

Abstract
Purple acid phosphatases (PAPs) are a family of binuclear metalloenzymes that catalyze the hydrolysis of phosphoric acid esters and anhydrides. A PAP in sweet potato has a unique, strongly antiferromagnetically coupled Fe(III)–Mn(II) center and is distinguished from other PAPs by its increased catalytic efficiency for a range of activated and unactivated phosphate esters, its strict requirement for Mn(II), and the presence of a μ-oxo bridge at pH 4.90. This enzyme displays maximum catalytic efficiency (kcat/Km) at pH 4.5, whereas its catalytic rate constant (kcat) is maximal at near-neutral pH, and, in contrast to other PAPs, its catalytic parameters are not dependent on the pKa of the leaving group. The crystal structure of the phosphate-bound Fe(III)–Mn(II) PAP has been determined to 2.5-Å resolution (final Rfree value of 0.256). Structural comparisons of the active site of sweet potato, red kidney bean, and mammalian PAPs show several amino acid substitutions in the sweet potato enzyme that can account for its increased catalytic efficiency. The phosphate molecule binds in an unusual tripodal mode to the two metal ions, with two of the phosphate oxygen atoms binding to Fe(III) and Mn(II), a third oxygen atom bridging the two metal ions, and the fourth oxygen pointing toward the substrate binding pocket. This binding mode is unique among the known structures in this family but is reminiscent of phosphate binding to urease and of sulfate binding to λ protein phosphatase. The structure and kinetics support the hypothesis that the bridging oxygen atom initiates hydrolysis.
Keywords: binuclear metal center, phosphate coordination
Purple acid phosphatases (PAPs) belong to a diverse group of binuclear metallohydrolases that use metal ion centers to catalyze the hydrolysis of amides and esters of carboxylic and phosphoric acids (1, 2). PAPs have been observed in a wide range of animals, plants, and fungi. Genome database searches imply that a limited number of bacteria also possess PAP or PAP-like genes (3). In mammals, biological roles for PAPs have been suggested in iron transport (4), bone resorption by oste-oclasts (5, 6), dephosphorylation of erythrocyte phosphoproteins (7), and the production of hydroxyradicals and reactive oxygen species (8, 9). In plants, a role in the release of phosphate from organophosphates has been proposed for PAPs from Arabidopsis thaliana and tomato (11, §). Furthermore, these enzymes also have been shown to have alkaline peroxidase activity, implying a role for plant PAPs in defense against pathogen infection (11–13).
The active site of PAPs consists of a center with seven invariant amino acid residues coordinating to the two metal ions (14, 15). The characteristic purple color associated with these enzymes is attributed to a charge transfer transition from a tyrosine ligand to the Fe(III) in the binuclear center (16–18). In active PAPs, the second metal ion is always divalent, but its identity depends on the source of the enzyme. For example, in red kidney bean and soybean Fe(III)–Zn(II) PAPs have been isolated (19, 20), whereas all active mammalian PAPs characterized to date have Fe(III)–Fe(II) centers. Oxidation to the diferric form reversibly inactivates the mammalian enzymes, leading to the suggestion that PAP may be regulated by changes in redox potential within animal cells (21).
The three-dimensional structure of red kidney bean PAP showed that it is a 110-kDa homodimer, each subunit consisting of an N-terminal (residues 1–120) and a C-terminal (residues 121–432) domain. The latter contains the catalytic site, including the binuclear Fe(III)–Zn(II) center (22). The structures of the enzyme–phosphate and enzyme–tungstate complexes also have been reported (23). More recently, the crystal structures of pig PAP complexed with phosphate to 1.55-Å resolution (24) and two crystal forms of rat PAP have been determined to 2.2 and 2.8-Å resolution (25, 26). The mammalian enzymes are smaller monomeric proteins of ≈35 kDa, but their overall structure resembles that of the C-terminal domain observed in the red kidney bean enzyme. In plants, a second type of PAP has been identified that has a molecular weight and amino acid sequence closely related to those of the mammalian enzymes (15). The physicochemical properties of this form, including its metal ion content, are yet to be determined.
In sweet potato, both low and high molecular weight types of PAPs have been cloned and sequenced (15, 20, 27). Two isoforms of the high molecular mass (≈110 kDa) protein have been isolated to date. One of these contains an Fe(III)–Zn(II) center with kinetic and spectroscopic properties similar to those of red kidney bean PAP (27). In contrast, the other isoform contains a strongly antiferromagnetically coupled Fe(III)–Mn(II) center, the first of its kind in any biological system (20, 28). Compared with other PAPs, this isoform from sweet potato is further distinguished by its increased catalytic efficiency toward a broad range of both activated and unactivated phosphate esters (20) and its strict requirement for manganese (28). In contrast, in other PAPs the native divalent metal ion can be replaced by other divalent metal ions without major loss of activity (29–31).
The mechanism proposed for PAP-catalyzed reactions generally involves a nucleophilic attack of a metal-bound hydroxide on the phosphorus atom of the substrate, leading to hydrolytic cleavage of the phosphate-ester bond and the release of the leaving group (32). The precise identity of the nucleophile has been the subject of extensive debate. Measurements of the pH dependence of kinetic parameters of pig PAP have led to the proposition that a terminal, Fe(III)-bound hydroxide acts as the nucleophile during catalysis (32, 33). More recently, metal ion replacement studies using bovine PAP (31, 34) and spectroscopic characterization of pig PAP by using x-ray absorption (35) and electron-nuclear double resonance (36) support a model where the hydroxide bridging the two metal ions is the attacking nucleophile.
Spectroscopic and multifield saturation magnetization data imply that the manganese-dependent isoform of sweet potato PAP contains a μ-oxo instead of a μ-hydroxo bridge at pH 4.90 (28). Thus, if the attacking nucleophile in this enzyme at this pH is the bridging ligand, it would be the μ-oxo species rather than a μ-hydroxo species. Here, the pH dependence of kinetic parameters and the inhibition by inorganic phosphate of the Fe–Mn sweet potato enzyme are compared with other PAPs. Furthermore, the dependence of the catalytic rate on the leaving group pKa is assessed in combination with the three-dimensional crystal structure of the phosphate-bound complex. These data allow us to discuss mechanistic features that appear at present to be unique to this PAP. In the process of structure determination an unusual coordination mode for phosphate has been observed, reminiscent of the phosphate complex of the binuclear nickel enzyme urease from Bacillus pasteurii (37).
Materials and Methods
Materials. All reagents were analytical grade and, unless otherwise stated, from Sigma. Benzyl phosphate, 3-nitrophenyl phosphate, and 3-chlorophenyl phosphate were synthesized according to the method of Zhang and van Etten (38).
Protein Purification and Characterization. PAP was extracted from sweet potato (Cultivar: Golden) as described in ref. 20. The purified enzyme was stored at 4°C in 0.1 M acetate buffer (pH 4.9) and concentrated to 22 mg/ml. Samples for kinetic measurements and crystallization were >95% pure, as judged by SDS/PAGE analysis and N-terminal amino acid sequencing. Metal analysis was carried out by using inductively coupled plasma-MS and indicated the presence of 1.04 Fe, 0.75 Mn, and trace amounts of Zn and Cu per active site. Red kidney bean and pig PAPs were isolated as described in refs. 19 and 39.
Activity Assays and pH Dependence of Kinetic Parameters. Kinetic parameters were determined by using standard assays as described in refs. 19, 20, 27, and 28–34 (see also Supporting Materials and Methods, which is published as supporting information on the PNAS web site).
Protein Crystallization. Crystals were grown as described in ref. 40. The well solution consisted of 0.1 M citric acid (pH 3.5–4.0), 7.5% polyethylene glycol 6000, 10% isopropanol, 50 mM phosphate, and 10% glycerol. For cryoprotection, crystals were transferred into a solution where the glycerol concentration was increased to 20%.
Data Collection and Structure Determination. Diffraction images were processed by using denzo and scalepack (41). The red kidney bean PAP dimer (4RKB) was used as the starting model for molecular replacement. Solutions for the rotation and translation searches were found by using the program x-plor v3.851 (42). The two largest peaks in the rotation function were symmetry equivalent solutions. One of these solutions was used in translation search calculations in P6522 and P6122. A single clear peak was obtained in both space groups, but in P6522 the top peak was 20.5σ above the background, whereas the peak in space group P6122 was only 5σ above background. R factors were 0.38 and 0.45 for the two space groups. Based on these results, the space group P6522 was assigned to this crystal. Model building and refinement were performed by using the programs o (43) and cns (44). procheck (45) was used to calculate Ramachandran plot statistics. Data collection and refinement statistics are listed in Table 2, which is published as supporting information on the PNAS web site. The refined coordinates and observed structure factors have been deposited in the Protein Data Bank (PDB ID code 1XZW). Figures were generated by using molscript (Version 2.0.1; ref. 46), raster 3d (Version 2; ref. 47), and setor (48).
Results and Discussion
Effect of pH on the Kinetic Parameters of Sweet Potato PAP. In Table 1, the kinetic parameters of sweet potato PAP are compared with those of pig PAP at various pH values. Sweet potato PAP is active over a pH range of 3.0–8.5; however, its kcat value is maximal close to neutral pH. In contrast, bovine PAP (49) and the FeZn PAPs from red kidney bean and sweet potato (27) display a maximum rate at a pH of ≈6 and pig PAP at a pH of ≈5 (50). However, like the pig enzyme, the FeMn sweet potato PAP reaches optimal catalytic efficiency (kcat/Km) at an acidic pH, which is mainly due to a reduced affinity for the substrate (4-nitrophenol phosphate) at higher pH values (Table 1). Over the pH range, sweet potato PAP has considerably higher turnover values than other known PAPs (Table 1 and ref. 20).
Table 1. Comparison of the pH dependence of the kinetic parameters of sweet potato and pig PAP.
| Sweet potato PAP
|
Pig PAP*
|
|||||
|---|---|---|---|---|---|---|
| pH | kcat, s-1 | Km, μM | kcat/Km, s-1·mM-1 | kcat, s-1 | Km, μM | kcat/Km, s-1·mM-1 |
| 3.5 | 810 ± 77 | 44 ± 6 | 18,000 ± 3,100 | 88 ± 2 | 62 ± 5 | 1,400 ± 120 |
| 4 | 1,700 ± 120 | 68 ± 5 | 26,000 ± 2,600 | 180 ± 3 | 200 ± 14 | 880 ± 60 |
| 4.5 | 2,100 ± 170 | 68 ± 10 | 30,000 ± 5,100 | 280 ± 17 | 390 ± 31 | 730 ± 73 |
| 5 | 2,200 ± 140 | 93 ± 12 | 23,000 ± 3,400 | 380 ± 12 | 750 ± 70 | 510 ± 50 |
| 6 | 2,400 ± 160 | 330 ± 14 | 7,400 ± 590 | 240 ± 3 | 1,700 ± 70 | 140 ± 6 |
| 6.5 | 3,200 ± 210 | 930 ± 33 | 3,500 ± 260 | 150 ± 4 | 3,900 ± 200 | 38 ± 2 |
| 7 | 3,600 ± 290 | 1,800 ± 120 | 5,000 ± 200 | 5 ± 2 | 37,000 ± 27,000 | 0.14 ± 0.12 |
| 8 | 950 ± 67 | 5,000 ± 450 | 190 ± 22 | n.d. | n.d. | n.d. |
When measuring the pH dependence of the kinetic parameters, 4-nitrophenol phosphate was used as a substrate. n.d., not determined.
See ref. 50
Inhibition of Sweet Potato PAP by Inorganic Phosphate. Inorganic phosphate is known to be a moderate competitive inhibitor of PAPs. Ki values have been determined previously (generally at a pH of ≈5) for PAPs from bovine (130–340 μM) (51), human (750 μM) (8), and soybean (700 μM) (52) sources. The reported Ki for the pig enzyme is ≈2 mM (53). A similar value has been determined for the Fe(III)–Zn(II) derivative of bovine PAP at pH 6.5 (49). Here, the Ki for sweet potato PAP was determined to be 196 ± 7 μM, 310 ± 28 μM, and 2.9 ± 0.7 mM at pH 3.5, 4.9, and 7.0, respectively. Based on these values it appears that the sweet potato enzyme has moderate to weak bonding interactions with phosphate similar to those of other PAPs, and it is thus unlikely that phosphoryl-enzyme intermediates are formed during catalysis. This finding is in agreement with a study on pig PAP that failed to show any phosphoryl transfer to the enzyme (54). In contrast, in alkaline phosphatases, where a nucleophilic serine residue is phosphorylated during catalysis, the corresponding inhibition constants are in the low micromolar range (55).
Leaving Group Dependence for PAP-Catalyzed Reactions. The kinetic parameters of sweet potato, red kidney bean, and pig PAP for the hydrolysis of a range of substrates with leaving group pKa values ranging from 7.14 to 15.44 were measured (Table 3, which is published as supporting information on the PNAS web site). Generally, for both red kidney bean and pig PAP, reactivity declines as the leaving group pKa increases. No such trend is apparent for the sweet potato enzyme (Fig. 1). Fitting the data in Table 3 to the equations (i) logkcat = βlgpKlg + constant or (ii) logkcat/Km = βlgpKlg + constant (where pKlg represents the pKa of the leaving group) results in the following βlg values for kcat and kcat/Km, respectively: sweet potato PAP, 0.06 and 0.007; red kidney bean PAP, -0.47 and -0.62; and pig PAP, -0.27 and -0.39. The small values of βlg for the sweet potato enzyme compared with the other PAPs indicate the lack of significant charge development in the transition state of the former.
Fig. 1.

Effect of leaving group pKa on kcat (A) and kcat/Km (B) of sweet potato (•), red kidney bean (○), and pig PAP (▴). The substrates and their corresponding leaving group pKa values are listed in Table 3. The lines were drawn using linear regression.
Overall Structure of Sweet Potato PAP and Comparison with Red Kidney Bean PAP. Sweet potato PAP consists of two identical polypeptides of 435-aa residues (20). Its crystal structure with bound phosphate was solved by molecular replacement with a final Rfree value of 0.264 for data to 2.5-Å resolution (Table 2). The two polypeptides in the asymmetric unit are structurally indistinguishable with a rms deviation of 0.15 Å for all Cα atoms (Fig. 5, which is published as supporting information on the PNAS web site). The average temperature factor for all atoms in both polypeptides is 45 Å2. The metal ions are well resolved in both subunits with temperature factors of 63.1 Å2 (subunit A) and 50.9 Å2 (subunit B) for Fe and 40.8 Å2 (subunit A) and 36.6 Å2 (subunit B) for Mn. Two phosphate molecules are observed in the structure as “islands” of electron density coordinated to the binuclear center. Temperature factors for the phosphate atoms average 52.6 and 41.8 Å2 for subunits A and B, respectively.
Although the overall structures of red kidney bean (22) and sweet potato PAP are highly conserved (Fig. 5), there are several regions where the structures are substantially different. The most significant difference in the backbone structure occurs at E365 (Y364 in red kidney bean PAP), which is located centrally within the active site (Fig. 2). In the superimposed sweet potato and red kidney bean structures, the equivalent Cα atoms in this position are shifted by 5.8 Å relative to each other with the polypeptide folding toward the active site in the sweet potato enzyme and away from the active site in red kidney bean PAP. The polypeptide scaffold in the sweet potato enzyme allows E365 to be oriented in such a way that it can form a direct hydrogen bond with the bound phosphate (Fig. 2 and also Fig. 6, which is published as supporting information on the PNAS web site). Another structural difference in the active site lies close to the dimer interface. In sweet potato PAP, the side chain of Y258 reaches across from one subunit to form part of the active site pocket of the other subunit. In red kidney bean PAP, the equivalent amino acid residue is Ala. For this reason, the size of the active site pocket is considerably smaller in the sweet potato enzyme than that in red kidney bean PAP. The crystal structures of sweet potato and red kidney bean PAP also differ at their N termini. In the red kidney bean enzyme, the first eight amino acid residues are disordered, but in sweet potato PAP, all of the amino acid residues in the N-terminal tail are visible. Interestingly, the N-terminal nitrogen is only 12 Å from Fe(III). Thus, for both proteins it is possible that the N terminus may play a role in substrate binding or in modulating specificity, particularly for protein substrates.
Fig. 2.

Stereodiagram of the active-site residues in sweet potato PAP. Side chains that coordinate directly to the metal ions are shown as stick models. Side chains that form the active site are shown as ball-and-stick models. Carbon atoms colored gray represent side chains that are conserved between red kidney bean and sweet potato PAP. Carbon atoms colored yellow are nonconserved, and the name of the equivalent residue in red kidney bean PAP is in brackets. Primed amino acid residues are from the neighboring subunit.
Active Site and Binuclear Metal Center. The active site consists of the two metal ions [Fe(III) and Mn(II)] separated by an average (between subunits A and B) of 3.26 Å (Fig. 3 and also Table 4, which is published as supporting information on the PNAS web site). Similar metal-to-metal distances have been observed in other PAPs. For example, the Fe(III)–Fe(III) distance in pig PAP is 3.31 Å (24), whereas in red kidney bean the distance is 3.26 Å for the Fe(III)–Zn(II) combination (23). Both metal ions are six coordinate. The coordination environment of the iron site is made up of a His nitrogen (H324 Nε2; 2.69 Å), a deprotonated Tyr oxygen (Y166 O; 1.88 Å), and two Asp oxygen atoms (D163 Oδ2 and D136 Oδ2; 2.23 and 2.35 Å, respectively), where the former bridges the two metal ions. The manganese is coordinated to two His nitrogen atoms (H285 Nε2 and H322 Nδ1; 2.13 and 2.10 Å, respectively), an Asn oxygen atom (N200 Oδ1; 2.27 Å), and the bridging Asp oxygen (D163 Oδ2; 2.27 Å; Fe–O–Mn, 93.6°). One oxygen atom of the bound phosphate anion (O3) acts as a bridge between the two metal ions (Fe(III), 2.01 Å; Mn(II), 2.36 Å; Fe–O–Mn, 87.0°), whereas O1 and O2 complete the coordination environment of the two metal ions (Fe–O1, 2.59 Å and 1.85 Å in subunits A and B, respectively; Mn-O2, 2.69 Å and 2.03 Å in subunits A and B, respectively). The protonation state of the triply coordinated phosphate [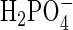 at pH 4 (crystallization conditions)] is defined by H-bonds that are distributed differently in the two subunits (Fig. 6); a similar H-bond pattern and phosphate protonation state is observed in the binuclear Ni(II) enzyme urease (see above and ref. 37).
at pH 4 (crystallization conditions)] is defined by H-bonds that are distributed differently in the two subunits (Fig. 6); a similar H-bond pattern and phosphate protonation state is observed in the binuclear Ni(II) enzyme urease (see above and ref. 37).
Fig. 3.

Stereodiagram of the binuclear metal center in the sweet potato PAP–phosphate complex. Overlayed in blue is the difference electron density, contoured at 3.0 σ, before phosphate was built into the model. Also shown are the phosphate (yellow sticks) and the μ–oxo (yellow ball) of pig PAP after superimposition of the metal ions in sweet potato and pig PAP. It was not possible to fit the phosphate in an equivalent conformation as observed in pig PAP.
The observed 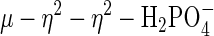 coordination mode in sweet potato PAP is unique among PAP structures. In the crystal structure of red kidney bean PAP complexed with phosphate (at 2.7 Å) (23), the anion binds symmetrically to the two metal ions through two oxygen atoms, displacing putative terminal solvent molecules on each metal ion in the phosphate free, resting enzyme. The bound phosphate forms fewer hydrogen bonds than in sweet potato PAP. The structure of the phosphate complex in the Fe(III)–Fe(III) rat PAP, determined to 2.7 Å, indicates that phosphate is coordinated in a bidentate fashion, approximately equidistant from the two Fe sites, although limited resolution precluded the establishment of the proper orientation of the phosphate (26). Perhaps the most relevant comparison is that to pig PAP in the oxidized Fe(III)–Fe(III) form, crystallized in the presence of inorganic phosphate (24). Like red kidney bean and rat PAP the phosphate ion bridges the two metal ions symmetrically (
coordination mode in sweet potato PAP is unique among PAP structures. In the crystal structure of red kidney bean PAP complexed with phosphate (at 2.7 Å) (23), the anion binds symmetrically to the two metal ions through two oxygen atoms, displacing putative terminal solvent molecules on each metal ion in the phosphate free, resting enzyme. The bound phosphate forms fewer hydrogen bonds than in sweet potato PAP. The structure of the phosphate complex in the Fe(III)–Fe(III) rat PAP, determined to 2.7 Å, indicates that phosphate is coordinated in a bidentate fashion, approximately equidistant from the two Fe sites, although limited resolution precluded the establishment of the proper orientation of the phosphate (26). Perhaps the most relevant comparison is that to pig PAP in the oxidized Fe(III)–Fe(III) form, crystallized in the presence of inorganic phosphate (24). Like red kidney bean and rat PAP the phosphate ion bridges the two metal ions symmetrically (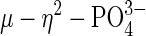 ). One of the two remaining, uncoordinated phosphate oxygen atoms resides between the two metal ions (Fe... O4, 3.81, 4.05 Å) and is hydrogen bonded to a neighboring histidine (H195 Nε2; 2.76 Å). Notably, O4 is located directly above the oxygen atom of the μ-hydroxo bridge. It is interesting to note that a rotation of the
). One of the two remaining, uncoordinated phosphate oxygen atoms resides between the two metal ions (Fe... O4, 3.81, 4.05 Å) and is hydrogen bonded to a neighboring histidine (H195 Nε2; 2.76 Å). Notably, O4 is located directly above the oxygen atom of the μ-hydroxo bridge. It is interesting to note that a rotation of the 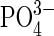 anion, or a nucleophilic attack of the μ-hydroxo on the P atom in the active form of this di-iron enzyme would lead, ultimately, to the conversion of the μ–η2 to the
anion, or a nucleophilic attack of the μ-hydroxo on the P atom in the active form of this di-iron enzyme would lead, ultimately, to the conversion of the μ–η2 to the 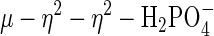 bonding mode seen in the Fe(III)–Mn(II) complex of sweet potato PAP.
bonding mode seen in the Fe(III)–Mn(II) complex of sweet potato PAP.
This μ–η2–η2-geometry observed for FeMn sweet potato PAP, although unusual in other PAP structures, is not without precedence in both metalloprotein and model systems. The structure of the B. pasteurii urease shows a similar 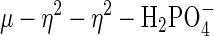 mode of coordination (37). On the basis of the observed structure, the authors proposed a bridging hydroxide nucleophile for enzymatic catalysis. Bimetallic Co(III) systems, designed to model the active site of metallophosphatases, also have been proposed as intermediate
mode of coordination (37). On the basis of the observed structure, the authors proposed a bridging hydroxide nucleophile for enzymatic catalysis. Bimetallic Co(III) systems, designed to model the active site of metallophosphatases, also have been proposed as intermediate 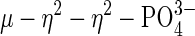 species resulting from deprotonation of μ–OH species, the latter acting as a nucleophile for cleaving a bridged phosphate monoester (56, 57). Additionally, this coordination geometry has been observed in other systems involving the coordination of similar small molecules, for example, carbonate and sulfate. Thus,
species resulting from deprotonation of μ–OH species, the latter acting as a nucleophile for cleaving a bridged phosphate monoester (56, 57). Additionally, this coordination geometry has been observed in other systems involving the coordination of similar small molecules, for example, carbonate and sulfate. Thus, 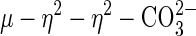 complexes of iron, cobalt, nickel, copper, and zinc (58–61), as well as the
complexes of iron, cobalt, nickel, copper, and zinc (58–61), as well as the 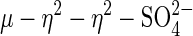 complex of the Mn(II)–Mn(II) bacteriophage λ protein phosphatase (PP) (62), have been characterized. However, sweet potato PAP is the only heterobinuclear and heterovalent system with such coordination geometry.
complex of the Mn(II)–Mn(II) bacteriophage λ protein phosphatase (PP) (62), have been characterized. However, sweet potato PAP is the only heterobinuclear and heterovalent system with such coordination geometry.
Mechanistic Implications for Sweet Potato PAP. A number of mechanistic studies, including isotope labeling (63) and measurements of kinetic and solvent isotope effects (64, 65) as well as site-directed mutagenesis (66) on PAPs and the closely related Ser/Thr PPs have led to the proposition of a catalytic mechanism that invokes a direct transfer of the phosphoryl group of the substrate to a metal-coordinated, nucleophilic solvent molecule. This mechanism also is consistent with the apparent lack of a burst of product formation and the absence of enzyme-catalyzed transphosphorylation (54). It generally is accepted that the initial step in catalysis is a rapid binding of the substrate to one of the metal ions in the active site, the divalent one in the case of the PAPs (33, 67) (Fig. 7, which is published as supporting information on the PNAS web site). However, proposals on the subsequent catalytic steps are more diverse. For instance, for both pig and red kidney bean PAP it has been suggested that an Fe(III)-bound hydroxide acts as the initiating nucleophile, attacking the phosphorus atom of the incoming substrate, leading to the release of the product alcohol and the formation of a μ–phosphate complex in the active site (23, 33). Although recent electron-nuclear double resonance measurements of resting pig PAP do not indicate the presence of an Fe(III)-bound solvent molecule (36), the high-resolution structure of this enzyme in the presence of phosphate (1.55 Å) shows that upon formation of a μ–phosphate adduct, the only solvent molecule remaining in the active site is that bridging the two metal ions (24). Because phosphate is both a reaction product and a competitive enzyme inhibitor, it is unclear whether this structure is a good model for a possible reaction intermediate or whether it represents a product-inhibited state. Nonetheless, the fact that the bridging solvent molecule is still present in this structure implies its lack of involvement in catalysis. Similar μ–phosphate complexes also are observed in PAPs from red kidney bean (23) and rat (26), as well as in the Ser/Thr PP calcineurin (68). Furthermore, similar coordination geometries were obtained for tungstate-bound (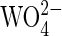 ) complexes of red kidney bean PAP (23) and human PP1 (69).
) complexes of red kidney bean PAP (23) and human PP1 (69).
The FeMn sweet potato PAP is clearly distinct from the above-mentioned complexes. The phosphate anion binds in a tridendate fashion to the two metal ions with one of its oxygen atoms occupying a position equivalent to the solvent bridge observed in pig PAP (Fig. 3). No structure of the resting active site of the sweet potato enzyme is yet available. However, spectroscopic studies indicate that at pH 4.90 the two metal ions are linked via a μ–oxo bridge (28). Hence, the combined spectroscopic and crystallographic data of sweet potato PAP are consistent with a role of the oxygen bridge in catalysis as a nucleophile (Figs. 4 and 7). Based on the equilibrium constants of phosphate binding (see above), an involvement of an amino acid as nucleophile is not likely. A bridging solvent molecule also has been postulated as the initiating nucleophile in λ PP (62) and B. pasteurii urease (37). In the former, sulfate is found to bind to the active site in either a terminal, monodentate or a tridentate arrangement. In the monodentate case a bridging solvent molecule is present, and this molecule is displaced by an oxygen atom of sulfate in the tridentate structure (62). Urease is the only known protein other than sweet potato PAP in which phosphate adopts a tridentate conformation. It is believed that this structure is an analogue for the activated complex in the nucleophilic attack (37). The H-bond interactions between phosphate and active site residues in urease and sweet potato PAP are similar, which may help restrain these two systems in such an unusual coordination geometry.
Fig. 4.

The role of H295 and E365 in the orientation of the substrate and the stabilization of the transition state. At low pH, E365 acts as proton donor for the leaving group; at higher pH, H295 occupies this role. The oxygen atom located between the two metal ions originates from the bridging oxygen, the proposed nucleophile (see Figs. 6 and 7).
Of interest is the observation that the H-bonds between phosphate and active site residues are different in the two subunits of sweet potato PAP (Fig. 6). Notably, O4 (the non-coordinated oxygen atom of phosphate) forms a H-bond with E365 in subunit A, whereas in subunit B it is H-bonded to H295. Although H295 is conserved amongst PAPs (20, 24), there is no residue corresponding to E365 in animal PAPs, and the latter is substituted to Tyr in the red kidney bean enzyme (20). In pig and red kidney bean PAP, H295 has been proposed to act as the proton donor for the leaving group (23, 24, 67). Clearly, a similar role for this residue is possible in sweet potato PAP. From the structural data it is evident that E365 is equally well located to donate its proton to the leaving group, which may be of particular importance at low pH values. Thus, in sweet potato PAP the identity of the proton donor may depend on the pH (Figs. 6 and 7). This hypothesis is consistent with the observation that this enzyme is active over a large pH range (Table 1).
For pig PAP the chemical step (hydrolysis) is proposed to be rate-limiting (54). This hypothesis is consistent with the Brønsted correlation shown in Fig. 1; the negative slope indicates that in the activated complex the oxygen atom of the leaving group has accumulated negative charge, implying bond deformation between this atom and the phosphorus atom before the formation of a bond between phosphorus and the nucleophile. The accumulation of negative charge can be accommodated more easily by leaving groups with lower pKa values. Thus, it is anticipated that an increase in leaving group pKa will result in a reduction of kcat, as observed for pig PAP (Fig. 1). The red kidney bean enzyme is likely to employ a similar mechanism. In contrast, the lack of a dependence of its kinetic parameters on the leaving group pKa at pH 4.90 may indicate that sweet potato PAP has an associative transition state; i.e., bond formation by the nucleophile is more significant than bond breaking to release the leaving group. In this respect the mechanism used by sweet potato PAP resembles that proposed for a binuclear Co(III) complex shown by kinetic isotope effects to use the bridging oxo group as nucleophile (70, 71). It was suggested that the transition state displays a considerable degree of bond formation as evidenced by the modest charge development on the leaving group (70). The spatial proximity of E365 and H295 (Figs. 2, 4, 6, and 7) assists in the optimal orientation of the substrate in the active site. At lower pH values, E365 may act as the proton donor for the leaving group. This hypothesis is consistent with the much faster kcat (Table 1) of sweet potato PAP at low pH compared with the pig enzyme that uses the histidine residue corresponding to H295 as donor (24). The associative transition state is stabilized by means of the effective charge neutralization by the tripodal coordination of the oxyphosphorane. The absence of a Brønsted correlation may be interpreted alternatively in terms of a rate-limiting step that follows the hydrolytic step, i.e., the dissociation of the phosphate product. However, the observations of (i) a large difference in kcat measured for α- and β-naphthyl phosphate (Table 3), (ii) similar Ki values for the competitive inhibition by phosphate for all PAPs studied to date, and (iii) the absence of a Brønsted correlation at pH 3.5 (data not shown) make this explanation seem less likely.
The observation that among PAPs only the sweet potato enzyme hydrolyzes both activated and unactivated esters with similar efficiency (Table 1 and ref. 20) is not only indicative of a more potent nucleophile and an increased stabilization of the transition state but also may reflect an optimal orientation of the substrates in the active site. Here an important role for Y258 (Fig. 2), a residue unique to sweet potato PAP, can be envisaged. Its presence reduces the size of the substrate-binding pocket, and its location at the subunit interface may make it susceptible to conformational change upon substrate binding. Small structural rearrangements can lead to a significant increase in catalytic efficiency of hydrolysis as has been shown for the mitogen-activated phosphatase MKP3, where substrate binding leads to a conformation change and a concomitant ≈35-fold increase in activity (10).
In summary, compared with other PAPs, the sweet potato enzyme has evolved into a highly efficient catalyst. The combined kinetic and structural data support the role of the bridging oxygen as nucleophile (Fig. 7). Furthermore, the presence of Y258 in the substrate binding pocket provides a novel means to optimize the orientation of the substrate in the active site. These structural studies imply that a phosphate oxygen atom of the substrate is wedged between H295 and E365 (Figs. 4, 6, and 7). This feature allows a direct attack by the bridging oxygen on the phosphorus atom, enabling proton donation by E365 (at low pH) or H295 (at higher pH). After hydrolysis and the release of the leaving group, the phosphate remains coordinated to the metal ions in an unusual tridentate geometry. Although the precise mechanism by which the resting form of the active site is regenerated remains unknown, it is probable that two water molecules replace the phosphate (Fig. 7). The Fe(III)-bound water is likely to be deprotonated and thus can attack the divalent metal ion, forming an oxygen bridge. Although speculative, this mechanism for the regeneration of the resting site would result in a five-coordinate Fe(III) site, consistent with the observations of an electron-nuclear double resonance study of pig PAP, which did not find any evidence of a solvent-derived ligand terminally bound to Fe(III) (36). The precise biological function for sweet potato PAP is unknown, but because of its high efficiency and broad range of utilizable substrates, a role in phosphate scavenging is likely. Its unique coordination geometry, combined with its favorable kinetic properties, make this enzyme a suitable system for the design and synthesis of model complexes mimicking phosphorolytic reactions.
Supplementary Material
Acknowledgments
We thank Dr. Harry Tong for assistance at beamline 14BM-C (Advanced Photon Source, Argonne National Laboratory). The use of the BioCARS Sector (Consortium for Advanced Radiation Sources) was supported by the Australian Synchrotron Research Program, which is funded by the Commonwealth of Australia under the Major National Research Facilities Program. Use of the BioCARS Sector 14 also was supported by National Institutes of Health/National Center for Research Resources Grant RR07707. Use of the Advanced Photon Source was supported by U.S. Department of Energy, Basic Energy Sciences, Office of Energy Research Contract W-31-109-Eng-38.
Author contributions: G.S., L.R.G., S.E.H., J.d.J., and L.W.G. designed research; G.S., L.E.C., N.M., M.V., and L.W.G. performed research; G.S., L.R.G., N.M., and L.W.G. analyzed data; and G.S., L.R.G., and L.W.G. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: PAP, purple acid phosphatase; PP, protein phosphatase.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 1XZW).
Footnotes
Patel, K., Lockless, S., Thomas, B. & McKnight, T. (1997) Plant Physiol. III, Suppl., 81 (abstr.).
References
- 1.Dismukes, G. C. (1996) Chem. Rev. 96, 2909-2926. [DOI] [PubMed] [Google Scholar]
- 2.Wilcox, D. E. (1996) Chem. Rev. 96, 2435-2458. [DOI] [PubMed] [Google Scholar]
- 3.Schenk, G., Korsinczky, M. L. J., Hume, D. A., Hamilton, S. & de Jersey, J. (2000) Gene 255, 419-424. [DOI] [PubMed] [Google Scholar]
- 4.Ketcham, C. M., Baumach, G. A., Bazer, F. W. & Roberts, R. M. (1985) J. Biol. Chem. 260, 5768-5776. [PubMed] [Google Scholar]
- 5.Reinholt, F. P., Hultenby, K. H., Oldberg, Å. & Heinegård, D. (1990) Proc. Natl. Acad. Sci. USA 87, 4473-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clarke, S. A., Armbrose, W. W., Anderson, T. R., Terrell, R. S. & Toverud, S. U. (1989) J. Bone Miner. Res. 4, 399-405. [DOI] [PubMed] [Google Scholar]
- 7.Schindelmeiser, J., Munstermann, D. & Witzel, H. (1987) Histochemistry 87, 13-19. [DOI] [PubMed] [Google Scholar]
- 8.Hayman, A. R. & Cox, T. M. (1994) J. Biol. Chem. 269, 1294-1300. [PubMed] [Google Scholar]
- 9.Kaija, H., Alatalo, S. L., Halleen, J. M., Lindqvist, Y., Schneider, G., Väänänen, H. K. & Vihko, P. (2002) Biochem. Biophys. Res. Commun. 292, 128-132. [DOI] [PubMed] [Google Scholar]
- 10.Fjeld, C. C., Rice, A. E., Kim, Y., Gee, K. R. & Denu, J. M. (2000) J. Biol. Chem. 275, 6749-6757. [DOI] [PubMed] [Google Scholar]
- 11.Bozzo, G. G., Raghothama, K. G. & Plaxton, W. C. (2002) Eur. J. Biochem. 269, 6278-6286. [DOI] [PubMed] [Google Scholar]
- 12.del Pozo, J. C., Allona, I., Rubio, V., Leyva, A., de la Pena, A., Aragoncillo, C. & Paz-Ares, J. (1999) Plant J. 19, 579-589. [DOI] [PubMed] [Google Scholar]
- 13.Bozzo, G. G, Raghothama, K. G. & Plaxton, W. C. (2004) Biochem. J. 377, 419-428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klabunde, T. & Krebs, B. (1997) Struct. Bonding 89, 177-198. [Google Scholar]
- 15.Schenk, G., Guddat, L. W., Ge, Y., Carrington, L. E., Hume, D. A., Hamilton, S. & de Jersey, J. (2000) Gene 250, 117-125. [DOI] [PubMed] [Google Scholar]
- 16.Antanaitis, B. C., Strekas, T. & Aisen, P. (1982) J. Biol. Chem. 257, 3766-3770. [PubMed] [Google Scholar]
- 17.Averill, B. A., Davis, J. C., Burman, S., Zirino, T., Sanders-Loehr, J., Loehr, T. M., Sage, T. & Debrunner, P. G. (1987) J. Am. Chem. Soc. 109, 3760-3767. [Google Scholar]
- 18.Yang, Y. S., McCormick, J. M. & Solomon, E. I. (1997) J. Am. Chem. Soc. 119, 11832-11842. [Google Scholar]
- 19.Beck, J. L., McConaghie, L. A., Summors, A. C., Arnold, W. N., de Jersey, J. & Zerner, B. (1986) Biochim. Biophys. Acta 869, 61-68. [Google Scholar]
- 20.Schenk, G. S., Ge, Y., Carrington, L. E., Wynne, C. J., Searle, I. R., Carroll, B. J., Hamilton, S. & de Jersey, J. (1999) Arch. Biochem. Biophys. 370, 183-189. [DOI] [PubMed] [Google Scholar]
- 21.Wang, D. L., Holz, R. C., David, S. S., Que, L., Jr., & Stankovich, M. T. (1991) Biochemistry 30, 8187-8194. [DOI] [PubMed] [Google Scholar]
- 22.Sträter, N., Klabunde, T., Tucker, P., Witzel, H. & Krebs, B. (1995) Science 268, 1489-1492. [DOI] [PubMed] [Google Scholar]
- 23.Klabunde, T., Sträter, N., Fröhlich, R., Witzel, H. & Krebs, B. (1996) J. Mol. Biol. 259, 737-748. [DOI] [PubMed] [Google Scholar]
- 24.Guddat., L. W., McAlpine, A. S., Hume, D., Hamilton, S., de Jersey, J. & Martin, J. L. (1999) Structure (London) 7, 757-767. [DOI] [PubMed] [Google Scholar]
- 25.Lindqvist, Y., Johansson, E., Kaija, H., Vihko, P. & Schneider, G. (1999) J. Mol. Biol. 291, 135-147. [DOI] [PubMed] [Google Scholar]
- 26.Uppenberg, J., Lindqvist, F., Svensson, C., Ek-Rylander, B. & Andersson, G. (1999) J. Mol. Biol. 290, 201-211. [DOI] [PubMed] [Google Scholar]
- 27.Durmus, A., Eicken, C., Sift, B. H., Kratel, A., Kappl, R., Hüttermann, J. & Krebs, B. (1999) Eur. J. Biochem. 260, 709-716. [DOI] [PubMed] [Google Scholar]
- 28.Schenk, G., Boutchard, C. L., Carrington, L. E., Noble, C. J., Moubaraki, B., Murray, K. S., de Jersey, J., Hanson, G. R. & Hamilton, S. (2001) J. Biol. Chem. 276, 19084-19088. [DOI] [PubMed] [Google Scholar]
- 29.Beck, J. L., Keough, D. T., de Jersey, J. & Zerner, B. (1984) Biochim. Biophys. Acta 791, 357-363. [DOI] [PubMed] [Google Scholar]
- 30.Beck, J. L., McArthur, M. J., de Jersey, J. & Zerner, B. (1988) Inorg. Chim. Acta 153, 39-44. [Google Scholar]
- 31.Merkx, M. & Averill, B. A. (1998) Biochemistry 37, 8490-8497. [DOI] [PubMed] [Google Scholar]
- 32.Twitchett, M. B. & Sykes, A. G. (1999) Eur. J. Inorg. Chem., 2105-2115.
- 33.Aquino, M. A. S., Lim, J. S. & Sykes, A. G. (1994) J. Chem. Soc. Dalton Trans., 429-436.
- 34.Merkx, M. & Averill, B. A. (1999) J. Am. Chem. Soc. 121, 6683-6689. [Google Scholar]
- 35.Wang, X., Randall, C. R., True, A. E. & Que, L., Jr. (1996) Biochemistry 35, 13946-13954. [DOI] [PubMed] [Google Scholar]
- 36.Smoukov, S. K., Quaroni, L., Wang, X., Doan, P. E., Hoffman, B. M. & Que, L., Jr. (2002) J. Am. Chem. Soc. 124, 2595-2603. [DOI] [PubMed] [Google Scholar]
- 37.Benini, S., Rypniewski, W. R., Wilson, K. S., Ciurli, S. & Mangani, S. (2001) J. Biol. Inorg. Chem. 6, 778-790. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, Z. Y. & van Etten, R. L. (1991) Biochemistry 30, 8954-8959. [DOI] [PubMed] [Google Scholar]
- 39.Campbell, H. D., Dionysius, D. A., Keough, D. T., Wilson, B. E., de Jersey, J. & Zerner, B. (1978) Biochem. Biophys. Res. Commun. 82, 615-620. [DOI] [PubMed] [Google Scholar]
- 40.Schenk, G., Carrington, L. E., Hamilton, S. E., de Jersey, J. & Guddat, L.W. (1999) Acta Crystallogr. D 55, 2051-2052. [DOI] [PubMed] [Google Scholar]
- 41.Otwinowski, Z. & Minor, W. (1997) Methods Enzymol. 276, 307-326. [DOI] [PubMed] [Google Scholar]
- 42.Brünger, A. T. (1992) x-plor: A System for X-ray Crystallography and NMR (Yale Univ. Press, New Haven, CT).
- 43.Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991) Acta Crystallogr. A 47, 110-119. [DOI] [PubMed] [Google Scholar]
- 44.Brünger, A. T., Adams, P. D., Clore, G. M., Delano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., et al. (1998) Acta Crystallogr. D 54, 905-921. [DOI] [PubMed] [Google Scholar]
- 45.Laskowski, R. A. (1993) J. Appl. Crystallogr. 26, 283-291. [Google Scholar]
- 46.Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946-950. [Google Scholar]
- 47.Merritt, E. A. & Bacon, D. J. (1997) Methods Enzymol. 277, 505-524. [DOI] [PubMed] [Google Scholar]
- 48.Evans, S. V. (1993) J. Mol. Graphics 11, 134-138. [DOI] [PubMed] [Google Scholar]
- 49.Merkx, M., Pinkse, M. W. H. & Averill, B. A. (1999) Biochemistry 38, 9914-9923. [DOI] [PubMed] [Google Scholar]
- 50.Valizadeh, M., Schenk, G., Nash, K., Oddie, G. W., Guddat, L. W., Hume, D. A., de Jersey, J., Burke, T. R., Jr., & Hamilton, S. (2004) Arch. Biochem. Biophys. 424, 154-162. [DOI] [PubMed] [Google Scholar]
- 51.Davis, J. C. & Averill, B. A. (1982) Proc. Natl. Acad. Sci. USA 79, 4623-4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LeBansky, B. R., McKnight, T. D. & Griffing, L. R. (1992) Plant Physiol. 99, 391-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keough, D. T., Beck, J. L., de Jersey, J. & Zerner, B. (1982) Biochem. Biophys. Res. Commun. 108, 1643-1648. [DOI] [PubMed] [Google Scholar]
- 54.Wynne, C. J., Hamilton, S. E., Dionysius, D. A., Beck, J. L. & de Jersey, J. (1995) Arch. Biochem. Biophys. 319, 133-141. [DOI] [PubMed] [Google Scholar]
- 55.O'Brian, P. J. & Herschlag, D. (2002) Biochemistry 41, 3207-3225. [DOI] [PubMed] [Google Scholar]
- 56.Humphry, T., Forconi, M., Williams, N. H. & Hengge, A. C. (2002) J. Am. Chem. Soc. 114, 14860-14861. [DOI] [PubMed] [Google Scholar]
- 57.Forconi, M. & Williams, N. H. (2002) Angew. Chem. Int. Ed. 41, 849-852. [DOI] [PubMed] [Google Scholar]
- 58.Katajima, N., Hikichi, S., Tanaka, M. & Moro-oka, Y. (1993) J. Am. Chem. Soc. 115, 5496-5508. [Google Scholar]
- 59.Mao, Z. W., Heinemann, F. W., Liehr, G. & van Eldik, R. (2001) J. Chem. Soc. Dalton Trans., 3652-3662.
- 60.Adams, H., Bradshaw, D. & Fenton, D. E. (2001) J. Chem. Soc. Dalton Trans., 3407-3409.
- 61.Youngme, S., Chaichit, N., Kongsaeree, P., van Albada G. A. & Reedijk, J. (2001) Inorg. Chim. Acta 324, 232-240. [Google Scholar]
- 62.Voegtli, W. C., White, D. J., Reiter, N. J., Rusnak, F. & Rosenzweig, A. C. (2000) Biochemistry 39, 15365-15374. [DOI] [PubMed] [Google Scholar]
- 63.Mueller, E. G., Crowder, M. A., Averill, B. A. & Knowles, J. R. (1993) J. Am. Chem. Soc. 115, 2974-2975. [Google Scholar]
- 64.Martin, B. L. & Graves, D. J. (1986) J. Biol. Chem. 261, 14545-14550. [PubMed] [Google Scholar]
- 65.Martin, B. L. & Graves, D. J. (1994) Biochim. Biophys. Acta 1206, 136-142. [DOI] [PubMed] [Google Scholar]
- 66.Mertz, P., Yu, L., Sikkink, R. & Rusnak, F. (1997) J. Biol. Chem. 272, 21296-21302. [DOI] [PubMed] [Google Scholar]
- 67.Twitchett, M. B., Schenk, G., Aquino, M. A. S., Yiu, D. T. Y., Lau, T. C. & Sykes, A. G. (2002) Inorg. Chem. 41, 5787-5794. [DOI] [PubMed] [Google Scholar]
- 68.Griffith, J. P., Kim, J. L., Kim, E. E., Sintchak, M. D., Thomson, J. A., Fitzgibbon, M. J., Fleming, M. A., Caron, P. R., Hsiao, K. & Navia, M. A. (1995) Cell 82, 507-522. [DOI] [PubMed] [Google Scholar]
- 69.Egloff, M. P., Cohen, P. T. W., Reinemer, P. & Barford, D. (1995) J. Mol. Biol. 254, 942-959. [DOI] [PubMed] [Google Scholar]
- 70.Williams, N. H., Lebuis, A. M. & Chin, J. (1999) J. Am. Chem. Soc. 121, 3341-3348. [Google Scholar]
- 71.Humphry, T., Forconi, M., Williams, N. H. & Hengge, A. C. (2004) J. Am. Chem. Soc. 126, 11864-11869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.