

Abstract
Reverse cholesterol transport (transfer of macrophage-cholesterol in the subendothelial space of the arterial wall to the liver) is terminated by selective high density lipoprotein (HDL)-cholesteryl ester (CE) uptake, mediated by scavenger receptor class B, type 1 (SR-B1). We tested the validity of two models for this process: “gobbling,” i.e. one-step transfer of all HDL-CE to the cell and “nibbling,” multiple successive cycles of SR-B1-HDL association during which a few CEs transfer to the cell. Concurrently, we compared cellular uptake of apoAI with that of apoAII, which is more lipophilic than apoAI, using HDL-[3H]CE labeled with [125I]apoAI or [125I]apoAII. The studies were conducted in CHO-K1 and CHO-ldlA7 cells (LDLR−/−) with (CHO-SR-B1) and without SR-B1 overexpression and in human Huh7 hepatocytes. Relative to CE, both apoAI and apoAII were excluded from uptake by all cells. However, apoAII was more highly excluded from uptake (2–4×) than apoAI. To distinguish gobbling versus nibbling mechanisms, media from incubations of HDL with CHO-SR-B1 cells were analyzed by non-denaturing PAGE, size-exclusion chromatography, and the distribution of apoAI, apoAII, cholesterol, and phospholipid among HDL species as a function of incubation time. HDL size gradually decreased, i.e. nibbling, with the concurrent release of lipid-free apoAI; apoAII was retained in an HDL remnant. Our data support an SR-B1 nibbling mechanism that is similar to that of streptococcal serum opacity factor, which also selectively removes CE and releases apoAI, leaving an apoAII-rich remnant.
Keywords: apolipoprotein, atherosclerosis, cholesterol, high-density lipoprotein (HDL), lipid, scavenger receptor, apolipoprotein AII, reverse cholesterol transport, scavenger receptor B1, selective lipid uptake
Introduction
Reverse cholesterol transport (RCT)3 is the putative mechanism that relieves the cholesterol burden within the arterial wall. RCT begins with free cholesterol (FC) efflux via macrophage ATP-binding cassette transporter A1 (ABCA1) in the subendothelial space of the arterial wall to apoAI and early forms of high density lipoprotein (HDL). The FC in the HDL so-formed is esterified to cholesteryl ester (CE) by plasma lecithin:cholesterol acyltransferase, giving a mature HDL from which the CE is extracted by the terminal RCT step, selective uptake via the HDL receptor, hepatic SR-B1. Although high plasma levels of HDL cholesterol correlate with lower risk for cardiovascular disease, the relative importance of SR-B1 and RCT in atheroprotection has been debated (1–4). According to a metabolic study of radiolabeled FC and CE in humans, very-low-density (VLDL), intermediate-density (IDL), and low-density (LDL) lipoproteins contribute to net CE hepatocyte disposal, whereas little is derived from HDL, suggesting that selective HDL-CE uptake is a minor pathway that contributes little to RCT in humans (5). Like all such studies, there were no direct measures of metabolism, and the conclusions were model-dependent.
The strongest early evidence supporting an important role for SR-B1 in RCT came from gain and loss of SR-B1 function studies in mice. Despite having low plasma HDL-C levels, SR-B1-overexpressing mice were athero-resistant (6–8); the converse was found in mice with SR-B1 deficiency (9–12). The importance of SR-B1 in human RCT is further supported by recent genetic studies. A homozygote for a loss-of-function SR-B1 variant (P376L) was identified by analysis of lipid-modifying genes in individuals with very high plasma HDL-cholesterol levels. The P376L variant in transfected cells and in mice exhibits impaired selective HDL-CE uptake, and human heterozygous P376L carriers have increased plasma HDL-cholesterol levels and have a distinct HDL phenotype and increased risk of coronary heart disease (13). Thus, the prominence of SR-B1 in HDL-determined atherogenesis has grown.
The HDL receptor SR-B1 mediates selective CE uptake from lipoproteins into liver and steroidogenic tissues and cholesterol efflux from macrophages to HDL (14). Selective hepatic uptake of HDL-CE is a key and likely most important step in reverse cholesterol transport because it is required for the eventual transfer of cholesterol to the bile and then to the feces for disposal. The activity of SR-B1 versus HDL is distinct from that of the low density lipoprotein receptor (LDLR). In the latter, holo particle uptake is succeeded by steps that include lysosomal breakdown of apoB-100, CE hydrolysis, and metabolism or recycling of cholesterol and the LDLR. HDL-CE uptake occurs without whole particle uptake or the concomitant uptake of the major HDL protein, apoAI (15). The relative SR-B1-mediated selective uptakes of HDL-CE, free cholesterol, triglycerides, and phosphatidylcholine, are 1, 1.6, 0.7, and 0.2, respectively, suggesting the formation of a nonpolar channel between SR-B1 and the HDL particle that better solubilizes the more neutral lipids (15, 16).
Here we address two mechanistic questions about SR-B1-mediated selective extraction of lipids from HDL. First, given the order of lipid exclusion from SR-B1-mediated uptake, which increases from least lipophilic to most lipophilic, is the rate of apoAII uptake greater than that of apoAI? According to multiple criteria, apoAII is more lipophilic than apoAI: apoAI but not apoAII is shed by HDL during centrifugation (17), physicochemical perturbation displaces apoAI but not apoAII (18), and apoAII displaces apoAI from HDL (19). Many proteins that modify HDL structure do so with the concomitant release of lipid-free apoAI but not apoAII (20–24). Given its greater lipophilicity versus apoAI, we hypothesized that like CE, the most lipophilic of the lipids, a greater fraction of apoAII, the most lipophilic apo, would partition into cells via SR-B1. Second, does SR-B1 selectively remove a few CEs from many HDLs (nibbling) or many if not all CEs from a few HDLs (gobbling)? Herein, we address these two questions using complementary experimental approaches.
Results
Kinetics of cellular HDL uptake according to the components of HDL
The rate of uptake of the radiolabeled CE and apolipoprotein components of HDL were compared in the three Chinese hamster ovary (CHO) cell types and in Huh7 hepatocytes. The CHO-SR-B1 cell line was derived by transfection of the CHO-ldlA7 line to overexpress SR-B1 in the absence of LDL receptor (14, 25). The LDL receptor in the CHO-ldlA7 line was shown to be non-functional. As a control, the wild-type CHO-K1 cell line was also used. Human hepatocyte Huh7 cells were used, as these are more physiologically relevant and express high amounts of endogenous SR-B1 as well as LDLR (26, 27). These data show that the rates of CE and apolipoprotein uptake were linear over 180 min and differed within and between cell types (Fig. 1; Table 1). As reported by others (14, 28), the cellular uptake for [3H]CE was much faster than that of [125I]apoAI, an effect that was most profound for the CHO-SR-B1 cells (Fig. 1; Table 1). However, when the two apolipoproteins were compared, [125I]apoAI was taken up by all four cell types at rates that were faster than those for [125I]apoAII. The differences in the rates of CE and apolipoprotein uptake were more apparent when the rates of apoAI and apoAII uptake versus CE uptake were compared (Fig. 2). These data reveal similarities and differences. First, the rate of CE uptake correlated linearly with those of apoAI and apoAII. Second, based on the slopes of the curves, apoAI uptake is greater than that of apoAII uptake, i.e. molar ratios of 2.1, 2.3, 3.6, and 1.8 times that of apoAII uptake for CHO K1, CHO ldlA7, CHO-SR-B1, and Huh7, respectively. Third, as expected, the rate of CE versus apolipoprotein uptake is greatest for CHO-SR-B1 cells for which the observed uptake occurred in molar ratios of 750 CE/apoAI and 2700 CE/apoAII. There was also significant selective uptake in the Huh7 hepatocytes, i.e. 274 CE/apoAI and 535 CE/apoAII as well as in the CHO cell lines with only endogenous SR-B1, 150–175 CE/apoAI, and 320–420 CE/apoAII.
Figure 1.
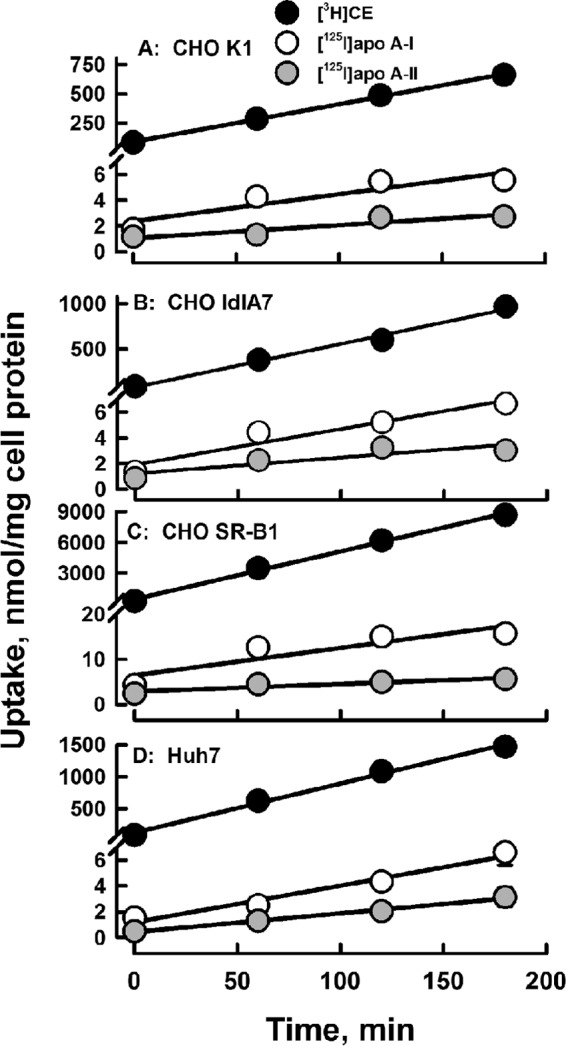
Kinetics of apolipoprotein and CE uptake are linear with rates increasing as HDL-[3H]CE ≫ HDL-[125I]apoAI > HDL-[125I]apoAII in all four cell lines as labeled. Data points are the mean ± S.D. for n = 6 for CE and n = 3 for apolipoprotein uptake. The error bars are not visible because they are smaller than the symbols.
Table 1.
Rate of HDL-analyte uptake by various cell types
Rates were calculated from the slopes of the curves in Fig. 1.
| Analyte/cell type | HDL analyte uptake |
|||
|---|---|---|---|---|
| CHO K1 | CHO ldlA7 | CHO SR-B1 | Huh7 | |
| nmol/min/mg cell protein | ||||
| CE | 3.20 ± 0.08 | 4.8 ± 0.3 | 46.7 ± 2.0 | 7.7 ± 0.4 |
| ApoAI | 0.021 ± 0.006 | 0.028 ± 0.005 | 0.06 ± 0.02 | 0.028 ± 0.004 |
| ApoAII | 0.010 ± 0.003 | 0.012 ± 0.004 | 0.017 ± 0.004 | 0.014 ± 0.001 |
Figure 2.
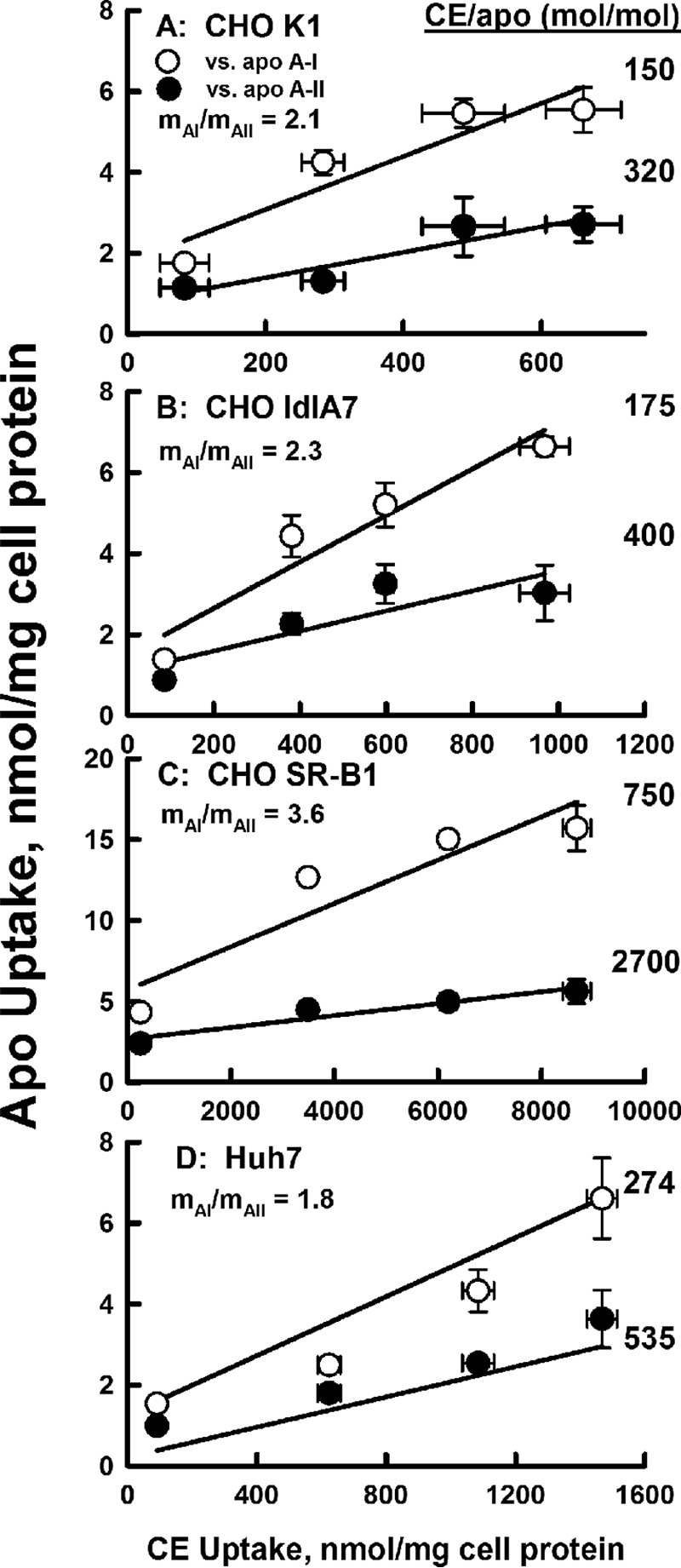
Selectivity of uptake. ApoAI (white symbols) and apoAII (black symbols) uptake is plotted as a function of CE uptake from the kinetic response data of Fig. 1. The molar ratio of CE to apolipoprotein uptake is calculated as the inverse of the slope of the linear fit to the data points. The relative uptake of apoAI to apoAII (mAI/mAII) is the ratio of the two slopes for a given cell type. The data shown are representative of two independent experiments, each done in triplicate.
The time course studies were corroborated by kinetics studies of CE and apolipoprotein uptake as a function of HDL concentrations (Fig. 3) from which a Vmax and a catalytic efficiency (E = Vmax/Km) were calculated; kinetic constants are given in Table 2. As with the rate data of Fig. 1, these data show that according to Vmax and catalytic efficiency, CE uptake is greatest for CHO-SR-B1 cells and that these same constants decrease in the order CE ⋙ apoAI > apoAII. The rates of uptake of all three HDL analytes from Fig. 1 are compared in Fig. 4A, where the data on the ordinate are plotted as a log function so that apoAI and apoAII kinetics could be compared on the same scale as CE. The Vmax and catalytic efficiencies were also compared (Fig. 4, B and C); again, the ordinate of the latter is plotted on a log scale. Above each bar in Fig. 4C we inserted the catalytic efficiency for each analyte with CE uptake normalized to 100. These data confirm the high selective lipid uptake by CHO-SR-B1 cells, with the exclusion of apoAI, and even more profoundly, of apoAII.
Figure 3.
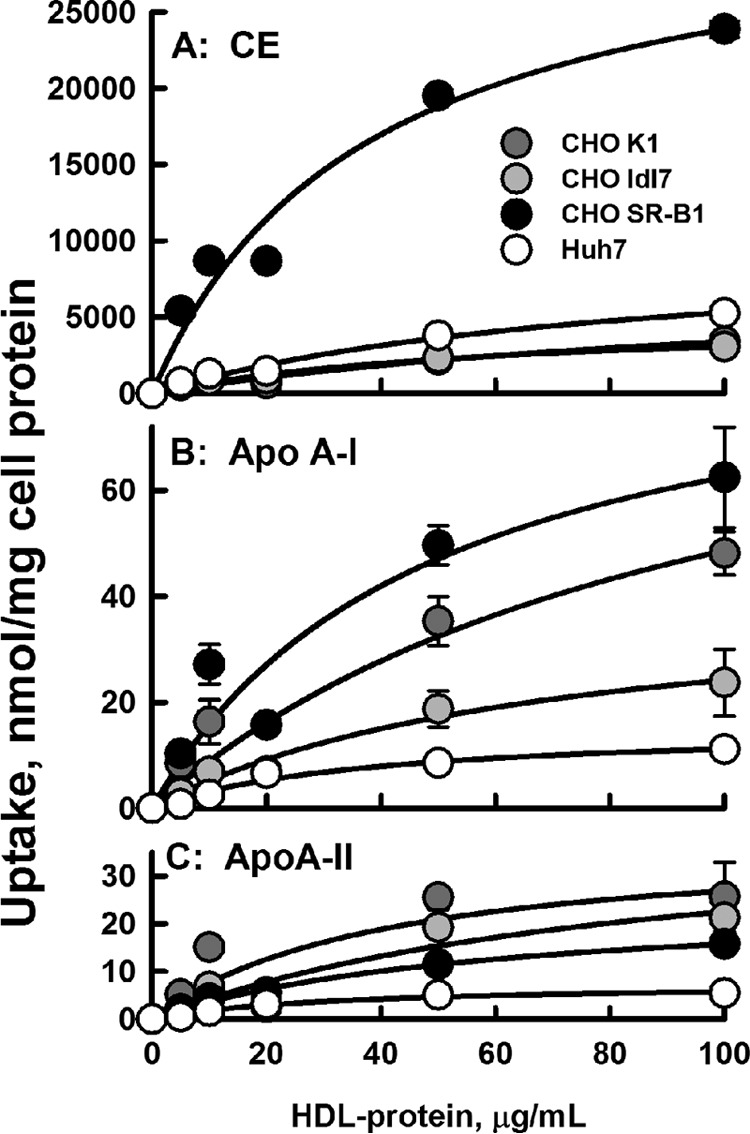
Dose-response kinetics of uptake. A, HDL-[3H]CE. B, HDL-[125I]apoAI. C, HDL- [125I]apoAII. Dose-response data were fitted to a hyperbolic function, i.e. y = ax/(b + x), where x is HDL-protein concentration, y is the radiolabel uptake, a is the maximum uptake (Vmax), and b is Km, the HDL-protein concentration at 50% of Vmax. The catalytic efficiency E = (Vmax/Km). Data points are the mean ± S.D. for n = 6 for CE and n = 3 for apolipoprotein uptake. All data points have error bars, but some are not visible because they are smaller than the symbols.
Table 2.
Kinetic constants of HDL-analyte uptake by various cell types
Kinetic constants ± S.E. were calculated from the fit of the dose-response curves in Fig. 3 to a two-parameter hyperbolic function, i.e., y = ax/(b + x), where x is HDL protein concentration, y is the radiolabel uptake, a is the maximum uptake (Vmax), and b is Km, the HDL-protein concentration at 50% of Vmax; catalytic efficiency (E) = (Vmax/ Km).
| Cell type | HDL analyte | Vmax | Km | Vmax/Km |
|---|---|---|---|---|
| nmol/mg cell protein | μg HDL-protein/ ml | |||
| CHO K1 | HDL-[3H]CE | 7,740 ± 3,500 | 127 ± 91 | 60.70 |
| HDL-[125I]apoAI | 97 ± 51 | 99 ± 89 | 0.98 | |
| HDL-[125I]apoAII | 38 ± 19 | 40 ± 45 | 0.94 | |
| CHO ldlA7 | HDL- [3H]CE | 4,900 ± 780 | 59 ± 19 | 82.70 |
| HDL-[125I]apoAI | 41 ± 9 | 68 ± 28 | 0.60 | |
| HDL-[125I]apoAII | 43 ± 25 | 90 ± 91 | 0.48 | |
| CHO SR-B1 | HDL- [3H]CE | 32,800 ± 4,800 | 37 ± 13 | 882.00 |
| HDL-[125I]apoAI | 93 ± 27 | 48 ± 31 | 1.92 | |
| HDL-[125I]apoAII | 25 ± 2 | 60 ± 10 | 0.42 | |
| Huh7 | HDL- [3H]CE | 9,730 ± 1,750 | 84 ± 27 | 116.00 |
| HDL-[125I]apoAI | 15 ± 2 | 38 ± 14 | 0.41 | |
| HDL-[125I]apoAII | 7.6 ± 0.9 | 32 ± 9 | 0.24 |
Figure 4.
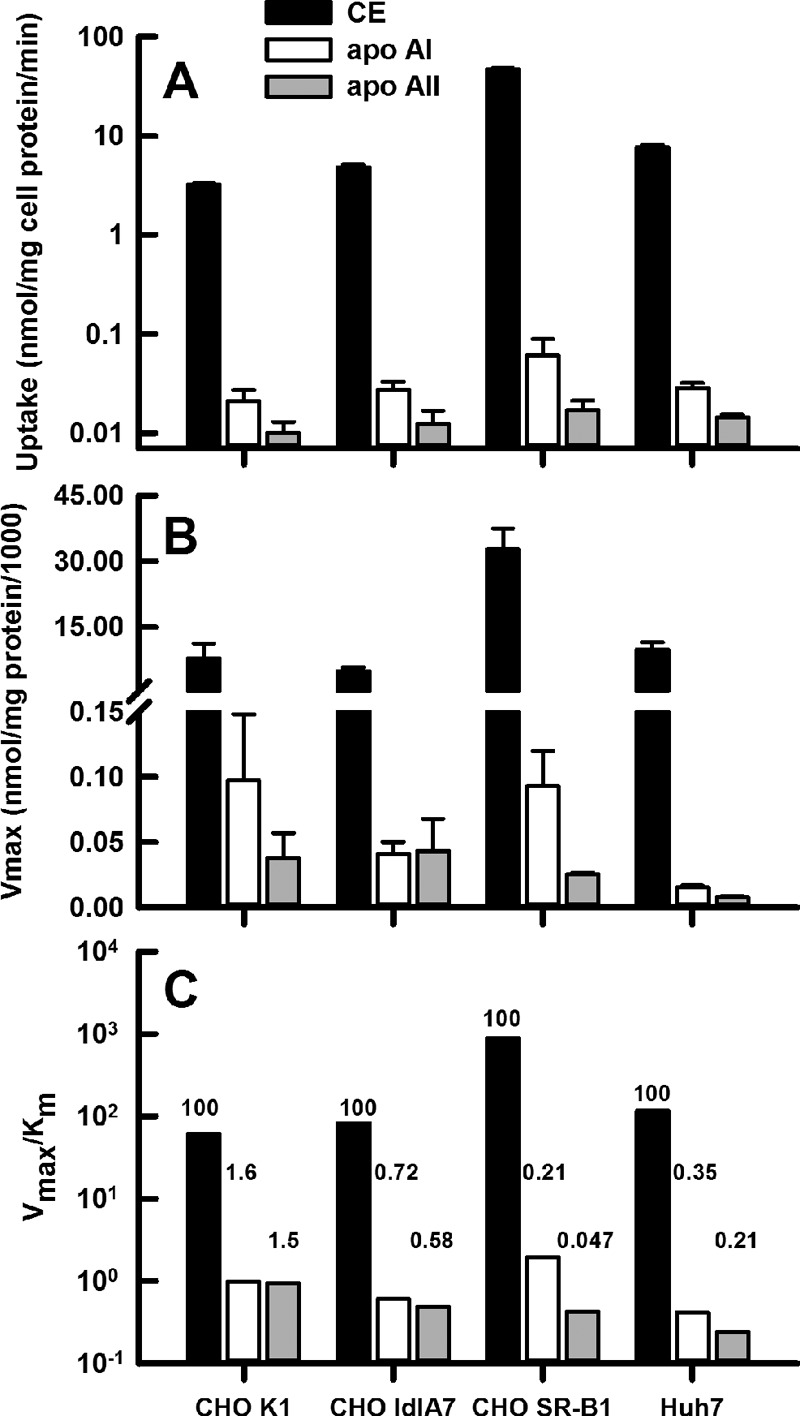
Kinetic constants for cell uptake of HDL CE (black bars), apoAI (white bars), and apoAII (gray bars). Cell lines are indicated on the x axis. A, rates of uptake, from time course data of Fig. 1. B, Vmax from dose-response data of Fig. 3. C, catalytic efficiency, Vmax/Km, from dose-response data of Fig. 3. Comparison of the molar efficiency of uptake for CE versus apoAI and apoAII are indicated by the numbers above the bars, with efficiency of CE uptake = 100 and the relative apoAI and apoAII uptake as shown for each cell line.
SR-B1 reduces HDL size while forming remnants and lipid-free apoAI
To test our two mechanistic models for selective uptake, HDL was incubated with CHO-SR-B1 cells for various times; aliquots of concentrated media were analyzed by size-exclusion chromatography (SEC) during which fractions were collected for analysis. The changes in the SEC profile as a function of time of incubation with CHO-SR-B1 cells were determined according to the elution of protein (absorbance280 nm) and apoAI and apoAII according to immunoblotting; cholesterol and phospholipid concentrations were determined by enzyme-based assays. HDL, which was purified by flotation followed by preparative SEC, eluted as a single symmetrical peak (Fig. 5A, 0 h). During incubation with CHO-SR-B1 cells, the main HDL peak shifted to later elution volumes (EVs), which corresponds to a time-dependent reduction in particle size. This change in molecular weight corresponds to a loss of 8 CE/HDL particle/h. The magnitude of the new peak, which corresponds to a smaller particle, increased with incubation time. The incremental loss of large HDL and appearance of a new protein-containing species is apparent in the difference between the SEC profiles at 1 and 6 h (Fig. 5A, bottom dashed curve). These data were corroborated by analysis of the fractions from SEC by immunoblotting for apoAI (Fig. 5B). At 4 h the apoAI distribution curve was a poorly resolved bimodal distribution of protein. At 6 h the peak SEC fraction was shifted by 1 ml, and the distribution showed an asymmetric trailing profile that extends to 33–34 ml, which is the EV for lipid-free apoAI. Smaller shifts were observed in the SEC profiles for cholesterol and phospholipid (Fig. 5, C and D). The total cholesterol eluting from the SEC columns declined over time, consistent with cellular CE uptake (Fig. 5C, top inset), whereas total phospholipid was little changed (Fig. 5D, top inset).
Figure 5.

Change in SEC profile of various HDL analytes in culture media over time during incubation of HDL with CHO-SR-B1 cells. Media were concentrated and passed over Superose HR6 columns, and the absorbance (280 nm) was measured (A), and fractions were collected and analyzed for apoAI by immunoblotting (B), total cholesterol (C), and phospholipid (D). Time of incubation is indicated on each chromatogram. TC, total cholesterol (sum of CE + FC). The plot at the bottom of panel A is the difference in the SEC absorbance profiles at t = 1 and 6 h; arrows denote the EVs of BSA and apoAI. The inset in A shows the shift in the peak EV over time. Immunoblots in panel B are overlaid on the plots of their band intensities in the same top-to-bottom order. Dashed vertical gray lines denote prominent peaks at initial t = 0 or 6 h. Insets at the tops of C and D are the changes in the total media cholesterol and phospholipid over time. Dashed vertical lines are to facilitate comparison of curves.
To better compare the changes in media HDL-apoAI, cholesterol, and phospholipid, we analyzed the SEC data according to the percent of the total analyte in each fraction, measured as its EV, over the 6-h incubation time. For apoAI, these data (Fig. 6A) reveal that the percent of the total apoAI in the early eluting HDL fractions (EV = 29 and 30) decreased over the 6-h incubation time, whereas the percent in the later eluting fractions (EV = 32 and 33) increased over the same time interval; there was little change in the apoAI concentrations in the intermediate fraction (EV = 31). Cholesterol distribution among SEC fractions showed that the % total cholesterol in the earliest eluting peak (EV = 29) declined as well, but those of the two successive fractions increased, whereas those of the final two fractions were virtually unchanged over 6 h (Fig. 6B). As the majority of cholesterol in HDL is CE (75–80% of the total cholesterol by weight 29), the total cholesterol values are a good indicator of the trend in CE content of the remnant HDL. The major change in the distribution of phospholipid was its reduction in the earliest-eluting fraction (EV = 29); there was no change in the next fraction (EV = 30), and the successive fractions showed modest to little increases (EV = 31–33). In contrast to the significant shift in apoAI distribution to later-eluting fractions over time, the distribution of apoAII among SEC fractions did not change significantly over 6 h (supplemental Fig. 1).
Figure 6.

HDL-analyte changes in SEC fractions over time. Data from the SEC chromatogram profiles in Fig. 5 are plotted as percent of total analyte/SEC fraction, to quantitate the amount of redistribution of each analyte across the SEC fractions. Shown are apoAI (A), total cholesterol (B), and phospholipid (C) concentrations. Symbol shade indicates that the amount of analyte in the respective fraction decreased with time (black circles), increased with time (white circles), or remained ∼neutral with time (gray circles). SEC fractions are indicated by their respective EVs, 29 to 33 ml, as indicated for each plot. Slopes m and standard errors were calculated from a linear fit to the data.
The kinetics of HDL remodeling by CHO-SR-B1 cells were also evaluated by non-denaturing PAGE followed by immunoblot analysis for apoAI and apoAII (Fig. 7). The non-denaturing gel images (Fig. 7A) and scans of the gels (Fig. 7B) reveal changes in the relative immunoblot signal intensity for gel bands as a function of time. These changes are similar to those reported for in vivo remnant HDL formation in apoAI−/− mice expressing high hepatic SR-B1 levels (30). According to the slopes of the curves of immuno-signal intensity versus time (Fig. 7C), apoAI disappeared from the main HDL bands (Bands 1 and 2, negative slopes) but appeared in remnant HDL and lipid-free apoAI bands (positive slopes). Purified lipid-free apoAI migrated at the position of Band 5 on the non-denaturing gels (Fig. 7A, Lane S). The slopes for Bands 1 and 2, which correspond to HDL, have negative slopes, i.e. loss of apoAI mass, whereas Bands 4 and 5, which are generated during remnant HDL formation, have positive slopes, i.e. gain of apoAI mass. According to similar analyses of apoAII, most associated with Bands 1, 2, and 3, and the gel scans and slopes of band intensity versus time show that its distribution did not change over the 6-h incubation. Unlike apoAI, apoAII distribution was not profoundly shifted toward Bands 4 and 5 (Fig. 7C).
Figure 7.
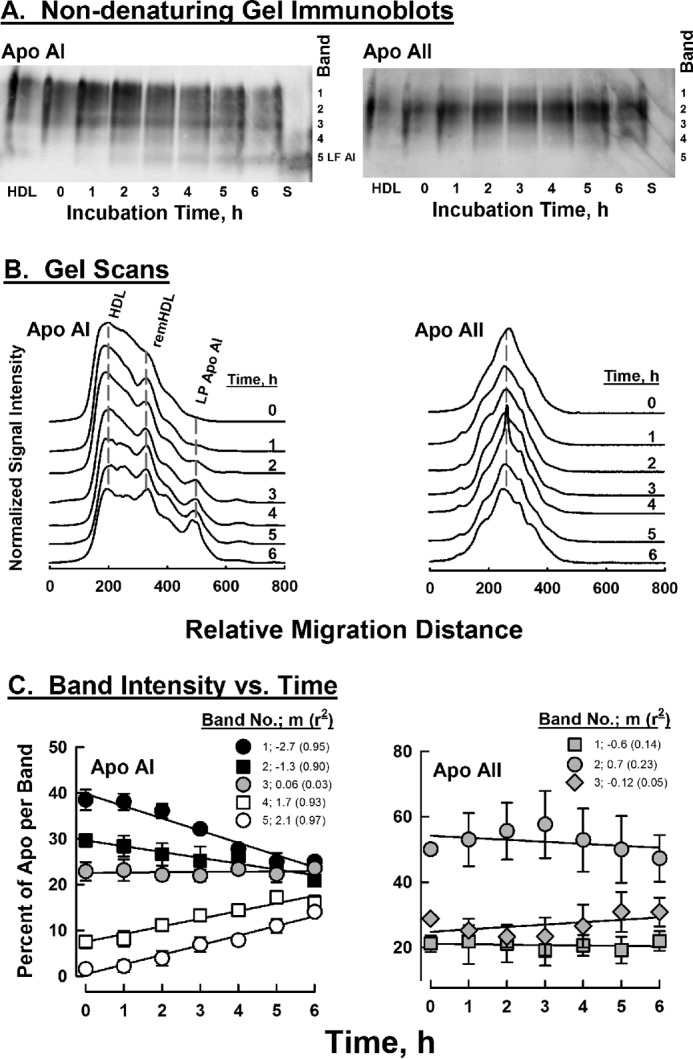
Size distributions of apoAI- and AII-associated HDL as a function of time of incubation of HDL with CHO-SR-B1 cells according to immunoblot intensities of non-denaturing PAGE. Left and right panels, respectively, contain data for apolipoproteins AI and AII. A, images of the non-denaturing gels according to immunoblot intensities; the right ordinate denotes the locations of bands that were quantified by image analysis. Lanes HDL and S contain starting HDL and purified lipid-free apoAI, respectively. B, scans of immunoblots. The incubation time for each sample is indicated on the respective scan. C, apolipoprotein distribution among non-denaturing gel bands as a function of time. Negative and positive slopes (m) correspond to the respective losses and gains of apolipoproteins over time; r2 = correlation coefficient. ApoAI shows a significant redistribution, whereas apoAII does not.
Discussion
Profound exclusion of apoAII from cellular uptake
Our data confirm the data of others (14, 28) showing that HDL-CE uptake far exceeds that of apoAI and that the stoichiometry is little affected by LDLR deletion (Figs. 1–4; Tables 1 and 2). We observed that HDL-CE uptake by CHO-SR-B1 cells is ∼15-, ∼10-, and ∼6-fold greater than wild-type CHO-K1 cells, LDLR-deficient CHO-ldlA7 cells, and Huh7 cells, respectively. The relatively higher amount of HDL-CE uptake by Huh7 cells may due to expression of SR-B1 at amounts ∼20% of CHO-SR-B1 cells (27). Among all four cell types, HDL-CE uptake greatly exceeded that of apoAI. However, apoAI uptake, although small compared with CE uptake, was consistently greater than that of apoAII, the opposite of our hypothesis in the Introduction. Thus, the exclusion of apolipoproteins AI and AII does not follow the rule of lipids in which more CE, the most lipophilic lipid, is transferred to the cell than phospholipid or triglyceride. This may be due to the much greater phospholipophilicity of the apoAII compared with apoAI; e.g. apoAI but not apoAII is discharged from HDL by chaotropes, heat, and prolonged ultracentrifugation (17, 18, 31). Moreover, the free energy of activation for the transfer of apoAI from HDL to water is ∼95 kJ (24, 31, 32). In contrast, the transfer of apoAII from lipid surfaces to water is not readily measurable (33). Alternatively, because the molar ratio of AI to AII in HDL is ∼ 2/1, an uptake ratio of 2/1 may reflect a small amount of HDL holoparticle uptake (34) that occurs concurrently with the specific lipid uptake that excludes protein. Still, the apoAI/apoAII uptake ratio in CHO-SR-B1 cells was greater, 3.6, suggesting a more profound exclusion of apoAII versus apoAI by SR-B1. These differences in apoAI and AII provoked studies to determine whether HDL apolipoproteins and lipids have distinct extracellular SR-B1-mediated itineraries.
SR-B1 segregates HDL-proteins and lipids
Although apoAI and apoAII belong to the same gene family (35), they have different physicochemical properties. Many physicochemical and biological modulators of HDL structure readily release apoAI, whereas apoAII remains bound to HDL. Our SEC data and our analyses (Figs. 5–7 and supplemental Fig. S1) support the hypothesis that SR-B1 is another physicochemical modulator of HDL structure that segregates the processing of apoAI and apoAII as well as those of cholesterol and phospholipid. The differences are most apparent in Fig. 6; during incubation of HDL with CHO-SR-B1 cells, apoAI transfers from early SEC fractions corresponding to native HDL to later fractions corresponding to the EV of HDL remnants (Fig. 5, A and B; EV ∼ 31–32 ml) and lipid-free apoAI (EV ∼ 33 ml). The redistribution of apoAI from larger HDLs to smaller remnant HDLs and release as lipid-poor apoAI is confirmed by the non-denaturing gel immunoblots (Fig. 7). In contrast, apoAII was not detected in the lipid-poor apolipoprotein region but remained primarily associated with mid-size HDLs (EV ∼ 30–31, supplemental Fig. S1 and Band 2 in Fig. 7). Due to its greater phospholipophilicity, more apoAII than apoAI remains with the mid-size HDL peak, which also retains most of its cholesterol and phospholipid throughout the 6-h incubation.
HDL-cholesterol and -phospholipid processing by SR-B1 are distinct from those of apoAI and apoAII and from each other. As expected, over the 6-h incubation with CHO-SR-B1 cells, much more cholesterol than phospholipid is transferred from the media to the cells (Fig. 5, C and D, upper curves); this confirms the findings of others (15). The SR-B1-mediated itineraries of cholesterol and phospholipid are also distinct. Although the early fraction (EV = 29) lost both lipids, the percent of the total cholesterol increased in the middle fractions (EV 30, 31) but not in the later fractions, i.e. the smaller HDL remnant fraction (EV 32) and the lipid-poor fraction (EV 33). The distribution of phospholipid shifts further to the smaller HDL (Fig. 5D) and the percent of total phospholipid increased in the remnant HDL fractions EV = 31 and 32 but not in the lipid-poor EV = 33 (Fig. 6C). Thus, compared with cholesterol, the smaller remnant particles are phospholipid-enriched.
SR-B1, a gobbler or nibbler?
We then addressed the question, Does SR-B1 extract all CE from a few HDLs, i.e. gobbling, or a few CE from many HDL, i.e. nibbling? If a gobbling mechanism were operative, one would expect a bimodal distribution of HDL in which over time HDL remains the same size but declines in abundance, whereas a new peak for the remnant would appear and grow. In contrast, the nibbling mechanism would show the gradual appearance of a remnant peak, whereas the size of the main HDL peak gradually decreases; our data support the nibbling mechanism. The SEC data show a gradual shift in HDL-apoAI, cholesterol, and phospholipid during the 6-h incubation with CHO-SR-B1 cells (Fig. 5). This effect is most apparent in the analysis of the SEC profiles according to absorbance at 280 nm, which better distinguishes changes because it is a continuous rather than fraction-wise analysis and is reproducible ± 15 μl (24). Absorbance reflects changes in HDL-protein profiles. These data show a gradual shift in the A280 nm peak for the HDL-protein profile from 28.9 ml at t = 0 h to 29.3 ml at t = 6 h (Fig. 5A, inset) corresponding to a loss of 8 CE/HDL particle/h. The SEC data are corroborated by kinetic studies in which the size distribution of HDL particles with apoAI and apoAII during incubation with CHO-SR-B1 cells was followed by non-denaturing PAGE; apoAI but not apoAII transferred from the main HDL peak to faster migrating remnant HDL and lipid-free apoAI (Fig. 7).
ApoAI−/− mice expressing high hepatic SR-B1 levels remodel human HDL2 into apoAI plus apoAII remnants that are converted back to HDL2-sized particles as well as smaller apoAI-only remnants that are catabolized (30, 36). This process produces a trimodal distribution of HDL, initially large HDL2 that is followed by a slightly smaller and then a much smaller remnant; this observation is consistent with a gobbling mechanism (37). However, in vivo and ex vivo studies showed that this effect is dependent on the activities of plasma enzymes (30). Collective consideration of those studies and our results supports the hypothesis that SR-B1 is an HDL nibbler but that in vivo coexisting plasma activities likely process the HDL into discrete particles during nibbling.
Since first reported (28), considerable progress has been made on refinement of the mechanism underlying selective HDL-CE uptake. Past studies support a model of high affinity binding of HDL to the dimeric SR-B1 extracellular domains, which hold it in close proximity to the cell surface (38, 39) wherefrom HDL-CE diffuses into the plasma membrane bilayer via a nonaqueous channel within SR-B1; this process is likely driven by a CE concentration gradient (16). Fig. 8 is a refined model for this process that incorporates our new data featuring apoAI release and CE nibbling. ApoAI release is observed at the earliest time point (Fig. 5B, 1 h), consistent with its higher lability than apoAII, which remains with the HDL remnant (supplemental Fig. 1). During the transient association of HDL with SR-B1, one or a few CE molecules transfer through a nonaqueous channel into the plasma membrane, and a smaller, CE-depleted, HDL remnant desorbs into the surrounding aqueous phase. We speculate that the driving force for HDL release is CE saturation of the plasma membrane, which can accommodate only 2–3 mol % CE (40). When the plasma membrane is once again CE-depleted by CE diffusion or hydrolysis, HDL, including those from which some CE has already been extracted, re-enter the SR-B1 pathway. Further refinement of the mechanism for SR-B1-mediated selective lipid uptake could be the basis for the design of new therapies for its potentiation.
Figure 8.
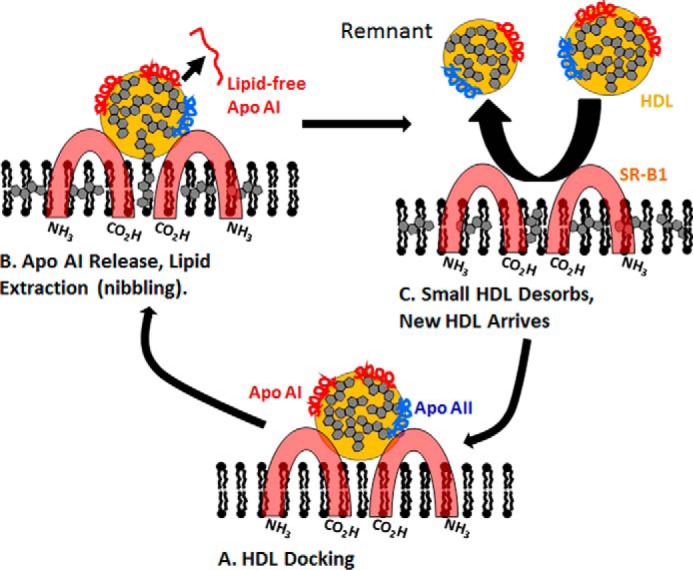
Mechanistic model of SR-B1 nibbling. HDL docks to dimeric SR-B1 (A). During or after docking, a molecule of apoAI is released into the aqueous phase, and one or a few CE molecules enter the non-aqueous channel between SR-B1 dimers after which the CE diffuses through the channel and into the hydrophobic region of the membrane-phospholipid bilayer (B). During this step, the CE content of the bilayer increases, whereas the size of HDL is incrementally reduced (size reduction is exaggerated to emphasize this process), and the smaller, CE-depleted HDL remnant desorbs from the cell and HDL or an HDL remnant, respectively, docks or re-docks to initiate another cycle of CE transfer (C). This model is based on the model of Connelly and Williams (39).
The nibbling mechanism may extend to other protein activities that remove cholesterol from HDL. One of these is streptococcal serum opacity factor (SOF), which profoundly disrupts HDL into three products that include a remnant, neo-HDL (24). Analysis of the SOF reaction by electron cryomicroscopy and SEC clearly shows the gradual disappearance of HDL and the formation of a remnant when the HDL has been totally consumed by SOF (41), i.e. nibbling. However, as with SR-B1 (30, 36, 37), <1% of the remnant is observed in vivo (42). Studies of families with a deficiency of cholesteryl ester transfer protein, which transfers the CE of HDL to other lipoproteins, suggest that this transfer protein also reduces HDL size; cholesteryl ester transfer protein-deficient patients carry markedly increased plasma concentrations of HDL that are larger than those from CETP-competent persons (43–45). A CETP nibbling mechanism is supported by electron cryo-microscopic studies of the transfer of CE from HDL to LDL, showing a gradual reduction of HDL diameter by ∼25%, which corresponds to a volume reduction of ∼60% (46). Another candidate for which there are little data include cholesteryl esterase activity, which reduces HDL-CE content.
Experimental procedures
Materials
Human HDL was isolated by sequential flotation of plasma (Houston Methodist Hospital Blood Donor Center) adjusted to d = 1.063 and 1.21 g/ml by the addition of KBr (47), which was removed by dialysis against Tris-buffered saline (TBS = 10 mm Tris, 100 mm NaCl, 1 mm EDTA, pH = 7.4). TBS was used throughout except where noted. Lipoprotein purity was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and SEC over two Superose HR 6 columns (GE Healthcare) in tandem (24).
Radiolabeled HDL
Lipid-free apoAI and apoAII were isolated from human plasma HDL as described (48) and labeled with 125I (PerkinElmer Life Sciences) by N-chlorobenzenesulfonamide immobilized on polystyrene beads (Pierce Iodination Beads, Thermo Scientific) according to the vendor's instructions. The specific activities of [125I]apoAI and [125I]apoAII were 416,000 and 391,000 cpm/μg, respectively. HDL-[3H]CE was prepared from HDL and [3H]cholesterol (PerkinElmer Life Sciences) by biological labeling as described (24). Aliquots of [3H]CE-labeled HDL (1 mg of protein) were mixed with [125I]apoAI or [125I]apoAII (12 μg of protein) to yield doubly labeled HDL-[3H]CE with [125I]apoAI or [125I]apoAII, respectively. SEC of the doubly labeled HDL revealed co-elution of 125I and [3H]CE and showed that 60 ± 8 and 90 ± 6% of the [125I]apoAI and [125I]AII radiolabel, respectively, incorporated into HDL. Specific activities of the doubly labeled HDL-[3H]CE-[125I]apoAI or HDL-[3H]CE-[125I]apoAII were 810 cpm/nmol CE, 79,000 cpm/nmol apoAI, and 127,000 cpm/nmol apoAII. Protein concentration was determined using the DC Protein Kit (Bio-Rad). Cholesterol and phospholipid were determined enzymatically (Wako Life Sciences).
Cell culture
Cell culture methods have been previously described (49). Wild-type CHO cells, i.e. CHO-K1, were from American Type Culture Collection (Manassas, VA). CHO-ldlA7 cells (LDLR-negative) and CHO-ldlA7 cells transfected to overexpress SR-B1 (CHO-SR-B1) were provided by Dr. Monty Krieger (25). CHO cell lines were cultured in Ham's F-12 with 5% FBS, 1 mm sodium pyruvate, and penicillin-streptomycin antibiotics (10 units/ml, 10 μg/ml respectively). The CHO-SR-B1 medium also contained G418 (300 μg/ml). Human hepatocyte Huh7 cells were the generous gift of Drs. Yumin Xu and Boris Yoffe (50) and were cultured as described (51) in minimal essential medium (MEM) with 10% FBS, 1 mm sodium pyruvate, and penicillin-streptomycin antibiotics (10 units/ml, 10 μg/ml, respectively). Huh7 cell DNA was analyzed by the Human Cell Line Authentication Service, DNA Analysis Facility of Yale University with a 94.4% match for human Huh7 hepatocytes. All cell lines tested negative for mycoplasma (LookoutR Mycoplasma PCR Detection Kit MP0035 (Sigma)). Tissue culture reagents were from Invitrogen (Thermo Fisher Scientific, Carlsbad, CA).
Cellular CE uptake
The CE uptake was assayed as described (14, 49). Our calculation of the relative uptakes of HDL apolipoproteins and CEs assumed an HDL3 with 2 and 1 apoAI and apoAII per particle (52), respectively, so apolipoprotein mass/particle = 2 × 28,300 Da + 17,400 Da = ∼74,000 Da. HDL3 is 55% protein so that the particle mass is 74,000/0.55 = ∼135,000. Of this, 13% is CE, which corresponds to 28 CE molecules/HDL3 resulting in CE/apoAI and CE/apoAII molar ratios of 14/1 and 28/1, respectively. Although an approximation, these ratios are far lower than those observed for CE/apolipoprotein uptake ratios for apoAI and apoAII by all four cell types studied.
Uptake was initiated by incubation of serum-free cell culture medium + 0.5% fatty-acid free bovine serum albumin containing various aliquots of [125I]HDL-[3H]CE (1 mg/ml; final volume = 1 ml) for 0–3 h at 37 °C in a 5% CO2 incubator. CE uptake was terminated by placing the cells on ice; cells were washed, and [3H]CE was extracted with isopropyl alcohol and β-counted. Cell-associated 125I was determined by γ-counting the hydrolysate obtained by the addition of 0.1 m NaOH to the cells. Uptake was expressed as 100% × cell-associated radiolabel/total radiolabel per incubation. Time course uptake experiments comprised incubations of [125I]HDL-[3H]CE (20 μg/ml) for 0, 60, 120, and 180 min; these data were fitted to a linear polynomial in Sigma Plot (Systat Software Inc.). Dose-response curves were based on incubations of [125I]HDL-[3H]CE at 5, 10, 20, 50, and 100 μg/ml with cells for 180 min; these data were fitted to a hyperbolic function having the form y = ax/(b + x) in Sigma Plot where y is the radiolabel uptake, a is the maximum uptake (Vmax), and b is Km, the HDL-protein concentration at 50% of Vmax.
HDL remnant isolation and analysis
To identify the mechanism for the initial association of HDL with SR-B1, remnants were produced by incubating HDL (20 μg/ml protein) for various times with CHO-SR-B1 cells. To facilitate detection of small changes in HDL size, human plasma HDL was purified by SEC, and the middle three peak fractions were pooled as the starting HDL for remnant formation. Analytical SEC showed this HDL had a normal bell-shaped chromatographic profile (see Fig. 5A, 0 h chromatogram). The low HDL concentration (∼10−7 m) was used to obviate the fusion of early remnants with co-existing HDL (36). The media for each time point were pooled and concentrated. Aliquots were analyzed by non-denaturing PAGE (Novex Wedgewell 4–20% Tris-glycine gels, Invitrogen), protein staining, and immunoblotting for apolipoproteins AI, AII, and E (anti-apolipoprotein antibodies from Academy Bio-Medical, Houston, TX). Antibody catalogue numbers and dilutions are given in supplemental Table S1. Aliquots of the concentrated media were analyzed by SEC; 1-ml fractions were collected and analyzed by SDS-PAGE (Novex Wedgewell 16% Tris-glycine gels, Invitrogen) followed by protein staining and immunoblotting for apolipoproteins AI, AII, and E, as above, and assayed for protein, total cholesterol, and phospholipid. Immunoblot apolipoprotein band intensities were quantified using ImageQuant analysis software (GE Healthcare).
Author contributions
H. J. P. developed the broad hypotheses and prepared the first draft of the paper. B. K. G. designed and executed the cell studies and their interpretation and helped write the paper. G. R. B. participated in the selective uptake experiments. C. R. and A. M. G. reviewed the paper as it was developed. All authors edited and approved the final submitted paper.
Supplementary Material
Acknowledgments
We thank Bingqing Xu for purification of HDL and Dedipya Yelamanchili and Dr. Perla J. Rodriguez for technical assistance with PAGE and immunoblotting.
This work was supported by National Institutes of Health Grant HL-56865 (to H. J. P.) and HL-129767 (to H. J. P. and C. R.). This work was also supported by the Bass Foundation (to A. M. G.) and Houston Methodist Foundation funding from Charif Souki, Patrick Studdert, and the Jerold B. Katz family. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Fig. S1.
- RCT
- reverse cholesterol transport
- FC
- cholesterol
- CE
- cholesteryl ester
- CHO-ldlA7
- HDL3, major fraction of human HDL, density 1.125–1.210 g/ml; LDLR-null CHO
- CHO-SR-B1
- SR-B1-overexpressing LDLR-null CHO
- SR-B1
- scavenger receptor class B member 1
- LDLR
- low density lipoprotein receptor
- SEC
- size-exclusion chromatography
- SOF
- serum opacity factor
- EV
- elution volume.
References
- 1. Pownall H. J., and Gotto A. M. Jr. (2016) New insights into the high-density lipoprotein dilemma. Trends Endocrinol. Metab. 27, 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Levinson S. S., and Wagner S. G. (2015) Implications of reverse cholesterol transport: recent studies. Clin. Chim. Acta 439, 154–161 [DOI] [PubMed] [Google Scholar]
- 3. Eyvazian V. A., and Frishman W. H. (2017) Evacetrapib: another CETP inhibitor for dyslipidemia with no clinical benefit. Cardiol. Rev. 25, 43–52 [DOI] [PubMed] [Google Scholar]
- 4. Rader D. J., and Tall A. R. (2012) The not-so-simple HDL story: is it time to revise the HDL cholesterol hypothesis? Nat. Med. 18, 1344–1346 [DOI] [PubMed] [Google Scholar]
- 5. Schwartz C. C., VandenBroek J. M., and Cooper P. S. (2004) Lipoprotein cholesteryl ester production, transfer, and output in vivo in humans. J. Lipid Res. 45, 1594–1607 [DOI] [PubMed] [Google Scholar]
- 6. Ueda Y., Gong E., Royer L., Cooper P. N., Francone O. L., and Rubin E. M. (2000) Relationship between expression levels and atherogenesis in scavenger receptor class B, type I transgenics. J. Biol. Chem. 275, 20368–20373 [DOI] [PubMed] [Google Scholar]
- 7. Kozarsky K. F., Donahee M. H., Glick J. M., Krieger M., and Rader D. J. (2000) Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 20, 721–727 [DOI] [PubMed] [Google Scholar]
- 8. Arai T., Wang N., Bezouevski M., Welch C., and Tall A. R. (1999) Decreased atherosclerosis in heterozygous low density lipoprotein receptor-deficient mice expressing the scavenger receptor BI transgene. J. Biol. Chem. 274, 2366–2371 [DOI] [PubMed] [Google Scholar]
- 9. Van Eck M., Twisk J., Hoekstra M., Van Rij B. T., Van der Lans C. A., Bos I. S., Kruijt J. K., Kuipers F., and Van Berkel T. J. (2003) Differential effects of scavenger receptor BI deficiency on lipid metabolism in cells of the arterial wall and in the liver. J. Biol. Chem. 278, 23699–23705 [DOI] [PubMed] [Google Scholar]
- 10. Trigatti B., Rayburn H., Viñals M., Braun A., Miettinen H., Penman M., Hertz M., Schrenzel M., Amigo L., Rigotti A., and Krieger M. (1999) Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc. Natl. Acad. Sci. U.S.A. 96, 9322–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huszar D., Varban M. L., Rinninger F., Feeley R., Arai T., Fairchild-Huntress V., Donovan M. J., and Tall A. R. (2000) Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arterioscler. Thromb. Vasc. Biol. 20, 1068–1073 [DOI] [PubMed] [Google Scholar]
- 12. Braun A., Trigatti B. L., Post M. J., Sato K., Simons M., Edelberg J. M., Rosenberg R. D., Schrenzel M., and Krieger M. (2002) Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ. Res. 90, 270–276 [DOI] [PubMed] [Google Scholar]
- 13. Zanoni P., Khetarpal S. A., Larach D. B., Hancock-Cerutti W. F., Millar J. S., Cuchel M., DerOhannessian S., Kontush A., Surendran P., Saleheen D., Trompet S., Jukema J. W., De Craen A., Deloukas P., et al. (2016) Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 351, 1166–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Acton S., Rigotti A., Landschulz K. T., Xu S., Hobbs H. H., and Krieger M. (1996) Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520 [DOI] [PubMed] [Google Scholar]
- 15. Thuahnai S. T., Lund-Katz S., Williams D. L., and Phillips M. C. (2001) Scavenger receptor class B, type I-mediated uptake of various lipids into cells: influence of the nature of the donor particle interaction with the receptor. J. Biol. Chem. 276, 43801–43808 [DOI] [PubMed] [Google Scholar]
- 16. Rodrigueza W. V., Thuahnai S. T., Temel R. E., Lund-Katz S., Phillips M. C., and Williams D. L. (1999) Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J. Biol. Chem. 274, 20344–20350 [DOI] [PubMed] [Google Scholar]
- 17. Kunitake S. T., and Kane J. P. (1982) Factors affecting the integrity of high density lipoproteins in the ultracentrifuge. J. Lipid Res. 23, 936–940 [PubMed] [Google Scholar]
- 18. Mehta R., Gantz D. L., and Gursky O. (2003) Human plasma high-density lipoproteins are stabilized by kinetic factors. J. Mol. Biol. 328, 183–192 [DOI] [PubMed] [Google Scholar]
- 19. Edelstein C., Halari M., and Scanu A. M. (1982) On the mechanism of the displacement of apolipoprotein A-I by apolipoprotein A-II from the high density lipoprotein surface. Effect of concentration and molecular forms of apolipoprotein A-II. J. Biol. Chem. 257, 7189–7195 [PubMed] [Google Scholar]
- 20. Silver E. T., Scraba D. G., and Ryan R. O. (1990) Lipid transfer particle-induced transformation of human high density lipoprotein into apolipoprotein A-I-deficient low density particles. J. Biol. Chem. 265, 22487–22492 [PubMed] [Google Scholar]
- 21. Liang H. Q., Rye K. A., and Barter P. J. (1996) Remodelling of reconstituted high density lipoproteins by lecithin: cholesterol acyltransferase. J. Lipid Res. 37, 1962–1970 [PubMed] [Google Scholar]
- 22. Settasatian N., Duong M., Curtiss L. K., Ehnholm C., Jauhiainen M., Huuskonen J., and Rye K. A. (2001) The mechanism of the remodeling of high density lipoproteins by phospholipid transfer protein. J. Biol. Chem. 276, 26898–26905 [DOI] [PubMed] [Google Scholar]
- 23. Rao R., Albers J. J., Wolfbauer G., and Pownall H. J. (1997) Molecular and macromolecular specificity of human plasma phospholipid transfer protein. Biochemistry 36, 3645–3653 [DOI] [PubMed] [Google Scholar]
- 24. Gillard B. K., Courtney H. S., Massey J. B., and Pownall H. J. (2007) Serum opacity factor unmasks human plasma high-density lipoprotein instability via selective delipidation and apolipoprotein A-I desorption. Biochemistry 46, 12968–12978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nieland T. J., Penman M., Dori L., Krieger M., and Kirchhausen T. (2002) Discovery of chemical inhibitors of the selective transfer of lipids mediated by the HDL receptor SR-BI. Proc. Natl. Acad. Sci. U.S.A. 99, 15422–15427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gillard B. K., Raya J. L., Ruiz-Esponda R., Iyer D., Coraza I., Balasubramanyam A., and Pownall H. J. (2013) Impaired lipoprotein processing in HIV patients on antiretroviral therapy: aberrant high-density lipoprotein lipids, stability, and function. Arterioscler. Thromb. Vasc. Biol. 33, 1714–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rodriguez P. J., Gillard B. K., Barosh R., Gotto A. M. Jr, Rosales C., and Pownall H. J. (2016) Neo high-density lipoprotein produced by the streptococcal serum opacity factor activity against human high-density lipoproteins is hepatically removed via dual mechanisms. Biochemistry 55, 5845–5853 [DOI] [PubMed] [Google Scholar]
- 28. Glass C., Pittman R. C., Weinstein D. B., and Steinberg D. (1983) Dissociation of tissue uptake of cholesterol ester from that of apoprotein A-I of rat plasma high density lipoprotein: selective delivery of cholesterol ester to liver, adrenal, and gonad. Proc. Natl. Acad. Sci. U.S.A. 80, 5435–5439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Havel R. J., Goldstein J. L., and Brown M. S. (1980) Lipoproteins and lipid transport. In The Metabolic Control of Disease (Bondy P. K., and Rosenberg L. E., eds) pp. 393–494, W. B. Saunders Co., Philadelphia, PA [Google Scholar]
- 30. Webb N. R., Cai L., Ziemba K. S., Yu J., Kindy M. S., van der Westhuyzen D. R., and de Beer F. C. (2002) The fate of HDL particles in vivo after SR-BI-mediated selective lipid uptake. J. Lipid Res. 43, 1890–1898 [DOI] [PubMed] [Google Scholar]
- 31. Pownall H. J., Hosken B. D., Gillard B. K., Higgins C. L., Lin H. Y., and Massey J. B. (2007) Speciation of human plasma high-density lipoprotein (HDL): HDL stability and apolipoprotein A-I partitioning. Biochemistry 46, 7449–7459 [DOI] [PubMed] [Google Scholar]
- 32. Handa D., Kimura H., Oka T., Takechi Y., Okuhira K., Phillips M. C., and Saito H. (2015) Kinetic and thermodynamic analyses of spontaneous exchange between high-density lipoprotein-bound and lipid-free apolipoprotein A-I. Biochemistry 54, 1123–1131 [DOI] [PubMed] [Google Scholar]
- 33. Pownall H., Pao Q., Hickson D., Sparrow J. T., Kusserow S. K., and Massey J. B. (1981) Kinetics and mechanism of association of human plasma apolipoproteins with dimyristoylphosphatidylcholine: effect of protein structure and lipid clusters on reaction rates. Biochemistry 20, 6630–6635 [DOI] [PubMed] [Google Scholar]
- 34. Pagler T. A., Rhode S., Neuhofer A., Laggner H., Strobl W., Hinterndorfer C., Volf I., Pavelka M., Eckhardt E. R., van der Westhuyzen D. R., Schütz G. J., and Stangl H. (2006) SR-BI-mediated high density lipoprotein (HDL) endocytosis leads to HDL resecretion facilitating cholesterol efflux. J. Biol. Chem. 281, 11193–11204 [DOI] [PubMed] [Google Scholar]
- 35. Li W. H., Tanimura M., Luo C. C., Datta S., and Chan L. (1988) The apolipoprotein multigene family: biosynthesis, structure, structure-function relationships, and evolution. J. Lipid Res. 29, 245–271 [PubMed] [Google Scholar]
- 36. Webb N. R., de Beer M. C., Asztalos B. F., Whitaker N., van der Westhuyzen D. R., and de Beer F. C. (2004) Remodeling of HDL remnants generated by scavenger receptor class B type I. J. Lipid Res. 45, 1666–1673 [DOI] [PubMed] [Google Scholar]
- 37. de Beer M. C., van der Westhuyzen D. R., Whitaker N. L., Webb N. R., and de Beer F. C. (2005) SR-BI-mediated selective lipid uptake segregates apoA-I and apoA-II catabolism. J. Lipid Res. 46, 2143–2150 [DOI] [PubMed] [Google Scholar]
- 38. Connelly M. A., Klein S. M., Azhar S., Abumrad N. A., and Williams D. L. (1999) Comparison of class B scavenger receptors, CD36 and scavenger receptor BI (SR-BI), shows that both receptors mediate high density lipoprotein-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J. Biol. Chem. 274, 41–47 [DOI] [PubMed] [Google Scholar]
- 39. Connelly M. A., and Williams D. L. (2003) SR-BI and cholesterol uptake into steroidogenic cells. Trends Endocrinol. Metab. 14, 467–472 [DOI] [PubMed] [Google Scholar]
- 40. Hamilton J. A., and Small D. M. (1982) Solubilization and localization of cholesteryl oleate in egg phosphatidylcholine vesicles. A carbon 13 NMR study. J. Biol. Chem. 257, 7318–7321 [PubMed] [Google Scholar]
- 41. Han M., Gillard B. K., Courtney H. S., Ward K., Rosales C., Khant H., Ludtke S. J., and Pownall H. J. (2009) Disruption of human plasma high-density lipoproteins by streptococcal serum opacity factor requires labile apolipoprotein A-I. Biochemistry 48, 1481–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rosales C., Tang D., Gillard B. K., Courtney H. S., and Pownall H. J. (2011) Apolipoprotein E mediates enhanced plasma high-density lipoprotein cholesterol clearance by low-dose streptococcal serum opacity factor via hepatic low-density lipoprotein receptors in vivo. Arterioscler. Thromb. Vasc. Biol. 31, 1834–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brown M. L., Inazu A., Hesler C. B., Agellon L. B., Mann C., Whitlock M. E., Marcel Y. L., Milne R. W., Koizumi J., and Mabuchi H. (1989) Molecular basis of lipid transfer protein deficiency in a family with increased high-density lipoproteins. Nature 342, 448–451 [DOI] [PubMed] [Google Scholar]
- 44. Bisgaier C. L., Siebenkas M. V., Brown M. L., Inazu A., Koizumi J., Mabuchi H., and Tall A. R. (1991) Familial cholesteryl ester transfer protein deficiency is associated with triglyceride-rich low density lipoproteins containing cholesteryl esters of probable intracellular origin. J. Lipid Res. 32, 21–33 [PubMed] [Google Scholar]
- 45. Koizumi J., Mabuchi H., Yoshimura A., Michishita I., Takeda M., Itoh H., Sakai Y., Sakai T., Ueda K., and Takeda R. (1985) Deficiency of serum cholesteryl-ester transfer activity in patients with familial hyperalphalipoproteinaemia. Atherosclerosis 58, 175–186 [DOI] [PubMed] [Google Scholar]
- 46. Zhang L., Yan F., Zhang S., Lei D., Charles M. A., Cavigiolio G., Oda M., Krauss R. M., Weisgraber K. H., Rye K. A., Pownall H. J., Qiu X., and Ren G. (2012) Structural basis of transfer between lipoproteins by cholesteryl ester transfer protein. Nat. Chem. Biol. 8, 342–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Havel R. J., Eder H. A., and Bragdon J. H. (1955) The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 34, 1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brewer H. B. Jr, Ronan R., Meng M., and Bishop C. (1986) Isolation and characterization of apolipoproteins A-I, A-II, and A-IV. Methods Enzymol. 128, 223–246 [DOI] [PubMed] [Google Scholar]
- 49. Gillard B. K., Rosales C., Pillai B. K., Lin H. Y., Courtney H. S., and Pownall H. J. (2010) Streptococcal serum opacity factor increases the rate of hepatocyte uptake of human plasma high-density lipoprotein cholesterol. Biochemistry 49, 9866–9873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xie Q., Khaoustov V. I., Chung C. C., Sohn J., Krishnan B., Lewis D. E., and Yoffe B. (2002) Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 36, 592–601 [DOI] [PubMed] [Google Scholar]
- 51. Gillard B. K., Lin H. Y., Massey J. B., and Pownall H. J. (2009) Apolipoproteins A-I, A-II and E are independently distributed among intracellular and newly secreted HDL of human hepatoma cells. Biochim. Biophys. Acta 1791, 1125–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cheung M. C., and Albers J. J. (1977) The measurement of apolipoprotein A-I and A-II levels in men and women by immunoassay. J. Clin. Invest. 60, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.