

Abstract
Fundamental cell signaling mechanisms that regulate dynamic remodeling of the extracellular matrix (ECM) in mechanically loaded tissues are not yet clearly understood. Trabecular meshwork (TM) tissue in the eye is under constant mechanical stress and continuous remodeling of ECM is crucial to maintain normal aqueous humor drainage and intraocular pressure (IOP). However, excessive ECM remodeling can cause fibrosis of the TM as in primary open-angle glaucoma (POAG) patients, and is characterized by increased resistance to aqueous humor drainage, elevated IOP, optic nerve degeneration and blindness. Increased levels of active transforming growth factor-β2 (TGF-β2) in the aqueous humor is the main cause of fibrosis of TM in POAG patients. Herein, we report a novel finding that, in TM cells, TGF-β-induced increase in collagen expression is associated with phosphorylation of phosphatase and tensin homolog (PTEN) at residues Ser380/Thr382/383. Exogenous overexpression of a mutated form of PTEN with enhanced phosphatase activity prevented the TGF-β-induced collagen expression by TM cells. We propose that rapid alteration of PTEN activity through changes in its phosphorylation status could uniquely regulate the continuous remodeling of ECM in the normal TM. Modulating PTEN activity may have high therapeutic potential to alleviating the fibrosis of TM in POAG patients.
Introduction
Dynamic remodeling of the extracellular matrix (ECM) is necessary for development, wound healing and maintenance of normal tissue homeostasis1. A breakdown in dynamic remodeling of the ECM can result in fibrosis which is characterized by excess deposition of ECM molecules that destroy the normal architecture of the tissue, leading to the impairment of organ function. It is estimated that loss of organ function due to fibrosis, as in diabetic nephropathy, pulmonary fibrosis and liver cirrhosis, contributes to one-third of natural deaths world-wide2. Currently there is no cure available for fibrotic diseases.
One of the factors that has a major impact on excess ECM deposition in many fibrotic diseases is transforming growth factor-beta (TGF-β)3. Thus, cell signaling mechanisms by which TGF-β induces excess ECM deposition are well studied4. Conversely, TGF-β in normal tissues has pleiotropic roles in the maintenance of tissue homeostasis5 and does not cause fibrosis. It is plausible that TGF-β-mediated signaling pathways that induce fibrosis are suppressed or balanced by TGF-β-induced increase in matrix metalloproteinase activity that degrades ECM6–9. Thus, further investigation are required to delineate fundamental regulatory feed-back signaling mechanisms that could prevent fibrotic actions of TGF-β without affecting its normal homeostatic roles in tissues. Such signaling mechanisms when identified may also serve as effective therapeutic targets to prevent disease-associated fibrosis.
TGF-β decreases the levels of Phosphatase and tensin homolog (PTEN) in several transformed cell lines, including HaCaT, PANC-1 and CAPAN-1, and also in primary glomerular mesangial cells10–13. Crucially, PTEN is capable of regulating TGF-β signaling as a co-factor for Smad2/3 phosphatase14 and is now considered a major regulator of ECM deposition15. Deletion of the Pten gene in dermal fibroblasts of mice induces excess collagen deposition/fibrosis in vivo, while in vitro overexpression of PTEN in dermal fibroblasts from scleroderma patients decreases collagen production, reversing the fibrotic phenotype16. Inhibition of PTEN activity in fibroblasts also increases collagen deposition17. Additionally, decrease in PTEN levels has been reported in many fibrotic diseases, including rheumatoid arthritis18 and pulmonary fibrosis19.
PTEN is a dual phosphatase and its major function is to dephosphorylate phosphatidylinositol 3,4,5-trisphosphate (PIP3) to phosphatidylinositol 4,5-bisphosphate20, thus inhibiting the PI3-kinase/AKT signaling pathway. PTEN is also known to dephosphorylate focal-adhesion kinase, and Src homology 2 domain containing transforming protein21. By regulating these signaling pathways, PTEN is able to modulate multiple cellular activities, including contractility, survival, apoptosis, migration and, cell-ECM interaction and signaling21. PTEN expression and activity is controlled by several mechanisms, including miRNAs, non-coding RNAs, phosphorylation, acetylation, oxidation, S-nitrosylation, and ubiquitylation22. These numerous and intricate modes of regulation of PTEN expression and activity indicate fundamental role of PTEN in regulating dynamic ECM remodeling in tissues.
Regular homeostatic ECM remodeling that occurs in most tissues1, 23 results in a rate of collagen turnover which is usually slow, with collagen half-life estimated to be 15 years in the skin and 117 years in the cartilage24. Conversely, under homeostatic conditions, accumulating evidence indicates that remodeling of ECM in the trabecular meshwork (TM) tissue is nearly continuous25. The TM, located at the irido-corneal angle, consists of TM cells and their porous extracellular matrix through which the aqueous humor, the clear fluid in the anterior segment of the eye, drains into the episcleral veins. Appropriate resistance to the drainage of aqueous humor through the TM is vital for homeostatic intraocular pressure (IOP). The TM tissue, thus occupies a high stress environment with fluctuations in mechanical and fluid shear forces26, 27. Continuous remodeling of ECM is crucial for appropriate resistance to aqueous humor drainage and maintenance of normal IOP. Indeed, it is expected that previously undescribed cell signaling mechanisms drive the dynamic and continuous ECM remodeling in the TM.
In contrast to the scenario in healthy eyes, fibrosis of the TM can result in primary open-angle glaucoma (POAG), the most common type of glaucoma which affects nearly 74% of the 70 million glaucoma patients worldwide28. Any increase in ECM deposition in the TM increases the resistance to aqueous humor drainage leading to an increase in IOP and is associated with optic nerve degeneration and blindness in POAG patients29–32. Lowering the IOP is the only medical strategy known to delay onset and slow progression of vision loss in glaucoma33, 34. One strategy to lower the IOP is to prevent the fibrosis of the TM; however, this has not yet been accomplished35.
Increased levels of active TGF-β2 in the aqueous humor of POAG patients36 are implicated in the fibrosis of the TM37. However, inhibiting TGF-β signaling is not a solution as TGF-β is also vital for maintaining the immune-privilege of the eye38. Moreover, TGF-β is present in the normal aqueous humor and TM cells both express and secrete this cytokine36, 39–42 and yet does not cause fibrosis of the TM. Indeed, induction of ECM deposition by TGF-β concurrently stimulates localized ECM digestion at invadosomes by increasing matrix metalloproteinase-2 activity in TM cells8. Thus, there is still a need to further delineate the elements in the TGF-β signaling pathway in normal and fibrotic TM, taking into account their potential to regulate the expeditious deposition or degradation of ECM in normal TM. While many of the mechanisms that drive ECM remodeling in TM are known43, there still could exist novel fundamental signaling mechanisms that enable pro-fibrotic TGF-β to expeditiously contribute to the continuous remodeling of ECM by TM cells.
In this study, we have investigated the modulation and regulation of PTEN levels/activity by TGF-β in human TM cells. We report that, in TM cells, TGF-β-induced increase in collagen expression is associated with the phosphorylation of PTEN at residues Ser380/Thr382/383. Phosphorylation of PTEN at these residues is known to suppress the activity of PTEN. Exogenous overexpression of mutant form of PTEN with enhanced phosphatase activity prevented TGF-β-induced collagen expression by TM cells. We propose rapid alteration of PTEN activity through changes in its phosphorylation status to be a unique mechanism that regulates the continuous remodeling of ECM in the normal TM. PTEN or elements in its signaling pathway could serve as a therapeutic target with high potential to alleviate fibrosis of the TM in glaucoma.
Results
TGF-β induces an increase in levels of PTEN and its phosphorylation
Levels of TGF-β2 in the aqueous humor range from ~0.5 ng/ml to ~8 ng/ml36, 42, whereas glaucomatous aqueous humor have higher levels of total as well as active TGF-β236. To explore the effect of TGF-β on ECM deposition, we treated TM cells with 1 ng/ml, 5 ng/ml, or 10 ng/ml of TGF-β2 to simulate physiological and pathological levels of TGF-β2 in the aqueous humor. Since TGF-β is a major inducer of fibrosis, addition of TGF-β to cultured human embryonic TM cells, as expected, increased collagen mRNA levels (Fig. 1A). Increase in COL1A1 mRNA is seen at 12 h, rising further to a 3.6 and 4.1 fold increase in mRNA levels at 24 h after 1 ng/ml and 5 ng/ml TGF-β treatment, respectively. A corresponding increase in collagen 1 protein levels are also seen in TM cells treated with TGF-β (Fig. 1C,D ). Surprisingly, TGF-β also significantly increased the levels of PTEN mRNA in TM cells, with a significant 2.2 fold increase after treatment with 5 ng/ml TGF-β for 24 h (Fig. 1B ). There is also an increase in PTEN protein levels in TM cells following TGF-β treatment for 24 h and 48 h; a nearly two fold increase is seen after 48 h of TGF-β treatment (Fig. 1E–G). However, the increase in PTEN protein levels was also accompanied by a concomitant increase in phosphorylation of residues Ser380/Thr382/383 in the PTEN tail region, with almost a 1.6 fold increase at 48 h (Fig. 1E,F,H). Phosphorylation of PTEN at these residues is indicative of inactivation of PTEN44–48. The increase in levels/phosphorylation of PTEN after TGF-β treatment was reproduced when TM cells were cultured on either tissue culture plastic or on collagen/pronectin-coated elastomer plates (data not shown).
Figure 1.
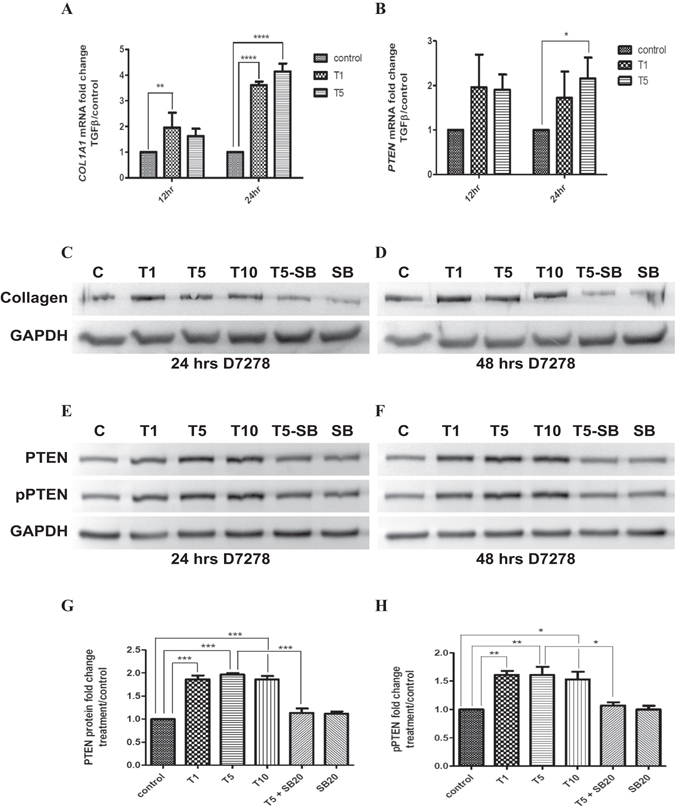
TGF-β induces an increase in levels of PTEN and its phosphorylation in human TM cells. (A,B) Real-time PCR analysis of COL1A1 and PTEN mRNA levels in TM cells treated with TGF-β2 (1 or 5 ng/ml). RNA was extracted at 12 and 24 h. mRNA expression levels were normalized to EUK18S. A two-way ANOVA with Bonferroni’s post-hoc was used to determine statistical significance. *p < 0.05, **p < 0.01, ****p < 0.0001. N = 3. (C,D) Western blot analysis of collagen levels in TM cells treated with TGF-β (T1, T5, T10 indicates 1, 5 or 10 ng/ml TGF-β2). Protein was extracted at 24 and 48 h. TGF-β receptor signaling was inhibited using the inhibitor SB431542 (SB; 20 μM). D7278- representative human donor. (E,F) Western blot analysis of PTEN levels and its phosphorylation (pPTEN- ser380/thr382/383) in TM cells treated with TGF-β and SB431542. Protein was extracted at 24 and 48 h. D7278- representative human donor. (G,H) Densitometry was performed on PTEN and pPTEN Western blot data at 48 h using ImageJ software. Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc. *p < 0.05, **p < 0.01, ***p < 0.001. N = 3. (I,J). TGF-β-mediated induction of PTEN levels and its phosphorylation was confirmed in TM cells from an adult human donor, aged 26 yrs (Adult 26Y).
Association of PI3-kinase and PTEN in TGF-β-mediated collagen expression
Non-canonical TGF-β type I receptor (ALK5)/Smad-independent signaling is associated with enhanced collagen deposition49–52. In our experiments, we assessed the PI3-kinase pathway not only because active PTEN prevents the PI3-kinase-mediated AKT signaling, but also because PI3-kinase is able to block PTEN activity53, 54. We found that addition of TGF-β induced the phosphorylation of AKT (Ser473) in TM cells, indicating again that the phosphorylation of PTEN following TGF-β treatment has inactivated its phosphatase activity. A 4.7 fold increase in phosphorylation of AKT is observed following 1 ng/ml TGF-β treatment for 24 h (Fig. 2A,B). Inhibition of PI3-kinase signaling by ZSTK474 and LY294002 almost completely eliminated the TGF-β-induced phosphorylation of AKT. Both PI3-kinase signaling inhibitors also significantly suppressed TGF-β-induced collagen expression. We found a corresponding attenuation of PTEN and its phosphorylation at residues Ser380/Thr382/383 to levels similar to that of controls (Fig. 3A,B).
Figure 2.
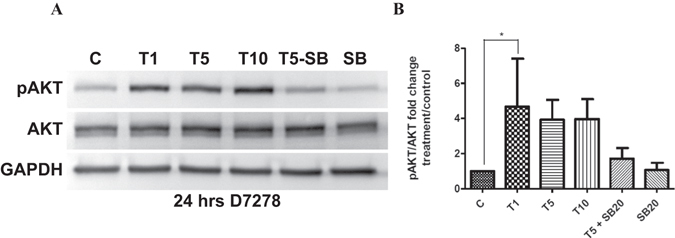
TGF-β induces phosphorylation of AKT in human TM cells. (A) Western blot analysis of AKT and its phosphorylation (pAKT- ser473) in TM cells treated with TGF-β2 (1, 5 or 10 ng/ml). Protein was extracted at 24 h. TGF-β receptor signaling was inhibited using SB431542 (SB; 20 μM). D7278- representative human donor. (B) Densitometry was performed on AKT and pAKT Western blot data using ImageJ software. Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc. *p < 0.05. N = 3.
Figure 3.
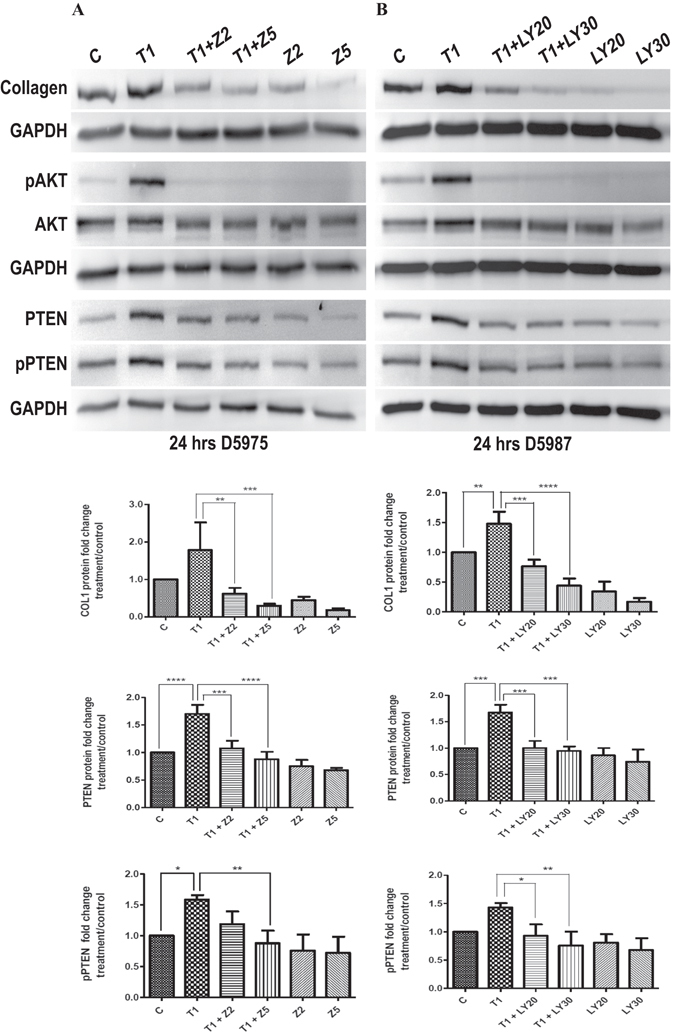
Inhibition of PI3-kinase/AKT signaling prevents TGF-β-induced increase in levels of collagen and PTEN/phosphorylation in human TM cells. (A,B) Human TM cells were treated with TGF-β2 (1 ng/ml) and PI3-kinase signaling inhibited using ZSTK474 (Z2, Z5- 2 μM or 5 μM) or LY294002 (LY20, LY30- 20 μM or 30 μM). Protein was extracted at 24 h and collagen, PTEN, pPTEN (ser380/thr382/383), AKT and pAKT detected by Western blot. Densitometry was performed on collagen, PTEN and pPTEN Western blots using ImageJ software and shown as graphs. Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. N = 3. D5975, D5987- representative human donors.
Inhibition of PTEN and overexpression of PTEN in TM cells modulates collagen expression
Since our results are consistent with the hypothesis that TGF-β regulates collagen deposition by modulating PTEN/AKT levels and phosphorylation, we further investigated whether inhibition of PTEN activity and its overexpression affected collagen expression by TM cells. PTEN activity was inhibited using VO-OHpic55; in TM cells application of 5 µM VO-OHpic increased the expression of collagen by nearly 4 fold after 48 h of treatment. VO-OHpic-induced increase in collagen levels was also accompanied by an increase in PTEN levels and its phosphorylation (Fig. 4). This further reinforces that changes in levels of PTEN/phosphorylation affects collagen expression. We also assessed the effect of mutant PTEN on collagen expression by transfecting TM cells with enhanced PTEN (ePTEN) which has an approximately eightfold increased ability to suppress PIP3 signaling, and by also transfecting PTEN with defective lipid phosphatase activity (C124SPTEN)56. Overexpression of ePTEN completely prevented TGF-β mediated upregulation of collagen expression by TM cells while decreasing PTEN/phosphorylation to levels similar to that of controls. Overexpression of C124SPTEN enhanced TGF-β-induced collagen expression by nearly 1.5 fold and enhanced the levels of PTEN/phosphorylation (Fig. 5). Preliminary experiments also indicate that overexpression of ePTEN and C124SPTEN has similar effects on the induction of fibronectin by TGF-β (Supplementary data Fig. 1); regulation of fibronectin expression by PTEN has been previously reported57.
Figure 4.
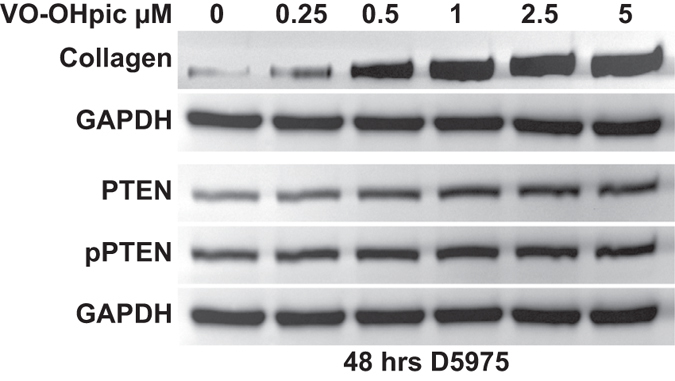
Inhibition of PTEN increases levels of collagen in human TM cells. Human TM cells were treated with VO-OHpic, a potent inhibitor of PTEN at varying concentrations. VO-OHpic at 5 μM caused a nearly 4-fold (N = 3) increase in collagen levels at 48 h. A corresponding increase in the levels of PTEN and phosphorylated PTEN was noted. D5975- representative human donor.
Figure 5.
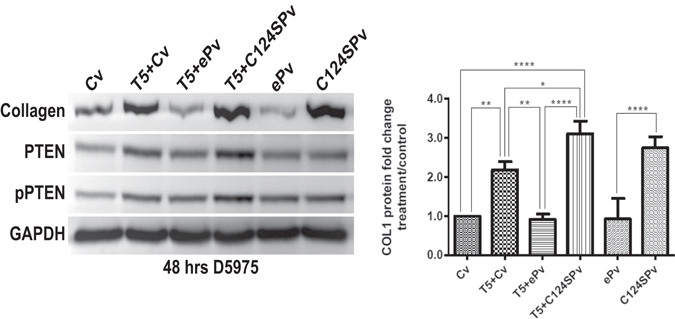
PTEN activity modulates collagen expression in human TM cells. TM cells were treated with TGF-β2 (5 ng/ml) and transfected with control GFP-vector (Cv), enhanced PTEN-GFP vector (ePv) or C124SPTEN-GFP vector (C124SPv). Enhanced PTEN has eight-fold increased ability to suppress PIP3 signaling, while C124SPTEN has defective lipid phosphatase activity. Densitometry was performed on collagen Western blots using ImageJ software. Statistical significance was determined using one-way ANOVA with Tukey’s post-hoc. *p < 0.05, **p < 0.01, ****p < 0.0001. N = 3. D5975- representative human donor. Transfection with enhanced PTEN decreased the levels of TGF-β-induced collagen to that of controls, while reducing the levels of PTEN and its phosphorylation. Transfection with C124SPTEN enhanced TGF-β induced increase in collagen levels and this was accompanied by an increase in the levels of PTEN and phosphorylated PTEN.
Discussion
TGF-β is a well-established inducer of collagen deposition by activation of canonical Smads and through non-canonical PI3-kinase and p38 MAPK signaling pathways49–52, 58, 59. However, the regulation of PTEN by TGF-β10, 11, 13, 60 and the increasingly key role of PTEN in the modulation of ECM deposition are recent findings14, 17, 61, 62. The role of PTEN in TGF-β signaling becomes crucial as it regulates TGF-β-induced collagen deposition by modulating Smad signaling14. Nuclear PTEN protects protein phosphatase, Mg2+/Mn2+ dependent 1 A (PPM1A, a Smad2/3 phosphatase) from degradation induced by TGF-β signaling. Any decrease in PTEN is thought to destabilize PPM1A, allowing Smad2/3 to remain phosphorylated in the nucleus, thus promoting TGF-β signaling14. We chose collagen I as the ECM protein to investigate as it is present in the striated collagen fibrils of the trabecular core, the basement membrane of the trabecular beams and in the loose aggregates in the juxtacanalicular tissue63 and is expressed by TM cells in culture64. Collagen I expression is also induced by TGF-β in TM cells65 and its accumulation causes increased IOP and progressive loss of optic nerve axons66, 67.
In this study we report that TGF-β treatment of TM cells induced expression of collagen despite an increase in the levels of PTEN. This is contrary to previous reports that an increase in the levels of PTEN reduced collagen deposition17, 61, 68. Our data suggest that TGF-β induced the expression of collagen by TM cells because the increase in PTEN levels is antagonized by an increase in its phosphorylation at Ser380/Thr382/383 in the C-terminal tail region. Phosphorylation at these residues is known to reduce PTEN activity48 and causes folding of the C-terminal tail on to the membrane-binding region, thus preventing translocation of PTEN to cell membrane, suppressing its enzymatic activity44–48. The inactivation of PTEN due to phosphorylation and its association with increase in collagen expression by TM cells is further supported by previous reports that Pten gene deletion or PTEN inhibition increased deposition of collagen17, 61.
The continuous remodeling of ECM in the TM reflects its adaptation to a high-stress mechanical stress environment and the necessity to maintain normal IOP, despite the persistent fluctuations of IOP26. Expeditious activation and deactivation of PTEN through alteration of phosphorylation status could be one of the fundamental signaling mechanisms that allow for continuous remodeling of ECM in TM tissue. Our study demonstrates for the first time that treatment with TGF-β is associated with phosphorylation of PTEN. Hitherto, TGF-β has only been known to decrease PTEN levels10–13, 60 in experiments mostly on transformed cell lines, representing tissues that do not need continuous ECM remodeling.
Reversible control of phosphorylation allows cells to respond rapidly to fluctuating changes in its environment69. Other factors present in the aqueous humor could balance the TGF-β-induced phosphorylation of PTEN. For example, Hepatocyte growth factor (HGF) is present in the normal aqueous humor70. HGF is known to inhibit TGF-β-induced decrease in PTEN levels71 and also antagonize the fibrotic actions of TGF-β72. Alternative isoform of TGF-β receptor II in TM cells could also explain the difference in TGF-β signaling mechanism in TM cells73. In this context, it is noteworthy that, in TM cells, TGF-β not only increases ECM deposition, but concurrently induces localized ECM digestion at invadosomes by increasing matrix metalloproteinase-2 activity8.
The increase in PTEN levels at 24 and 48 hours that we found following TGF-β treatment, could represent a feed-back mechanism to compensate for an early decrease in PTEN activity due to phosphorylation. Our preliminary experiments (Supplementary data Fig. 2) indicate an early increase in PTEN phosphorylation at 15 to 30 minutes after TGF-β treatment. This increase appears to attenuate at one hour after TGF-β treatment. The early increase in PTEN phosphorylation coincides with known peak phosphorylation of Smad3 at 30 minutes after TGF-β treatment74, 75, and is relevant as PTEN is known to regulate Smad signaling through Smad2/3 phosphatase PPM1A14. Moreover, being a highly regulated protein22, activation and recruitment to the membrane is known to rapidly degrade PTEN44, while phosphorylation at Ser380/Thr382/Thr383 increases its stability48. Thus, the increase in PTEN levels could also be a mechanism that promptly compensates for any activity-induced degradation of PTEN.
We further showed that, in TM cells, the increase in collagen after TGF-β treatment is associated with an increase in phosphorylation of AKT at Ser473. Since PTEN inhibits the PI3-kinase/AKT signaling pathway76, the increase in pAKT yet again indicates a reduction in PTEN activity. Conversely, we demonstrate that inhibition of the PI3-kinase pathway resulted in almost complete elimination of AKT phosphorylation by TGF-β, and also prevented TGF-β-induced increase in collagen levels. The decrease in AKT phosphorylation and collagen expression following inhibition of PI3-kinase pathway indicates an increase in PTEN activity. Activation of PTEN with inhibition of PI3-kinase pathway has been previously reported53, 77, 78. Moreover, inhibition of PI3-kinase pathway reduced TGF-β-induced increase in PTEN levels and phosphorylation. This again indicates a rapid degradation of PTEN following its activation and recruitment to the membrane44.
Additionally, we show that PTEN inhibition increased collagen production by TM cells. Furthermore, overexpression of ePTEN, with approximately eight-fold increased ability to suppress PIP3 signaling56, prevented TGF-β-induced collagen and fibronectin expression. In contrast, overexpression of PTEN with defective lipid phosphatase activity56 further enhanced TGF-β-induced collagen and fibronectin expression in TM cells. Considering that even a 20% reduction in PTEN levels induces early lethality and cancer susceptibility79, it is highly plausible that even a modest therapeutic increase in PTEN activity could effectively reduce fibrosis of TM in glaucoma.
New therapeutic approaches are critical, as progressive vision loss is common among glaucoma patients despite effective current treatments80. However, no new drug classes have been approved worldwide to treat glaucoma for nearly two decades81, 82. Several new drugs are currently in clinical trials but they have varying efficacies and side-effect profiles and strategies of treatment are still evolving81, 82. Our results indicate that PTEN has high potential to emerge as an effective therapeutic treatment for glaucoma. Several therapeutic drugs that indirectly enhance PTEN activity are currently available83, and may be used to prevent PTEN phosphorylation, increase PTEN activity or modulate elements associated with its signaling to alleviate fibrosis of TM.
Methods
Cell culture
For all experiments human trabecular meshwork cells at passage 4 were used. Donor cells 7278, 5975, and 5987 were of embryonic origin (ScienCell Research Laboratories, Carlsbad, CA). Phenotype of donor cells was further confirmed by immunostaining for von-Willebrand factor (data not shown) and by detection of myocilin after dexamethasone treatment (Supplementary data Fig. 3). Cells were cultured in low glucose DMEM (Life Technologies, Burlington, ON, Canada) supplemented with 10% FBS with 1% Penicillin-Streptomycin (Life Technologies) on either tissue culture-treated plastic or on Bioflex pronectin/collagen-coated (Flexcell International Corporation, Burlington, NC) plates. Cells were in low serum medium (0.5% FBS) for at least 6–12 h and nearly confluent when treated with human recombinant TGF-β2 (R&D Systems, Inc, Minneapolis, MN). When small molecule inhibitors of cell signaling were used [SB431542, VO-OHpic (Tocris Bioscience, Minneapolis, MN), LY294002, or ZSTK474 (Selleck Chemicals, Houston, TX)] the cells were pretreated for 30 min to 1 h.
Real-time PCR
Total RNA was isolated using the RNeasy RNA extraction kit (Qiagen, Mississauga, ON) and quantity and integrity of RNA sample was checked using a Nanodrop 2000 (Thermo Scientific, Waltham, MA). 25 ng of total RNA was reverse-transcribed and amplified using one-step RT-qPCR master mix (qScript XLT One-step RT-qPCR ToughMix, ROX; Quanta Biosciences, Beverly, MA) and detected with ABI Prism 7900HT (Applied Biosystems/Thermo Scientific). TaqMan Assay-on-Demand primers (Applied Biosystems/Thermo Scientific) were used and samples were run in triplicate. Levels of PTEN and COL1A1 mRNA expression in samples were normalized to values of control 18S and fold change determined using the 2−ΔΔCt method.
Western blots
Since increase in PTEN expression was surprisingly accompanied by an increase in its phosphorylation, Western blots to probe the expression of PTEN and phosphorylated PTEN were initially conducted separately (Supplementary data Fig. 4). This was to avoid any technical problem associated with stripping and reprobing the blots. Once the concomitant increase in both PTEN and phosphorylated PTEN was established, further experiments probed the same blot, first for phosphorylated PTEN and then followed by PTEN. Following detection of phosphorylated PTEN, blots were stripped and checked for any residual HRP-conjugated secondary antibody before being probed for PTEN.
Proteins were extracted using IP lysis buffer (Pierce; Thermo Scientific) and protein estimation was performed using the Micro BCA Protein Assay kit (Thermo Scientific). Proteins were resolved on polyacrylamide gels and blotted on to nitrocellulose membranes and probed with anti-PTEN rabbit pAb (AF847) (R&D Systems, Inc), anti-phosphoPTEN Ser380/Thr382/383 rabbit mAb (44A7) (Cell Signaling Technologies, Danvers, MA), anti-Akt (pan) rabbit mAb (C67E7) (Cell Signaling Technologies), anti-phosphoAkt Ser473 (D9E XP) rabbit mAb (Cell Signaling Technologies), anti-collagen type 1 rabbit mAb (EPR7785) (Abcam, Cambridge, UK) and anti-GAPDH mouse mAb (9B3: sc-66163) (Santa Cruz Biotechnology, Santa Cruz, CA). HRP conjugated anti-mouse and anti-rabbit antibodies were used in conjunction with an ECL imaging system. Densitometric analysis of western blot bands was performed using ImageJ software.
Overexpression of PTEN
TM cells were transfected using the Polymag Neo Magnetofection Kit (OZ Biosciences Inc, San Diego, CA) according to manufacturer’s instructions. TM cells were transfected with 1 µg of either IRES GFP84 (Addgene, Cambridge, USA), ePTEN-GFP or C124SPTEN-GFP56 in pcDNA3.1 backbone, followed by addition of 5 ng/ml TGF-β2, 20–24 h after transfection. Protein was extracted at 24 and 48 h after addition of TGF-β to transfected TM cells.
Statistical analysis
Two-way ANOVA with Bonferroni’s post-hoc test or one-way ANOVA with Tukey’s post hoc test was applied to determine the statistical significance in differences between treatment groups. Differences were considered statistically significant when P < 0.05. GraphPad Prism software (version 6.0, GraphPad Software, San Diego, CA) was used for all statistical analyses.
Electronic supplementary material
Acknowledgements
This work was made possible by grants from Glaucoma Research Society of Canada, Canadian National Institute for the Blind, Internal Research Fund-Lawson Health Research Institute, Medical and Health Sciences Research Board-University of Western Ontario and Pilot Funds- Department of Ophthalmology, UWO. N.T. has received the Ontario Graduate Scholarship and the Dr. Wm. P. McGrath Scholarship for Research in Ophthalmology. Undergraduate students Deelan Patel and Wei Lin Su were involved in some of the preliminary experiments. We also acknowledge Dr. Martin Duennwald for constructive criticisms of the manuscript.
Author Contributions
N.T.: performed the experiments, analyzed data, graphs and statistics; J.C.B.: performed preliminary experiments; A.C.T., C.H. and M.M.: clinical perspective, discussions and clinical samples; A.L.: drafted manuscript; H.L.: technical assistance; M.I.: designed experiments and PTEN mutant constructs; S.K.P.: conceived and designed experiments, performed preliminary experiments, analyzed data, drafted manuscript and prepared digital images.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-00845-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeisberg M, Kalluri R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am J Physiol Cell Physiol. 2013;304:C216–225. doi: 10.1152/ajpcell.00328.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pohlers D, et al. TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 5.Fleisch MC, Maxwell CA, Barcellos-Hoff MH. The pleiotropic roles of transforming growth factor beta in homeostasis and carcinogenesis of endocrine organs. Endocr Relat Cancer. 2006;13:379–400. doi: 10.1677/erc.1.01112. [DOI] [PubMed] [Google Scholar]
- 6.Kim ES, Sohn YW, Moon A. TGF-beta-induced transcriptional activation of MMP-2 is mediated by activating transcription factor (ATF)2 in human breast epithelial cells. Cancer Lett. 2007;252:147–156. doi: 10.1016/j.canlet.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 7.Munshi HG, et al. Differential regulation of membrane type 1-matrix metalloproteinase activity by ERK 1/2- and p38 MAPK-modulated tissue inhibitor of metalloproteinases 2 expression controls transforming growth factor-beta1-induced pericellular collagenolysis. J Biol Chem. 2004;279:39042–39050. doi: 10.1074/jbc.M404958200. [DOI] [PubMed] [Google Scholar]
- 8.Han H, Kampik D, Grehn F, Schlunck G. TGF-beta2-induced invadosomes in human trabecular meshwork cells. PLoS One. 2013;8:e70595. doi: 10.1371/journal.pone.0070595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim ES, Kim MS, Moon A. TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004;25:1375–1382. [PubMed] [Google Scholar]
- 10.Chow JY, et al. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis. 2007;28:2321–2327. doi: 10.1093/carcin/bgm159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881–889. doi: 10.1038/ncb1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ebert MP, et al. Reduced PTEN expression in the pancreas overexpressing transforming growth factor-beta 1. Br J Cancer. 2002;86:257–262. doi: 10.1038/sj.bjc.6600031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 14.Bu S, Kapanadze B, Hsu T, Trojanowska M. Opposite effects of dihydrosphingosine 1-phosphate and sphingosine 1-phosphate on transforming growth factor-beta/Smad signaling are mediated through the PTEN/PPM1A-dependent pathway. J Biol Chem. 2008;283:19593–19602. doi: 10.1074/jbc.M802417200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trojanowska M. Noncanonical transforming growth factor beta signaling in scleroderma fibrosis. Curr Opin Rheumatol. 2009;21:623–629. doi: 10.1097/BOR.0b013e32833038ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parapuram, S. K. et al. Loss of PTEN Expression by Dermal Fibroblasts Causes Skin Fibrosis. J Invest Dermatol (2011). [DOI] [PubMed]
- 17.White ES, et al. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10) Am J Respir Crit Care Med. 2006;173:112–121. doi: 10.1164/rccm.200507-1058OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pap T, et al. Activation of synovial fibroblasts in rheumatoid arthritis: lack of Expression of the tumour suppressor PTEN at sites of invasive growth and destruction. Arthritis Res. 2000;2:59–64. doi: 10.1186/ar69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White ES, et al. Integrin alpha4beta1 regulates migration across basement membranes by lung fibroblasts: a role for phosphatase and tensin homologue deleted on chromosome 10. Am J Respir Crit Care Med. 2003;168:436–442. doi: 10.1164/rccm.200301-041OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 21.Tamura M, Gu J, Tran H, Yamada KM. PTEN gene and integrin signaling in cancer. J Natl Cancer Inst. 1999;91:1820–1828. doi: 10.1093/jnci/91.21.1820. [DOI] [PubMed] [Google Scholar]
- 22.Shi Y, Paluch BE, Wang X, Jiang X. PTEN at a glance. J Cell Sci. 2012;125:4687–4692. doi: 10.1242/jcs.093765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Molnar JA, Alpert N, Burke JF, Young VR. Synthesis and degradation rates of collagens in vivo in whole skin of rats, studied with 1802 labelling. Biochem J. 1986;240:431–435. doi: 10.1042/bj2400431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verzijl N, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275:39027–39031. doi: 10.1074/jbc.M006700200. [DOI] [PubMed] [Google Scholar]
- 25.Keller KE, Acott TS. The Juxtacanalicular Region of Ocular Trabecular Meshwork: A Tissue with a Unique Extracellular Matrix and Specialized Function. J Ocul Biol. 2013;1:3. [PMC free article] [PubMed] [Google Scholar]
- 26.Downs JC, et al. 24-hour IOP telemetry in the nonhuman primate: implant system performance and initial characterization of IOP at multiple timescales. Invest Ophthalmol Vis Sci. 2011;52:7365–7375. doi: 10.1167/iovs.11-7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.WuDunn D. Mechanobiology of trabecular meshwork cells. Exp Eye Res. 2009;88:718–723. doi: 10.1016/j.exer.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 28.Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boland MV, Quigley HA. Risk factors and open-angle glaucoma: classification and application. J Glaucoma. 2007;16:406–418. doi: 10.1097/IJG.0b013e31806540a1. [DOI] [PubMed] [Google Scholar]
- 30.Gottanka J, Johnson DH, Martus P, Lutjen-Drecoll E. Severity of optic nerve damage in eyes with POAG is correlated with changes in the trabecular meshwork. J Glaucoma. 1997;6:123–132. doi: 10.1097/00061198-199704000-00009. [DOI] [PubMed] [Google Scholar]
- 31.Knepper PA, Samples JR, Yue BY. Biomarkers of primary open-angle glaucoma. Expert Rev Ophthalmol. 2010;5:731–742. doi: 10.1586/eop.10.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tektas OY, Lutjen-Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. 2009;88:769–775. doi: 10.1016/j.exer.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 33.Leskea MC, Heijl A, Hyman L, Bengtsson B, Komaroff E. Factors for progression and glaucoma treatment: the Early Manifest Glaucoma Trial. Curr Opin Ophthalmol. 2004;15:102–106. doi: 10.1097/00055735-200404000-00008. [DOI] [PubMed] [Google Scholar]
- 34.Lichter PR, et al. Interim clinical outcomes in the Collaborative Initial Glaucoma Treatment Study comparing initial treatment randomized to medications or surgery. Ophthalmology. 2001;108:1943–1953. doi: 10.1016/S0161-6420(01)00873-9. [DOI] [PubMed] [Google Scholar]
- 35.Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008;86:543–561. doi: 10.1016/j.exer.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res. 1994;59:723–727. doi: 10.1006/exer.1994.1158. [DOI] [PubMed] [Google Scholar]
- 37.Gottanka J, Chan D, Eichhorn M, Lutjen-Drecoll E, Ethier CR. Effects of TGF-beta2 in perfused human eyes. Invest Ophthalmol Vis Sci. 2004;45:153–158. doi: 10.1167/iovs.03-0796. [DOI] [PubMed] [Google Scholar]
- 38.Denniston AK, et al. Endogenous cortisol and TGF-beta in human aqueous humor contribute to ocular immune privilege by regulating dendritic cell function. J Immunol. 2011;186:305–311. doi: 10.4049/jimmunol.1001450. [DOI] [PubMed] [Google Scholar]
- 39.Liton PB, Liu X, Challa P, Epstein DL, Gonzalez P. Induction of TGF-beta1 in the trabecular meshwork under cyclic mechanical stress. J Cell Physiol. 2005;205:364–371. doi: 10.1002/jcp.20404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tripathi RC, Chan WF, Li J, Tripathi BJ. Trabecular cells express the TGF-beta 2 gene and secrete the cytokine. Exp Eye Res. 1994;58:523–528. doi: 10.1006/exer.1994.1046. [DOI] [PubMed] [Google Scholar]
- 41.Tripathi RC, Li J, Borisuth NS, Tripathi BJ. Trabecular cells of the eye express messenger RNA for transforming growth factor-beta 1 and secrete this cytokine. Invest Ophthalmol Vis Sci. 1993;34:2562–2569. [PubMed] [Google Scholar]
- 42.Jampel HD, Roche N, Stark WJ, Roberts AB. Transforming growth factor-beta in human aqueous humor. Curr Eye Res. 1990;9:963–969. doi: 10.3109/02713689009069932. [DOI] [PubMed] [Google Scholar]
- 43.Vranka JA, Kelley MJ, Acott TS, Keller KE. Extracellular matrix in the trabecular meshwork: intraocular pressure regulation and dysregulation in glaucoma. Exp Eye Res. 2015;133:112–125. doi: 10.1016/j.exer.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 46.Rahdar M, et al. A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vazquez F, et al. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem. 2001;276:48627–48630. doi: 10.1074/jbc.C100556200. [DOI] [PubMed] [Google Scholar]
- 48.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/MCB.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J Biol Chem. 2004;279:2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 50.Asano Y, et al. Phosphatidylinositol 3-kinase is involved in alpha2(I) collagen gene expression in normal and scleroderma fibroblasts. J Immunol. 2004;172:7123–7135. doi: 10.4049/jimmunol.172.11.7123. [DOI] [PubMed] [Google Scholar]
- 51.Jiang W, et al. Role of cross-talk between the Smad2 and MAPK pathways in TGF-beta1-induced collagen IV expression in mesangial cells. Int J Mol Med. 2010;26:571–576. doi: 10.3892/ijmm_00000501. [DOI] [PubMed] [Google Scholar]
- 52.Meyer-Ter-Vehn T, et al. p38 inhibitors prevent TGF-beta-induced myofibroblast transdifferentiation in human tenon fibroblasts. Invest Ophthalmol Vis Sci. 2006;47:1500–1509. doi: 10.1167/iovs.05-0361. [DOI] [PubMed] [Google Scholar]
- 53.Papakonstanti EA, Ridley AJ, Vanhaesebroeck B. The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J. 2007;26:3050–3061. doi: 10.1038/sj.emboj.7601763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagata Y, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6:117–127. doi: 10.1016/j.ccr.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 55.Mak LH, Vilar R, Woscholski R. Characterisation of the PTEN inhibitor VO-OHpic. J Chem Biol. 2010;3:157–163. doi: 10.1007/s12154-010-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen HN, et al. Engineering ePTEN, an enhanced PTEN with increased tumor suppressor activities. Proc Natl Acad Sci USA. 2014;111:E2684–2693. doi: 10.1073/pnas.1409433111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dey N, Ghosh-Choudhury N, Kasinath BS, Choudhury GG. TGFbeta-stimulated microRNA-21 utilizes PTEN to orchestrate AKT/mTORC1 signaling for mesangial cell hypertrophy and matrix expansion. PLoS One. 2012;7:e42316. doi: 10.1371/journal.pone.0042316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. 2005;280:10055–10064. doi: 10.1074/jbc.M409381200. [DOI] [PubMed] [Google Scholar]
- 59.Pervan CL. Smad-independent TGF-beta2 signaling pathways in human trabecular meshwork cells. Exp Eye Res. 2016 doi: 10.1016/j.exer.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 60.Chow JY, et al. TGF-beta mediates PTEN suppression and cell motility through calcium-dependent PKC-alpha activation in pancreatic cancer cells. Am J Physiol Gastrointest Liver Physiol. 2008;294:G899–905. doi: 10.1152/ajpgi.00411.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parapuram SK, et al. Loss of PTEN expression by dermal fibroblasts causes skin fibrosis. J Invest Dermatol. 2011;131:1996–2003. doi: 10.1038/jid.2011.156. [DOI] [PubMed] [Google Scholar]
- 62.Parapuram SK, et al. Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2-dependent mechanism. Matrix Biol. 2015;43:35–41. doi: 10.1016/j.matbio.2015.01.017. [DOI] [PubMed] [Google Scholar]
- 63.Marshall GE, Konstas AG, Lee WR. Immunogold ultrastructural localization of collagens in the aged human outflow system. Ophthalmology. 1991;98:692–700. doi: 10.1016/S0161-6420(91)32232-2. [DOI] [PubMed] [Google Scholar]
- 64.Liton PB, Luna C, Challa P, Epstein DL, Gonzalez P. Genome-wide expression profile of human trabecular meshwork cultured cells, nonglaucomatous and primary open angle glaucoma tissue. Mol Vis. 2006;12:774–790. [PMC free article] [PubMed] [Google Scholar]
- 65.Inoue-Mochita M, et al. p38 MAP kinase inhibitor suppresses transforming growth factor-beta2-induced type 1 collagen production in trabecular meshwork cells. PLoS One. 2015;10:e0120774. doi: 10.1371/journal.pone.0120774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Aihara M, Lindsey JD, Weinreb RN. Ocular hypertension in mice with a targeted type I collagen mutation. Invest Ophthalmol Vis Sci. 2003;44:1581–1585. doi: 10.1167/iovs.02-0759. [DOI] [PubMed] [Google Scholar]
- 67.Mabuchi F, Lindsey JD, Aihara M, Mackey MR, Weinreb RN. Optic nerve damage in mice with a targeted type I collagen mutation. Invest Ophthalmol Vis Sci. 2004;45:1841–1845. doi: 10.1167/iovs.03-1008. [DOI] [PubMed] [Google Scholar]
- 68.Takashima M, et al. The tumor suppressor protein PTEN inhibits rat hepatic stellate cell activation. J Gastroenterol. 2009;44:847–855. doi: 10.1007/s00535-009-0073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wheeler-Jones CP. Cell signalling in the cardiovascular system: an overview. Heart. 2005;91:1366–1374. doi: 10.1136/hrt.2005.072280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Araki-Sasaki K, Danjo S, Kawaguchi S, Hosohata J, Tano Y. Human hepatocyte growth factor (HGF) in the aqueous humor. Jpn J Ophthalmol. 1997;41:409–413. doi: 10.1016/S0021-5155(97)00081-6. [DOI] [PubMed] [Google Scholar]
- 71.Iekushi K, et al. Hepatocyte growth factor attenuates transforming growth factor-beta-angiotensin II crosstalk through inhibition of the PTEN/Akt pathway. Hypertension. 2011;58:190–196. doi: 10.1161/HYPERTENSIONAHA.111.173013. [DOI] [PubMed] [Google Scholar]
- 72.Dai C, Liu Y. Hepatocyte growth factor antagonizes the profibrotic action of TGF-beta1 in mesangial cells by stabilizing Smad transcriptional corepressor TGIF. J Am Soc Nephrol. 2004;15:1402–1412. doi: 10.1097/01.ASN.0000130568.53923.FD. [DOI] [PubMed] [Google Scholar]
- 73.Wordinger RJ, Clark AF, Agarwal R, Lambert W, Wilson SE. Expression of alternatively spliced growth factor receptor isoforms in the human trabecular meshwork. Invest Ophthalmol Vis Sci. 1999;40:242–247. [PubMed] [Google Scholar]
- 74.Wang G, Matsuura I, He D, Liu F. Transforming growth factor-{beta}-inducible phosphorylation of Smad3. J Biol Chem. 2009;284:9663–9673. doi: 10.1074/jbc.M809281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zi Z, Chapnick DA, Liu X. Dynamics of TGF-beta/Smad signaling. FEBS Lett. 2012;586:1921–1928. doi: 10.1016/j.febslet.2012.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leslie NR, Dixon MJ, Schenning M, Gray A, Batty IH. Distinct inactivation of PI3K signalling by PTEN and 5-phosphatases. Adv Biol Regul. 2012;52:205–213. doi: 10.1016/j.advenzreg.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 77.Tzenaki N, et al. High levels of p110delta PI3K expression in solid tumor cells suppress PTEN activity, generating cellular sensitivity to p110delta inhibitors through PTEN activation. FASEB J. 2012;26:2498–2508. doi: 10.1096/fj.11-198192. [DOI] [PubMed] [Google Scholar]
- 78.Chow JY, Cabral JA, Chang J, Carethers JM. TGFbeta modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol Ther. 2008;7:1694–1699. doi: 10.4161/cbt.7.10.6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Carracedo A, Alimonti A, Pandolfi PP. PTEN level in tumor suppression: how much is too little? Cancer Res. 2011;71:629–633. doi: 10.1158/0008-5472.CAN-10-2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupta N, Yucel YH. Glaucoma as a neurodegenerative disease. Curr Opin Ophthalmol. 2007;18:110–114. doi: 10.1097/ICU.0b013e3280895aea. [DOI] [PubMed] [Google Scholar]
- 81.Vicente A, Prud’homme S, Ferreira J, Abegao Pinto L, Stalmans I. Open-Angle Glaucoma: Drug Development Pipeline during the Last 20 Years (1995–2015) Ophthalmic Res. 2017 doi: 10.1159/000453527. [DOI] [PubMed] [Google Scholar]
- 82.Schehlein EM, Novack GD, Robin AL. New classes of glaucoma medications. Curr Opin Ophthalmol. 2016;28:161–168. doi: 10.1016/j.joco.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 83.Boosani CS, Agrawal DK. PTEN modulators: a patent review. Expert Opin Ther Pat. 2013;23:569–580. doi: 10.1517/13543776.2013.768985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schaefer MR, et al. A novel trafficking signal within the HLA-C cytoplasmic tail allows regulated expression upon differentiation of macrophages. J Immunol. 2008;180:7804–7817. doi: 10.4049/jimmunol.180.12.7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.