‐2, IL‐13, IL‐6, MUC‐1, PTPRO, USP‐7, were shown to have elevated levels in locally confined early stage cancer specifically. Four of these, AGAP‐2, CDK‐2, PTPRO, and USP‐7, have not previously been measured in our microarray assay. CDK‐2, or Cyclin‐dependent kinase 2, is involved in controlling the cell cycle and the aberrant activation of the CDKs is a well‐known hallmark of many cancers, including PDAC (Feldmann et al., 2011). The remaining novel markers have not previously been associated with PDAC specifically, although they have been identified in other cancers. AGAP‐2, a GTPase‐activating protein for ARF1 and ARF5, has been shown to prevent apoptosis and promote cancer cell invasion and its cancer‐related over‐expression has also been demonstrated previously (Xia et al., 2003). Likewise, PTPRO, or protein tyrosine phosphatase receptor‐type O, has been identified as a tumor suppressor in a variety of cancers, including hepatocellular, lung and breast cancers (Hou et al., 2013; Huang et al., 2013; Motiwala et al., 2004). USP‐7, or Ubiquitin carboxyl‐terminal hydrolase 7, deubiquitinates several cancer‐associated target proteins, including p53 and PTEN (Song et al., 2008) and its over‐expression has been demonstrated for several cancers (Zhao et al., 2015). Other markers have been previously identified in PDAC (Ingvarsson et al., 2008; Wingren et al., 2012), including MUC‐1, which is overexpressed in 90% of PDAC cases (Winter et al., 2012), IL‐6 (Bellone et al., 2006), and IL‐13 (Gabitass et al., 2011), as well as the tyrosine kinase BTK. Albeit of interest, the role of these markers in early stage PDAC needs to be further analyzed and validated in larger early‐stage patient cohorts.
Differential diagnosis of PDAC vs. pancreatitis is sometimes difficult and addressed by us in a previous study where late stage PDAC could be discriminated from a combined control group of different pancreatic inflammatory indications, as well as healthy individuals (Wingren et al., 2012). Following this, an additional study was conducted on pancreatitis subtypes, where we identified protein signatures specific for acute, chronic and autoimmune pancreatitis (Sandstrom et al., 2012). The lack of pancreatitis samples is a limitation of the current study. However, the stage‐defined profiles identified here were significantly different from the inflammation associated signatures previously presented.
Although ethnic genetic diversity has, in addition to environmental factors, been coupled to the incidence and progression of cancer in different parts of the world (Gupta et al., 2014; Rastogi et al., 2004), the protein profiles observed in PDAC patients of Asian and Caucasian origin were similar. While biological heterogeneity has been described as a hurdle in the search for gene biomarkers (Gupta et al., 2014), our findings may indicate that protein biomarkers could be more robust and transferrable between different ethnicities, potentially due to less diversity on a whole protein level as compared to genetic mutations.
The biological diversity of tumors due to localization in the pancreas has been previously demonstrated (Ling et al., 2013). Tumors in the body and tail of pancreas are rarer than tumor in the head of pancreas (77% of PDAC) (Lau et al., 2010). Because of differences in e.g. blood supply and lymphatic and venous backflow, there are also differences in the disease presentation with body and tail tumors causing less jaundice, more pain, higher albumin and CEA levels and lower CA19‐9 levels (Eyigor et al., 2010; Watanabe et al., 2004). Body and tail tumors are often detected at a later stage and show higher rate of metastasis than head tumors. As the biological variances can result in different treatment efficiency (Wu et al., 2007), markers that can discriminate between these tumors could be of clinical relevance and aid personalized treatment strategies. Few differences have been found on a genetic level, with no significant variation in the overall number of mutations, deletions and amplifications, or in K‐ras point mutations (Ling et al., 2013). Although the SVM analysis showed only moderate discrimination, 37 antibodies were significantly differentially expressed in head vs. body/tail tumors, with a protein expression pattern that correlated well with a previous study. The strong concordance between the independent studies is encouraging for a future development of a blood protein biomarker signature discriminating body/tail and head tumors at an early disease stage.
In summary, this study demonstrated the technical transferability of a recombinant antibody microarray platform. Resectable (stage I/II) as well as locally advanced (stage III) and distant metastatic (stage IV) PDAC could be discriminated from normal controls, prerequisites for a test focusing on early diagnosis of PDAC. Next, the protein profiles identified here will be tested for use as biomarker signatures for PDAC diagnosis in larger cohorts, including relevant risk groups.
Competing interests
CB, CW and ASG are inventors of pending patent on pancreatic cancer biomarkers.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgements
For the support and professionalism in establishing valuable contacts in China we gratefully acknowledge Invest in Skåne. We also acknowledge the excellent laboratory assistance of Maria Walle and Ann‐Charlotte Olsson, as well as the laboratory support from CapitalBio Corp. (Beijing). This study was supported by grants from VINNOVA (20011‐03926), CREATE Health Translational Cancer Center, the Lund University Faculty of Engineering, EU FP7 Grant Agreement 241481 AFFINOMICS and BioCARE (20141023‐107717).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.07.001.
Gerdtsson Anna Sandström, Wingren Christer, Persson Helena, Delfani Payam, Nordström Malin, Ren He, Wen Xin, Ringdahl Ulrika, Borrebaeck Carl A.K., Hao Jihui, (2016), Plasma protein profiling in a stage defined pancreatic cancer cohort – Implications for early diagnosis, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.07.001.
Contributor Information
Anna Sandström Gerdtsson, Email: anna.sandstrom@immun.lth.se.
Christer Wingren, Email: christer.wingren@immun.lth.se.
Helena Persson, Email: helena.persson@immun.lth.se.
Payam Delfani, Email: payam.delfani@immun.lth.se.
Malin Nordström, Email: malin.nordstrom@immunovia.se.
He Ren, Email: renhe@tjmuch.com.
Xin Wen, Email: xinwen8685@163.com.
Ulrika Ringdahl, Email: ulrika.ringdahl@skane.se.
Carl A.K. Borrebaeck, Email: carl.borrebaeck@immun.lth.se
Jihui Hao, Email: haojihui@tjmuch.com.
References
- Alonzo, T.A. , Pepe, M.S. , Moskowitz, C.S. , 2002. Sample size calculations for comparative studies of medical tests for detecting presence of disease. Stat. Med. 21, 835–852. [DOI] [PubMed] [Google Scholar]
- Bauden, M. , Pamart, D. , Ansari, D. , Herzog, M. , Eccleston, M. , Micallef, J. , Andersson, B. , Andersson, R. , 2015. Circulating nucleosomes as epigenetic biomarkers in pancreatic cancer. Clin. Epigenetics 7, 106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone, G. , Smirne, C. , Mauri, F.A. , Tonel, E. , Carbone, A. , Buffolino, A. , Dughera, L. , Robecchi, A. , Pirisi, M. , Emanuelli, G. , 2006. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: implications for survival. Cancer Immunol. Immunother. 55, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrebaeck, C.A. , Wingren, C. , 2011. Recombinant antibodies for the generation of antibody arrays. Methods Mol. Biol. 785, 247–262. [DOI] [PubMed] [Google Scholar]
- Brand, R.E. , Nolen, B.M. , Zeh, H.J. , Allen, P.J. , Eloubeidi, M.A. , Goldberg, M. , Elton, E. , Arnoletti, J.P. , Christein, J.D. , Vickers, S.M. , Langmead, C.J. , Landsittel, D.P. , Whitcomb, D.C. , Grizzle, W.E. , Lokshin, A.E. , 2011. Serum biomarker panels for the detection of pancreatic cancer. Clin. Cancer Res. 17, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunger, S. , Laubert, T. , Roblick, U.J. , Habermann, J.K. , 2011. Serum biomarkers for improved diagnostic of pancreatic cancer: a current overview. J. Cancer Res. Clin. Oncol. 137, 375–389. [DOI] [PubMed] [Google Scholar]
- Carlsson, A. , Wingren, C. , Ingvarsson, J. , Ellmark, P. , Baldertorp, B. , Ferno, M. , Olsson, H. , Borrebaeck, C.A. , 2008. Serum proteome profiling of metastatic breast cancer using recombinant antibody microarrays. Eur. J. Cancer 44, 472–480. [DOI] [PubMed] [Google Scholar]
- Carlsson, A. , Wingren, C. , Kristensson, M. , Rose, C. , Ferno, M. , Olsson, H. , Jernstrom, H. , Ek, S. , Gustavsson, E. , Ingvar, C. , Ohlsson, M. , Peterson, C. , Borrebaeck, C.A. , 2011. Molecular serum portraits in patients with primary breast cancer predict the development of distant metastases. Proc. Natl. Acad. Sci. U. S. A 108, 14252–14257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson, A. , Wuttge, D.M. , Ingvarsson, J. , Bengtsson, A.A. , Sturfelt, G. , Borrebaeck, C.A. , Wingren, C. , 2011. Serum protein profiling of systemic lupus erythematosus and systemic sclerosis using recombinant antibody microarrays. Mol. Cell. Proteomics 10, M110 005033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Wu, W. , Zhen, C. , Zhou, H. , Yang, R. , Chen, L. , Hu, L. , 2013. Expression and clinical significance of complement C3, complement C4b1 and apolipoprotein E in pancreatic cancer. Oncol. Lett. 6, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon, K.C. , Klimstra, D.S. , Brennan, M.F. , 1996. Long-term survival after curative resection for pancreatic ductal adenocarcinoma. Clinicopathologic analysis of 5-year survivors. Ann. Surg. 223, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens, L.M. , Werb, Z. , 2002. Inflammation and cancer. Nature 420, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dexlin-Mellby, L. , Sandstrom, A. , Centlow, M. , Nygren, S. , Hansson, S.R. , Borrebaeck, C.A. , Wingren, C. , 2010. Tissue proteome profiling of preeclamptic placenta using recombinant antibody microarrays. Proteomics Clin. Appl. 4, 794–807. [DOI] [PubMed] [Google Scholar]
- Eyigor, C. , Karaca, B. , Kuzeyli-Yildirim, Y. , Uslu, R. , Uyar, M. , Coker, A. , 2010. Does the tumor localization in advanced pancreatic cancer have an influence on the management of symptoms and pain?. J. B.U.ON. 15, 543–546. [PubMed] [Google Scholar]
- Feldmann, G. , Mishra, A. , Bisht, S. , Karikari, C. , Garrido-Laguna, I. , Rasheed, Z. , Ottenhof, N.A. , Dadon, T. , Alvarez, H. , Fendrich, V. , Rajeshkumar, N.V. , Matsui, W. , Brossart, P. , Hidalgo, M. , Bannerji, R. , Maitra, A. , Nelkin, B.D. , 2011. Cyclin-dependent kinase inhibitor Dinaciclib (SCH727965) inhibits pancreatic cancer growth and progression in murine xenograft models. Cancer Biol. Ther. 12, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa, H. , Okada, S. , Saisho, H. , Ariyama, J. , Karasawa, E. , Nakaizumi, A. , Nakazawa, S. , Murakami, K. , Kakizoe, T. , 1996. Clinicopathologic features of small pancreatic adenocarcinoma. A collective study. Cancer 78, 986–990. [DOI] [PubMed] [Google Scholar]
- Gabitass, R.F. , Annels, N.E. , Stocken, D.D. , Pandha, H.A. , Middleton, G.W. , 2011. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 60, 1419–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangi, S. , Fletcher, J.G. , Nathan, M.A. , Christensen, J.A. , Harmsen, W.S. , Crownhart, B.S. , Chari, S.T. , 2004. Time interval between abnormalities seen on CT and the clinical diagnosis of pancreatic cancer: retrospective review of CT scans obtained before diagnosis. AJR. Am. J. Roentgenol 182, 897–903. [DOI] [PubMed] [Google Scholar]
- Gerdtsson, A.S. , Malats, N. , Säll, A. , Real, F.X. , Porta, M. , Skoog, P. , Persson, H. , Wingren, C. , Borrebaeck, C.A.K. , 2015. A multicenter trial defining a serum protein signature associated with pancreatic ductal adenocarcinoma. Int. J. Proteomics 2015, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S. , Venkatesh, A. , Ray, S. , Srivastava, S. , 2014. Challenges and prospects for biomarker research: a current perspective from the developing world. Biochim. Biophys. Acta 1844, 899–908. [DOI] [PubMed] [Google Scholar]
- Gustavsson, E. , Ek, S. , Steen, J. , Kristensson, M. , Algenas, C. , Uhlen, M. , Wingren, C. , Ottosson, J. , Hober, S. , Borrebaeck, C.A. , 2011. Surrogate antigens as targets for proteome-wide binder selection. New Biotechnol. 28, 302–311. [DOI] [PubMed] [Google Scholar]
- Haab, B.B. , Geierstanger, B.H. , Michailidis, G. , Vitzthum, F. , Forrester, S. , Okon, R. , Saviranta, P. , Brinker, A. , Sorette, M. , Perlee, L. , Suresh, S. , Drwal, G. , Adkins, J.N. , Omenn, G.S. , 2005. Immunoassay and antibody microarray analysis of the HUPO Plasma Proteome Project reference specimens: systematic variation between sample types and calibration of mass spectrometry data. Proteomics 5, 3278–3291. [DOI] [PubMed] [Google Scholar]
- Honda, K. , Okusaka, T. , Felix, K. , Nakamori, S. , Sata, N. , Nagai, H. , Ioka, T. , Tsuchida, A. , Shimahara, T. , Shimahara, M. , Yasunami, Y. , Kuwabara, H. , Sakuma, T. , Otsuka, Y. , Ota, N. , Shitashige, M. , Kosuge, T. , Buchler, M.W. , Yamada, T. , 2012. Altered plasma apolipoprotein modifications in patients with pancreatic cancer: protein characterization and multi-institutional validation. PLoS One 7, e46908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, J. , Xu, J. , Jiang, R. , Wang, Y. , Chen, C. , Deng, L. , Huang, X. , Wang, X. , Sun, B. , 2013. Estrogen-sensitive PTPRO expression represses hepatocellular carcinoma progression by control of STAT3. Hepatology 57, 678–688. [DOI] [PubMed] [Google Scholar]
- Huang, Y.T. , Li, F.F. , Ke, C. , Li, Z. , Li, Z.T. , Zou, X.F. , Zheng, X.X. , Chen, Y.P. , Zhang, H. , 2013. PTPRO promoter methylation is predictive of poorer outcome for HER2-positive breast cancer: indication for personalized therapy. J. Transl Med. 11, 245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingvarsson, J. , Larsson, A. , Sjoholm, A.G. , Truedsson, L. , Jansson, B. , Borrebaeck, C.A. , Wingren, C. , 2007. Design of recombinant antibody microarrays for serum protein profiling: targeting of complement proteins. J. Proteome Res. 6, 3527–3536. [DOI] [PubMed] [Google Scholar]
- Ingvarsson, J. , Wingren, C. , Carlsson, A. , Ellmark, P. , Wahren, B. , Engstrom, G. , Harmenberg, U. , Krogh, M. , Peterson, C. , Borrebaeck, C.A. , 2008. Detection of pancreatic cancer using antibody microarray-based serum protein profiling. Proteomics 8, 2211–2219. [DOI] [PubMed] [Google Scholar]
- Itakura, J. , Ishiwata, T. , Friess, H. , Fujii, H. , Matsumoto, Y. , Buchler, M.W. , Korc, M. , 1997. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin. Cancer Res. 3, 1309–1316. [PubMed] [Google Scholar]
- Jimenez-Vidal, M. , Srivastava, J. , Putney, L.K. , Barber, D.L. , 2010. Nuclear-localized calcineurin homologous protein CHP1 interacts with upstream binding factor and inhibits ribosomal RNA synthesis. J. Biol. Chem. 285, 36260–36266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Q. , Kong, B. , Yang, X. , Cui, B. , Wei, Y. , Yang, Q. , 2007. Overexpression of CHP2 enhances tumor cell growth, invasion and metastasis in ovarian cancer. In Vivo 21, 593–598. [PubMed] [Google Scholar]
- Johnson, W.E. , Li, C. , Rabinovic, A. , 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kristensson, M. , Olsson, K. , Carlson, J. , Wullt, B. , Sturfelt, G. , Borrebaeck, C.A. , Wingren, C. , 2012. Design of recombinant antibody microarrays for urinary proteomics. Proteomics Clin. Appl. 6, 291–296. [DOI] [PubMed] [Google Scholar]
- Lau, M.K. , Davila, J.A. , Shaib, Y.H. , 2010. Incidence and survival of pancreatic head and body and tail cancers: a population-based study in the United States. Pancreas 39, 458–462. [DOI] [PubMed] [Google Scholar]
- Ling, Q. , Xu, X. , Zheng, S.S. , Kalthoff, H. , 2013. The diversity between pancreatic head and body/tail cancers: clinical parameters and in vitro models. Hepatobiliary Pancreat Dis Int 12, 480–487. [DOI] [PubMed] [Google Scholar]
- Locker, G.Y. , Hamilton, S. , Harris, J. , Jessup, J.M. , Kemeny, N. , Macdonald, J.S. , Somerfield, M.R. , Hayes, D.F. , Bast, R.C. , 2006. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J. Clin. Oncol. 24, 5313–5327. [DOI] [PubMed] [Google Scholar]
- Melo, S.A. , Luecke, L.B. , Kahlert, C. , Fernandez, A.F. , Gammon, S.T. , Kaye, J. , LeBleu, V.S. , Mittendorf, E.A. , Weitz, J. , Rahbari, N. , Reissfelder, C. , Pilarsky, C. , Fraga, M.F. , Piwnica-Worms, D. , Kalluri, R. , 2015. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 523, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motiwala, T. , Kutay, H. , Ghoshal, K. , Bai, S. , Seimiya, H. , Tsuruo, T. , Suster, S. , Morrison, C. , Jacob, S.T. , 2004. Protein tyrosine phosphatase receptor-type O (PTPRO) exhibits characteristics of a candidate tumor suppressor in human lung cancer. Proc. Natl. Acad. Sci. U. S. A. 101, 13844–13849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pannala, R. , Basu, A. , Petersen, G.M. , Chari, S.T. , 2009. New-onset diabetes: a potential clue to the early diagnosis of pancreatic cancer. Lancet Oncol. 10, 88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauly, F. , Dexlin-Mellby, L. , Ek, S. , Ohlin, M. , Olsson, N. , Jirstrom, K. , Dictor, M. , Schoenmakers, S. , Borrebaeck, C.A. , Wingren, C. , 2013. Protein expression profiling of formalin-fixed paraffin-embedded tissue using recombinant antibody microarrays. J. Proteome Res. 12, 5943–5953. [DOI] [PubMed] [Google Scholar]
- Pauly, F. , Smedby, K.E. , Jerkeman, M. , Hjalgrim, H. , Ohlsson, M. , Rosenquist, R. , Borrebaeck, C.A. , Wingren, C. , 2014. Identification of B-cell lymphoma subsets by plasma protein profiling using recombinant antibody microarrays. Leuk. Res. 38, 682–690. [DOI] [PubMed] [Google Scholar]
- Pelaez-Luna, M. , Takahashi, N. , Fletcher, J.G. , Chari, S.T. , 2007. Resectability of presymptomatic pancreatic cancer and its relationship to onset of diabetes: a retrospective review of CT scans and fasting glucose values prior to diagnosis. Am. J. Gastroenterol. 102, 2157–2163. [DOI] [PubMed] [Google Scholar]
- Rastogi, T. , Hildesheim, A. , Sinha, R. , 2004. Opportunities for cancer epidemiology in developing countries. Nature reviews. Cancer 4, 909–917. [DOI] [PubMed] [Google Scholar]
- Säll, A. , Walle, M. , Wingren, C. , Müller, S. , Nyman, T. , Vala, A. , Ohlin, M. , Borrebaeck, C.A.K. , Persson, H. , 2016. Generation and analyses of human synthetic antibody libraries and their application for protein microarrays. Protein Eng Des Sel (in press) [DOI] [PubMed] [Google Scholar]
- Sandstrom, A. , Andersson, R. , Segersvard, R. , Lohr, M. , Borrebaeck, C.A. , Wingren, C. , 2012. Serum proteome profiling of pancreatitis using recombinant antibody microarrays reveals disease-associated biomarker signatures. Proteomics Clin. Appl. 6, 486–496. [DOI] [PubMed] [Google Scholar]
- Shaw, V.E. , Lane, B. , Jenkinson, C. , Cox, T. , Greenhalf, W. , Halloran, C.M. , Tang, J. , Sutton, R. , Neoptolemos, J.P. , Costello, E. , 2014. Serum cytokine biomarker panels for discriminating pancreatic cancer from benign pancreatic disease. Mol. Cancer 13, 114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, Y. , Yasui, K. , Matsueda, K. , Yanagisawa, A. , Yamao, K. , 2005. Small carcinoma of the pancreas is curable: new computed tomography finding, pathological study and postoperative results from a single institute. J. Gastroenterol. Hepatol. 20, 1591–1594. [DOI] [PubMed] [Google Scholar]
- Soderlind, E. , Strandberg, L. , Jirholt, P. , Kobayashi, N. , Alexeiva, V. , Aberg, A.M. , Nilsson, A. , Jansson, B. , Ohlin, M. , Wingren, C. , Danielsson, L. , Carlsson, R. , Borrebaeck, C.A. , 2000. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat. Biotechnol. 18, 852–856. [DOI] [PubMed] [Google Scholar]
- Sohn, T.A. , Yeo, C.J. , Cameron, J.L. , Koniaris, L. , Kaushal, S. , Abrams, R.A. , Sauter, P.K. , Coleman, J. , Hruban, R.H. , Lillemoe, K.D. , 2000. Resected adenocarcinoma of the pancreas-616 patients: results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 4, 567–579. [DOI] [PubMed] [Google Scholar]
- Song, M.S. , Salmena, L. , Carracedo, A. , Egia, A. , Lo-Coco, F. , Teruya-Feldstein, J. , Pandolfi, P.P. , 2008. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoevesandt, O. , Taussig, M.J. , 2012. European and international collaboration in affinity proteomics. New Biotechnol. 29, 511–514. [DOI] [PubMed] [Google Scholar]
- Surinova, S. , Schiess, R. , Huttenhain, R. , Cerciello, F. , Wollscheid, B. , Aebersold, R. , 2011. On the development of plasma protein biomarkers. J. Proteome Res. 10, 5–16. [DOI] [PubMed] [Google Scholar]
- Watanabe, I. , Sasaki, S. , Konishi, M. , Nakagohri, T. , Inoue, K. , Oda, T. , Kinoshita, T. , 2004. Onset symptoms and tumor locations as prognostic factors of pancreatic cancer. Pancreas 28, 160–165. [DOI] [PubMed] [Google Scholar]
- Wingren, C. , Ingvarsson, J. , Dexlin, L. , Szul, D. , Borrebaeck, C.A. , 2007. Design of recombinant antibody microarrays for complex proteome analysis: choice of sample labeling-tag and solid support. Proteomics 7, 3055–3065. [DOI] [PubMed] [Google Scholar]
- Wingren, C. , Sandstrom, A. , Segersvard, R. , Carlsson, A. , Andersson, R. , Lohr, M. , Borrebaeck, C.A. , 2012. Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res. 72, 2481–2490. [DOI] [PubMed] [Google Scholar]
- Winter, J.M. , Tang, L.H. , Klimstra, D.S. , Brennan, M.F. , Brody, J.R. , Rocha, F.G. , Jia, X. , Qin, L.X. , D'Angelica, M.I. , DeMatteo, R.P. , Fong, Y. , Jarnagin, W.R. , O'Reilly, E.M. , Allen, P.J. , 2012. A novel survival-based tissue microarray of pancreatic cancer validates MUC1 and mesothelin as biomarkers. PLoS One 7, e40157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, T.C. , Shao, Y.F. , Shan, Y. , Wu, J.X. , Zhao, P. , 2007. [Surgical effect of malignant tumor of body and tail of the pancreas: compare with pancreatic head cancer]. Zhonghua Wai Ke Za Zhi 45, 30–33. [PubMed] [Google Scholar]
- Xia, C. , Ma, W. , Stafford, L.J. , Liu, C. , Gong, L. , Martin, J.F. , Liu, M. , 2003. GGAPs, a new family of bifunctional GTP-binding and GTPase-activating proteins. Mol. Cell. Biol. 23, 2476–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida, S. , Jones, S. , Bozic, I. , Antal, T. , Leary, R. , Fu, B. , Kamiyama, M. , Hruban, R.H. , Eshleman, J.R. , Nowak, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Iacobuzio-Donahue, C.A. , 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Zeng, L. , Chen, Y. , Lian, G. , Qian, C. , Chen, S. , Li, J. , Huang, K. , 2016. Pancreatic cancer epidemiology, detection, and management. Gastroenterol. Res. Pract. 2016, 8962321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, G.Y. , Lin, Z.W. , Lu, C.L. , Gu, J. , Yuan, Y.F. , Xu, F.K. , Liu, R.H. , Ge, D. , Ding, J.Y. , 2015 Mar. USP7 overexpression predicts a poor prognosis in lung squamous cell carcinoma and large cell carcinoma. Tumour Biol. 36, (3) 1721–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data