

Abstract
1. Background
PIK3CA is the most frequent somatic mutated oncogene in estrogen receptor (ER) positive breast cancer. We previously observed an association between PIK3CA genotype and aromatase inhibitors (AI) treatment outcome. This study now evaluates whether expression of mRNAs and miRs are linked to PIK3CA genotype and are independently related to AI therapy response in order to define potential expressed biomarkers for treatment outcome.
2. Materials and methods
The miR and mRNA expression levels were evaluated for their relationship with the PIK3CA genotype in two breast tumor datasets, i.e. 286 luminal cancers from the TCGA consortium and our set of 84 ER positive primary tumors of metastatic breast cancer patients who received first line AI. BRB Array tools class comparison was performed to define miRs and mRNAs whose expression associate with PIK3CA exon 9 and 20 status. Spearman correlations established miR–mRNA pairs and mRNAs with related expression. Next, a third dataset of 25 breast cancer patients receiving neo‐adjuvant letrozole was evaluated, to compare expression levels of identified miRs and mRNAs in biopsies before and after treatment. Finally, to identify potential biomarkers miR and mRNA levels were related with overall survival (OS) and progression free survival (PFS) after first‐line AI therapy.
3. Results
Expression of 3 miRs (miR‐449a, miR‐205‐5p, miR‐301a‐3p) and 9 mRNAs (CCNO, FAM81B, LRG1, NEK10, PLCL1, PGR, SERPINA3, SORBS2, VTCN1) was related to the PIK3CA status in both datasets. All except miR‐301a‐3p had an increased expression in tumors with PIK3CA mutations. Validation in a publicly available dataset showed that LRG1, PGR, and SERPINA3 levels were decreased after neo‐adjuvant AI‐treatment. Six miR–mRNA pairs correlated significantly and stepdown analysis of all 12 factors revealed 3 mRNAs (PLCL1, LRG1, FAM81B) related to PFS. Further analyses showed LRG1 and PLCL1 expression to be unrelated with luminal subtype and to associate with OS and with PFS, the latter independent from traditional predictive factors.
4. Conclusion
We showed in two datasets of ER positive and luminal breast tumors that the expression of 3 miRs and 9 mRNAs associate with the PIK3CA status. Expression of LRG1 is independent of luminal (A or B) subtype, decreased after neo‐adjuvant AI‐treatment, and is proposed as potential biomarker for AI therapy outcome.
Keywords: PIK3CA mutations, Breast cancer, First line aromatase inhibitors therapy, LRG1
Highlights
Expression of 9 mRNAs and 3 miRs relates to PIK3CA genotype in 2 breast cancer cohorts.
All 9 mRNAs and 2 miRs were upregulated in tumors with PIK3CA mutations.
LRG1 and PLCL1 mRNA levels relate to PIK3CA status irrespective luminal subtype.
LRG1 and PLCL1 mRNA levels associate with aromatase inhibitor therapy outcome.
LRG1 expression is decreased after neo‐adjuvant letrozole treatment.
Abbreviations
- AI
aromatase inhibitors
- AKT
also known as Protein kinase B (PKB)
- BC
breast cancer
- ER
estrogen receptor
- FAM81B
family with sequence similarity, 81 member B
- HER2
human epidermal growth factor receptor 2
- LRG1
leucine rich alpha 2 glycoprotein 1
- MBC
metastatic breast cancer
- mTOR
mechanistic target of rapamycin
- PFS
progression free survival
- PLCL1
phospholipase C like 1
- SNP
single nucleotide polymorphism
1. Introduction
Breast cancer (BC) is a heterogeneous disease with different clinical, biological and phenotypical features (Koren and Bentires‐Alj, 2015). Targeted therapies against two critical pathways in BC, the ER and HER2, such as tamoxifen or aromatase inhibitors and trastuzumab respectively, are successful when BC patients are stratified based upon their ER and HER2 status. Unfortunately, not all patients respond (de novo resistance) while in the metastatic setting, patients who do respond will eventually relapse (acquired resistance).
Resistance to ER and HER2 targeted therapies may occur through activation of the PI3K pathway and/or their downstream targets AKT and mTOR (Clarke et al., 2015). Activation of the PI3K pathway was especially seen in ER negative tumors with predominantly amplified but hardly mutated (7%) PIK3CA, whereas mutations in this gene are most frequently found in ER positive tumors, up to 52% in luminal BC according to Ma et al. (2015) and The Cancer Genome Atlas Network (2012). Furthermore, primary tumors of BC patients with PIK3CA mutations were unexpected associated with favorable prognosis (Volinia and Croce, 2013) and clinical benefit from endocrine treatment (Ramirez‐Ardila et al., 2013). Interestingly, PIK3CA mRNA was the most prominent gene in a prognostic signature of 30 mRNAs and 7 miRs (Volinia and Croce, 2013) established in one of two genome wide integrated transcriptome studies performed in BC until now (Buffa et al., 2011; Volinia and Croce, 2013). The prognostic value of the mRNA component of this signature was confirmed on eight BC cohorts and outperformed several well‐known RNA predictors, however, the predictive value of this signature remains to be established.
The aim of our current study was to investigate mRNAs and miRs as potential predictive biomarkers correlated with PIK3CA genotype and AI therapy outcome. We evaluated the genome wide transcriptome of two BC datasets, and established overlapping mRNAs and miRs whose expression was related to the genotype of PIK3CA. The levels of these mRNAs and miRs were correlated with response to neo‐adjuvant and first‐line AI therapy.
2. Materials and methods
2.1. Patients and datasets
This study evaluated two previously published cohorts of breast cancer specimens for which PIK3CA genotype and mRNA expression profiles were available. It included a cohort of 286 luminal breast cancer patients (TCGA dataset) (The Cancer Genome Atlas Network, 2012) and a cohort of 84 ER positive breast cancer patients with metastatic disease who received first line aromatase inhibitors (AI dataset) (Ramirez‐Ardila et al., 2013). The TCGA transcriptome data for mRNA and miR were uploaded from their database portal (https://tcga‐data.nci.nih.gov/), and evaluated for only luminal specimens (based on PAM50 classification) with wildtype PIK3CA or with an exon 9 or exon 20 mutation for this gene.
The AI dataset included ER positive breast cancer patients from 3 institutes (Erasmus University Medical Center (EMC), Rotterdam; Netherlands Cancer Institute (NKI), Amsterdam; Sint Augustinus Hospital, Antwerpen). Patient characteristics, medical ethics board approval, code of conduct, report criteria, and therapy response criteria for this cohort have been described previously (Ramirez‐Ardila et al., 2013). Briefly, the patients received either steroidal (15 exemestane) or non‐steroidal AI (43 anastrozole, 26 letrozole). PIK3CA status and mRNA expression profiles were established by SnaPshot® multiplex assays (Life technologies) (Ramirez‐Ardila et al., 2013) and 44k mRNA oligoarrays (Agilent Technologies) (Jansen et al., 2013), respectively. The miR expression profiles were obtained for 768 probes (671 unique miRs) with Human MicroRNA Array v2.0 fluidic cards (Taqman Low Density Arrays, TLDAs) from Life Technologies according to the manufacturer's protocol. Expression data are deposited at NCBI GEO, with accession number GSE41994 for mRNAs (Jansen et al., 2013) and GSE78870 for miRs.
In addition, we evaluated a third dataset of 25 breast cancer patients who received neo‐adjuvant letrozole, available at NCBI GEO with accession code GSE59515 (Turnbull et al., 2015). This neo‐adjuvant dataset contains the transcriptome of primary tumor biopsies taken before and after 2 weeks and after 3 months treatment.
2.2. Data analyses
The TCGA set mRNA levels were median centered by gene while the mature/star miR strand levels were normalized to reads per million mapped miRNAs. The AI set mRNA levels were quantified and normalized using Agilent Feature Extraction software (Agilent, Santa Clara, CA, US). The miRNA TLDAs generated Ct values which were normalized against the median Ct value of the TLDA. All mRNA and miR expression data were log2 transformed and evaluated using BRB ArrayTools, Version 4.5.0‐Beta_1 (June 2015) (http://linus.nci.nih.gov/BRB‐ArrayTools.html). BRB class comparison was performed when at least 80% expression data points were available in the AI set. The expression data in the TCGA set were available for all the samples. A P value <0.005 and <0.05 after 100,000 permutations was considered significant for mRNAs and miRs, respectively. The integrated evaluation of miR–mRNA pairs was performed using the following databases: miRTarbase, mirSel, miRecords (V.4 – Apr 27‐2013), Ingenuity Pathway Analysis (IPA®) integrated with TarBase V.5.0. Pathway analyses were performed using (IPA®) and DAVID (https://david.ncifcrf.gov).
2.3. Statistics
Computations were performed with the STATA statistical package, release 13 (STATA Corp., College Station, TX). All P values were two sided and P < 0.05 was considered statistically significant. Bonferroni multiple test corrections were also used. Briefly, relationships between PIK3CA genotype, clinic‐pathological factors and miR/mRNA expression levels were investigated using nonparametric methods, i.e. Spearman rank correlations for continuous variables and Wilcoxon rank sum or Kruskal Wallis and chi square test for ordered variables. The Cox proportional hazard model was used in univariate analysis to compute the hazard ratio (HR) for PFS. The HR was presented with its 95% confidence intervals (95% CI). Survival curves were generated using the Kaplan–Meier method and a log rank test was applied to test for differences. PFS was defined as the time elapsed between initiation of first line AI therapy and the first detection of disease progression. miRs and mRNAs considered statistically significant in univariate analysis were included in a multivariate stepdown analysis to determine the strongest independent predictive biomarkers. Finally, these potential predictive biomarkers were added to the base model of traditional clinic‐pathological factors for metastatic breast cancer, i.e., age at start therapy (≤55, 56–70 or >70 years), disease‐free interval (0–24 or >24 months), dominant site of relapse (Local regional relapse vs bone vs visceral), PR‐status (negative or positive), and HER2‐status (negative or positive, based on Target‐Print HER2 mRNA classification).
3. Results
3.1. PIK3CA genotype and transcriptome profiles
The class comparison algorithm of BRB Array tools was used to identify differentially expressed mRNAs and miRs between PIK3CA wildtype and PIK3CA mutated ER positive (luminal) primary breast cancer specimens. This analysis revealed 119 and 111 unique differentially expressed mRNAs (P < 0.005) in the TCGA and AI dataset, respectively (Figures 1, Supplemental Table S1). The majority of genes showed increased expression in tumors with a PIK3CA mutation in both datasets, i.e. 69% (82/119) in the TCGA dataset and 84% (93/111) in the AI dataset. Likewise, 17 and 31 unique miRs were differentially expressed between PIK3CA wild type and mutated tumors (P < 0.05) in the TCGA and AI datasets (Figures 1, Supplemental Table S2). About half of the miRs showed increased expression in PIK3CA mutated tumors for the TCGA, i.e. 9 miRs in TCGA [53% (9/17)] and 16 miRs [52% (16/31)] in the AI dataset were upregulated.
Figure 1.
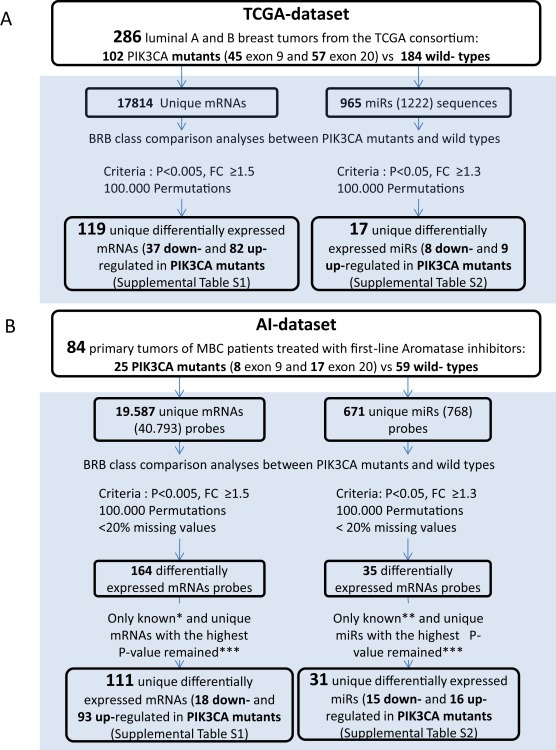
Workflow used to discover differentially expressed mRNAs and miRs when analyzed by PIK3CA mutation status. A) TCGA dataset (publicly available data from the TCGA consortium). B) AI dataset. *Known mRNAs indicates mRNAs with gene names annotated according to platform GPL6480 updated in Nov 16, 2014 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL6480); **known miRs indicates miRs annotated according to miRbaseV20; ***unique mRNAs or miRs with the highest P‐value indicate those cases in which at least two probes for an annotated mRNA or miR name were found differentially expressed. We selected the probe with the highest P‐value.
The 119 and 111 identified mRNAs were examined with Ingenuity® Pathway Analysis (IPA) to establish regulated signaling pathways in both datasets. Not surprising IPA revealed Estrogen as the only common significant up stream regulator for a subset of genes both in the AI dataset (P value = 7.09E−03) and the TCGA dataset (P value = 1.43E−06) (Supplemental Figure S1). Additional pathways were not found to be enriched (based on the IPA and DAVID analyses).
3.2. Breast cancer PIK3CA mutation specific transcriptome
To obtain a PIK3CA mutation specific transcriptome independent of the cohort studied, we selected the overlapping potential biomarkers between both the TCGA‐ and AI‐dataset (Supplemental Figure S2). In total 9 mRNAs (CCNO, FAM81B, LRG1, NEK10, PGR, PLCL1, SERPINA3, SORBS2 and VTCN1; Supplemental Table S3) and 3 miRs (hsa‐miR‐205‐5p, hsa‐miR‐301a‐3p and hsa‐miR‐449a) were related with PIK3CA genotype in both datasets. Interestingly, all biomarkers except one (miR‐301a‐3p) showed increased expression in PIK3CA mutated ER positive (luminal) breast cancer (Figure 2). Since the TCGA dataset contains both luminal A and B samples, the 12 potential biomarkers associated with PIK3CA status were further evaluated for the relationship between their expression and luminal A or B subtype. The expression of LRG1, PLCL1, SERPINA3, CCNO and miR‐449a were confirmed after multiple testing correction to be independent of subtype (Table 1). Moreover, the levels of these 5 biomarkers were in both TCGA‐ and AI‐dataset not correlated with MKI67 levels, a gene significantly upregulated in luminal B compared to luminal A subtype. This again indicates that LRG1, PLCL1, SERPINA3, CCNO and miR‐449a expression are independent from luminal subtype.
Figure 2.
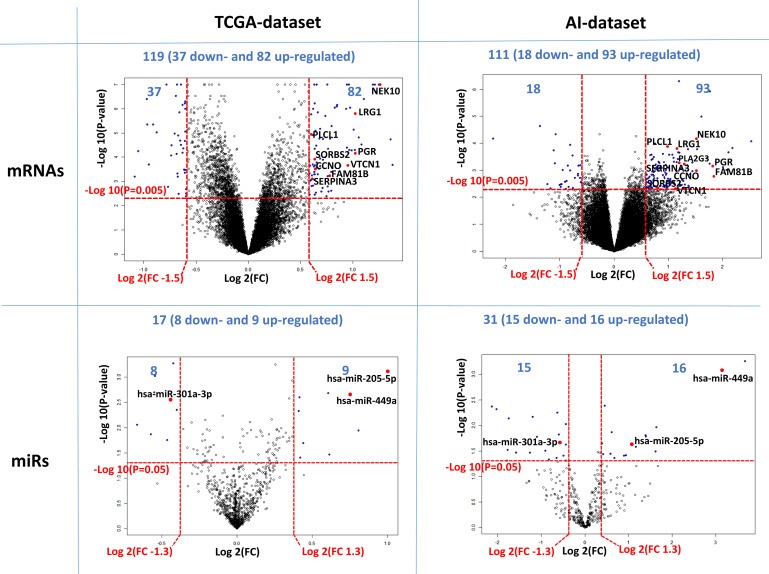
Differentially expressed mRNAs and miRs when analyzed by PIK3CA mutation status. Volcano plots of differentially expressed mRNAs (top side) and miRs (bottom side) in the TCGA dataset (left side) and in the AI dataset (right side) analyzed by PIK3CA genotype. The vertical dotted lines indicate the threshold for a relative expression fold change (FC) of breast cancer PIK3CA exon 9 or exon 20 mutated tumors compare to PIK3CA wild types. The horizontal dotted lines represent the threshold of a P value. The blue dots in the upper sides are significantly upregulated (on the right) and down regulated (on the left) in PIK3CA mutants. Overlapping mRNAs and miRs in both datasets are labeled and indicated in red dots. The volcano plots are based on number of probes (see Figure 1 for details).
Table 1.
Overview: Identification of potential biomarkers related to the PIK3CA status in relation to PFS after first line AI therapy.
| Factor | TCGA‐set | AI‐set | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Relationships with PIK3CA status and Luminal subtype for 286 patients | Relationships with overall survival available for 272 patients | Relationships with progression‐free survival in 75 patients who received first‐line AI therapy | |||||||||||||
| PIK3CA status | Subtype | Univariate analysis | Univariate analysis | Mutivariate analysis: each factor added to the base model of traditional factorsc | |||||||||||
| Wild‐type | Mutated | P | Luminal A | Luminal B | P | HR | 95% CI | P | HR | 95% CI | P | HR | 95% CI | P | |
| MWa | MWa | ||||||||||||||
| Median expression levels for: | |||||||||||||||
| LRG1 | 0.10 | 1.06 | <0.001 | 0.65 | 0.35 | 0.139 | 0.83 | 0.70–0.99 | 0.033 | 0.69 | 0.55–0.87 | 0.002 | 0.70 | 0.54–0.90 | 0.006 |
| PLCL1 | −0.11 | 0.76 | <0.001 | 0.45 | −0.03 | 0.013 | 0.71 | 0.52–0.97 | 0.029 | 0.61 | 0.46–0.81 | 0.001 | 0.58 | 0.43–0.79 | <0.001 |
| FAM81B | 0.27 | 1.64 | <0.001 | 1.19 | 0.00 | <0.001b | 0.88 | 0.71–1.08 | 0.216 | 0.82 | 0.73–0.92 | <0.001 | 0.83 | 0.73–0.94 | 0.004 |
| CCNO | 0.11 | 0.67 | <0.001 | 0.41 | 0.23 | 0.319 | 0.97 | 0.75–1.27 | 0.832 | 0.84 | 0.73–0.96 | 0.010 | 0.84 | 0.73–0.97 | 0.018 |
| NEK10 | 0.21 | 1.66 | <0.001 | 1.29 | −0.21 | <0.001b | 0.88 | 0.72–1.06 | 0.172 | 0.76 | 0.65–0.90 | 0.001 | 0.79 | 0.65–0.95 | 0.012 |
| PGR | 0.73 | 2.03 | <0.001 | 1.81 | 0.45 | <0.001b | 0.91 | 0.77–1.06 | 0.221 | 0.84 | 0.74–0.95 | 0.006 | 0.85 | 0.72–0.99 | 0.039 |
| SERPINA3 | 0.17 | 0.77 | 0.001 | 0.53 | 0.32 | 0.069 | 0.81 | 0.66–0.98 | 0.031 | 0.82 | 0.70–0.97 | 0.020 | 0.85 | 0.70–1.04 | NS |
| SORBS2 | −0.37 | 0.33 | <0.001 | 0.30 | −0.86 | <0.001b | 0.82 | 0.63–1.07 | 0.142 | 0.69 | 0.56–0.86 | 0.001 | 0.71 | 0.56–0.89 | 0.004 |
| VTCN1 | −0.90 | 0.37 | <0.001 | 0.30 | −1.44 | <0.001b | 0.91 | 0.77–1.08 | 0.284 | 0.84 | 0.73–0.97 | 0.021 | 0.87 | 0.73–1.03 | NS |
| hsa‐miR‐449a | 0.93 | 1.59 | 0.011 | 1.13 | 1.21 | 0.817 | 0.84 | 0.66–1.07 | 0.166 | 0.93 | 0.87–0.99 | 0.022 | 0.93 | 0.87–0.99 | 0.030 |
| hsa‐miR‐301a‐3p | 3.36 | 2.88 | 0.003 | 2.84 | 3.74 | <0.001b | 1.38 | 1.00–1.89 | 0.047 | 1.30 | 1.01–1.68 | 0.043 | 1.21 | 0.91–1.61 | NS |
| hsa‐miR‐205‐5p | 9.46 | 10.82 | <0.001 | 10.49 | 9.04 | <0.001b | 1.00 | 0.86–1.16 | 0.988 | 0.93 | 0.84–1.04 | 0.209 | |||
Mann Whitney (MW) nonparametric test was applied for the evaluation of the expression levels in relationship to PIK3CA status (wild type vs mutant) or luminal subtype (A vs B).
Significant after the Bonferroni correction for multiple testing (P = 0.05/12 = 0.004).
Each factor added to the base model of traditional factors including age, disease free interval, dominant site of relapse, PR and HER2 status.
3.3. Integrated analyses of common miRs and mRNAs
Since miRs can regulate expression of specific target genes, the expression of the common 9 mRNAs and 3 miRs were checked for their statistical relationship by spearman correlation in both the TCGA‐ and AI‐dataset. This exploratory analysis resulted in 14 miR–mRNA pairs with a P < 0.05, including 6 pairs still significant after multiple testing correction (Supplemental Table S4). These 6 miR–mRNA pairs were explored in different miR databases to identify putative mRNAs targeted by our miRs, and demonstrated only PGR as target of miR‐205‐5p in the miRSel database (Sempere et al., 2007). Highest correlations in the TCGA and AI dataset, i.e. rs = 0.69 and 0.78, were observed between subtype independent expression of CCNO and miR‐449a, possibly due to their chromosomal co‐localization on 5q11.
Additionally, expression levels of the 9 mRNAs were also correlated with each other (Supplemental Table S5), and showed coregulatory expression for 12 mRNA pairs in both datasets after multiple testing correction. Ingenuity identified TP73 for the LRG1‐SERPINA3 pair and STAT3 for the PGR‐SERPINA3 pair as their common transcription regulator, respectively.
3.4. PIK3CA mutation specific transcriptome before and after neo‐adjuvant AI therapy
Next, above identified potential biomarkers were analyzed on the third neo‐adjuvant dataset to establish whether their expression levels change after letrozole treatment (Figure 3). Expression in the before treatment biopsies were only significantly decreased for VTCN1 in the 8 clinical non‐responders when compared to the 17 clinical responders. On the other hand, expression levels for LRG1, PGR, and SERPINA3 were decreased after 2 weeks treatment and attenuated up to 3 months therapy when compared to the pre‐treatment levels (Figure 3A). The downregulation for these 3 genes during neo‐adjuvant letrozole therapy was only observed in patients with clinical response but not in those without clinical response, as exemplified for LRG1 (Figure 3B).
Figure 3.
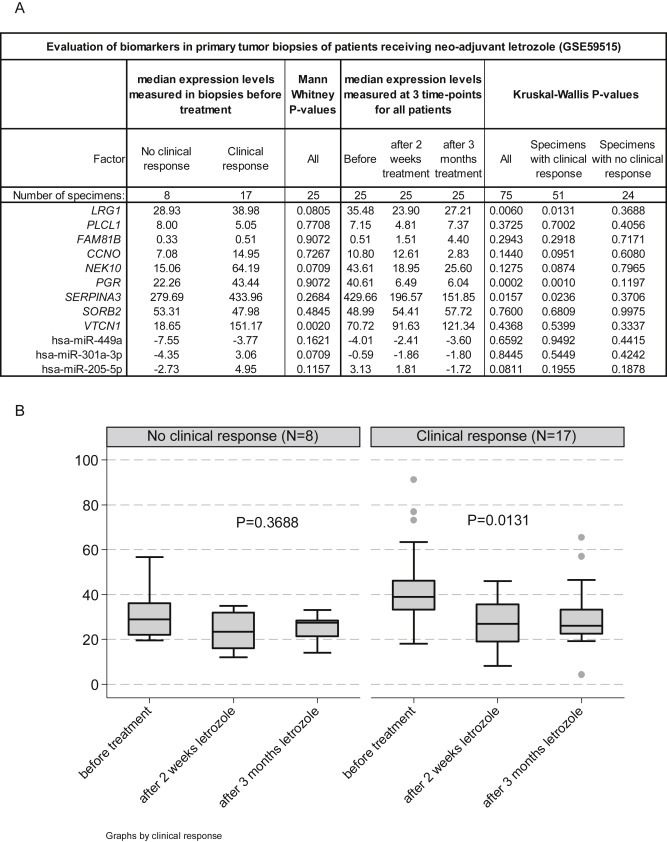
Gene expression alterations in primary tumor biopsies before and after AI treatment from patients who received neo‐adjuvant AI therapy. The figure illustrates the results after validation in the publicly available NCBI GEO‐dataset GSE59515 including 25 breast cancer patients who received neo‐adjuvant letrozole therapy. The cohort contained a subset of 17 patients with and a subset of 8 patients without clinical response after AI‐treatment. The dataset contains the expression profiles of primary tumor biopsies before and after 2 weeks and 3 months treatment from these patients. A presents the median expression levels and P‐values measured before treatment for the 2 patient subsets (Mann–Whitney P‐values) and before and after 2 weeks and 3 months AI‐treatment for all patients (Kruskal Wallis P‐values). The Kruskal Wallis test was also performed on each subset of patients separately. B is a boxplot illustrating LRG1 expression levels before and after treatment in both patients with and without clinical response to neo‐adjuvant letrozole.
3.5. PIK3CA mutation specific transcriptome and hormonal therapy outcome
The PIK3CA genotype linked potential biomarkers were further evaluated for their relationship with OS for 272 patients of the TCGA‐dataset and with PFS for 75 MBC patients who received first line AI and for whom both expression and clinical data were available. Expression of LRG1, PLCL1, SERPINA3 and miR‐301a‐3p was related with OS, based on all therapies that the patients had received (Table 1). In addition, expression levels of all biomarkers except miR‐205‐5p were significantly (P < 0.05) related to PFS (Figure 4A). Only the expression of miR‐301a‐3p was linked to therapy resistance whereas expression of all other biomarkers was associated with response to AI. The 11 potential biomarkers that associated with therapy outcome, were each further evaluated along with traditional predictive factors, the base model. In this multivariable analyses eight factors were independent from the base model (Table 1) and these were combined for a stepdown analysis. Since we know that PIK3CA mutations correlate with PFS in our cohort, as reported by us (Ramirez‐Ardila et al., 2013), a stepdown including PIK3CA status was analyzed to reveal variables that outperformed the PIK3CA status correlation with PFS, i.e. are more strongly correlated with PFS. Performing the stepdown analysis with and without PIK3CA status resulted in similar findings, i.e. only FAM81B (P < 0.001), LRG1 (P = 0.001) and PLCL1 (P = 0.013) were independent biomarkers associated with AI therapy outcome in MBC patients (Figure 4B). Interestingly, LRG1 and PLCL1 expression was shown to be related to PIK3CA independently of the luminal subtype. The observed association with PFS for these 2 potential biomarkers is illustrated in Kaplan Meier survival curves in Figure 4C–D. Finally, we investigated the expression of LRG1 and PLCL1 in our dataset of 101 MBC patients who received first line tamoxifen, however, no significant relationship with tamoxifen outcome was observed for both genes (data not shown).
Figure 4.
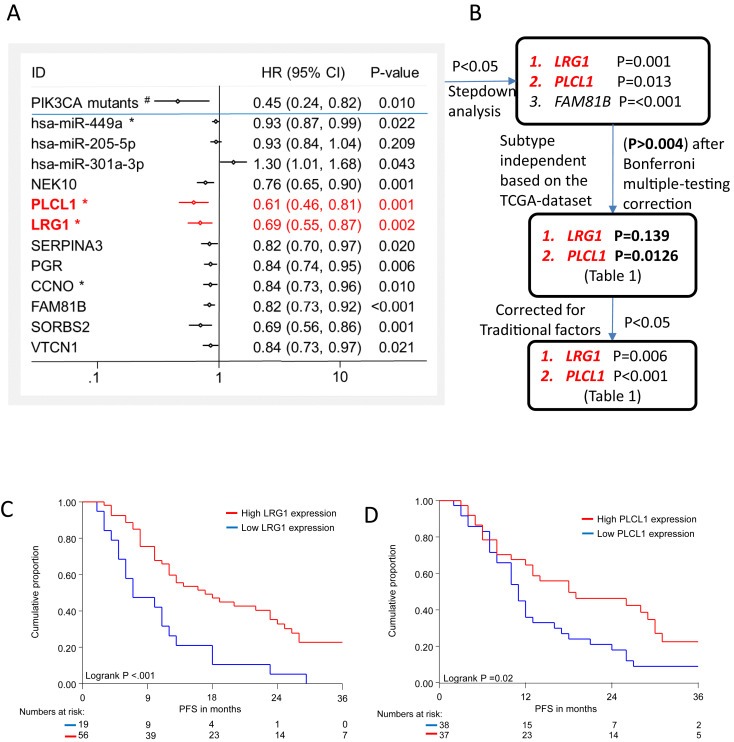
Relation to PFS after first line AI treatment. Forest plot summarizing univariate analyses in 75 MBC patients in relation to PFS after first line AI treatment. Traditional factors are not included here. The variables in red remained significant in multivariate analysis after stepdown analyses and corrected for traditional factors. (#) indicates dichotomized variable, all others are continuous variables. All variables are up‐regulated in PIK3CA mutants except for miR‐301a‐3p. (*) indicates variables which are subtype independent, i.e. had no significant relationship (P > 0.004) with subtypes after Bonferroni multiple‐testing correction. B) Steps followed to identify independent potential biomarkers related to PIK3CA status and independent of subtype C–D). PFS analyses in 75 ER positive breast cancer patients with advanced disease treated with first line AI as function of C) PLCL1 expression and D) LRG1 expression.
4. Discussion
In this study, the transcriptome of a TCGA cohort of 286 luminal A and B tumors from BC patients and our cohort of 84 ER positive primary breast tumors from patients who developed metastatic disease were stratified by PIK3CA genotype. In both datasets we demonstrated 1 miR with decreased and 2 miRs and 9 mRNAs with increased expression in tumors with a PIK3CA hotspot mutation. Evaluation of these biomarkers in the neo‐adjuvant dataset showed that expression of 3 mRNAs decreased after letrozole therapy. Multivariate analyses, including clinico‐pathological factors and molecular tumor subtypes, revealed LRG1 and PLCL1 as independent potential biomarkers for PFS after first line AI therapy. All our findings indicate that high LRG1 expression levels hallmark primary breast cancer with PIK3CA mutations and treatment response after neo‐adjuvant and first‐line AI‐therapy.
The majority of differentially expressed mRNAs in both datasets had an increased expression in PIK3CA mutated tumors. In addition, we found 6 miR–mRNA pairs with significant related expression levels in both datasets after Bonferroni multiple correction test. Negative correlations have been generally reported since miRs control gene expression by mRNA degradation and/or translation inhibition (Bartel, 2004). However, most of our miR–mRNA pairs showed a positive correlation. Dvinge et al. indicated that the BC miRNA–mRNA landscape was dominated by positive connotations, and suggested co‐transcriptional modules, especially in ER+ samples (Dvinge et al., 2013).
The established PIK3CA mutation specific potential biomarkers were previously linked to breast cancer, PI3K pathway and/or endocrine therapy. For example, PGR belonged to the regulated genes after PI3K inhibition (Bosch et al., 2015), whereas SERPINA3 was part of activated PI3K signatures (Loi et al., 2010). SERPINA3 was identified as biomarker for estrogen regulation in response to neoadjuvant AI therapy (Miller and Larionov, 2010), whereas SNPs for NEK10 (Milne et al., 2014) and VTCN1 (Tsai et al., 2015) were linked to BC susceptibility. Moreover, miR‐449a expression was increased in luminal BC compared to the other subtypes (Dvinge et al., 2013) and its tumor suppressor activity was regulated by PI3K (Liu et al., 2015). The upregulated expression of miR‐205 and miR‐449a in tumors with a PIK3CA mutation was shown to suppress the epithelial mesenchymal transition (EMT) in BC and liver cancer, respectively (De Cola et al., 2015) (Chen et al., 2015a). Interestingly, miR‐205‐5p directly targets HER3 receptor, involved as well in EMT, inhibiting the PI3K pathway activation respectively (De Cola et al., 2015). In contrast, the down regulated miR‐301a‐3p in tumors with PIK3CA mutation, was shown to increase the EMT in laryngeal neoplasm (Lu et al., 2015) and to target in BC especially genes involved in T cell related processes (Dvinge et al., 2013). Combining results in BC cell lines on PIK3CA status obtained from the COSMIC database (http://cancer.sanger.ac.uk/cell_lines) and on miR‐301a‐3p expression described by Ma et al. (2014) suggest that BC cell lines with PIK3CA mutations have decreased miR‐301a‐3p expression compared to wild types.
Although all 9 mRNAs and 2 miRs associated with PIK3CA genotype and PFS in MBC, only PLCL1, LRG1, CCNO, SERPINA3 and miR‐449a were not related with luminal subtypes A and B. Furthermore, mRNA expression of FAM81B, PLCL1, and LRG1 remained as independent potential biomarkers for PFS after first line AI treatment once stepdown analyses and multivariate analysis with well‐known clinical traditional factors are performed.
Thus, we propose PLCL1 and LRG1 as potential luminal subtype independent biomarkers that associate with PIK3CA genotype and first line AI treatment outcome.
PLCL1 and LRG1 have so far not been linked with BC and PIK3CA genotype. PLCL1 encodes for phospholipase C like 1, also known as PRIP 1, which has been suggested to be involved in insulin induced GABA(A) receptor functioning (Fujii et al., 2010) and in gonadotropin secretion (Matsuda et al., 2009). Interestingly, CA2+ and phospholipase C signaling was recently indicated as one of the over represented canonical networks in ER positive BC (Ellis et al., 2012). LRG1 (encodes for leucine rich alpha 2 glycoprotein 1) was proposed as a metastasis suppressor in hepatocellular carcinoma (HCC) since exogenous recombinant human LRG1 protein inhibited migration and invasion of HCC cells in vitro (Zhang et al., 2015). Moreover, LRG1 was shown as potential biomarker for detection of cancer in urine and/or serum such as epithelial ovarian, lung and colon cancer (Chen et al., 2015b; Ivancic et al., 2014; Smith et al., 2014; Wu et al., 2015). In endometrial carcinoma patients LRG1 expression was related to poor prognosis (Wen et al., 2014). Our transcriptome analyses of tumor biopsies before and after neo‐adjuvant letrozole showed decreased expression upon treatment for LRG1, PGR, and SERPINA3. These 3 genes were also reported within the top 150 differentially expressed genes between pre‐ and 2 weeks post anastrozole neo‐adjuvant treatment (Dunbier et al., 2013). All this suggest that alterations in LRG1 levels may be used as readout for a response to AI. Further studies in BC are needed to resolve the relationship of PLCL1 and LRG1 with PIK3CA mutation and AI therapy response.
In conclusion, we used mRNA and miR expression and identified 9 mRNAs and 3 miRs overlapping between ER positive and luminal A and B tumors in relation to PIK3CA mutation status. The expression of 2 genes, LRG1 and PLCL1, was shown to be independent of subtype and related to PFS after first line AI treatment in MBC patients. Expression levels of LRG1, but not PLCL1, were shown to decrease significantly after treatment in patients with clinical response to neo‐adjuvant letrozole. Since our analyses were based on PIK3CA genotype, and given the high frequency of PIK3CA mutations, this exploratory study provides potential novel biomarkers for PFS after first line AI treatment. These results need to be validated in future studies.
Funding
This work was supported in part by TI Pharma T3‐502 (EB, MJ) and ERACOL (DRA).
Conflict of interests
None of the authors has a conflict of interest.
Authors' contributions
DR, MJ and EB have made substantial contributions to conception and design of the study. KRR, JH, ML, LD were responsible for the acquisition and/or interpretation of data. The authors DR, MJ, EB were responsible for the study and wrote the manuscript. ML was responsible for statistical analyses whereas the authors LD and SL revised it on important contents. All authors have read and approved the final version of the manuscript and agree to be responsible for the work.
Supporting information
The following are the supplementary data related to this article:
Supplemental Figure S1 Overlapping and non‐overlapping mRNAs and miRs between the AI‐ and the TCGA datasets. Up‐regulated mRNAs and miRs are in red font whereas in green font are the down‐regulated mRNAs and miRs.
Supplemental Figure S2 Estrogen: Common up stream effector in the TCGA and AI datasets. Estrogen: Common up stream effector of significant differentially expressed genes in each and both datasets (TCGA and AI). PGR and SERPINA3 are the common genes in both datasets.
Supplemental Table S1 Differentially expressed mRNAs according to the PIK3CA mutation status in the TCGA and AI datasets.
Supplemental Table S2 Differentially expressed mature miRs according to the PIK3CA mutation status in the TCGA and AI datasets.
Supplemental Table S3 Extended information of the 9 common mRNAs in the TCGA and AI datasets.
Supplemental Table S4 Spearman correlation coefficient (r) between expression levels of miRs and mRNAs in the TCGA and AI datasets. This table only shows significant (P < 0.05) Spearman correlation coefficients (Spearman r) between the overlapping 9 mRNAs and 3 miRs in the two analyzed datasets [TCGA (N = 286) and AI (N = 84)] (Supplemental Tables S1 and S2). Bold font: Significant (r) after the Bonferroni correction for multiple testing (P < 0.05/27 = 0.0018). Coefficients highlighted in blue indicate that in the both datasets (TCGA and AI), the Spearman correlation is (P < 0.0018). Negative correlations are shown with a (−) preceding the Spearman (r) value.
Supplemental Table S5 Spearman correlation coefficient (r) between expression levels of mRNAs in the TCGA and AI datasets. This table only shows significant (P < 0.05) Spearman correlation coefficients (Spearman r) between the overlapping 9 mRNAs in the two analyzed datasets [TCGA (N = 286) and AI (N = 84)] (Supplemental Tables S1 and S2). The table presents also the relationship between the expression of MKI67 and the 9 mRNAs. Bold font: Significant (r) after the Bonferroni correction for multiple testing (P < 0.05/81 = 0.00062). Coefficients highlighted in blue indicate that in the both datasets (TCGA and AI), the Spearman correlation is (P < 0.00062). *Only 77 samples available were used for correlation.
Acknowledgements
The authors are especially grateful to Marcel Smid of Erasmus MC – Cancer Institute, department of Medical Oncology, Rotterdam, the Netherlands; Sabine C. Linn – Department of Medical Oncology, Netherlands Cancer Institute, Amsterdam, the Netherlands; and Iris Simon and Paul Roepman, former employees from Agendia NV, Amsterdam, The Netherlands.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.07.004.
Ramirez-Ardila Diana E., Ruigrok-Ritstier Kirsten, Helmijr Jean C., Look Maxime P., van Laere Steven, Dirix Luc, Berns Els M.J.J., Jansen Maurice P.H.M., (2016), LRG1 mRNA expression in breast cancer associates with PIK3CA genotype and with aromatase inhibitor therapy outcome, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.07.004.
Contributor Information
Diana E. Ramirez-Ardila, Email: d.ramirezardila@erasmusmc.nl
Kirsten Ruigrok-Ritstier, Email: k.ritstier@erasmusmc.nl.
Jean C. Helmijr, Email: j.helmijr@erasmusmc.nl
Maxime P. Look, Email: m.look@erasmusmc.nl
Steven van Laere, Email: s.vanlaere@gza.be.
Luc Dirix, Email: luc.dirix@gza.be.
Els M.J.J. Berns, Email: p.berns@erasmusmc.nl
Maurice P.H.M. Jansen, Email: m.p.h.m.jansen@erasmusmc.nl
References
- Bartel, D.P. , 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- Bosch, A. , Li, Z. , Bergamaschi, A. , Ellis, H. , Toska, E. , Prat, A. , Tao, J.J. , Spratt, D.E. , Viola-Villegas, N.T. , Castel, P. , Minuesa, G. , Morse, N. , Rodon, J. , Ibrahim, Y. , Cortes, J. , Perez-Garcia, J. , Galvan, P. , Grueso, J. , Guzman, M. , Katzenellenbogen, J.A. , Kharas, M. , Lewis, J.S. , Dickler, M. , Serra, V. , Rosen, N. , Chandarlapaty, S. , Scaltriti, M. , Baselga, J. , 2015. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci. Transl. Med. 7, 283ra251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffa, F.M. , Camps, C. , Winchester, L. , Snell, C.E. , Gee, H.E. , Sheldon, H. , Taylor, M. , Harris, A.L. , Ragoussis, J. , 2011. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 71, 5635–5645. [DOI] [PubMed] [Google Scholar]
- Chen, S.P. , Liu, B.X. , Xu, J. , Pei, X.F. , Liao, Y.J. , Yuan, F. , Zheng, F. , 2015. MiR-449a suppresses the epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma by multiple targets. BMC Cancer 15, 706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Z. , Liu, J. , Lin, L. , Xie, H. , Zhang, W. , Zhang, H. , Wang, G. , 2015. Analysis of differentially expressed proteome in urine from non-small cell lung cancer patients. Zhongguo Fei Ai Za Zhi 18, 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, R. , Tyson, J.J. , Dixon, J.M. , 2015. Endocrine resistance in breast cancer – an overview and update. Mol. Cell Endocrinol. 418, (Pt 3) 220–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cola, A. , Volpe, S. , Budani, M.C. , Ferracin, M. , Lattanzio, R. , Turdo, A. , D'Agostino, D. , Capone, E. , Stassi, G. , Todaro, M. , Di Ilio, C. , Sala, G. , Piantelli, M. , Negrini, M. , Veronese, A. , De Laurenzi, V. , 2015. miR-205-5p-mediated downregulation of ErbB/HER receptors in breast cancer stem cells results in targeted therapy resistance. Cell Death Dis. 6, e1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbier, A.K. , Ghazoui, Z. , Anderson, H. , Salter, J. , Nerurkar, A. , Osin, P. , A'Hern, R. , Miller, W.R. , Smith, I.E. , Dowsett, M. , 2013. Molecular profiling of aromatase inhibitor-treated postmenopausal breast tumors identifies immune-related correlates of resistance. Clin. Cancer Res. 19, 2775–2786. [DOI] [PubMed] [Google Scholar]
- Dvinge, H. , Git, A. , Graf, S. , Salmon-Divon, M. , Curtis, C. , Sottoriva, A. , Zhao, Y. , Hirst, M. , Armisen, J. , Miska, E.A. , Chin, S.F. , Provenzano, E. , Turashvili, G. , Green, A. , Ellis, I. , Aparicio, S. , Caldas, C. , 2013. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature 497, 378–382. [DOI] [PubMed] [Google Scholar]
- Ellis, M.J. , Ding, L. , Shen, D. , Luo, J. , Suman, V.J. , Wallis, J.W. , Van Tine, B.A. , Hoog, J. , Goiffon, R.J. , Goldstein, T.C. , Ng, S. , Lin, L. , Crowder, R. , Snider, J. , Ballman, K. , Weber, J. , Chen, K. , Koboldt, D.C. , Kandoth, C. , Schierding, W.S. , McMichael, J.F. , Miller, C.A. , Lu, C. , Harris, C.C. , McLellan, M.D. , Wendl, M.C. , DeSchryver, K. , Allred, D.C. , Esserman, L. , Unzeitig, G. , Margenthaler, J. , Babiera, G.V. , Marcom, P.K. , Guenther, J.M. , Leitch, M. , Hunt, K. , Olson, J. , Tao, Y. , Maher, C.A. , Fulton, L.L. , Fulton, R.S. , Harrison, M. , Oberkfell, B. , Du, F. , Demeter, R. , Vickery, T.L. , Elhammali, A. , Piwnica-Worms, H. , McDonald, S. , Watson, M. , Dooling, D.J. , Ota, D. , Chang, L.W. , Bose, R. , Ley, T.J. , Piwnica-Worms, D. , Stuart, J.M. , Wilson, R.K. , Mardis, E.R. , 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486, 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii, M. , Kanematsu, T. , Ishibashi, H. , Fukami, K. , Takenawa, T. , Nakayama, K.I. , Moss, S.J. , Nabekura, J. , Hirata, M. , 2010. Phospholipase C-related but catalytically inactive protein is required for insulin-induced cell surface expression of gamma-aminobutyric acid type A receptors. J. Biol. Chem. 285, 4837–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivancic, M.M. , Irving, A.A. , Jonakin, K.G. , Dove, W.F. , Sussman, M.R. , 2014. The concentrations of EGFR, LRG1, ITIH4, and F5 in serum correlate with the number of colonic adenomas in ApcPirc/+ rats. Cancer Prev. Res. (Phila) 7, 1160–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen, M.P. , Knijnenburg, T. , Reijm, E.A. , Simon, I. , Kerkhoven, R. , Droog, M. , Velds, A. , van Laere, S. , Dirix, L. , Alexi, X. , Foekens, J.A. , Wessels, L. , Linn, S.C. , Berns, E.M. , Zwart, W. , 2013. Hallmarks of aromatase inhibitor drug resistance revealed by epigenetic profiling in breast cancer. Cancer Res. 73, 6632–6641. [DOI] [PubMed] [Google Scholar]
- Koren, S. , Bentires-Alj, M. , 2015. Breast tumor heterogeneity: source of fitness, hurdle for therapy. Mol. Cell 60, 537–546. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Wang, Y. , Sun, X. , Mei, C. , Wang, L. , Li, Z. , Zha, X. , 2015. miR-449a promotes liver cancer cell apoptosis by down-regulation of Calpain6 and POU2F1. Oncotarget 7, 13491–13501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi, S. , Haibe-Kains, B. , Majjaj, S. , Lallemand, F. , Durbecq, V. , Larsimont, D. , Gonzalez-Angulo, A.M. , Pusztai, L. , Symmans, W.F. , Bardelli, A. , Ellis, P. , Tutt, A.N. , Gillett, C.E. , Hennessy, B.T. , Mills, G.B. , Phillips, W.A. , Piccart, M.J. , Speed, T.P. , McArthur, G.A. , Sotiriou, C. , 2010. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proc. Natl. Acad. Sci. U. S. A. 107, 10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. , Gao, W. , Zhang, C. , Wen, S. , Huangfu, H. , Kang, J. , Wang, B. , 2015. Hsa-miR-301a-3p acts as an oncogene in laryngeal squamous cell carcinoma via target regulation of Smad4. J. Cancer 6, 1260–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, F. , Zhang, J. , Zhong, L. , Wang, L. , Liu, Y. , Wang, Y. , Peng, L. , Guo, B. , 2014. Upregulated microRNA-301a in breast cancer promotes tumor metastasis by targeting PTEN and activating Wnt/beta-catenin signaling. Gene 535, 191–197. [DOI] [PubMed] [Google Scholar]
- Ma, C.X. , Reinert, T. , Chmielewska, I. , Ellis, M.J. , 2015. Mechanisms of aromatase inhibitor resistance. Nat. Rev. Cancer 15, 261–275. [DOI] [PubMed] [Google Scholar]
- Matsuda, M. , Tsutsumi, K. , Kanematsu, T. , Fukami, K. , Terada, Y. , Takenawa, T. , Nakayama, K.I. , Hirata, M. , 2009. Involvement of phospholipase C-related inactive protein in the mouse reproductive system through the regulation of gonadotropin levels. Biol. Reprod. 81, 681–689. [DOI] [PubMed] [Google Scholar]
- Miller, W.R. , Larionov, A. , 2010. Changes in expression of oestrogen regulated and proliferation genes with neoadjuvant treatment highlight heterogeneity of clinical resistance to the aromatase inhibitor, letrozole. Breast Cancer Res. 12, R52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne, R.L. , Burwinkel, B. , Michailidou, K. , Arias-Perez, J.I. , Zamora, M.P. , Menendez-Rodriguez, P. , Hardisson, D. , Mendiola, M. , Gonzalez-Neira, A. , Pita, G. , Alonso, M.R. , Dennis, J. , Wang, Q. , Bolla, M.K. , Swerdlow, A. , Ashworth, A. , Orr, N. , Schoemaker, M. , Ko, Y.D. , Brauch, H. , Hamann, U. , Network, G. , Andrulis, I.L. , Knight, J.A. , Glendon, G. , Tchatchou, S. , kConFab, I. , Australian Ovarian Cancer Study, G. , Matsuo, K. , Ito, H. , Iwata, H. , Tajima, K. , Li, J. , Brand, J.S. , Brenner, H. , Dieffenbach, A.K. , Arndt, V. , Stegmaier, C. , Lambrechts, D. , Peuteman, G. , Christiaens, M.R. , Smeets, A. , Jakubowska, A. , Lubinski, J. , Jaworska-Bieniek, K. , Durda, K. , Hartman, M. , Hui, M. , Yen Lim, W. , Wan Chan, C. , Marme, F. , Yang, R. , Bugert, P. , Lindblom, A. , Margolin, S. , Garcia-Closas, M. , Chanock, S.J. , Lissowska, J. , Figueroa, J.D. , Bojesen, S.E. , Nordestgaard, B.G. , Flyger, H. , Hooning, M.J. , Kriege, M. , van den Ouweland, A.M. , Koppert, L.B. , Fletcher, O. , Johnson, N. , dos-Santos-Silva, I. , Peto, J. , Zheng, W. , Deming-Halverson, S. , Shrubsole, M.J. , Long, J. , Chang-Claude, J. , Rudolph, A. , Seibold, P. , Flesch-Janys, D. , Winqvist, R. , Pylkas, K. , Jukkola-Vuorinen, A. , Grip, M. , Cox, A. , Cross, S.S. , Reed, M.W. , Schmidt, M.K. , Broeks, A. , Cornelissen, S. , Braaf, L. , Kang, D. , Choi, J.Y. , Park, S.K. , Noh, D.Y. , Simard, J. , Dumont, M. , Goldberg, M.S. , Labreche, F. , Fasching, P.A. , Hein, A. , Ekici, A.B. , Beckmann, M.W. , Radice, P. , Peterlongo, P. , Azzollini, J. , Barile, M. , Sawyer, E. , Tomlinson, I. , Kerin, M. , Miller, N. , Hopper, J.L. , Schmidt, D.F. , Makalic, E. , Southey, M.C. , Hwang Teo, S. , Har Yip, C. , Sivanandan, K. , Tay, W.T. , Shen, C.Y. , Hsiung, C.N. , Yu, J.C. , Hou, M.F. , Guenel, P. , Truong, T. , Sanchez, M. , Mulot, C. , Blot, W. , Cai, Q. , Nevanlinna, H. , Muranen, T.A. , Aittomaki, K. , Blomqvist, C. , Wu, A.H. , Tseng, C.C. , Van Den Berg, D. , Stram, D.O. , Bogdanova, N. , Dork, T. , Muir, K. , Lophatananon, A. , Stewart-Brown, S. , Siriwanarangsan, P. , Mannermaa, A. , Kataja, V. , Kosma, V.M. , Hartikainen, J.M. , Shu, X.O. , Lu, W. , Gao, Y.T. , Zhang, B. , Couch, F.J. , Toland, A.E. , Tnbcc, Yannoukakos, D. , Sangrajrang, S. , McKay, J. , Wang, X. , Olson, J.E. , Vachon, C. , Purrington, K. , Severi, G. , Baglietto, L. , Haiman, C.A. , Henderson, B.E. , Schumacher, F. , Le Marchand, L. , Devilee, P. , Tollenaar, R.A. , Seynaeve, C. , Czene, K. , Eriksson, M. , Humphreys, K. , Darabi, H. , Ahmed, S. , Shah, M. , Pharoah, P.D. , Hall, P. , Giles, G.G. , Benitez, J. , Dunning, A.M. , Chenevix-Trench, G. , Easton, D.F. , 2014. Common non-synonymous SNPs associated with breast cancer susceptibility: findings from the Breast Cancer Association Consortium. Hum. Mol. Genet. 23, 6096–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Ardila, D.E. , Helmijr, J.C. , Look, M.P. , Lurkin, I. , Ruigrok-Ritstier, K. , van Laere, S. , Dirix, L. , Sweep, F.C. , Span, P.N. , Linn, S.C. , Foekens, J.A. , Sleijfer, S. , Berns, E.M. , Jansen, M.P. , 2013. Hotspot mutations in PIK3CA associate with first-line treatment outcome for aromatase inhibitors but not for tamoxifen. Breast Cancer Res. Treat 139, 39–49. [DOI] [PubMed] [Google Scholar]
- Sempere, L.F. , Christensen, M. , Silahtaroglu, A. , Bak, M. , Heath, C.V. , Schwartz, G. , Wells, W. , Kauppinen, S. , Cole, C.N. , 2007. Altered MicroRNA expression confined to specific epithelial cell subpopulations in breast cancer. Cancer Res. 67, 11612–11620. [DOI] [PubMed] [Google Scholar]
- Smith, C.R. , Batruch, I. , Bauca, J.M. , Kosanam, H. , Ridley, J. , Bernardini, M.Q. , Leung, F. , Diamandis, E.P. , Kulasingam, V. , 2014. Deciphering the peptidome of urine from ovarian cancer patients and healthy controls. Clin. Proteomics 11, 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network, 2012. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, S.M. , Wu, S.H. , Hou, M.F. , Yang, H.H. , Tsai, L.Y. , 2015. The immune regulator VTCN1 gene polymorphisms and its impact on susceptibility to breast cancer. J. Clin. Lab. Anal. 29, 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull, A.K. , Arthur, L.M. , Renshaw, L. , Larionov, A.A. , Kay, C. , Dunbier, A.K. , Thomas, J.S. , Dowsett, M. , Sims, A.H. , Dixon, J.M. , 2015. Accurate prediction and validation of response to endocrine therapy in breast cancer. J. Clin. Oncol. 33, 2270–2278. [DOI] [PubMed] [Google Scholar]
- Volinia, S. , Croce, C.M. , 2013. Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer. Proc. Natl. Acad. Sci. U. S. A. 110, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen, S.Y. , Zhang, L.N. , Yang, X.M. , Zhang, Y.L. , Ma, L. , Ge, Q.L. , Jiang, S.H. , Zhu, X.L. , Xu, W. , Ding, W.J. , Yang, B.Q. , Zhang, Z.G. , Teng, Y.C. , 2014. LRG1 is an independent prognostic factor for endometrial carcinoma. Tumour Biol. 35, 7125–7133. [DOI] [PubMed] [Google Scholar]
- Wu, J. , Yin, H. , Zhu, J. , Buckanovich, R.J. , Thorpe, J.D. , Dai, J. , Urban, N. , Lubman, D.M. , 2015. Validation of LRG1 as a potential biomarker for detection of epithelial ovarian cancer by a blinded study. PLoS One 10, e0121112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Luo, Q. , Wang, N. , Hu, F. , Jin, H. , Ge, T. , Wang, C. , Qin, W. , 2015. LRG1 suppresses the migration and invasion of hepatocellular carcinoma cells. Med. Oncol. 32, 146 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplemental Figure S1 Overlapping and non‐overlapping mRNAs and miRs between the AI‐ and the TCGA datasets. Up‐regulated mRNAs and miRs are in red font whereas in green font are the down‐regulated mRNAs and miRs.
Supplemental Figure S2 Estrogen: Common up stream effector in the TCGA and AI datasets. Estrogen: Common up stream effector of significant differentially expressed genes in each and both datasets (TCGA and AI). PGR and SERPINA3 are the common genes in both datasets.
Supplemental Table S1 Differentially expressed mRNAs according to the PIK3CA mutation status in the TCGA and AI datasets.
Supplemental Table S2 Differentially expressed mature miRs according to the PIK3CA mutation status in the TCGA and AI datasets.
Supplemental Table S3 Extended information of the 9 common mRNAs in the TCGA and AI datasets.
Supplemental Table S4 Spearman correlation coefficient (r) between expression levels of miRs and mRNAs in the TCGA and AI datasets. This table only shows significant (P < 0.05) Spearman correlation coefficients (Spearman r) between the overlapping 9 mRNAs and 3 miRs in the two analyzed datasets [TCGA (N = 286) and AI (N = 84)] (Supplemental Tables S1 and S2). Bold font: Significant (r) after the Bonferroni correction for multiple testing (P < 0.05/27 = 0.0018). Coefficients highlighted in blue indicate that in the both datasets (TCGA and AI), the Spearman correlation is (P < 0.0018). Negative correlations are shown with a (−) preceding the Spearman (r) value.
Supplemental Table S5 Spearman correlation coefficient (r) between expression levels of mRNAs in the TCGA and AI datasets. This table only shows significant (P < 0.05) Spearman correlation coefficients (Spearman r) between the overlapping 9 mRNAs in the two analyzed datasets [TCGA (N = 286) and AI (N = 84)] (Supplemental Tables S1 and S2). The table presents also the relationship between the expression of MKI67 and the 9 mRNAs. Bold font: Significant (r) after the Bonferroni correction for multiple testing (P < 0.05/81 = 0.00062). Coefficients highlighted in blue indicate that in the both datasets (TCGA and AI), the Spearman correlation is (P < 0.00062). *Only 77 samples available were used for correlation.