

Abstract
Patients with metastatic bladder cancer have a median survival of only 13–14 months. Precision medicine using targeted therapy may improve survival. Here we investigated spatial and temporal tumour evolution and tumour heterogeneity in order to evaluate the potential use of targeted treatment of metastatic bladder cancer. We performed a proof‐of‐concept study by whole exome sequencing of multiple tumour regions (n = 22) from three patients with metastatic bladder cancer. DNA from primary and metastatic tumour biopsies was analysed for mutations using Mutect and potential therapeutic targets were identified. We identified 256, 265 and 378 somatic mutations per patient, encompassing mutations with an estimated functional impact in 6–12 known disease driver genes per patient. Disease driver mutations present in all tumour regions could be identified in all cases, however, over time metastasis specific driver mutations emerged. For each patient we identified 6–10 potentially therapeutic targets, however very few targets were present in all regions. Low mutational allele frequencies were observed in most regions suggesting a complex mixture of different cancer cells with no spatial demarcation of subclones. In conclusion, primary bladder tumours and metastatic lesions showed heterogeneity at the molecular level, but within the primary tumour the heterogeneity appeared low. The observed lack of potential therapeutic targets common to all cancer cells in primary tumours and metastases emphasizes the challenges in designing rational targeted therapy solely based on analysis of the primary tumours.
Keywords: Bladder cancer, Clonal evolution, Intra tumour heterogeneity, Metastasis, Targeted therapy, Whole exome sequencing
Highlights
Low intra‐tumour genetic diversity in primary tumour.
High genetic diversity between primary tumours and metastatic lesions.
Few potential therapeutic targets appear shared by all tumour cells.
APOBEC‐related mutagenesis observed in aggressive primary tumours.
Abbreviations
- FF
fresh frozen
- FFPE
formalin fixed paraffin embedded
- LMD
laser micro dissection
- ITH
intra tumour heterogeneity
- WES
whole exome sequencing
1. Introduction
Bladder cancer is the 5th most common cancer in the Western world. About 90% of the cases are urothelial carcinomas originating from the urothelium. Approximately 75% of patients are diagnosed with non‐muscle invasive bladder cancer (NMIBC) and 10–15% eventually progress to muscle‐invasive bladder cancer (MIBC) (Babjuk et al., 2013; Knowles and Hurst, 2015). The remaining 25% of patients are initially diagnosed with MIBC. Standard treatment for MIBC is radical cystectomy and lymphadenectomy preceded by neo‐adjuvant chemotherapy in selected cases, whereas the benefit of adjuvant chemotherapy remains controversial and is seldom used (Hermans et al., 2016; Witjes et al., 2014). Metastatic bladder cancer is a highly aggressive disease and if untreated, the survival time is only 3–6 months on average. Patients with distant metastases are offered cisplatin based chemotherapy, which may prolong median survival to 13–14 months with a response rate of 50% (von der Maase, 2003).
Patients with metastatic disease may benefit from targeted therapy and optimal effect may be obtained by targeting clonal alterations. However, cancer evolution during progression and metastasis needs to be better understood in order to offer patients optimal targeted treatment. The efficacy of targeted therapy may depend on the level of intra tumour heterogeneity (ITH) and the clonal status of disease driver alterations – if these are the actionable targets. Optimal effect of personalized targeted treatment may be obtained by targeting clonal alterations (shared by all tumour cells) in order to eradicate the entire tumour mass. Alternatively, the lethal metastatic lesions may be targeted; however, these may be genetically different from the primary tumour (Gerlinger et al., 2014, 2012).
ITH has long been acknowledged in tumour biology at the morphological level, though before the advances in sequencing technologies it has not been feasible to distinguish tumour cells at a genetic or epigenetic level (Swanton, 2012). Since then great effort has been put into characterizing ITH in several cancer types by means of next generation sequencing (NGS) methods. Exome and full genome sequencing have previously been applied to multiple topological selected tumour biopsies from primary tumour as well as associated lymph node and distant metastases in renal clear cell carcinomas. These studies allowed tracking of the aggressive clone and pinpointed the need for multiple sampling for identifying critical subpopulations. Further, it was found that the majority of the disease driver mutations were subclonal (Gerlinger et al., 2014, 2012). On the other hand, to identify known disease driver events Zhang and colleagues found that single biopsies could prove adequate in localized lung adenocarcinomas since the known driver mutations were found to be early events mapping to the trunk of phylogenetic trees (Zhang et al., 2014). This is in concordance with a study in non‐small cell lung cancer, where the cancer driver mutations with highest driver‐confidence proved to be significantly more clonal compared to non‐driver mutations (de Bruin et al., 2014). In recent years, mutational signatures reflecting the exposure of endogenous and exogenous carcinogens have emerged. Especially, the APOBEC mutational signature derived from APOBEC enzymes acting on single stranded DNA causing C to T or G mutations in either a TCT or TCA context has been documented in bladder cancer (Kim et al., 2015; Nordentoft et al., 2014; Roberts et al., 2013). APOBEC mutagenesis has been speculated to contribute to ITH and tumour progression (Swanton et al., 2015) and found to be associated with high risk NMIBC (Hedegaard et al., 2016).
The origin of recurrent bladder tumours has been shown to be mono‐ or oligo‐clonal, indicating a common field from which the tumours evolve (Hartmann et al., 2000; Hoglund, 2007; Nordentoft et al., 2014). However, ITH and the clonality of disease driver mutations and possible therapeutic targets have not yet been thoroughly investigated in bladder cancer at the genomic level. Here we performed a pilot study of whole exome sequencing (WES) of multiple small regions from paired primary tumours and metastatic lesions from three patients with metastatic bladder cancer.
2. Material and methods
2.1. Selection of patients and tissue samples
Patients were treated at Aarhus University Hospital, Denmark and Hospital of West Jutland, Holstebro, Denmark. All patients gave their written informed consent and the study was approved by the National Committees on Health Research Ethics (#1300174). Patients where tissue samples from both primary and metastatic lesions were available, were selected. Patients were treatment naive, and the disease was uni‐focal in patients 1 and 2, and multi‐focal with T1 tumours for patient 3. None of the patients were previously diagnosed with bladder cancer. Fresh frozen (FF) samples where used when available, otherwise formalin fixed and paraffin embedded (FFPE) where used (applies to primary tumour from patient 2 along with distant metastases included in the study). FF samples were embedded in TissueTek® OCT™ Compound (Sakura Finetek, Vaerloese, Denmark) and snap frozen in liquid nitrogen before placed in our biobank at −80°. FFPE samples were fixed and embedded at the respective pathology departments at Aarhus University Hospital and Hospital of West Jutland, Holstebro in Denmark. Blood samples and adjacent normal sample were taken at the initial visit. Blood was frozen at −80° in EDTA tubes.
2.2. DNA extraction and laser micro dissection
Laser micro dissection (LMD) was performed using the Veritas Microdissection Instrument (Arcturus Bioscience) and Capsure Macro LCM caps (Life Technologies). From FF tumour biopsies a 4 μm section was cut, stained with haematoxylin and eosin and examined by an experienced pathologist (SH). Additional 15 sections of 7 μm for LMD were mounted onto Arcturus PEN membrane glass slides (Life Technologies, Naerum, Denmark) and stained with Histogene® LCM Frozen Section Staining Kit (Life Technologies) using manufacturers protocol. We traced small cellular regions across 15 sections for LMD by following histological features. To verify that the same area had been traced across all sections another 4 μm section was stained with haematoxylin and eosin (see Supplementary Figure S1). Genomic DNA was extracted from LMD (Arcturus Biosciences, Mountain View, CA, US) of 22 small cellular regions with a mean area of 6.48 mm2 (range 1.5–10.35 mm2) over 15 sections. Genomic DNA from FFPE samples was obtained from punctures with a core size of 1.5 mm. A detailed description of DNA extraction procedures is found in the Supplementary data.
2.3. Whole exome sequencing and data processing
Library construction was performed using Kapa Hyper Library Prep kit (Kapa Biosystems) followed by whole exome capture with the SeqCap EZ Exome v3 Capture kit (Nimblegene) or using the Nextera Rapid Capture Expanded Exome kit (illumina). Input material for the Nextera protocol was 50 ng of gDNA and 25–100 ng for the Kapa Hyper Library Prep kit protocol. Libraries were sequenced using illumina HiSeq2000 or NextSeq500 platforms. Reads were mapped using BWA (Li and Durbin, 2009) against HG19. The mapped reads were marked for duplicates, and the alignments were recalibrated and realigned using the Picard (http://picard.sourceforge.net/) and GATK suites (DePristo et al., 2011). Analysis of ITH has only been performed on the overlapping genomic regions between the two bait sets.
2.4. Deep targeted sequencing
Deep targeted sequencing was performed using illumina TruSeq Custom Amplicon Low Input Prep Kit (illumina). PCR amplicons were designed using DesignStudio software v1.5 (illumina) for all mutations estimated to have a functional impact (tier 0 and 1 mutations – see Supplementary data). Libraries were generated from 10 to 40 ng DNA and sequenced using the NextSeq500 platform. Reads were mapped as described for the whole exome analysis. To avoid including sequencing errors when applying deep sequencing, we subdivided the number of reads for all positions in all samples into eight bins with increasing read depth. In each bin we defined an error rate by taking the mean to the number of times the bases different from the reference and the mutated base was encountered. We then set the requirement that the mutated base should be present more than twice than the error rate for each bin otherwise we regarded the mutation as not present.
2.5. Phylogenetic analysis
We evaluated the presence (2+ reads) or absence (0–1 read) of all category 1 and 2 mutations called in a patient in all the samples. We then used the status of each mutation in all the samples to find the most probable phylogenetic tree by scoring all configurations and possible tree topologies involving the same number of samples. A phylogenetic tree was then drawn where the length of the branches is proportional to the number of mutations supporting it. A detailed description is available in the Supplementary data.
2.6. APOBEC mutational signature
For each sample we counted the number of times a given mutation (C→T or G) was observed in an APOBEC context (TCT or TCA) as described earlier (Burns et al., 2013) An APOBEC score was assigned to each sample similar to previously described (Nordentoft et al., 2014). In brief, the ratio between C→T or G in an APOBEC context against C→T or G in a non‐APOBEC context was calculated. Samples were grouped into high, intermediate, and low APOBEC scores reflecting the strength of the APOBEC signature in the samples.
2.7. Analysis of actionable targets
To identify potential therapeutic targets we searched the Drug Gene Interaction database (DGIdb version 2.0) (Griffith et al., 2013), the Target database (Tumour Alteration Relevant for Genomics‐Driven Therapy, version 3.0) available from the Broad Institute, the IntOGen database (Gonzalez‐Perez et al., 2013; Rubio‐Perez et al., 2015), the Personalized Cancer Therapy database available from MD Anderson Cancer Center, and lastly we used the Qiagen Clinical Insight software. FDA approved drugs available were identified by the Personalized Cancer Therapy database, the Qiagen Clinical Insight software or by search in DrugBank, version 4.3 (Law et al., 2014; Wishart et al., 2006).
3. Results
3.1. Patient samples and DNA sequencing
Samples from three patients who underwent radical cystectomy and lymphadenectomy were used (Figure 1 and Supplementary Table S1). DNA was extracted from LMD of 22 small cellular regions (Supplementary Figure S1–S3) and WES was performed on all samples including matched germline DNA. The mean target coverage obtained for tumour samples was 68X (range 27–215X) (Figure 2) and 167X (95–301X) for germline samples. We identified a total of 256, 265 and 378 somatic mutations in the tumour DNA from the three patients using MuTect (Cibulskis et al., 2013), respectively. Mutations were classified based on call confidence (categories 1–3) and based on functional impact (tiers 0–2) as previously described (Nordentoft et al., 2014) (see Supplementary data).
Figure 1.
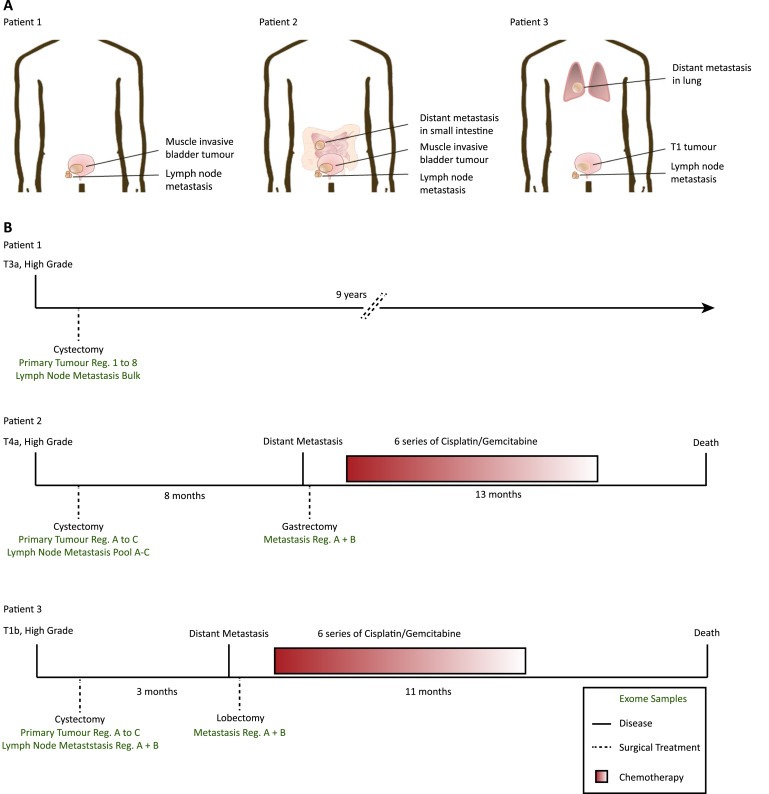
Patient disease courses and patient samples analysed. (A) Biopsy sites in the three selected patients. (B) Sample and treatment timeline. Samples procured for whole exome sequencing are indicated in green below each timeline. Patient 1: 8× Tumour Regions, 1× Lymph Node Bulk. Patient 2: 3× Tumour Regions, 1× Lymph Node Pool, 2× Distant Metastasis Regions. Patient 3: 3× Tumour Regions, 2× Lymph Node Regions, 2× Distant Metastasis Regions. Bulk: DNA extracted from 5 to 10 cross sections of the biopsy, Reg.: DNA extracted from cellular regions procured by LMD or puncture, Pool: pool of exomes performed on cellular regions A–C.
Figure 2.
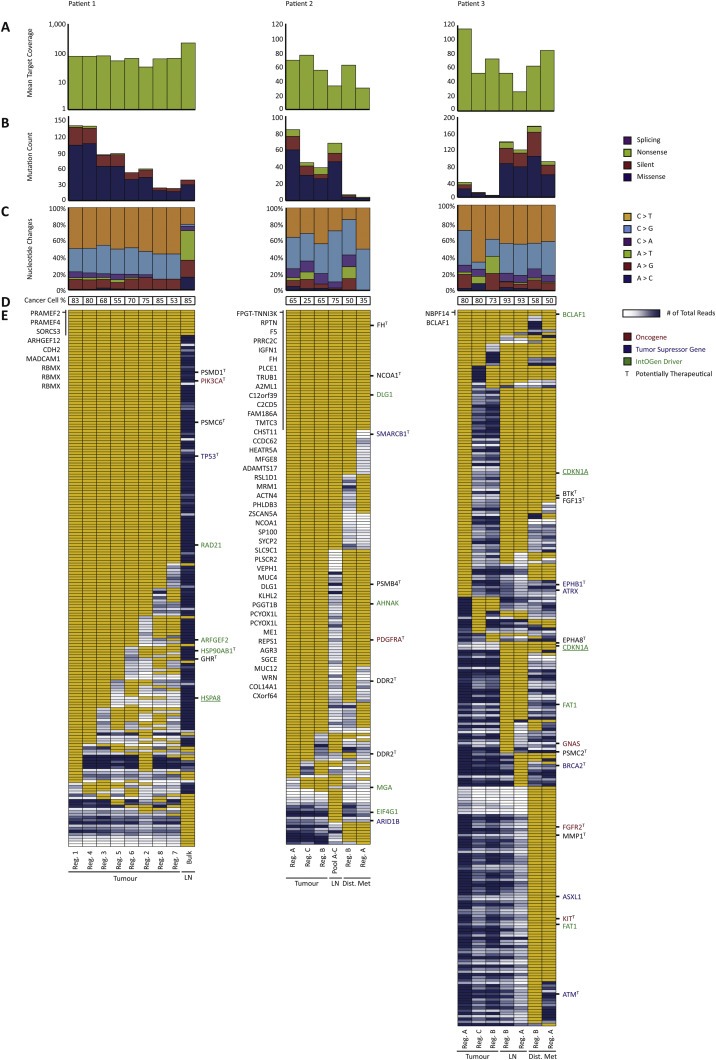
Mutational landscape in primary tumours and metastatic lesions. (A) Mean target coverage obtained in whole exome sequencing (note that data for patient 1 is shown on log‐scale). (B) Mutation count and the distribution within missense, silent, nonsense, or splicing (including tiers 0–2, categories 1–2 mutations). (C) Nucleotide change distribution (including tiers 0–2, categories 1–2 mutations). (D) Cancer cell percentage. (E) Heat map of whole exome sequencing data in a dichotomized format (including tiers 0–1 mutations, called with a category score of 1 or 2 in at least one of the samples). Yellow: mutation present, white: less than 10 reads in the given position, blue range: mutation not present (presented with number of reads ranging from 11 to 1026 reads excluding the lower and upper 10% quartiles) with dark blue equalling high amount of total reads. Known oncogenes (red), tumour suppressor genes (blue), and IntOGen driver genes (green) are annotated to the right of each heat map. Underlined genes are of high impact having either gained or lost a stop or start site. To the left of the heat map, shared mutated genes are listed. Potentially therapeutic genes are marked with a T to the right of each heat map. Samples are ordered in descending number of mutations within primary tumour, the lymph node metastasis, and the distant metastasis in each patient. Lastly, if a mutation was called by Mutect with a category score of 1 or 2 in any of the samples within a patient, the remaining samples within the patient were manually investigated for the presence of the mutation. Thus, the number of mutations depicted in E does not necessarily reflect the numbers of mutations depicted in B. Reg.: Region, LN: Lymph node metastasis, Bulk: Exome on bulk DNA, Pool A–C: Pool of exomes, Dist. Met: Distant metastasis.
To validate mutations and heterogeneity measures we performed deep targeted sequencing (3200 × mean coverage) of all tier 0–1 mutations from patient 1. A high concordance between the exome analysis and the deep targeted sequencing was found (Supplementary Figure S4). The validation rate for mutations called using WES was 91% (633/696) when requiring >100 reads for a given nucleotide position across all samples. Furthermore, applying higher sequence coverage we observed mutations in 28% (51/184) of the positions called as non‐mutated using WES. Consequently, some mutations are only present when applying deep sequencing, indicating that the mutated allele may only be present in very few cells within the tumour region sequenced. In conclusion, we find the obtained read depth from WES may be adequate to explore ITH but deep sequencing may be needed to determine the actual clonal compositions.
3.2. Spatial and temporal heterogeneity
To compare the mutational architecture between small cellular regions we first analysed mutations with functional impact (tiers 0–1 mutations, categories 1–2). Known oncogenes, tumour suppressor genes (Vogelstein et al., 2013) as well as IntOGen driver genes were identified (Figure 2E). IntOGen driver genes are disease driver genes identified for bladder cancer by use of the Cancer Driver database (available at the IntOGen web discovery platform (Gonzalez‐Perez et al., 2013; Rubio‐Perez et al., 2015)). We defined mutations as: shared, if present in all regions; regional, if present only in some regions; tissue specific, if only present in regions for a given tissue; or private, if present in one region only. A mutation was classified as clonal if the allele frequency was 0.4 or higher, taking normal cell contamination into account.
For patient 1 we identified 193 mutations which most likely had a functional impact based on SnpEff (Cingolani et al., 2012). Specifically, we identified tissue specific mutations in the disease driver genes PIK3CA (p.E545K), TP53, and RAD21 in the primary tumour. Regional mutations were identified in ARFGEF2, HSP90AB1, and HSPA8. In general we observed low spatial ITH. None of the known driver mutations were detected in the lymph node metastasis despite a high carcinoma cell content of 85% and mean target coverage of 215X.
For patient 2 we identified 192 mutations in total with a functional impact. Assessing mutations in disease driver genes we identified a shared mutation in DLG1, whereas mutations in AHNAK and PDGFRA were regionally mutated since only mutated in samples from the primary tumour as well as in the distant metastasis. The AHNAK and PDGFRA mutations thus characterized the clone seeding the distant metastasis. A SMARCB1 mutation was present in all samples except in region A of the distant metastasis. However, as only seven reads were covering this position in the region A sample the mutation may be present in this sample as well. The lymph node metastasis had acquired mutations in EIF4G1 and ARID1B, which seem tissue specific to the lymph node metastasis. The mutational pattern could indicate that different clones seeded the lymph node metastasis and the distant metastasis, respectively. Lastly, parts of the primary tumour probably acquired a regional mutation in MGA not detected in the metastases. Although we only examined three samples from the primary tumour in patient 2, we observed low spatial ITH.
For patient 3 we identified 257 mutations. We found a remarkable similarity between region A of the primary tumour and the metastases. Regions B and C of the primary tumour may represent early dysplastic cells due to the low number of mutations (Supplementary Figure S3). Region A of the primary tumour had a stop codon in CDKN1A, which was shared with the metastases. We observed a high degree of acquired mutations being tissue specific for the metastases. Another CDKN1A mutation causing loss of a start codon was shared only between the metastases; however, region A of the primary tumour had low coverage in this particular position and by manual inspection 13 reference alleles and 1 alternate allele were observed. Thus, this CDKN1A mutation could potentially be shared with region A of the primary tumour as well. Other mutations were found solely shared between the metastases and not detected in the primary tumour indicating that the distant metastasis may have been seeded from the lymph node metastasis.
3.3. Models of disease evolution
To further assess disease evolution in the three patients we generated phylogenetic trees using both synonymous and non‐synonymous mutations (Figure 3A, 3C, and 3E). Shared mutations that fell in the ancestral branch could be described as the field disease mutations. The number of shared mutations (defined by length) varied across the three patients, being very short in patient 3 where the primary tumour was most heterogeneous and the disease course very rapid. Cellular regions from the primary tumour in patient 1 appeared homogeneous as seen by the short length of the branches. The lymph node and distant metastasis for patient 2 showed many private mutations following disease spread. The distant metastasis appeared to be seeded by at least two clonally different cells since the distant metastasis region B shared a greater number of mutations with the primary tumour than the distant metastasis region A. Lastly, primary tumour regions B and C in patient 3 clustered closely together due to very few mutations in these regions. It appeared that the lymph node metastasis and distant metastasis were seeded by two individual clones both originating from the related region A of the primary tumour and both having developed multiple private mutations. This finding contradicts the observation of shared mutations between lymph node metastasis and distant metastasis seen in Figure 2E; however, the phylogenetic analysis represents a best fit approach to model cancer evolution, and some shared mutations may have been discarded.
Figure 3.
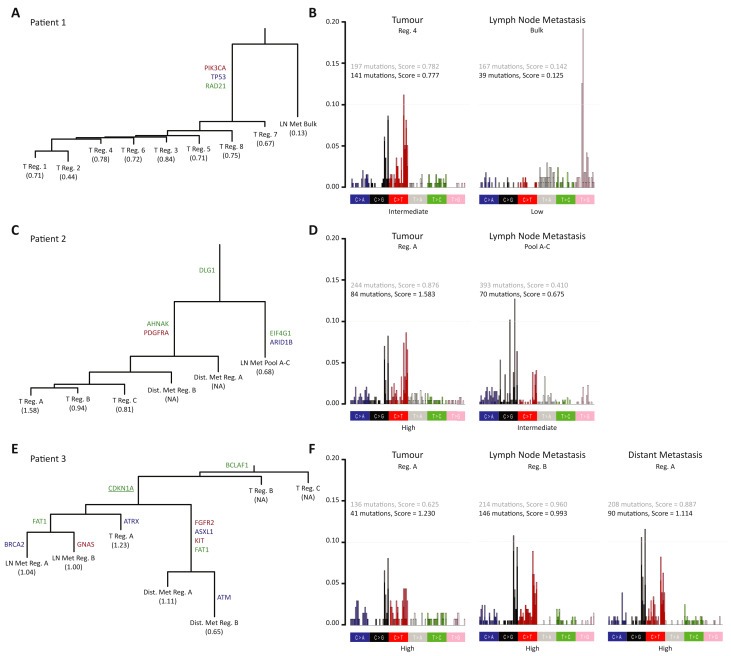
Mapping of patient specific tumour evolution by phylogenetic analysis. (A) Phylogenetic trees based on all mutations (tiers 0–2, categories 1–2). Relative branch lengths present the number of mutations in each branch. Oncogenes (red), tumour suppressor genes (blue) and IntOGen drivers (green) are annotated. APOBEC mutational score (based on tiers 0–2 categories 1–2) is listed for each sample with NA for samples with too few mutations. (B) Representative plots of tri‐nucleotide contexts and associated APOBEC scores from primary tumour, lymph node metastasis and distant metastasis (not applicable for patient 2). Bars are differentiated with the dark part of the bars including category 1 and 2 mutations and the light part of the bars adding category 3 mutations to the analysis. APOBEC scores are given in black based on category 1 and 2 mutations and in grey adding category 3 mutations to the analysis. APOBEC scores are categorised as low, intermediate or high (see Supplementary Figure S5–S7 for full analysis). Reg.: Region, LN Met: Lymph node metastasis, Bulk: Exome on bulk DNA, Pool A–C: Pool of exomes, Dist. Met: Distant metastasis.
3.4. Nucleotide changes and APOBEC related mutagenesis
Next we assessed the context of the mutations called in the three patients including both synonymous and non‐synonymous mutations. The distribution of the nucleotide changes appeared different within the patients (Figure 2B and 2C). The observed differences in nucleotide distribution may be caused by different mutational mechanism such as APOBEC. We next assessed the proportion of APOBEC related mutagenesis (Supplementary Figure S5–S7). By analysing regions from primary tumour, lymph node metastasis, and distant metastasis we did not find APOBEC related mutagenesis to be related to tumour metastasis; rather we found the signature to be a large contributor within the primary tumours for patients 1 and 2 (Figure 3B and 3D, Supplementary Figure S5 and S6). In patient 1 we found APOBEC‐related mutagenesis in all eight primary tumour regions and no APOBEC contribution seen in the lymph node metastasis. Likewise, we found a similar contribution across the three primary tumour samples and a lower contribution in the lymph node metastasis in patient 2. This may be observed as only the APOBEC related mutations in the few cells that seed the metastasis are measured in the metastasis. In patient 3 (Figure 3F and Supplementary Figure S7) we found an equal contribution of APOBEC related mutagenesis in the metastases compared to the primary tumour excluding regions B and C from the primary tumour from the analysis.
3.5. Identification of therapeutic targets
We found the shared mutations to constitute up to 41% of all mutations identified and we observed up to 34% of the mutations to be tissue specific for the metastatic sites (Supplementary Figure S8). To investigate potential therapeutic targets we looked up all mutated genes in drug databases (see Materials and Methods). Including FDA approved drugs and drugs currently being tested in clinical trials we identified six, six, and ten potential therapeutic targets for the three patients, respectively (Figure 2E and Supplementary Table S2). Only few of the therapeutic targets were found in the primary tumour, lymph node metastases, and distant metastases simultaneously. The majority of the drugs supposedly target genes that are only tissue specifically mutated or only regionally mutated. No therapeutic targets were identified in the lymph node metastasis in patient 1. Two shared therapeutic targets were found in patient 2 (FH and NCOA1). No therapeutic targets were found shared in patient 3, since no therapeutic targets were identified in regions B and C of the primary tumour. Two therapeutic targets (BTK and FGF13) were identified in all other regions. Overall, the analysis revealed remarkably few shared mutations that can be targeted and the analysis further shows the important necessity for analysis of both primary tumours and metastatic lesions before selection of targeted therapy.
3.6. Model of clonal development and disease spread in bladder cancer
In order to gain further insight into clonal development we next focused on mutation allele frequencies. Overall we observed low allele frequencies across all samples – including the allele frequencies for shared mutations (Figure 4B). Correlations between the individual samples within the primary tumour were assessed for patient 1 where eight samples were compared. In general we found high correlations between primary tumour regions (R scores >0.67) indicating that all samples contained the same variants at similar frequencies (Figure 4A). However, the vast majority of the variants were observed at low frequency. Accordingly very few mutations were present in all tumour cells, and mutations that appeared clonal were not clonal in all samples within the primary tumours (e.g. TP53 mutation appeared clonal in four out of eight sampled regions). We also observed mutations of low frequency in the metastatic lesions in patient 1 and patient 2 pointing towards parallel seeding of the metastases by different clones. Metastatic seeding by fewer clones may explain the higher allele frequencies as observed for patient 3.
Figure 4.
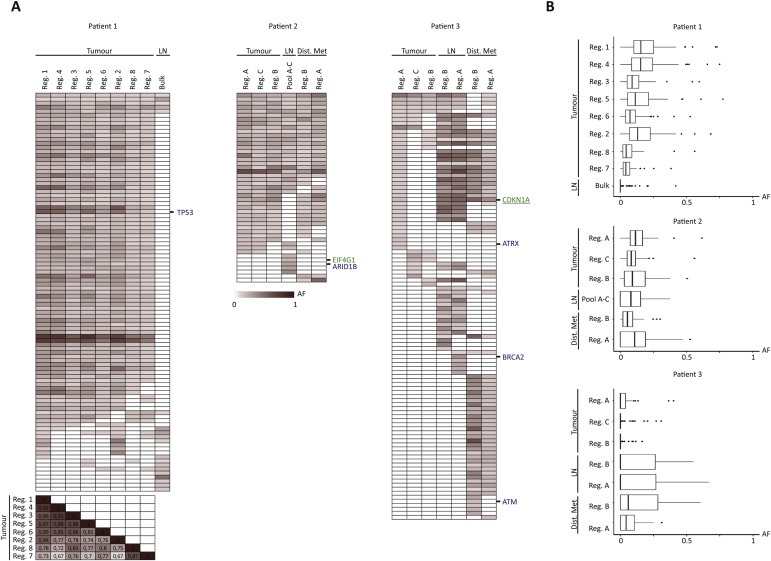
Distribution of allele frequencies across all samples. (A) Heat maps of allele frequencies ranging from 0 (white) to 1 (dark red) (including tiers 0–1 mutations, called with a category score of 1 or 2 in at least one of the samples). We further excluded mutations where we did not reach 30 total reads in the given position for all samples. Known tumour suppressor genes and IntOGen driver genes are annotated in blue and green, respectively. No oncogenes were left after filtering. For patient 1, a correlation matrix of r scores is shown for the 8 primary tumour regions for comparison of the individual samples. (B) Boxplots summarising the distribution of the allele frequencies for the given samples after exclusion of mutations with less than 30 total reads. Samples are ordered in descending number of mutations within primary tumour, lymph node metastasis, and distant metastasis in each patient. AF: Allele frequency. Reg.: Region, LN: Lymph node metastasis, Bulk: Exome on bulk DNA, Pool A–C: Pool of exomes, Dist. Met: Distant metastasis.
4. Discussion
We performed WES on multiple small cellular regions from both primary tumour and metastatic lesions from three patients with metastatic bladder cancer. A strikingly large genetic diversity between primary tumours and metastatic lesions was observed together with a low spatial heterogeneity in primary tumours. In addition, low allele frequencies were observed that may be caused by an intermix of multiple cellular subclones. This may have important implications for targeted therapy.
Studies of heterogeneity in renal clear cell carcinoma revealed a requirement for multiple sampling (Gerlinger et al., 2014, 2012). Whether larger ITH would be observed including multiple biopsies from the primary tumours requires further analysis as e.g. superficial and deep parts of the tumours may be genetically different and show larger heterogeneity. Further, it cannot be excluded, that the mutations identified in the metastatic lesions were present in other regions of the primary tumour. Again, multiregional sequencing studies compared to metastatic lesions will be required.
Drugs targeting commonly mutated genes are thought to be most optimal to obtain clinically efficacious therapy. Most targeted therapies as single agents or in combination with chemotherapy tested in bladder cancer have to date proved inefficacious (Jordan and Iyer, 2015). In this study, we found very few therapeutic targets to be present in all samples in all tissues in the three patients analysed (Figure 2E and Supplementary Table S2). Focussing on drugs primarily targeting the metastatic lesions may prove to prolong survival, since in 90% of the cases the metastatic lesions is the cause of death (Gupta and Massague, 2006). However, the therapeutic targets do not appear clonal in the samples which question the possibility of complete eradication. Tumour subclone inter‐dependence has been proposed where eradicating one or more clones causes complete tumour collapse (Marusyk et al., 2014), and it may be advantageous to aim for such a bystander effect when applying targeted therapy. Whether, the drug–gene interactions are valid for the given variant of the gene observed in the patients would require further investigation regarding specific mutation type and drug efficiency, and is beyond the scope of this retrospective analysis.
Recently, Sottoriva et al. (2015) hypothesized that tumour development may include early intermixing of subclones. The data from patients 1 and 2 could be described by this model where intermixes of genetically different cells results in measurements of low frequency mutations across all cellular regions. Similar intermixes were recently published in breast cancers for larger tumours (Yates et al., 2015). The low frequencies could also be explained by non‐Darwinian evolution of the tumours as recently described in a patient with hepatocellular carcinoma (Ling et al., 2015).
The APOBEC mutational signature has recently been studied intensively (Burns et al., 2013). An increase in APOBEC related mutations has been observed over time in lung cancer (de Bruin et al., 2014) and APOBEC mutations may be responsible for driving tumour progression (Swanton et al., 2015). Here we found a substantial contribution of APOBEC related mutagenesis in three patients with metastatic disease; however, whether APOBEC related mutagenesis drives metastatic development cannot be determined from this study. A low APOBEC score for the metastatic lesion may reflect a purifying selection from many subclones in the primary tumour with widespread APOBEC related mutagenesis to one/few clones in the metastatic lesion where APOBEC related mutations will appear clonal and thus less frequent. A rise in APOBEC score in the metastatic lesions may reflect continued tumour heterogeneity and acquisition of de novo mutations in an APOBEC context after dissemination. To fully establish the role of APOBEC related mutagenesis during metastasis analysis of multiple patients is required.
We only assessed the heterogeneity within single biopsies and thus local heterogeneity for three patients in total. It is possible, that the lymph node metastasis in patient 1 was seeded from a different part of the tumour and the shared mutations identified reflect the field disease/cancerization associated with bladder cancer (Hoglund, 2007; Nordentoft et al., 2014). This emphasizes the importance of analysing both primary tumours and metastatic lesions in order not to miss potential actionable targets of the metastatic lesion. Since bladder cancer has the highest mutation rate only preceded by melanomas and lung cancer (TCGA, 2014) we expected to observe a high level of heterogeneity. We however observed low regional heterogeneity within the primary tumours. Whether, larger differences as well as regional clonal sweeps would be observed if more distant areas are compared or if early stage tumours show lower heterogeneity is presently not known and requires further multiregional sequencing studies. Also, since patient 3 was multi‐focal, we cannot rule out parallel seeding of the metastases from multiple tumours, however, due to the high number of shared mutations between region A of the investigated primary tumour and the metastatic lesions, we find it likely, that this tumour seeded the metastases. Furthermore, we only focused on the distribution of known disease driver mutations (Vogelstein et al., 2013) and IntOGen driver mutations identified in bladder cancer (Rubio‐Perez et al., 2015). The IntOGen database also includes 459 potential pan‐cancer driver genes not investigated in this study. These and others may also be involved in the malignant process. Finally, this study includes only three patients and more patients with metastatic bladder cancer should be investigated to fully establish the ITH in this group of patients.
4.1. Conclusions
We identified known cancer disease driver genes to be both shared and regionally mutated and many disease driver mutations to be acquired during the metastatic process. Our data demonstrates that sequencing of both primary tumour and metastases may be needed to identify mutated genes and potential therapeutic targets. Targeted therapy could be offered in combination with chemotherapy, or as 2nd line therapy following chemotherapy failure.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgements
We thank Louise Nielsen, Margaret Gellet, Hanne Steen, Pamela Celis, Gitte Glistrup Nielsen, Anita Roest, and Lone Andersen for technical assistance. This work was supported by the Danish Cancer Society, the Health Research Fund of Central Denmark Region, The Danish Cancer Biobank, and the Danish Association for Cancer Research. Grant numbers not applicable.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.08.003.
Thomsen Mathilde B.H., Nordentoft Iver, Lamy Philippe, Høyer Søren, Vang Søren, Hedegaard Jakob, Borre Michael, Jensen Jørgen B., Ørntoft Torben F., Dyrskjøt Lars, (2016), Spatial and temporal clonal evolution during development of metastatic urothelial carcinoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.08.003.
References
- Babjuk, M. , Burger, M. , Zigeuner, R. , Shariat, S.F. , van Rhijn, B.W. , Comperat, E. , Sylvester, R.J. , Kaasinen, E. , Bohle, A. , Palou Redorta, J. , Roupret, M. , 2013. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2013. Eur. Urol. 64, 639–653. [DOI] [PubMed] [Google Scholar]
- Burns, M.B. , Lackey, L. , Carpenter, M.A. , Rathore, A. , Land, A.M. , Leonard, B. , Refsland, E.W. , Kotandeniya, D. , Tretyakova, N. , Nikas, J.B. , Yee, D. , Temiz, N.A. , Donohue, D.E. , McDougle, R.M. , Brown, W.L. , Law, E.K. , Harris, R.S. , 2013. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature. 494, 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis, K. , Lawrence, M.S. , Carter, S.L. , Sivachenko, A. , Jaffe, D. , Sougnez, C. , Gabriel, S. , Meyerson, M. , Lander, E.S. , Getz, G. , 2013. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang le, L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. , Lu, X. , Ruden, D.M. , 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin, E.C. , McGranahan, N. , Mitter, R. , Salm, M. , Wedge, D.C. , Yates, L. , Jamal-Hanjani, M. , Shafi, S. , Murugaesu, N. , Rowan, A.J. , Gronroos, E. , Muhammad, M.A. , Horswell, S. , Gerlinger, M. , Varela, I. , Jones, D. , Marshall, J. , Voet, T. , Van Loo, P. , Rassl, D.M. , Rintoul, R.C. , Janes, S.M. , Lee, S.M. , Forster, M. , Ahmad, T. , Lawrence, D. , Falzon, M. , Capitanio, A. , Harkins, T.T. , Lee, C.C. , Tom, W. , Teefe, E. , Chen, S.C. , Begum, S. , Rabinowitz, A. , Phillimore, B. , Spencer-Dene, B. , Stamp, G. , Szallasi, Z. , Matthews, N. , Stewart, A. , Campbell, P. , Swanton, C. , 2014. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science (New York, N.Y.). 346, 251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. , del Angel, G. , Rivas, M.A. , Hanna, M. , McKenna, A. , Fennell, T.J. , Kernytsky, A.M. , Sivachenko, A.Y. , Cibulskis, K. , Gabriel, S.B. , Altshuler, D. , Daly, M.J. , 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger, M. , Horswell, S. , Larkin, J. , Rowan, A.J. , Salm, M.P. , Varela, I. , Fisher, R. , McGranahan, N. , Matthews, N. , Santos, C.R. , Martinez, P. , Phillimore, B. , Begum, S. , Rabinowitz, A. , Spencer-Dene, B. , Gulati, S. , Bates, P.A. , Stamp, G. , Pickering, L. , Gore, M. , Nicol, D.L. , Hazell, S. , Futreal, P.A. , Stewart, A. , Swanton, C. , 2014. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 46, 225–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger, M. , Rowan, A.J. , Horswell, S. , Larkin, J. , Endesfelder, D. , Gronroos, E. , Martinez, P. , Matthews, N. , Stewart, A. , Tarpey, P. , Varela, I. , Phillimore, B. , Begum, S. , McDonald, N.Q. , Butler, A. , Jones, D. , Raine, K. , Latimer, C. , Santos, C.R. , Nohadani, M. , Eklund, A.C. , Spencer-Dene, B. , Clark, G. , Pickering, L. , Stamp, G. , Gore, M. , Szallasi, Z. , Downward, J. , Futreal, P.A. , Swanton, C. , 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl. J. Med. 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez, A. , Perez-Llamas, C. , Deu-Pons, J. , Tamborero, D. , Schroeder, M.P. , Jene-Sanz, A. , Santos, A. , Lopez-Bigas, N. , 2013. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods. 10, 1081–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith, M. , Griffith, O.L. , Coffman, A.C. , Weible, J.V. , McMichael, J.F. , Spies, N.C. , Koval, J. , Das, I. , Callaway, M.B. , Eldred, J.M. , Miller, C.A. , Subramanian, J. , Govindan, R. , Kumar, R.D. , Bose, R. , Ding, L. , Walker, J.R. , Larson, D.E. , Dooling, D.J. , Smith, S.M. , Ley, T.J. , Mardis, E.R. , Wilson, R.K. , 2013. DGIdb: mining the druggable genome. Nat. Methods. 10, 1209–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, G.P. , Massague, J. , 2006. Cancer metastasis: building a framework. Cell. 127, 679–695. [DOI] [PubMed] [Google Scholar]
- Hartmann, A. , Rosner, U. , Schlake, G. , Dietmaier, W. , Zaak, D. , Hofstaedter, F. , Knuechel, R. , 2000. Clonality and genetic divergence in multifocal low-grade superficial urothelial carcinoma as determined by chromosome 9 and p53 deletion analysis. Lab. Invest. 80, 709–718. [DOI] [PubMed] [Google Scholar]
- Hedegaard, J. , Lamy, P. , Nordentoft, I. , Algaba, F. , Hoyer, S. , Ulhoi, B.P. , Vang, S. , Reinert, T. , Hermann, G.G. , Mogensen, K. , Thomsen, M.B. , Nielsen, M.M. , Marquez, M. , Segersten, U. , Aine, M. , Hoglund, M. , Birkenkamp-Demtroder, K. , Fristrup, N. , Borre, M. , Hartmann, A. , Stohr, R. , Wach, S. , Keck, B. , Seitz, A.K. , Nawroth, R. , Maurer, T. , Tulic, C. , Simic, T. , Junker, K. , Horstmann, M. , Harving, N. , Petersen, A.C. , Calle, M.L. , Steyerberg, E.W. , Beukers, W. , van Kessel, K.E. , Jensen, J.B. , Pedersen, J.S. , Malmstrom, P.U. , Malats, N. , Real, F.X. , Zwarthoff, E.C. , Orntoft, T.F. , Dyrskjot, L. , 2016 Jul 11. Comprehensive transcriptional analysis of early-stage urothelial carcinoma. Cancer Cell. 30, (1) 27–42. 10.1016/j.ccell.2016.05.004 [Epub 2016 Jun 16] [DOI] [PubMed] [Google Scholar]
- Hermans, T.J. , Fransen van de Putte, E.E. , Horenblas, S. , Lemmens, V. , Aben, K. , van der Heijden, M.S. , Beerepoot, L.V. , Verhoeven, R.H. , van Rhijn, B.W. , 2016. Perioperative treatment and radical cystectomy for bladder cancer – a population based trend analysis of 10,338 patients in The Netherlands. Eur. J. Cancer. 54, 18–26. [DOI] [PubMed] [Google Scholar]
- Hoglund, M. , 2007. On the origin of syn- and metachronous urothelial carcinomas. Eur. Urol. 51, 1185–1193. discussion 1193 [DOI] [PubMed] [Google Scholar]
- Jordan, E.J. , Iyer, G. , 2015. Targeted therapy in advanced bladder cancer: what have we learned?. Urol. Clin. North Am. 42, 253–262. ix [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Akbani, R. , Creighton, C.J. , Lerner, S.P. , Weinstein, J.N. , Getz, G. , Kwiatkowski, D.J. , 2015. Invasive bladder cancer: genomic insights and therapeutic promise. Clin. Cancer Res. 21, 4514–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles, M.A. , Hurst, C.D. , 2015. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat. Rev. Cancer. 15, 25–41. [DOI] [PubMed] [Google Scholar]
- Law, V. , Knox, C. , Djoumbou, Y. , Jewison, T. , Guo, A.C. , Liu, Y. , Maciejewski, A. , Arndt, D. , Wilson, M. , Neveu, V. , Tang, A. , Gabriel, G. , Ly, C. , Adamjee, S. , Dame, Z.T. , Han, B. , Zhou, Y. , Wishart, D.S. , 2014. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 42, D1091–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Durbin, R. , 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England). 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling, S. , Hu, Z. , Yang, Z. , Yang, F. , Li, Y. , Lin, P. , Chen, K. , Dong, L. , Cao, L. , Tao, Y. , Hao, L. , Chen, Q. , Gong, Q. , Wu, D. , Li, W. , Zhao, W. , Tian, X. , Hao, C. , Hungate, E.A. , Catenacci, D.V. , Hudson, R.R. , Li, W.H. , Lu, X. , Wu, C.I. , 2015. Extremely high genetic diversity in a single tumor points to prevalence of non-Darwinian cell evolution. Proc. Natl. Acad. Sci. U. S. A. 112, E6496–E6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marusyk, A. , Tabassum, D.P. , Altrock, P.M. , Almendro, V. , Michor, F. , Polyak, K. , 2014. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature. 514, 54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordentoft, I. , Lamy, P. , Birkenkamp-Demtroder, K. , Shumansky, K. , Vang, S. , Hornshoj, H. , Juul, M. , Villesen, P. , Hedegaard, J. , Roth, A. , Thorsen, K. , Hoyer, S. , Borre, M. , Reinert, T. , Fristrup, N. , Dyrskjot, L. , Shah, S. , Pedersen, J.S. , Orntoft, T.F. , 2014. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 7, 1649–1663. [DOI] [PubMed] [Google Scholar]
- Roberts, S.A. , Lawrence, M.S. , Klimczak, L.J. , Grimm, S.A. , Fargo, D. , Stojanov, P. , Kiezun, A. , Kryukov, G.V. , Carter, S.L. , Saksena, G. , Harris, S. , Shah, R.R. , Resnick, M.A. , Getz, G. , Gordenin, D.A. , 2013. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 45, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez, C. , Tamborero, D. , Schroeder, M.P. , Antolin, A.A. , Deu-Pons, J. , Perez-Llamas, C. , Mestres, J. , Gonzalez-Perez, A. , Lopez-Bigas, N. , 2015. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 27, 382–396. [DOI] [PubMed] [Google Scholar]
- Sottoriva, A. , Kang, H. , Ma, Z. , Graham, T.A. , Salomon, M.P. , Zhao, J. , Marjoram, P. , Siegmund, K. , Press, M.F. , Shibata, D. , Curtis, C. , 2015. A Big Bang model of human colorectal tumor growth. Nat. Genet. 47, 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, C. , 2012. Intratumor heterogeneity: evolution through space and time. Cancer Res. 72, 4875–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanton, C. , McGranahan, N. , Starrett, G.J. , Harris, R.S. , 2015. APOBEC enzymes: mutagenic fuel for cancer evolution and heterogeneity. Cancer Discov. 5, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TCGA, 2014. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 507, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , Papadopoulos, N. , Velculescu, V.E. , Zhou, S. , Diaz, L.A. , Kinzler, K.W. , 2013. Cancer genome landscapes. Science (New York, N.Y.). 339, 1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Maase, H. , 2003. Gemcitabine in transitional cell carcinoma of the urothelium. Expert Rev. Anticancer Ther. 3, 11–19. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. , Knox, C. , Guo, A.C. , Shrivastava, S. , Hassanali, M. , Stothard, P. , Chang, Z. , Woolsey, J. , 2006. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34, D668–D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjes, J.A. , Comperat, E. , Cowan, N.C. , De Santis, M. , Gakis, G. , Lebret, T. , Ribal, M.J. , Van der Heijden, A.G. , Sherif, A. , 2014. EAU guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2013 guidelines. Eur. Urol. 65, 778–792. [DOI] [PubMed] [Google Scholar]
- Yates, L.R. , Gerstung, M. , Knappskog, S. , Desmedt, C. , Gundem, G. , Van Loo, P. , Aas, T. , Alexandrov, L.B. , Larsimont, D. , Davies, H. , Li, Y. , Ju, Y.S. , Ramakrishna, M. , Haugland, H.K. , Lilleng, P.K. , Nik-Zainal, S. , McLaren, S. , Butler, A. , Martin, S. , Glodzik, D. , Menzies, A. , Raine, K. , Hinton, J. , Jones, D. , Mudie, L.J. , Jiang, B. , Vincent, D. , Greene-Colozzi, A. , Adnet, P.Y. , Fatima, A. , Maetens, M. , Ignatiadis, M. , Stratton, M.R. , Sotiriou, C. , Richardson, A.L. , Lonning, P.E. , Wedge, D.C. , Campbell, P.J. , 2015. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 21, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Fujimoto, J. , Zhang, J. , Wedge, D.C. , Song, X. , Zhang, J. , Seth, S. , Chow, C.W. , Cao, Y. , Gumbs, C. , Gold, K.A. , Kalhor, N. , Little, L. , Mahadeshwar, H. , Moran, C. , Protopopov, A. , Sun, H. , Tang, J. , Wu, X. , Ye, Y. , William, W.N. , Lee, J.J. , Heymach, J.V. , Hong, W.K. , Swisher, S. , Wistuba, II , Futreal, P.A. , 2014. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science (New York, N.Y.). 346, 256–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data